

Antimycobacterial Drugs as a Novel Strategy to Inhibit Pseudomonas aeruginosa Virulence Factors and Combat Antibiotic Resistance: A Molecular Simulation Study


Abstract
1. Introduction
2. Materials and Methods
2.1. Preparation of Library of Antimycobacterial Drugs
2.2. Prediction of the ADMET Properties Using pkCSM
2.3. Pharmacokinetics and Drug Likeliness Assessment
2.4. Selection of the Target Proteins of Pseudomonas aeruginosa
2.5. Molecular Docking Studies
2.6. Molecular Dynamics Simulations
3. Results and Discussion
3.1. Prediction of Toxicity of the Antimycobacterial Drugs
3.2. Prediction of ADME Properties of the Antimycobacterial Drugs
3.3. Evaluation of Drug-Like and Pharmacokinetic Parameters
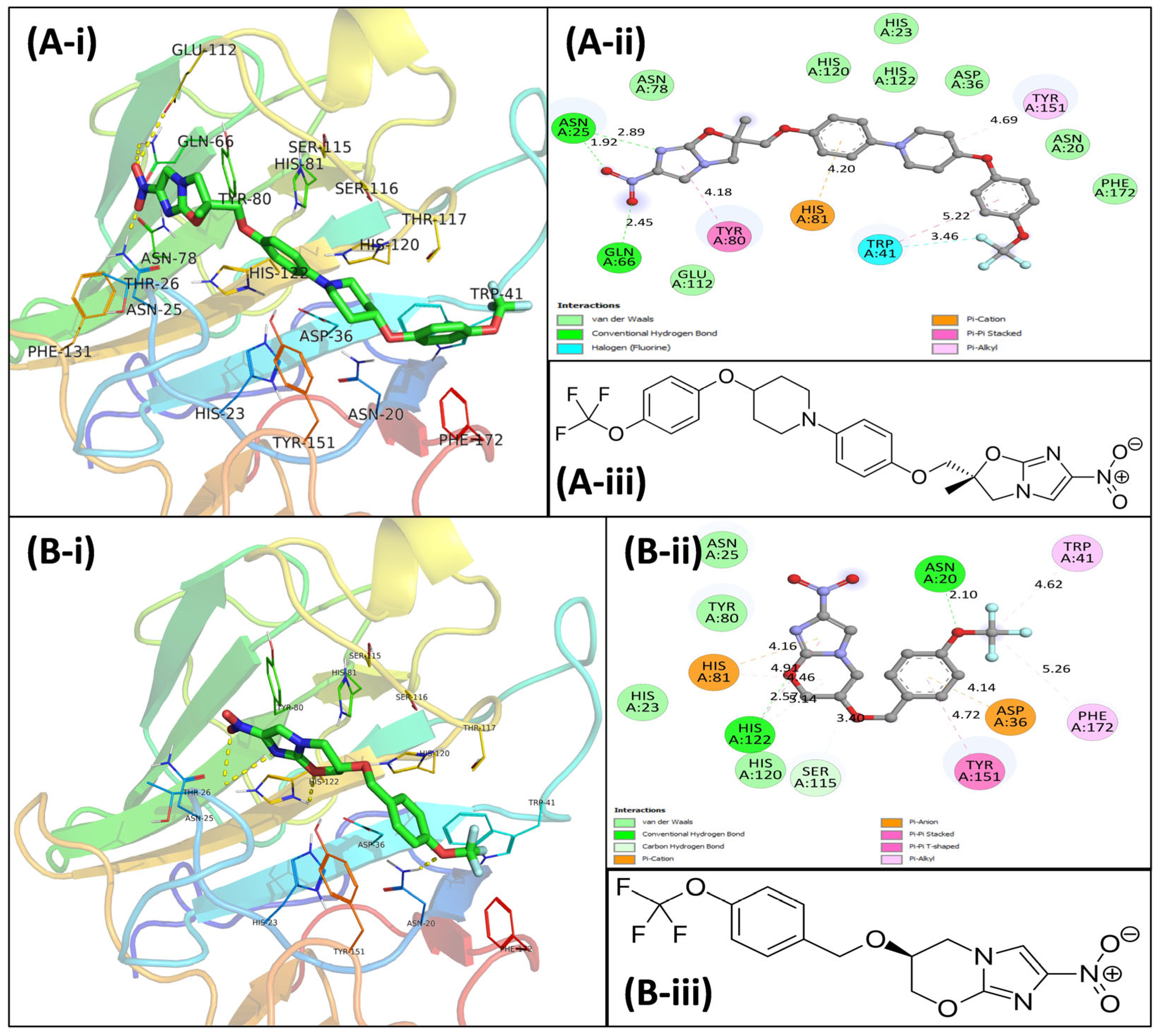
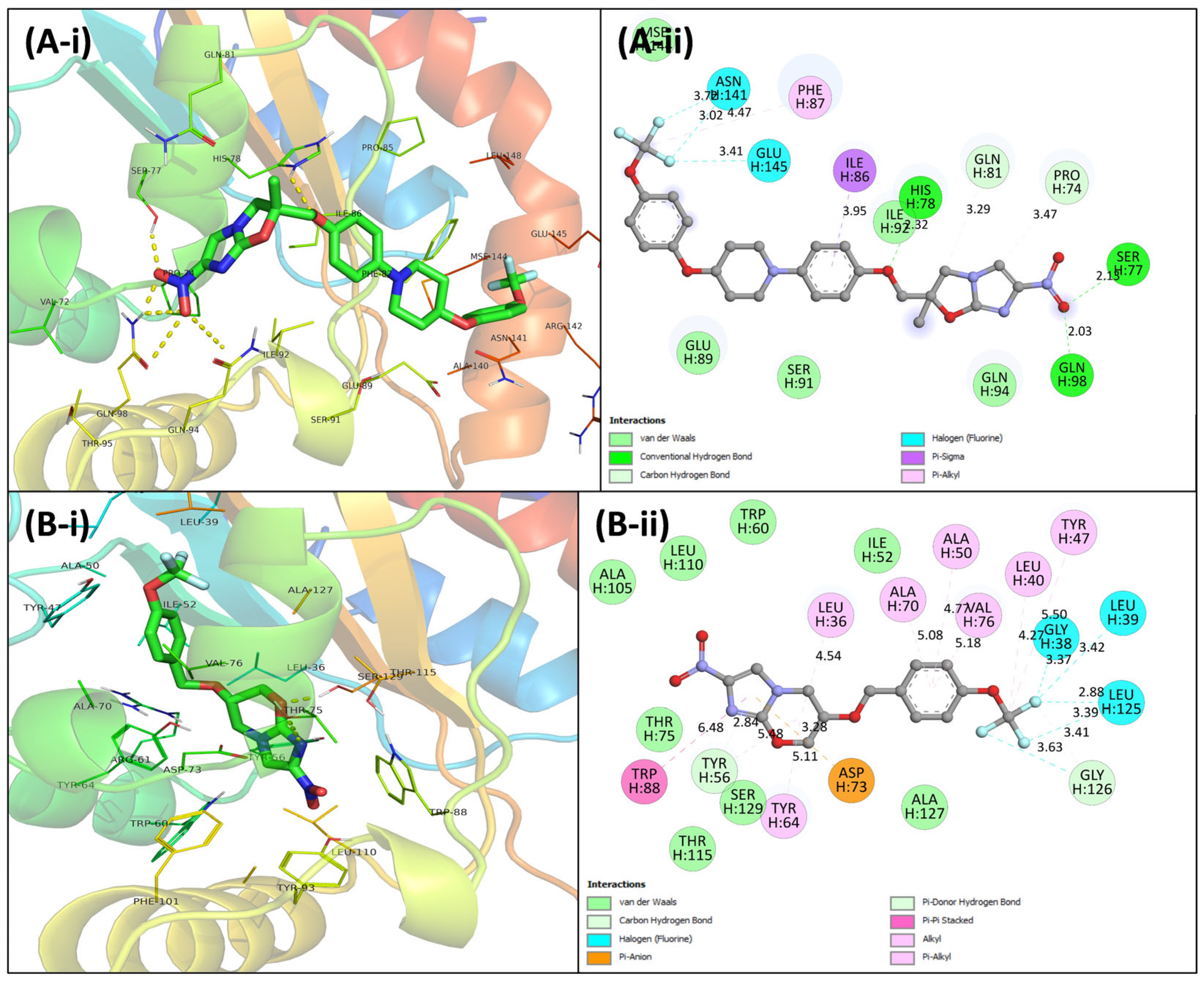
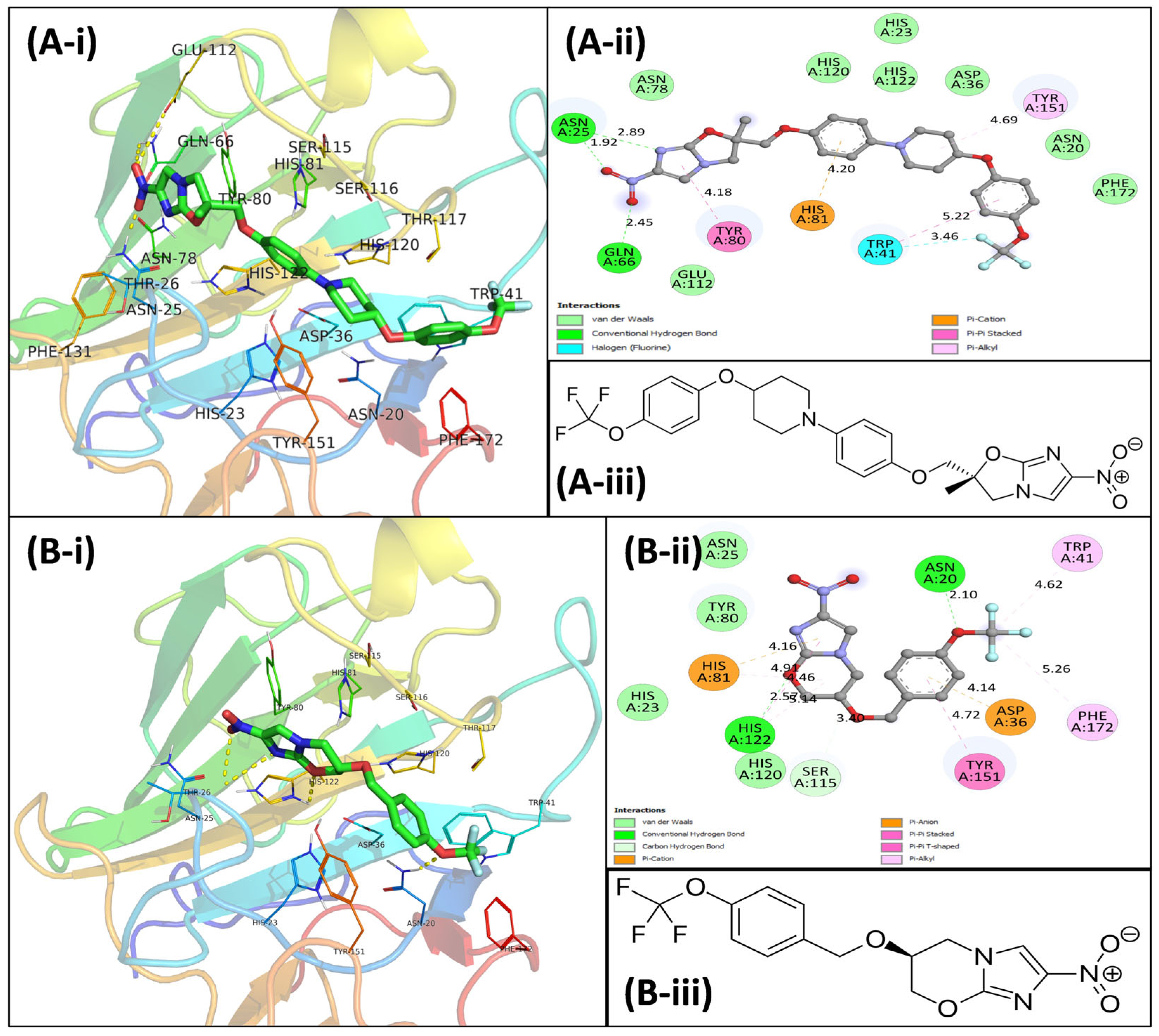
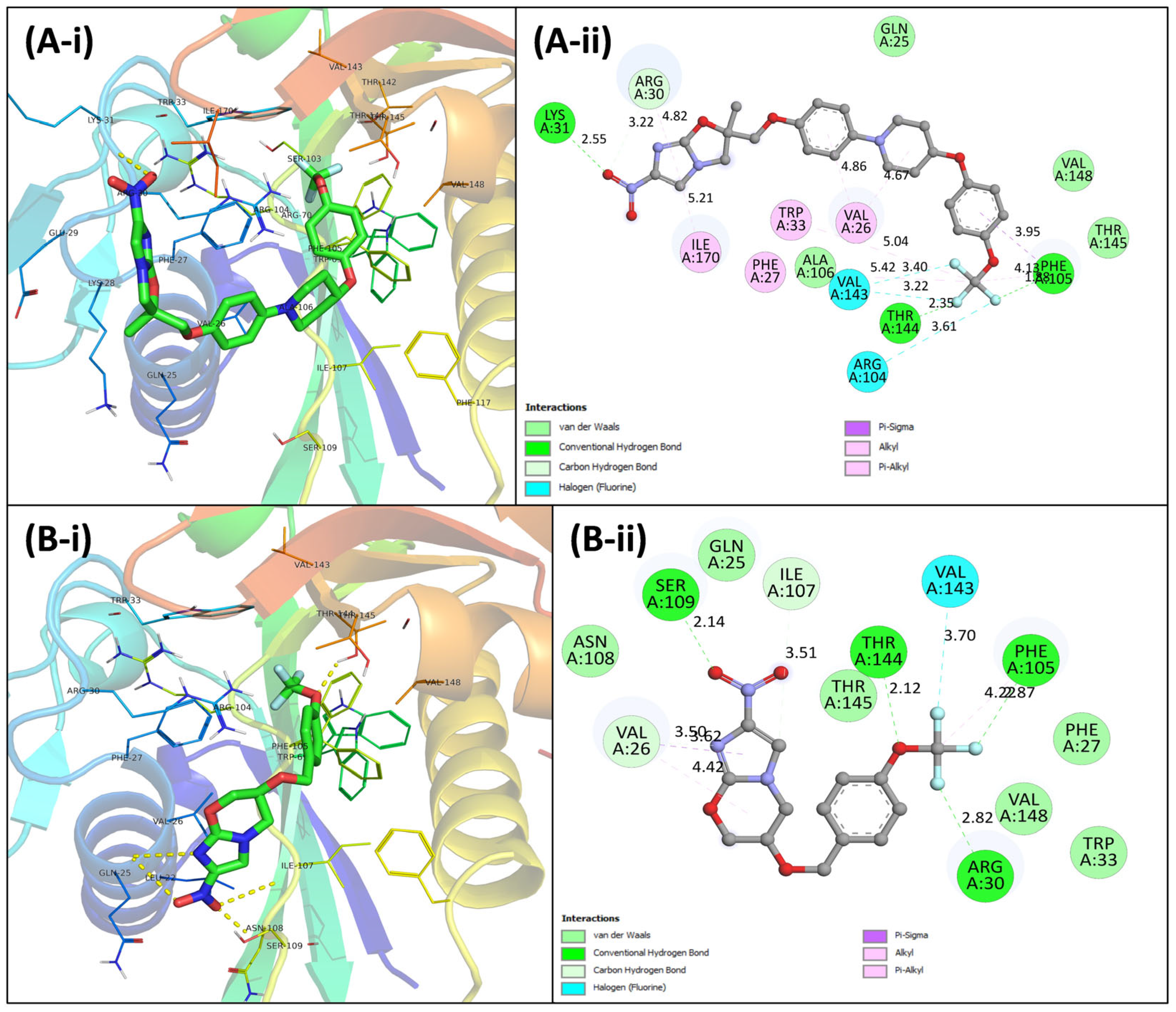
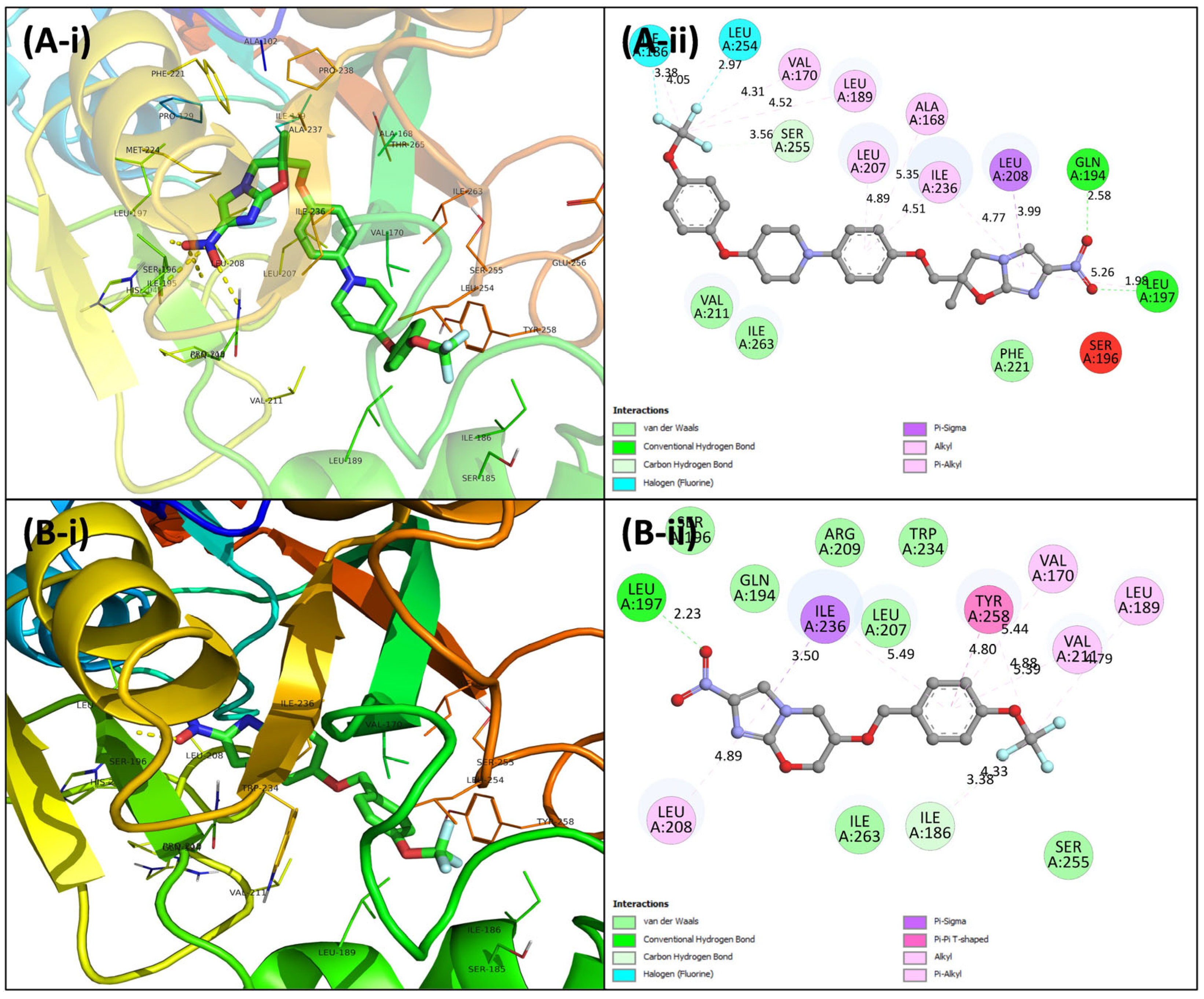
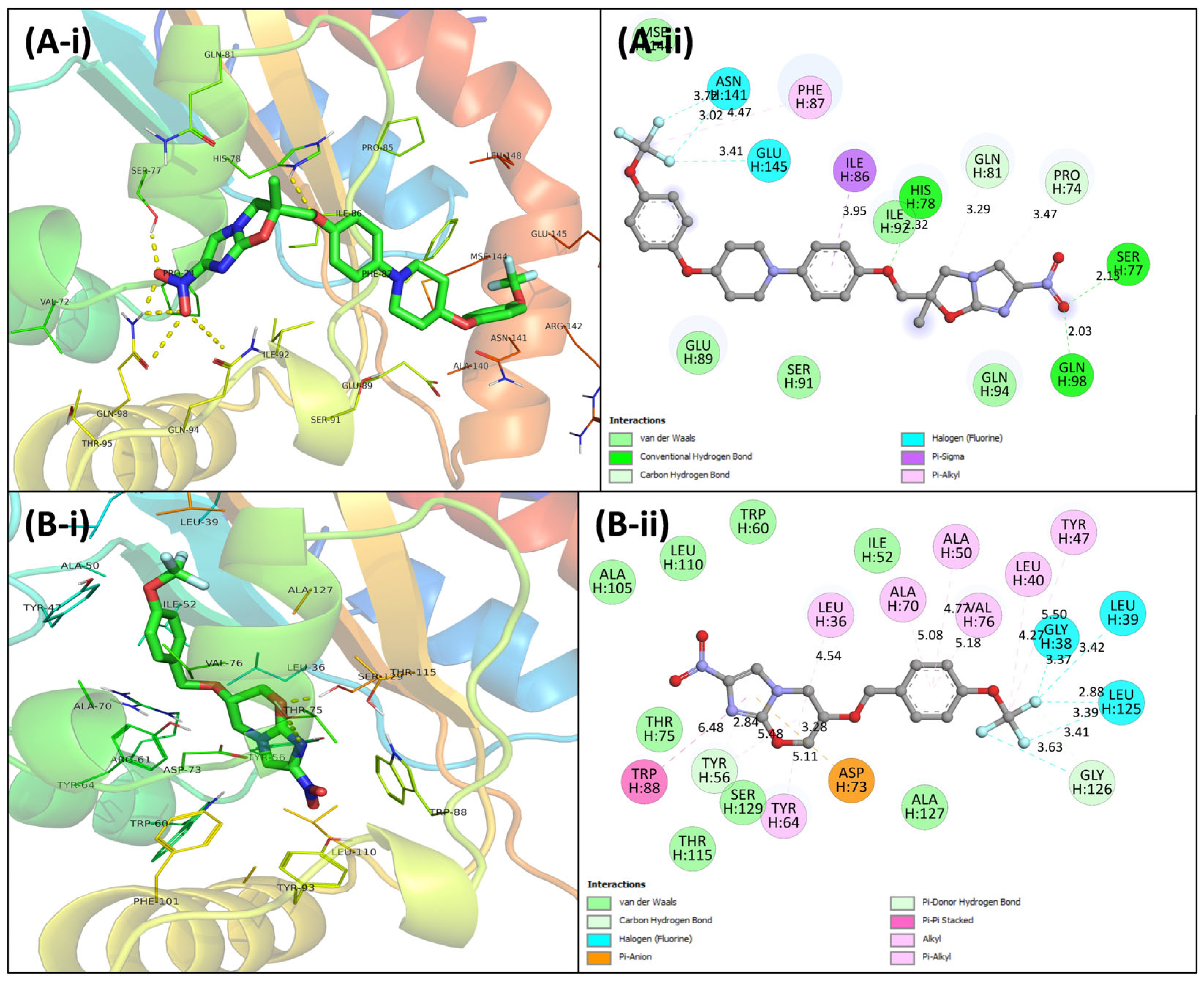
3.4. Virtual Screening Using Molecular Docking
3.5. Molecular Dynamics Simulations
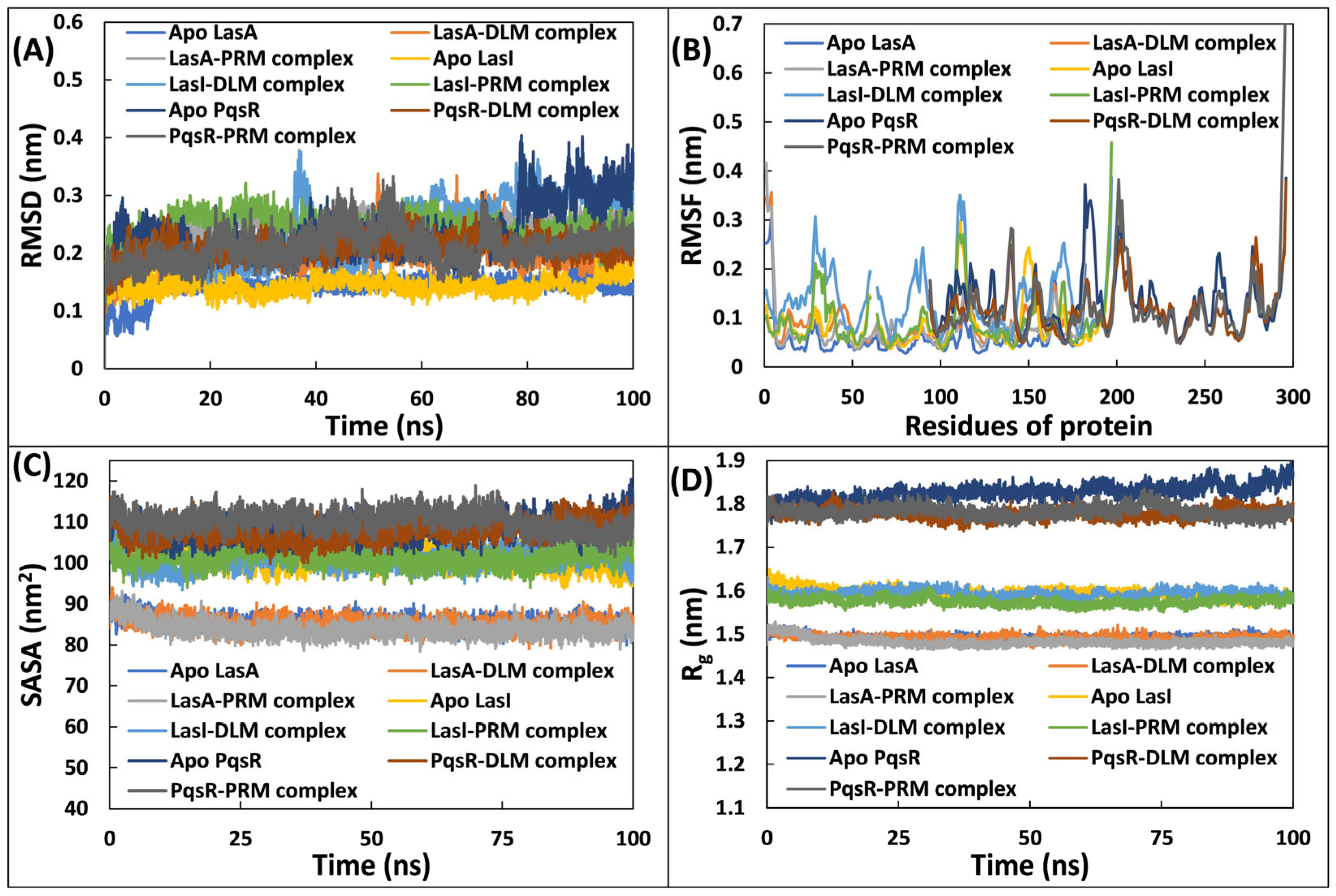
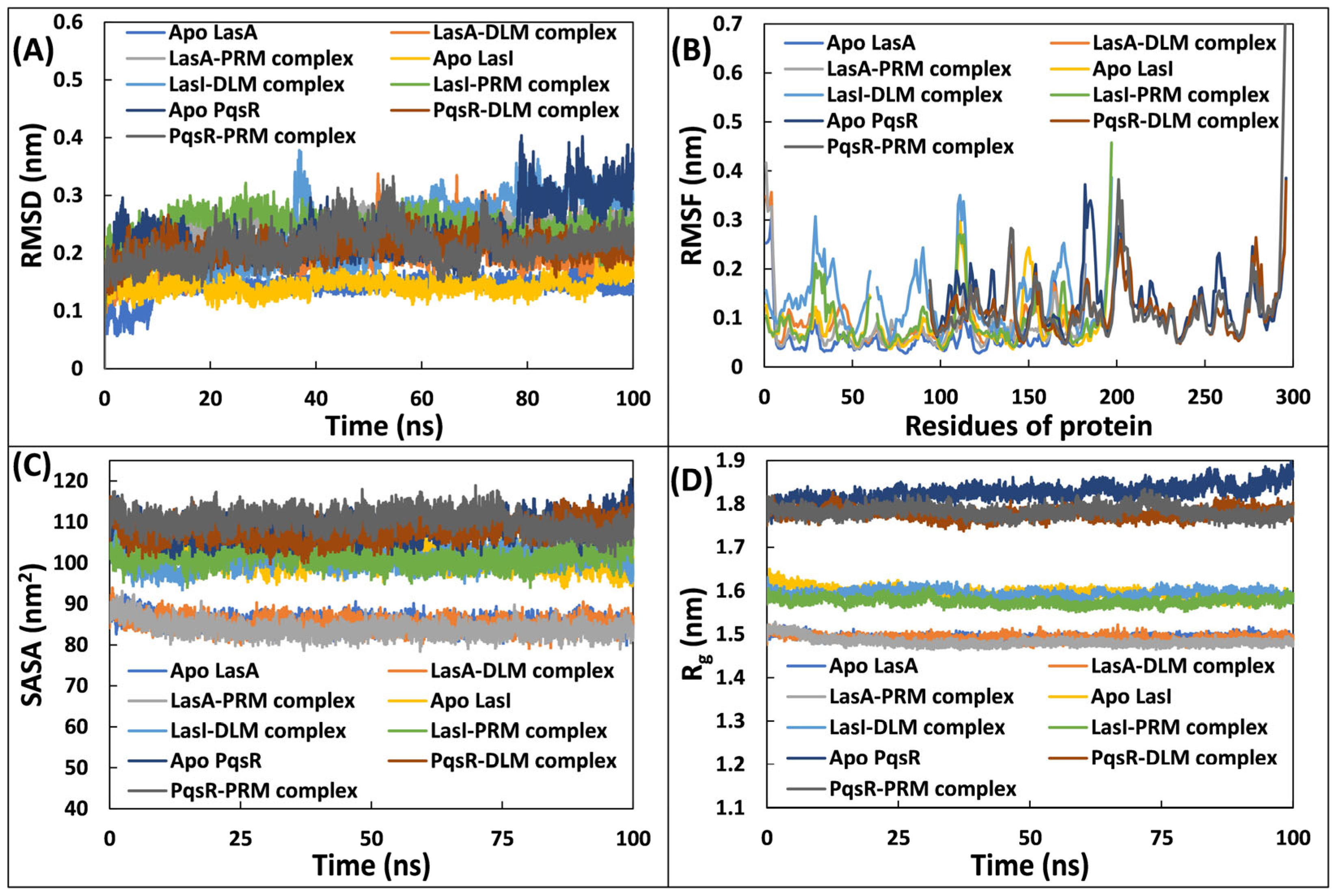
3.5.1. Examination of Deviations and Fluctuations
3.5.2. Examination of and Physicochemical Parameters Structural Compactness
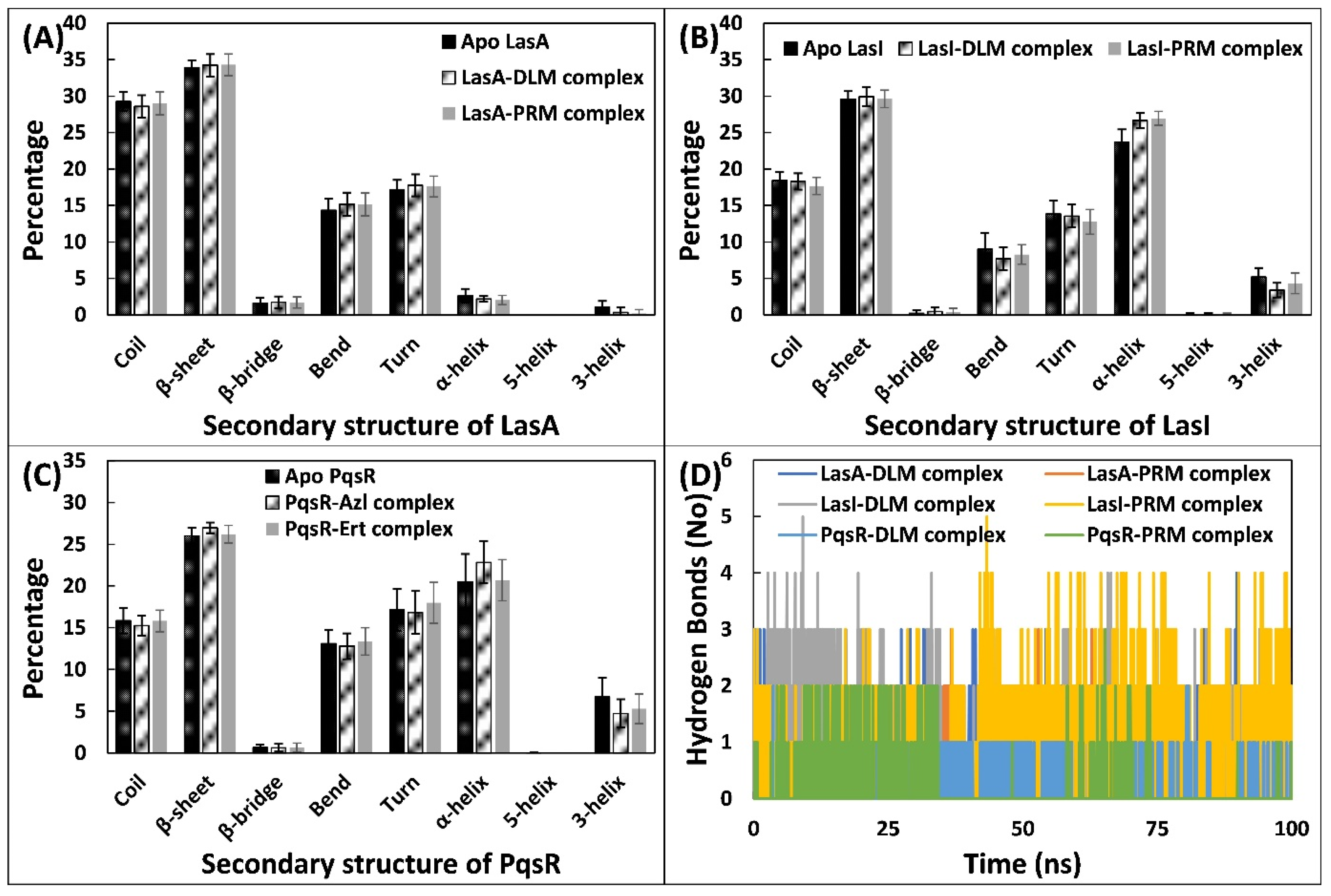
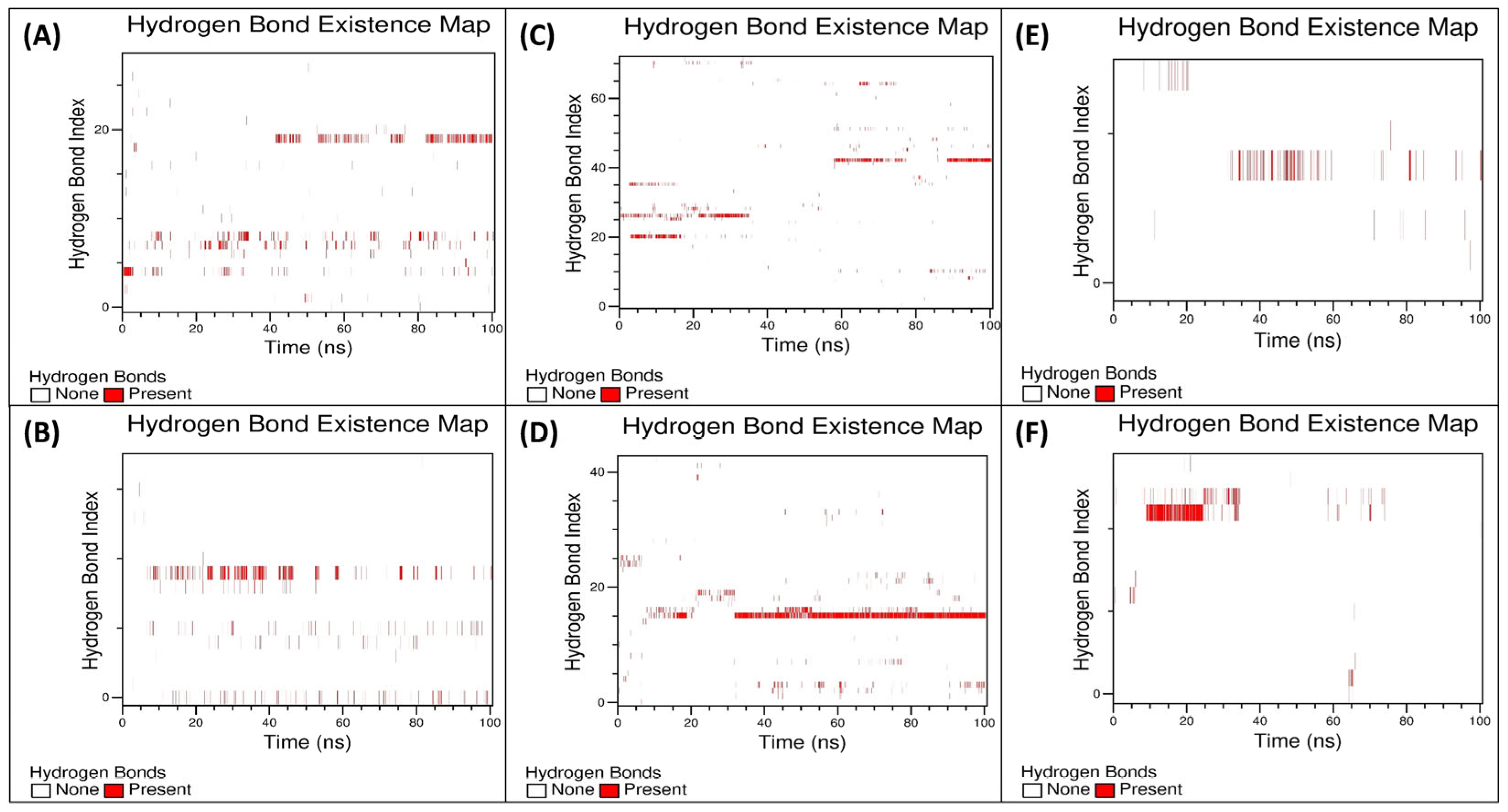
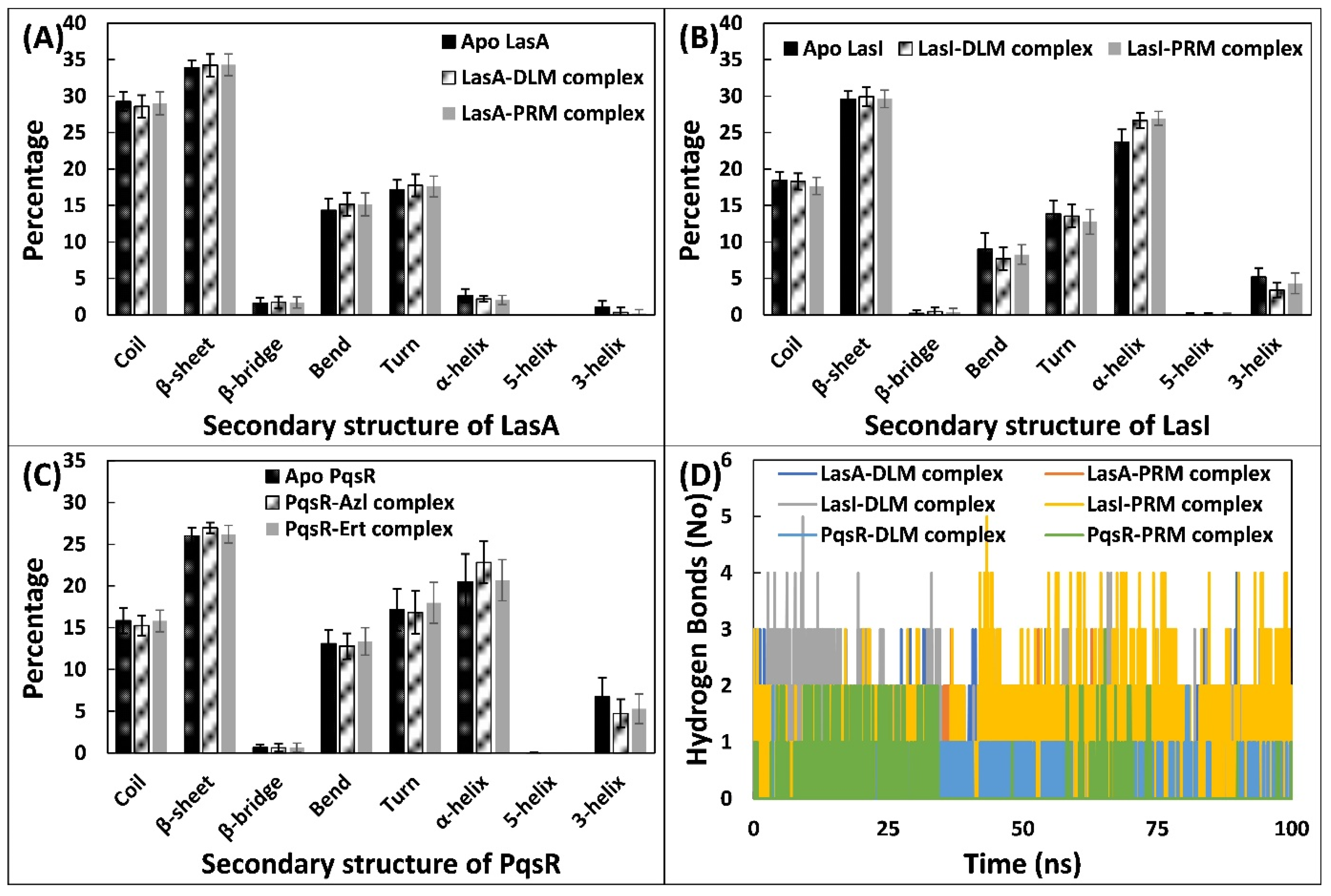
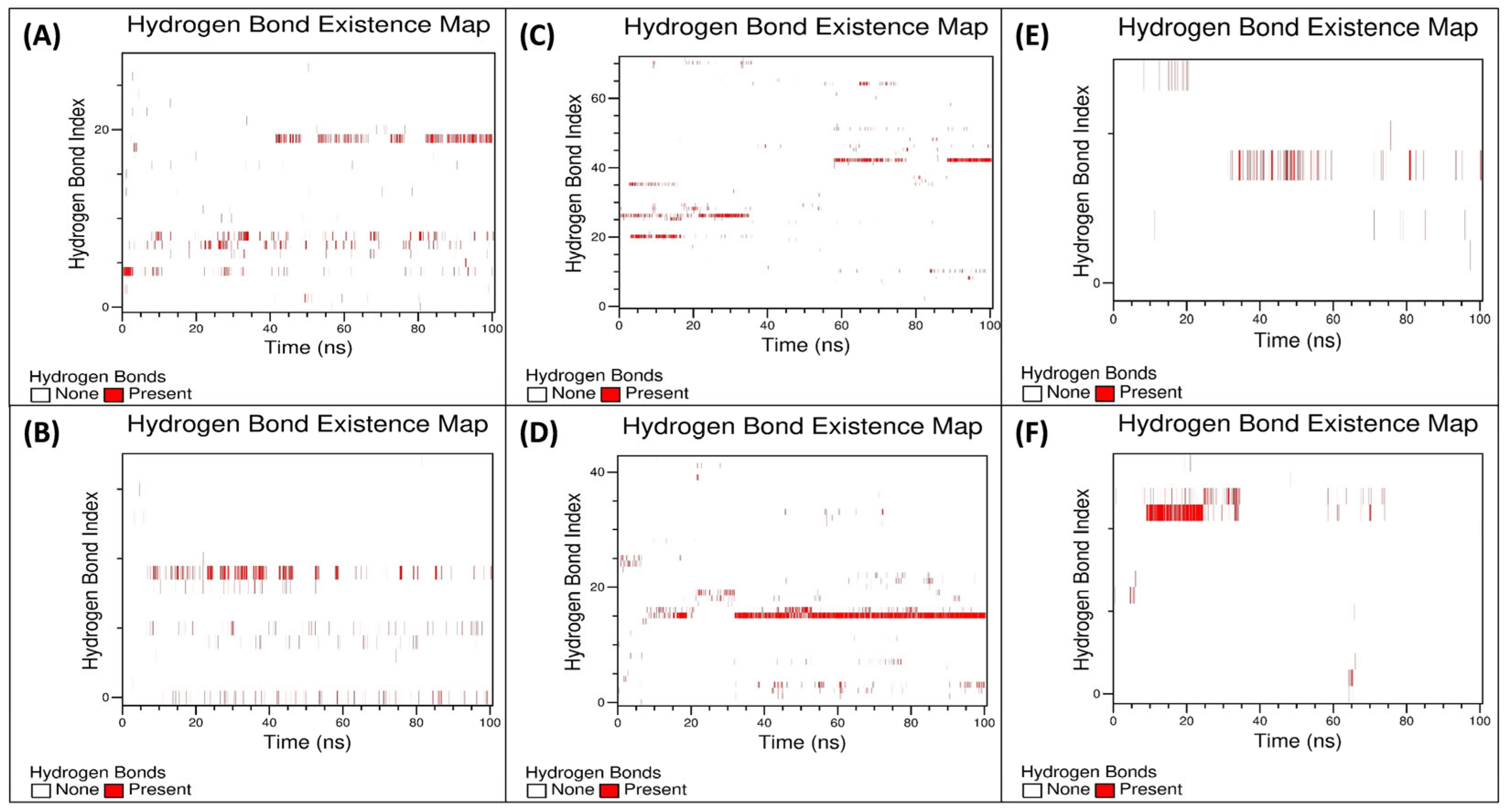
3.5.3. Examination of the Secondary Structural Components and Hydrogen Bonds
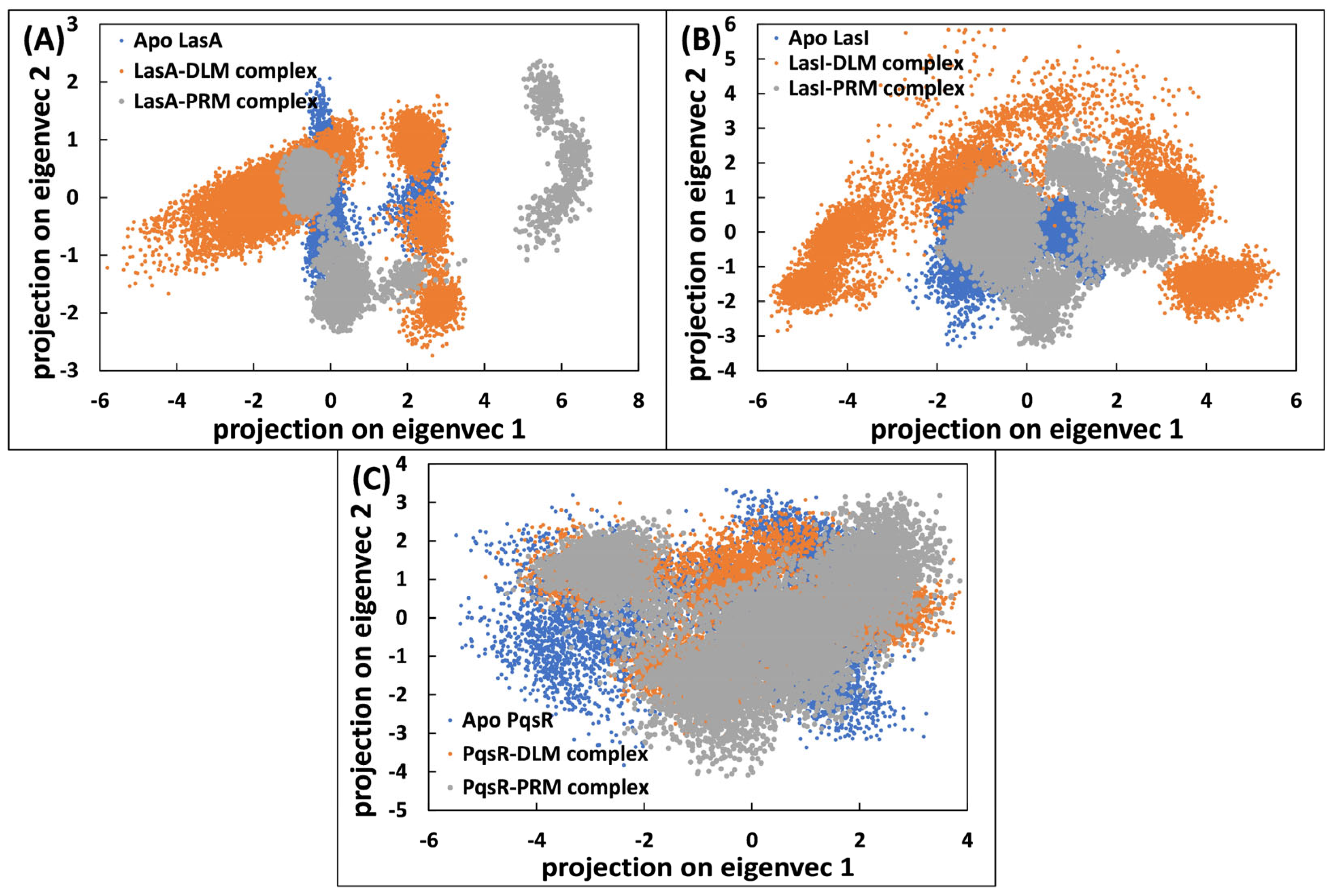
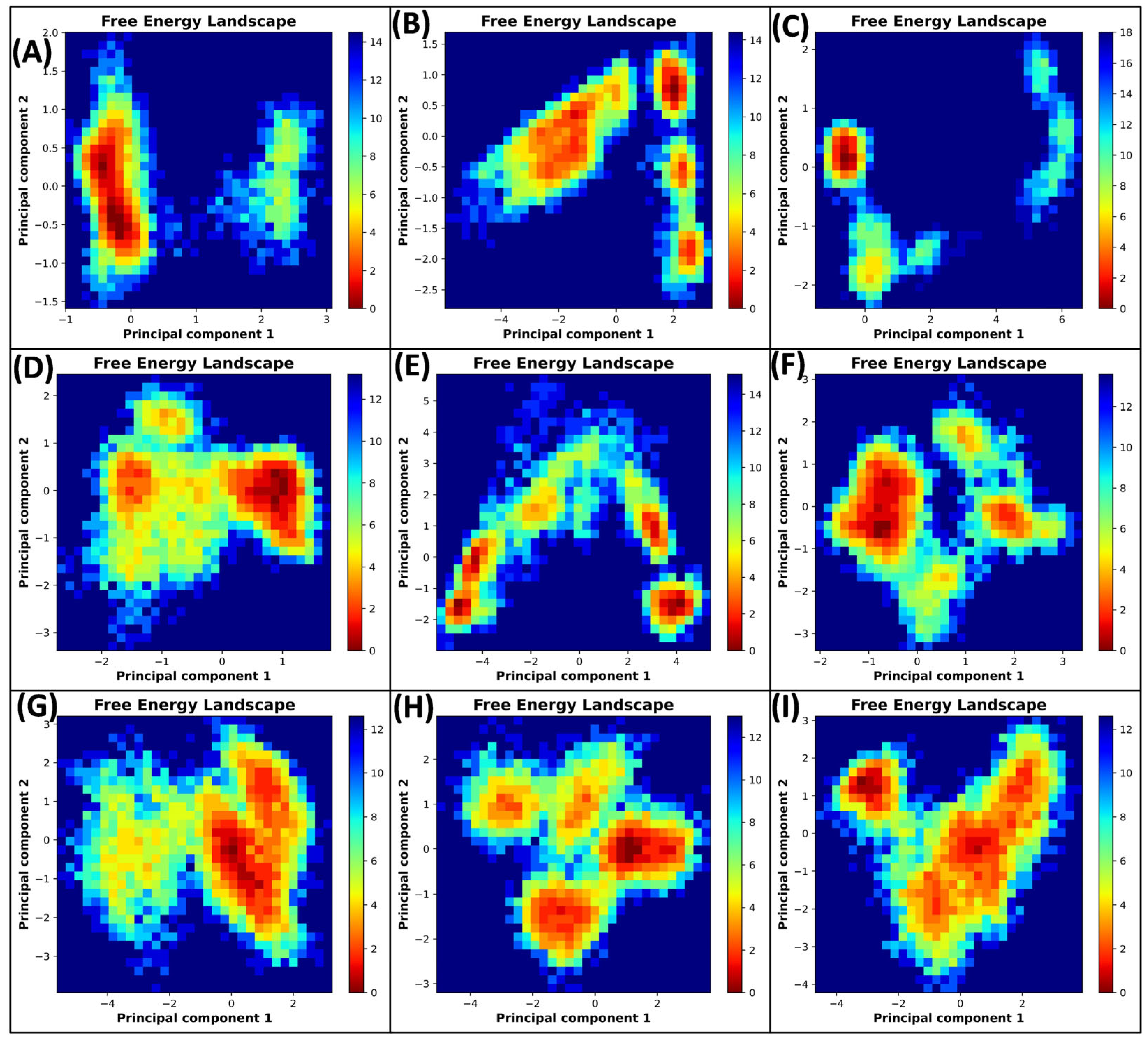
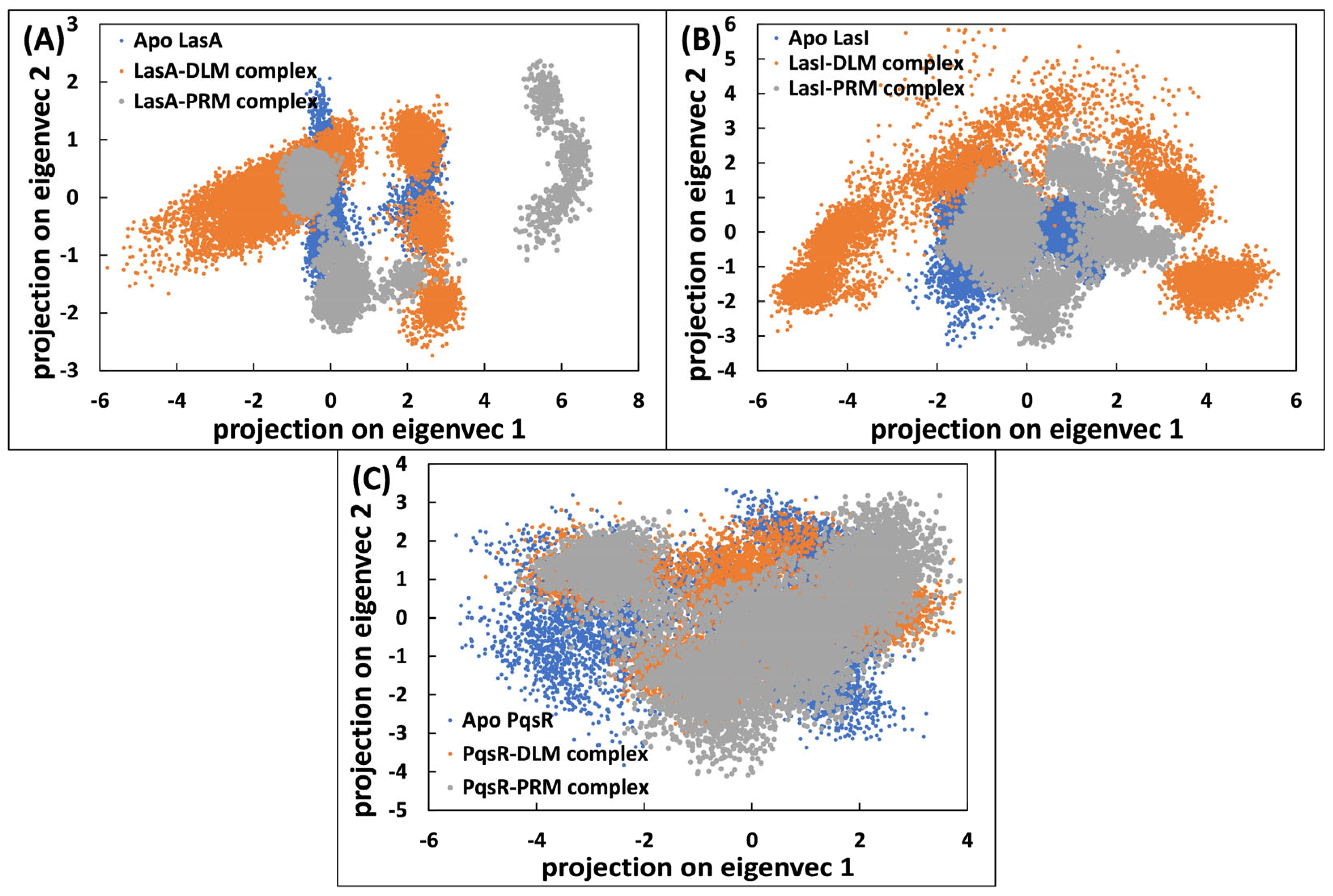
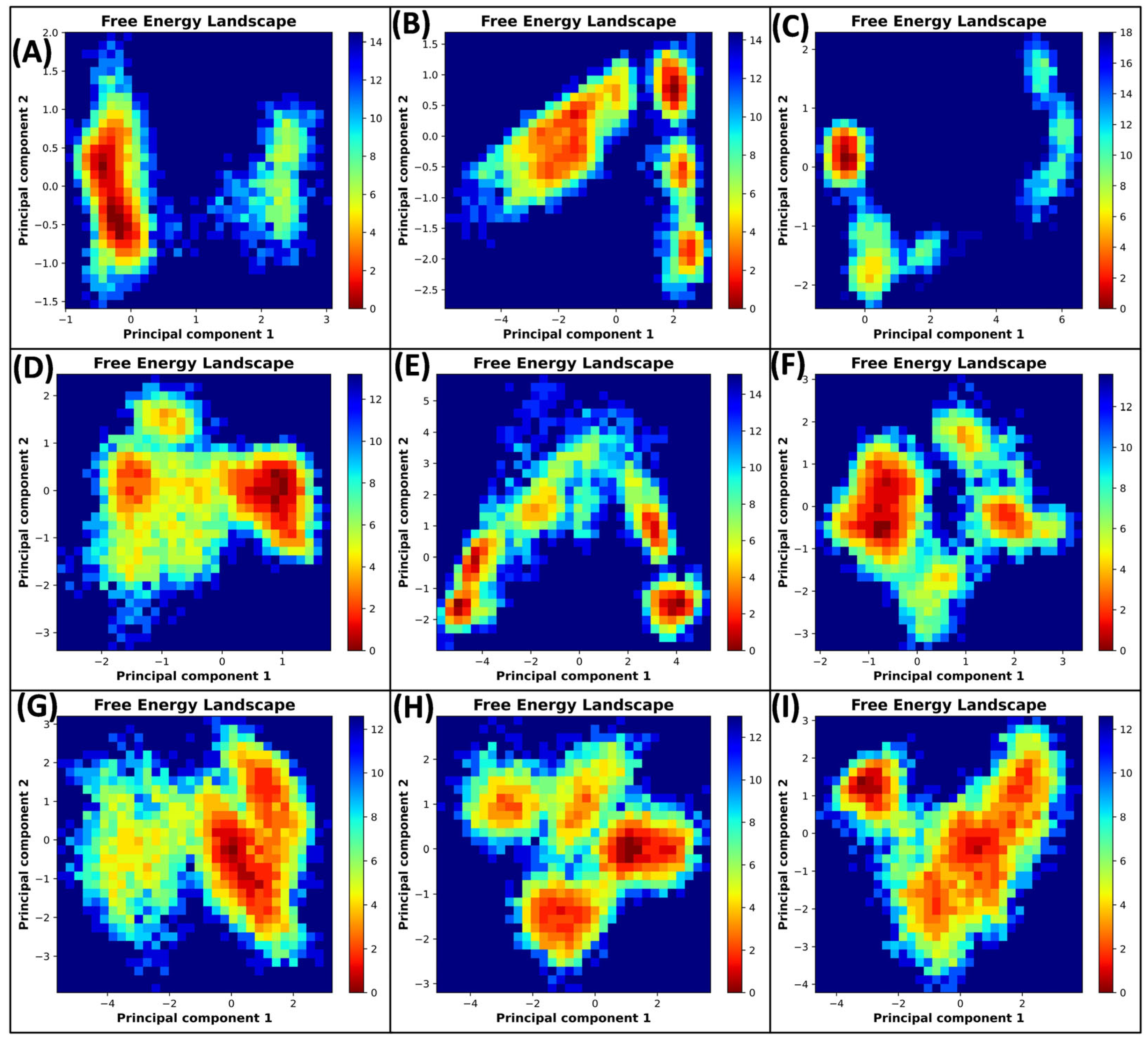
3.5.4. Principal Component Analysis
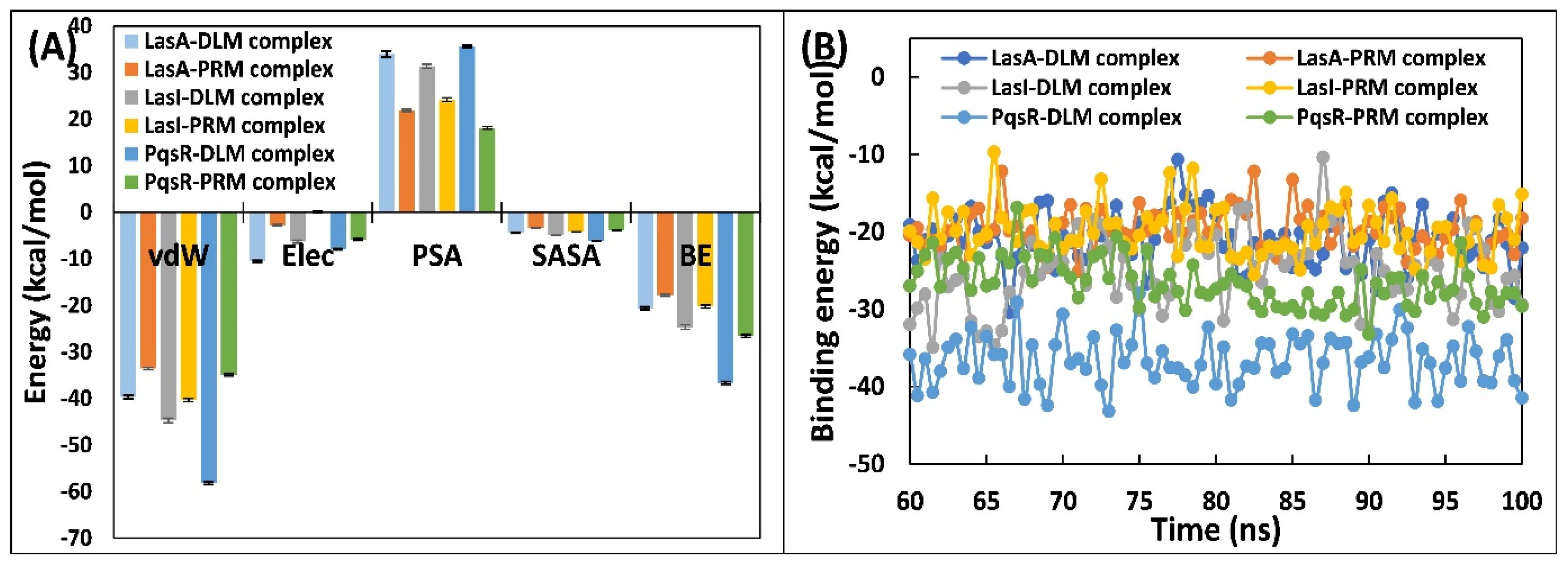
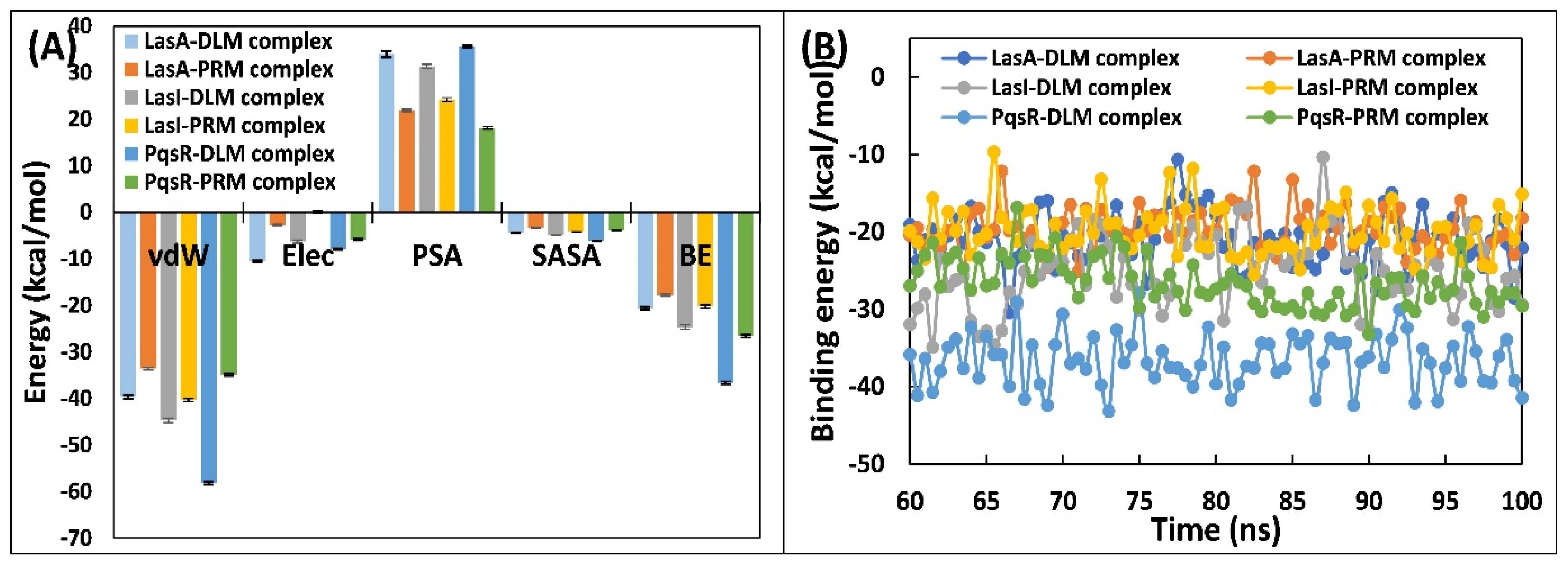
3.5.5. Examination of Binding Energies
4. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Darby, E.M.; Trampari, E.; Siasat, P.; Gaya, M.S.; Alav, I.; Webber, M.A.; Blair, J.M.A. Molecular Mechanisms of Antibiotic Resistance Revisited. Nat. Rev. Microbiol. 2023, 21, 280–295. [Google Scholar] [CrossRef]
- Akram, F.; Imtiaz, M.; Haq, I.L. Emergent Crisis of Antibiotic Resistance: A Silent Pandemic Threat to 21st Century. Microb. Pathog. 2023, 174, 105923. [Google Scholar] [CrossRef]
- Kumar, S.; Adithan, C.; Harish, B.; Roy, G.; Malini, A.; Sujatha, S. Antimicrobial Resistance in India: A Review. J. Nat. Sci. Biol. Med. 2013, 4, 286. [Google Scholar] [CrossRef] [PubMed]
- Nathan, C. Resisting Antimicrobial Resistance. Nat. Rev. Microbiol. 2020, 18, 259–260. [Google Scholar] [CrossRef] [PubMed]
- Rehman, S. A Parallel and Silent Emerging Pandemic: Antimicrobial Resistance (AMR) amid COVID-19 Pandemic. J. Infect. Public Health 2023, 16, 611–617. [Google Scholar] [CrossRef]
- Nathan, C.; Cars, O. Antibiotic Resistance—Problems, Progress, and Prospects. N. Engl. J. Med. 2014, 371, 1761–1763. [Google Scholar] [CrossRef]
- Azimi, S.; Klementiev, A.D.; Whiteley, M.; Diggle, S.P. Bacterial Quorum Sensing During Infection. Annu. Rev. Microbiol. 2020, 74, 201–219. [Google Scholar] [CrossRef] [PubMed]
- Whiteley, M.; Diggle, S.P.; Greenberg, E.P. Progress in and Promise of Bacterial Quorum Sensing Research. Nature 2017, 551, 313–320. [Google Scholar] [CrossRef]
