
Serum PCR Diagnosis of Brucella melitensis Infection in Rev. 1 Vaccinated Sheep

Abstract
1. Introduction
2. Materials and Methods
2.1. Bacterial DNA Isolation
2.2. Serum Samples and DNA Purification
2.3. DNA Preparation
2.4. PCR
2.5. DNA Sequencing
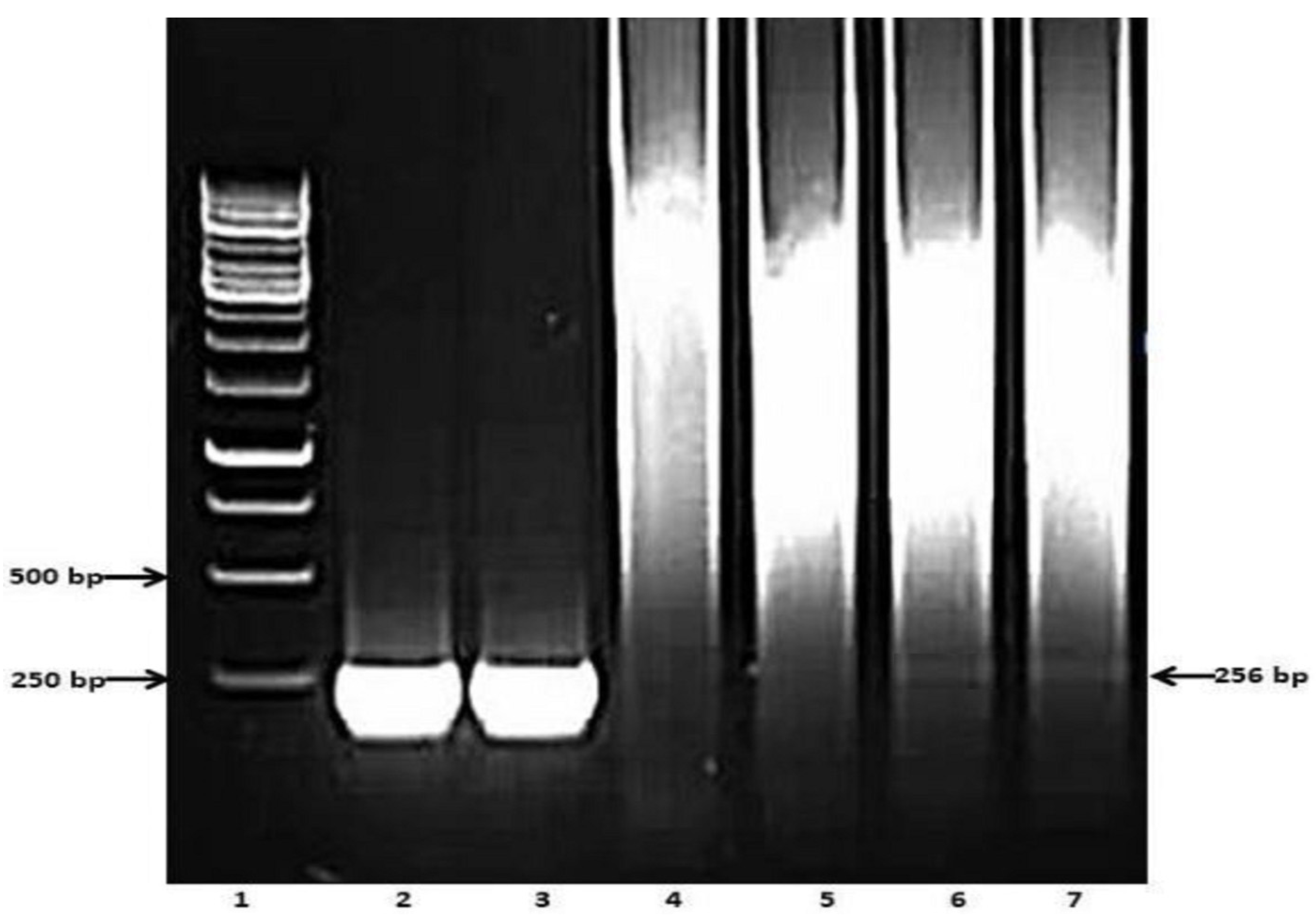
3. Results
3.1. Design of a DIVA Method


3.2. Preliminary Analyses of the Specificity and Reproducibility of the Technique
3.3. Determining Analytical Sensitivity
3.4. Validating Serum PCR
3.5. A Case Study
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Corbel, M.J. Brucellosis: An overview. Emerg. Infect. Dis. 1997, 3, 213–221. [Google Scholar] [CrossRef]
- Scholz, H.C.; Banai, M.; Cloeckaert, A.; Kämpfer, P.; Whatmore, A.M. Brucella. In Bergey’s Manual of Systematics of Archaea and Bacteria; Whitman, W.B., Rainey, F., Kämpfer, P., Trujillo, M., Chun, J., DeVos, P., Hedlund, B., Dedysh, S., Eds.; John Wiley & Sons Inc.: Hoboken, NJ, USA, 2018. [Google Scholar]
- Verger, J.M.; Grimont, F.; Grimont, P.A.D.; Grayon, M. Brucella, a monospecific genus as shown by deoxyribonucleic acid hybridization. Int. J. Syst. Evol. Microbiol. 1985, 35, 292–295. [Google Scholar] [CrossRef]
- Maes, D.G.D.; Dewulf, J.; Piñeiro, C.; Edwards, S.; Kyriazakis, I. A critical reflection on intensive pork production with an emphasis on animal health and welfare. J. Animal Sci. 2019, 98 (Suppl. S1), S15–S26. [Google Scholar] [CrossRef]
- Van Straten, M.; Bardenstein, S.; Keningswald, G.; Bana, M. Brucella abortus S19 vaccine protects dairy cattle against natural infection with Brucella melitensis. Vaccine 2016, 34, 5837–5839. [Google Scholar] [CrossRef]
- Mathur, S.; Banai, M.; Cohen, D. Natural Brucella melitensis infection and Rev. 1 vaccination induce specific Brucella O-Polysaccharide antibodies involved in complement mediated Brucella cell killing. Vaccines 2022, 10, 317. [Google Scholar] [CrossRef]