- West, S.A.; Griffin, A.S.; Gardner, A.; Diggle, S.P. Social Evolution Theory for Microorganisms. Nat. Rev. Microbiol. 2006, 4, 597–607. [Google Scholar] [CrossRef]
- Schuster, M.; Joseph Sexton, D.; Diggle, S.P.; Peter Greenberg, E. Acyl-Homoserine Lactone Quorum Sensing: From Evolution to Application. Annu. Rev. Microbiol. 2013, 67, 43–63. [Google Scholar] [CrossRef]
- Warrier, A.; Satyamoorthy, K.; Murali, T.S. Quorum-Sensing Regulation of Virulence Factors in Bacterial Biofilm. Future Microbiol. 2021, 16, 1003–1021. [Google Scholar] [CrossRef]
- Rahme, L.G.; Stevens, E.J.; Wolfort, S.F.; Shao, J.; Tompkins, R.G.; Ausubel, F.M. Common Virulence Factors for Bacterial Pathogenicity in Plants and Animals. Science 1995, 268, 1899–1902. [Google Scholar] [CrossRef] [PubMed]
- Turner, K.H.; Everett, J.; Trivedi, U.; Rumbaugh, K.P.; Whiteley, M. Requirements for Pseudomonas Aeruginosa Acute Burn and Chronic Surgical Wound Infection. PLoS Genet. 2014, 10, e1004518. [Google Scholar] [CrossRef] [PubMed]
- Zegans, M.E.; DiGiandomenico, A.; Ray, K.; Naimie, A.; Keller, A.E.; Stover, C.K.; Lalitha, P.; Srinivasan, M.; Acharya, N.R.; Lietman, T.M. Association of Biofilm Formation, Psl Exopolysaccharide Expression, and Clinical Outcomes in Pseudomonas Aeruginosa Keratitis. JAMA Ophthalmol. 2016, 134, 383. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Bandara, R.; Conibear, T.C.R.; Thuruthyil, S.J.; Rice, S.A.; Kjelleberg, S.; Givskov, M.; Willcox, M.D.P. Pseudomonas Aeruginosa with LasI Quorum-Sensing Deficiency during Corneal Infection. Investig. Opthalmology Vis. Sci. 2004, 45, 1897. [Google Scholar] [CrossRef] [PubMed]
- Mowat, E.; Paterson, S.; Fothergill, J.L.; Wright, E.A.; Ledson, M.J.; Walshaw, M.J.; Brockhurst, M.A.; Winstanley, C. Pseudomonas Aeruginosa Population Diversity and Turnover in Cystic Fibrosis Chronic Infections. Am. J. Respir. Crit. Care Med. 2011, 183, 1674–1679. [Google Scholar] [CrossRef] [PubMed]
- Bjarnsholt, T.; Jensen, P.Ø.; Fiandaca, M.J.; Pedersen, J.; Hansen, C.R.; Andersen, C.B.; Pressler, T.; Givskov, M.; Høiby, N. Pseudomonas Aeruginosa Biofilms in the Respiratory Tract of Cystic Fibrosis Patients. Pediatr. Pulmonol. 2009, 44, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Pesci, E.C.; Milbank, J.B.J.; Pearson, J.P.; McKnight, S.; Kende, A.S.; Greenberg, E.P.; Iglewski, B.H. Quinolone Signaling in the Cell-to-Cell Communication System of Pseudomonas Aeruginosa. Proc. Natl. Acad. Sci. USA 1999, 96, 11229–11234. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Jiménez, A.; Llamas, M.A.; Marcos-Torres, F.J. Transcriptional Regulators Controlling Virulence in Pseudomonas Aeruginosa. Int. J. Mol. Sci. 2023, 24, 11895. [Google Scholar] [CrossRef]
- Miranda, S.W.; Asfahl, K.L.; Dandekar, A.A.; Greenberg, E.P. Pseudomonas Aeruginosa Quorum Sensing; Springer International Publishing: Cham, Switzerland, 2022; pp. 95–115. [Google Scholar]
- Sánchez-Jiménez, A.; Marcos-Torres, F.J.; Llamas, M.A. Mechanisms of Iron Homeostasis in Pseudomonas Aeruginosa and Emerging Therapeutics Directed to Disrupt This Vital Process. Microb. Biotechnol. 2023, 16, 1475–1491. [Google Scholar] [CrossRef]
- Reig, S.; Le Gouellec, A.; Bleves, S. What Is New in the Anti–Pseudomonas Aeruginosa Clinical Development Pipeline Since the 2017 WHO Alert? Front. Cell Infect. Microbiol. 2022, 12, 862. [Google Scholar] [CrossRef]
- Waller, N.J.E.; Cheung, C.-Y.; Cook, G.M.; McNeil, M.B. The Evolution of Antibiotic Resistance Is Associated with Collateral Drug Phenotypes in Mycobacterium Tuberculosis. Nat. Commun. 2023, 14, 1517. [Google Scholar] [CrossRef]
- Chauhan, A.; Kumar, M.; Kumar, A.; Kanchan, K. Comprehensive Review on Mechanism of Action, Resistance and Evolution of Antimycobacterial Drugs. Life Sci. 2021, 274, 119301. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. PkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Wang, Y.; Xing, J.; Xu, Y.; Zhou, N.; Peng, J.; Xiong, Z.; Liu, X.; Luo, X.; Luo, C.; Chen, K.; et al. In Silico ADME/T Modelling for Rational Drug Design. Q. Rev. Biophys. 2015, 48, 488–515. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated Docking Using a Lamarckian Genetic Algorithm and an Empirical Binding Free Energy Function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; van der Spoel, D.; van Drunen, R. GROMACS: A Message-Passing Parallel Molecular Dynamics Implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of Multiple Amber Force Fields and Development of Improved Protein Backbone Parameters. Proteins Struct. Funct. Bioinform. 2006, 65, 712–725. [Google Scholar] [CrossRef]
- Sousa da Silva, A.W.; Vranken, W.F. ACPYPE—AnteChamber PYthon Parser InterfacE. BMC Res. Notes 2012, 5, 367. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical Sampling through Velocity Rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic Transitions in Single Crystals: A New Molecular Dynamics Method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Lynn, A. G_mmpbsa—A GROMACS Tool for High-Throughput MM-PBSA Calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Sasi, P.; Rai, G.; Gupta, V.H.; Amarapurkar, D.; Wangikar, P.P. Studies on Toxicity of Antitubercular Drugs Namely Isoniazid, Rifampicin, and Pyrazinamide in an in Vitro Model of HepG2 Cell Line. Med. Chem. Res. 2011, 20, 1611–1615. [Google Scholar] [CrossRef]
- Tostmann, A.; Boeree, M.J.; Aarnoutse, R.E.; De Lange, W.C.M.; Van Der Ven, A.J.A.M.; Dekhuijzen, R. Antituberculosis Drug-induced Hepatotoxicity: Concise Up-to-date Review. J. Gastroenterol. Hepatol. 2008, 23, 192–202. [Google Scholar] [CrossRef] [PubMed]
- Agal, S.; Baijal, R.; Pramanik, S.; Patel, N.; Gupte, P.; Kamani, P.; Amarapurkar, D. Monitoring and Management of Antituberculosis Drug Induced Hepatotoxicity. J. Gastroenterol. Hepatol. 2005, 20, 1745–1752. [Google Scholar] [CrossRef] [PubMed]
- Mirlohi, M.-S.; Ekrami, A.; Shirali, S.; Ghobeishavi, M.; Pourmotahari, F. Hematological and Liver Toxicity of Anti-Tuberculosis Drugs. Electron. Physician 2016, 8, 3005–3010. [Google Scholar] [CrossRef] [PubMed]
- Illamola, S.M.; Hirt, D.; Tréluyer, J.M.; Urien, S.; Benaboud, S. Challenges Regarding Analysis of Unbound Fraction of Highly Bound Protein Antiretroviral Drugs in Several Biological Matrices: Lack of Harmonisation and Guidelines. Drug Discov. Today 2015, 20, 466–474. [Google Scholar] [CrossRef] [PubMed]
- Cervelli, M.J.; Russ, G.R. PPrinciples of Drug Therapy, Dosing, and Prescribing in Chronic Kidney Disease and Renal Replacement Therapy. In Comprehensive Clinical Nephrology; Elsevier: Amsterdam, The Netherlands, 2010; pp. 871–893. [Google Scholar]