- Ganesh, N.V.; Sadowska, J.M.; Sarkar, S.; Howells, L.; McGiven, J.; Bundle, D.R. Molecular recognition of Brucella A and M antigens dissected by synthetic oligosaccharide glycoconjugates leads to a disaccharide diagnostic for brucellosis. J. Am. Chem. Soc. 2014, 136, 16260–16269. [Google Scholar] [CrossRef]
- Baum, M.; Zamir, O.; Bergman-Rios, R.; Katz, E.; Beider, Z.; Cohen, A.; Banai, M. Comparative evaluation of microagglutination test and serum agglutination test as supplementary diagnostic methods for brucellosis. J. Clin. Microbiol. 1995, 33, 2166–2170. [Google Scholar] [CrossRef]
- Nielsen, K.H.; Kelly, L.; Gall, D.; Balsevicius, S.; Bosse, J.; Nicoletti, P.; Kelly, W. Comparison of enzyme immunoassays for the diagnosis of bovine brucellosis. Prev. Vet. Med. 1996, 26, 17–32. [Google Scholar] [CrossRef]
- Lukambagire, A.S.; Mendes, Â.J.; Bodenham, R.F.; McGiven, J.A.; Mkenda, N.A.; Mathew, C.; Rubach, M.P.; Sakasaka, P.; Shayo, D.D.; Maro, V.P.; et al. Performance characteristics and costs of serological tests for brucellosis in a pastoralist community of northern Tanzania. Sci. Rep. 2022, 11, 5480. [Google Scholar] [CrossRef]
- Batrinou, A.; Strati, I.F.; Tsantes, A.G.; Papaparaskevas, J.; Dimou, I.; Vourvidis, D.; Kyrma, A.; Antonopoulos, D.; Halvatsiotis, P.; Houhoula, D. The importance of complementary PCR analysis in addition to serological testing for the detection of transmission sources of Brucella spp. in Greek ruminants. Vet. Sci. 2022, 9, 193. [Google Scholar] [CrossRef]
- Bundle, D.R.; McGiven, J. Brucellosis: Improved diagnostics and vaccine insights from synthetic glycans. Acc. Chem. Res. 2017, 50, 2958–2967. [Google Scholar] [CrossRef]
- Mandal, S.S.; Duncombe, L.; Ganesh, N.V.; Sarkar, S.; Howells, L.; Hogarth, P.J.; Bundle, D.R.; McGiven, J. Novel solutions for vaccines and diagnostics to combat brucellosis. ACS Cent. Sci. 2017, 3, 224–231. [Google Scholar] [CrossRef]
- Zerva, L.; Bourantas, K.; Mitka, S.; Kansouzidou, A.; Legakis, N.J. Serum is the preferred clinical specimen for diagnosis of human brucellosis by PCR. J. Clin. Microbiol. 2001, 39, 1661–1664. [Google Scholar] [CrossRef]