- Ince, I.; Knibbe, C.A.J.; Danhof, M.; de Wildt, S.N. Developmental Changes in the Expression and Function of Cytochrome P450 3A Isoforms: Evidence from In Vitro and In Vivo Investigations. Clin. Pharmacokinet. 2013, 52, 333–345. [Google Scholar] [CrossRef]
- Kacevska, M.; Robertson, G.R.; Clarke, S.J.; Liddle, C. Inflammation and CYP3A4-Mediated Drug Metabolism in Advanced Cancer: Impact and Implications for Chemotherapeutic Drug Dosing. Expert. Opin. Drug Metab. Toxicol. 2008, 4, 137–149. [Google Scholar] [CrossRef]
- Zanger, U.M.; Schwab, M. Cytochrome P450 Enzymes in Drug Metabolism: Regulation of Gene Expression, Enzyme Activities, and Impact of Genetic Variation. Pharmacol. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead- and Drug-like Compounds: The Rule-of-Five Revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A Knowledge-Based Approach in Designing Combinatorial or Medicinal Chemistry Libraries for Drug Discovery. 1. A Qualitative and Quantitative Characterization of Known Drug Databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef]
- Toder, D.S.; Ferrell, S.J.; Nezezon, J.L.; Rust, L.; Iglewski, B.H. LasA and LasB Genes of Pseudomonas Aeruginosa: Analysis of Transcription and Gene Product Activity. Infect. Immun. 1994, 62, 1320–1327. [Google Scholar] [CrossRef] [PubMed]
- Spencer, J.; Murphy, L.M.; Conners, R.; Sessions, R.B.; Gamblin, S.J. Crystal Structure of the LasA Virulence Factor from Pseudomonas Aeruginosa: Substrate Specificity and Mechanism of M23 Metallopeptidases. J. Mol. Biol. 2010, 396, 908–923. [Google Scholar] [CrossRef] [PubMed]
- Qais, F.A.; Khan, M.S.; Ahmad, I.; Husain, F.M.; Al-kheraif, A.A.; Arshad, M.; Alam, P. Plumbagin Inhibits Quorum Sensing-Regulated Virulence and Biofilms of Gram-Negative Bacteria: In Vitro and in Silico Investigations. Biofouling 2021, 37, 724–739. [Google Scholar] [CrossRef]
- Gould, T.A.; Schweizer, H.P.; Churchill, M.E.A. Structure of the Pseudomonas Aeruginosa Acyl-Homoserinelactone Synthase LasI. Mol. Microbiol. 2004, 53, 1135–1146. [Google Scholar] [CrossRef] [PubMed]
- Qais, F.A.; Khan, M.S.; Ahmad, I. Broad-Spectrum Quorum Sensing and Biofilm Inhibition by Green Tea against Gram-Negative Pathogenic Bacteria: Deciphering the Role of Phytocompounds through Molecular Modelling. Microb. Pathog. 2019, 126, 379–392. [Google Scholar] [CrossRef] [PubMed]
- Qais, F.A.; Khan, M.S.; Ahmad, I.; Husain, F.M.; Khan, R.A.; Hassan, I.; Shahzad, S.A.; AlHarbi, W. Coumarin Exhibits Broad-Spectrum Antibiofilm and Antiquorum Sensing Activity against Gram-Negative Bacteria: In Vitro and In Silico Investigation. ACS Omega 2021, 6, 18823–18835. [Google Scholar] [CrossRef]
- Wade, D.S.; Calfee, M.W.; Rocha, E.R.; Ling, E.A.; Engstrom, E.; Coleman, J.P.; Pesci, E.C. Regulation of Pseudomonas Quinolone Signal Synthesis in Pseudomonas Aeruginosa. J. Bacteriol. 2005, 187, 4372–4380. [Google Scholar] [CrossRef]
- Qais, F.A.; Parveen, N.; Ahmad, I.; Husain, F.M.; Khan, A.; Adil, M. Multi-Targeting of Virulence Factors of P. Aeruginosa by β-Lactam Antibiotics to Combat Antimicrobial Resistance. J. Biomol. Struct. Dyn. 2023, 1–19. [Google Scholar] [CrossRef] [PubMed]
- McGrath, S.; Wade, D.S.; Pesci, E.C. Dueling Quorum Sensing Systems in Pseudomonas Aeruginosa Control the Production of the Pseudomonas Quinolone Signal (PQS). FEMS Microbiol. Lett. 2004, 230, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Qais, F.A.; Ahmad, I.; Husain, F.M.; Arshad, M.; Khan, A.; Adil, M. Umbelliferone Modulates the Quorum Sensing and Biofilm of Gram− ve Bacteria: In Vitro and in Silico Investigations. J. Biomol. Struct. Dyn. 2023, 1–14. [Google Scholar] [CrossRef]
- Medina, G.; Juarez, K.; Valderrama, B.; Soberon-Chavez, G. Mechanism of Pseudomonas Aeruginosa RhlR Transcriptional Regulation of the RhlAB Promoter. J. Bacteriol. 2003, 185, 5976–5983. [Google Scholar] [CrossRef] [PubMed]
- Al-Yousef, H.M.; Ahmed, A.F.; Al-Shabib, N.A.; Laeeq, S.; Khan, R.A.; Rehman, M.T.; Alsalme, A.; Al-Ajmi, M.F.; Khan, M.S.; Husain, F.M. Onion Peel Ethylacetate Fraction and Its Derived Constituent Quercetin 4′-O-β-D Glucopyranoside Attenuates Quorum Sensing Regulated Virulence and Biofilm Formation. Front. Microbiol. 2017, 8, 1675. [Google Scholar] [CrossRef]
- Packiavathy, I.I.A.S.V.I.; Agilandeswari, P.; Musthafa, K.K.S.K.; Pandian, S.S.K.S.; Ravi, A.A.V.; Sybiya Vasantha Packiavathy, I.A.; Agilandeswari, P.; Musthafa, K.K.S.K.; Karutha Pandian, S.; Veera Ravi, A.; et al. Antibiofilm and Quorum Sensing Inhibitory Potential of Cuminum Cyminum and Its Secondary Metabolite Methyl Eugenol against Gram Negative Bacterial Pathogens. Food Res. Int. 2012, 45, 85–92. [Google Scholar] [CrossRef]
- Yang, L.; Rybtke, M.T.; Jakobsen, T.H.; Hentzer, M.; Bjarnsholt, T.; Givskov, M.; Tolker-Nielsen, T. Computer-Aided Identification of Recognized Drugs as Pseudomonas Aeruginosa Quorum-Sensing Inhibitors. Antimicrob. Agents Chemother. 2009, 53, 2432–2443. [Google Scholar] [CrossRef]
- Qais, F.A.; Alomar, S.Y.; Imran, M.A.; Hashmi, M.A. In-Silico Analysis of Phytocompounds of Olea Europaea as Potential Anti-Cancer Agents to Target PKM2 Protein. Molecules 2022, 27, 5793. [Google Scholar] [CrossRef]
- Hosen, M.A.; Qais, F.A.; Chtita, S.; Rahman, I.A.; Almehdi, A.M.; Ali, F.; Almalki, F.A.; Hadda, T.B.; Laaroussi, H.; Kawsar, S.M.A. In Silico and POM Analysis for Potential Antimicrobial Agents of Thymidine Analogs by Using Molecular Docking, Molecular Dynamics and ADMET Profiling. Nucleosides Nucleotides Nucleic Acids 2023, 42, 877–918. [Google Scholar] [CrossRef]
- Samreen; Qais, F.A.; Ahmad, I. In Silico Screening and in Vitro Validation of Phytocompounds as Multidrug Efflux Pump Inhibitor against E. Coli. J. Biomol. Struct. Dyn. 2023, 41, 2189–2201. [Google Scholar] [CrossRef]
- Chtita, S.; Fouedjou, R.T.; Belaidi, S.; Djoumbissie, L.A.; Ouassaf, M.; Qais, F.A.; Bakhouch, M.; Efendi, M.; Tok, T.T.; Bouachrine, M.; et al. In Silico Investigation of Phytoconstituents from Cameroonian Medicinal Plants towards COVID-19 Treatment. Struct. Chem. 2022, 33, 1799–1813. [Google Scholar] [CrossRef]
- Liao, S.-Y.; Mo, G.-Q.; Chen, J.-C.; Zheng, K.-C. Exploration of the Binding Mode between (−)-Zampanolide and Tubulin Using Docking and Molecular Dynamics Simulation. J. Mol. Model. 2014, 20, 2070. [Google Scholar] [CrossRef]
- Riaz, S.; Siddiqui, S.; Qais, F.A.; Mateen, S.; Moin, S. Inhibitory Effect of Baicalein against Glycation in HSA: An in Vitro Approach. J. Biomol. Struct. Dyn. 2023, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, S.; Ameen, F.; Jahan, I.; Nayeem, S.M.; Tabish, M. A Comprehensive Spectroscopic and Computational Investigation on the Binding of the Anti-Asthmatic Drug Triamcinolone with Serum Albumin. N. J. Chem. 2019, 43, 4137–4151. [Google Scholar] [CrossRef]
- Ding, K.; Zhang, H.; Wang, H.; Lv, X.; Pan, L.; Zhang, W.; Zhuang, S. Atomic-Scale Investigation of the Interactions between Tetrabromobisphenol, A.; Tetrabromobisphenol, S. Bovine Trypsin by Spectroscopies and Molecular Dynamics Simulations. J. Hazard. Mater. 2015, 299, 486–494. [Google Scholar] [CrossRef]
- Siddiqui, S.; Ameen, F.; Kausar, T.; Nayeem, S.M.; Ur Rehman, S.; Tabish, M. Biophysical Insight into the Binding Mechanism of Doxofylline to Bovine Serum Albumin: An in Vitro and in Silico Approach. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2021, 249, 119296. [Google Scholar] [CrossRef] [PubMed]
- Rath, B.; Qais, F.A.; Patro, R.; Mohapatra, S.; Sharma, T. Design, Synthesis and Molecular Modeling Studies of Novel Mesalamine Linked Coumarin for Treatment of Inflammatory Bowel Disease. Bioorg Med. Chem. Lett. 2021, 41, 128029. [Google Scholar] [CrossRef]
- Qais, F.A.; Ahmad, I. Mechanism of Non-Enzymatic Antiglycation Action by Coumarin: A Biophysical Study. N. J. Chem. 2019, 43, 12823–12835. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antimycobacterial Drugs | AMES Toxicity | Max. Tol. Dose | hERG I Inhibitor | Skin Sensitisation | T. pyriformis Toxicity | Minnow Toxicity |
|---|---|---|---|---|---|---|
| Delamanid | No | 0.741 | No | No | 0.285 | −1.301 |
| Pretomanid | Yes | 1.071 | No | No | 0.285 | −0.032 |
| Clofazimine | Yes | 0.429 | No | No | 0.286 | −3.215 |
| Bedaquiline | No | 0.509 | No | No | 0.285 | 2.32 |
| Terizidone | No | 0.043 | No | No | 0.454 | 2.61 |
| Amithiozone | No | 0.714 | No | No | 0.136 | 1.98 |
| Dapsone | No | −0.126 | No | No | 0.612 | 1.846 |
| Morinamide | No | 0.685 | No | No | 0.083 | 2.797 |
| Protionamide | No | 0.845 | No | Yes | 0.27 | 1.361 |
| Isoniazid | No | 1.166 | No | No | −0.134 | 3.12 |
| Ethionamide | No | 0.902 | No | Yes | 0.084 | 1.643 |
| Pyrazinamide | No | 1.354 | No | No | −0.482 | 2.869 |
| Ethambutol | No | 0.987 | No | Yes | −0.362 | 3.107 |
| Cycloserine | No | 1.049 | No | No | 0.021 | 3.465 |
| Antimycobacterial Drugs | Absorption | Distribution | Metabolism | Excretion | ||||
|---|---|---|---|---|---|---|---|---|
| Int Abs | Water Sol | VDss | Frac Unb | CYP3A4 Substrate | CYP3A4 Inhibitor | Total Clear | R-OCT2 | |
| Delamanid | 100 | 0.251 | −0.113 | 0.058 | Yes | No | −0.047 | No |
| Pretomanid | 92.641 | 0.070 | −0.149 | 0.294 | No | No | 0.13 | Yes |
| Clofazimine | 94.188 | 0.002 | −0.401 | 0.082 | Yes | No | −0.036 | No |
| Bedaquiline | 94.384 | 0.0003 | −0.904 | 0.255 | Yes | No | 0.522 | No |
| Terizidone | 71.541 | 2.144 | −0.369 | 0.449 | No | No | 0.435 | No |
| Amithiozone | 78.815 | 0.396 | −0.185 | 0.401 | No | Yes | −0.215 | No |
| Dapsone | 94.391 | 0.563 | 0.289 | 0.167 | No | Yes | 0.484 | No |
| Morinamide | 85.029 | 4.100 | −0.157 | 0.659 | No | No | 0.912 | No |
| Protionamide | 95.125 | 0.732 | 0.092 | 0.534 | No | Yes | 0.055 | No |
| Isoniazid | 92.601 | 3.444 | −0.352 | 0.728 | No | No | 0.722 | No |
| Ethionamide | 99.428 | 1.831 | 0.021 | 0.582 | No | Yes | 0.035 | No |
| Pyrazinamide | 92.813 | 29.87 | −0.338 | 0.773 | No | No | 0.666 | No |
| Ethambutol | 66.168 | 22.04 | 0.29 | 0.851 | No | No | 1.234 | No |
| Cycloserine | 84.675 | 124.16 | 0.001 | 0.87 | No | No | 0.891 | No |
| Antimycobacterial Drugs | Drug Likeness | Pharmacokinetics | |||
|---|---|---|---|---|---|
| Lipinski Violations | Ghose Violations | GI Absorption | BBB Permeant | Pgp Substrate | |
| Delamanid | 1 | 3 | Low | No | No |
| Pretomanid | 0 | 0 | High | No | No |
| Clofazimine | 1 | 2 | Low | No | No |
| Bedaquiline | 2 | 3 | Low | No | Yes |
| Terizidone | 0 | 1 | High | No | No |
| Amithiozone | 0 | 0 | High | No | No |
| Dapsone | 0 | 0 | High | No | No |
| Morinamide | 0 | 1 | High | No | No |
| Protionamide | 0 | 0 | High | Yes | No |
| Isoniazid | 0 | 3 | High | No | No |
| Ethionamide | 0 | 0 | High | No | No |
| Pyrazinamide | 0 | 4 | High | No | No |
| Ethambutol | 0 | 0 | High | No | No |
| Cycloserine | 0 | 4 | Low | No | No |
| Antimycobacterial Drugs | LasI | LasR | LasA | PqsR | RhlR | Av BE | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BE | Kb | Ki | BS | BE | Kb | Ki | BS | BE | Kb | Ki | BS | BE | Kb | Ki | BS | BE | Kb | Ki | BS | ||
| Delamanid | −9.1 | 472.4 | 0.21 | AS | −8.3 | 122.3 | 0.82 | OS | −8.6 | 203.0 | 0.49 | AS | −9.6 | 1099.3 | 0.09 | AS | −7.3 | 22.6 | 4.42 | OS | −8.58 |
| Pretomanid | −7.9 | 62.27 | 1.61 | AS | −10.9 | 9876 | 0.01 | AS | −7.4 | 26.76 | 3.74 | AS | −7.8 | 52.59 | 1.90 | AS | −6.3 | 4.18 | 23.95 | OS | −8.06 |
| Clofazimine | −8.1 | 87.29 | 1.15 | AS | −7.3 | 22.60 | 4.42 | OS | −8.1 | 87.29 | 1.15 | AS | −9.4 | 784.2 | 0.13 | AS | −7.2 | 19.0 | 5.24 | OS | −8.02 |
| Bedaquiline | −8.9 | 337.05 | 0.30 | AS | −7.1 | 16.12 | 6.20 | OS | −7.3 | 22.60 | 4.42 | AS | −8.5 | 171.5 | 0.58 | AS | −6.3 | 4.18 | 23.95 | OS | −7.62 |
| Terizidone | −7.6 | 37.52 | 2.67 | AS | −9.7 | 1301 | 0.08 | AS | −7.5 | 31.69 | 3.16 | AS | −7 | 13.62 | 7.34 | OS | −5.8 | 1.79 | 55.72 | OS | −7.52 |
| Amithiozone | −6.5 | 5.85 | 17.08 | AS | −8.5 | 171.5 | 0.58 | AS | −5.7 | 1.52 | 65.97 | AS | −6.2 | 3.53 | 28.35 | AS | −6.8 | 9.72 | 10.29 | AS | −6.74 |
| Dapsone | −7.2 | 19.09 | 5.24 | AS | −8.3 | 122.3 | 0.82 | AS | −6.2 | 3.53 | 28.35 | AS | −6 | 2.52 | 39.75 | AS | −5.3 | 0.77 | 129.6 | OS | −6.6 |
| Morinamide | −6 | 2.52 | 39.75 | AS | −7.8 | 52.59 | 1.90 | AS | −6.1 | 2.98 | 33.57 | AS | −5.6 | 1.28 | 78.11 | OS | −6.1 | 2.98 | 33.57 | AS | −6.32 |
| Protionamide | −6 | 2.52 | 39.75 | AS | −6.4 | 4.94 | 20.23 | AS | −5.9 | 2.12 | 47.06 | OS | −5.7 | 1.52 | 65.97 | AS | −6.1 | 2.98 | 33.57 | AS | −6.02 |
| Isoniazid | −5.7 | 1.52 | 65.97 | AS | −6.7 | 8.21 | 12.19 | AS | −5.3 | 0.77 | 129.6 | AS | −5.9 | 2.12 | 47.06 | AS | −5.6 | 1.28 | 78.11 | AS | −5.84 |
| Ethionamide | −5.7 | 1.52 | 65.97 | AS | −6.1 | 2.98 | 33.57 | AS | −5.7 | 1.52 | 65.97 | OS | −5.7 | 1.52 | 65.97 | AS | −6 | 2.52 | 39.75 | AS | −5.84 |
| Pyrazinamide | −5.2 | 0.65 | 153.4 | AS | −6 | 2.52 | 39.75 | AS | −5 | 0.46 | 215.1 | AS | −4.8 | 0.33 | 301.6 | OS | −4.8 | 0.33 | 301.6 | AS | −5.16 |
| Ethambutol | −5.2 | 0.65 | 153.48 | AS | −5.8 | 1.79 | 55.72 | AS | −4.8 | 0.33 | 301.6 | AS | −4.3 | 0.14 | 701.7 | OS | −5.6 | 1.28 | 78.11 | AS | −5.14 |
| Cycloserine | −4.5 | 0.20 | 500.58 | AS | −4.9 | 0.39 | 254.74 | AS | −4.2 | 0.12 | 830.8 | AS | −4.7 | 0.28 | 357.1 | OS | −4.3 | 0.14 | 701.7 | AS | −4.52 |
| LasA–delamanid Complex | LasI–delamanid Complex | PqsR–delamanid Complex | |||
|---|---|---|---|---|---|
| Residues | Total Energy | Residues | Total Energy | Residues | Total Energy |
| His23 | −0.289 ± 0.025 | Leu22 | −0.692 ± 0.053 | Ala102 | −0.776 ± 0.043 |
| Asp40 | −0.191 ± 0.008 | Arg23 | −0.312 ± 0.023 | Ile149 | −0.916 ± 0.026 |
| Tyr79 | −0.240 ± 0.011 | Gln25 | −0.454 ± 0.118 | Ala168 | −0.721 ± 0.032 |
| Tyr80 | −2.143 ± 0.036 | Val26 | −2.120 ± 0.071 | Leu189 | −0.750 ± 0.030 |
| Asp83 | −0.323 ± 0.011 | Phe27 | −0.533 ± 0.026 | Leu208 | −1.457 ± 0.045 |
| Gly113 | −0.198 ± 0.029 | Glu29 | −0.339 ± 0.041 | Val211 | −1.280 ± 0.032 |
| Gly114 | −0.275 ± 0.042 | Glu40 | −0.339 ± 0.042 | Ile236 | −2.248 ± 0.035 |
| His122 | −0.258 ± 0.045 | Arg104 | −0.215 ± 0.057 | Ala237 | −1.076 ± 0.023 |
| Ser124 | −0.177 ± 0.039 | Ile107 | −0.824 ± 0.064 | Pro238 | −1.688 ± 0.029 |
| Leu126 | −0.236 ± 0.023 | Asn108 | −0.667 ± 0.062 | Glu259 | −0.763 ± 0.015 |
| Phe131 | −0.479 ± 0.022 | Gly110 | −0.834 ± 0.056 | Ile263 | −1.217 ± 0.029 |
| Tyr151 | −0.695 ± 0.047 | Val148 | −0.629 ± 0.030 | Asp264 | −0.726 ± 0.012 |
| LasA-pretomanid complex | LasI–pretomanid complex | PqsR–pretomanid complex | |||
| Residues | Total energy | Residues | Total energy | Residues | Total energy |
| His23 | −0.522 ± 0.046 | Lys21 | −0.327 ± 0.009 | Ile149 | −0.957 ± 0.033 |
| Ser24 | −0.438 ± 0.029 | Gln25 | −0.614 ± 0.042 | Glu151 | −0.698 ± 0.020 |
| Thr26 | −0.398 ± 0.015 | Val26 | −2.738 ± 0.052 | Ala168 | −0.743 ± 0.023 |
| Gly27 | −0.435 ± 0.015 | Phe27 | −0.639 ± 0.026 | Val170 | −0.500 ± 0.023 |
| Ser34 | −0.361 ± 0.032 | Glu29 | −0.306 ± 0.018 | Leu207 | −0.659 ± 0.068 |
| Arg60 | −0.247 ± 0.007 | Trp33 | −0.605 ± 0.045 | Leu208 | −1.160 ± 0.040 |
| Arg64 | −0.246 ± 0.008 | Ala106 | −1.414 ± 0.027 | Ile236 | −2.193 ± 0.048 |
| Tyr80 | −1.634 ± 0.049 | Ile107 | −0.591 ± 0.046 | Ala237 | −0.533 ± 0.026 |
| Gly113 | −0.520 ± 0.035 | Asn108 | −0.622 ± 0.037 | Pro238 | −1.073 ± 0.030 |
| Phe131 | −0.597 ± 0.022 | Phe117 | −0.690 ± 0.029 | Glu259 | −0.597 ± 0.032 |
| Asp152 | −0.290 ± 0.004 | Thr145 | −0.364 ± 0.046 | Ile263 | −1.518 ± 0.049 |
| Asp154 | −0.249 ± 0.002 | Val148 | −0.711 ± 0.039 | Asp264 | −0.530 ± 0.037 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anwer, R. Antimycobacterial Drugs as a Novel Strategy to Inhibit Pseudomonas aeruginosa Virulence Factors and Combat Antibiotic Resistance: A Molecular Simulation Study. Microbiol. Res. 2024, 15, 290-313. https://doi.org/10.3390/microbiolres15010020
Anwer R. Antimycobacterial Drugs as a Novel Strategy to Inhibit Pseudomonas aeruginosa Virulence Factors and Combat Antibiotic Resistance: A Molecular Simulation Study. Microbiology Research. 2024; 15(1):290-313. https://doi.org/10.3390/microbiolres15010020
Chicago/Turabian StyleAnwer, Razique. 2024. "Antimycobacterial Drugs as a Novel Strategy to Inhibit Pseudomonas aeruginosa Virulence Factors and Combat Antibiotic Resistance: A Molecular Simulation Study" Microbiology Research 15, no. 1: 290-313. https://doi.org/10.3390/microbiolres15010020
APA StyleAnwer, R. (2024). Antimycobacterial Drugs as a Novel Strategy to Inhibit Pseudomonas aeruginosa Virulence Factors and Combat Antibiotic Resistance: A Molecular Simulation Study. Microbiology Research, 15(1), 290-313. https://doi.org/10.3390/microbiolres15010020