- Queipo-Ortuño, M.I.; Colmenero, J.D.; Bravo, M.J.; García-Ordoñez, M.A.; Morata, P. Usefulness of a quantitative real-time PCR assay using serum samples to discriminate between inactive; serologically positive and active human brucellosis. Clin. Microbiol. Infect. 2008, 14, 1128–1134. [Google Scholar] [CrossRef]
- Debeaumont, C.; Falconnet, P.A.; Maurin, M. Real-time PCR for detection of Brucella spp. DNA in human serum samples. Eur. J. Clin. Microbiol. 2005, 24, 842–845. [Google Scholar] [CrossRef]
- Fatima, S.; Khan, I.; Nasi, A.; Younus, M.; Saqib, M.; Melzer, F.; Neubauer, H.; El-Adawy, H. Serological, molecular detection and potential risk factors associated with camel brucellosis in Pakistan. Trop. Anim. Health Prod. 2016, 48, 1711–1718. [Google Scholar] [CrossRef]
- Gwida, M.M.; El-Gohary, A.H.; Melzer, F.; Tomaso, H.; Rösler, U.; Wernery, U.; Wernery, R.; Elschner, M.C.; Khan, I.; Eickhoff, M.; et al. Comparison of diagnostic tests for the detection of Brucella spp. in Camel sera. BMC Res. Notes 2011, 4, 525. [Google Scholar] [CrossRef]
- Gu, W.; Miller, S.; Chiu, C.Y. Clinical metagenomic next-generation sequencing for pathogen detection. Annu. Rev. Pathol. 2019, 14, 319–338. [Google Scholar] [CrossRef]
- Edwards, K.; Johnstone, C.; Thompson, C. A simple and rapid method for the preparation of plant genomic DNA for PCR analysis. Nucleic Acids Res. 1991, 19, 1349. [Google Scholar] [CrossRef]
- DelVecchio, V.G.; Kapatral, V.; Redkar, R.J.; Patra, G.; Mujer, C.; Los, T.; Ivanova, N.; Anderson, I.; Bhattacharyya, A.; Lykidis, A.; et al. The genome sequence of the facultative intracellular pathogen Brucella melitensis. Proc. Natl. Acad. Sci. USA 2002, 99, 443–448. [Google Scholar] [CrossRef]
- Bardenstein, S.; Mandelboim, M.; Ficht, T.A.; Baum, M.; Banai, M. Identification of the Brucella melitensis vaccine strain Rev.1 in animals and humans in Israel by PCR analysis of the PstI site polymorphism of its omp2 gene. J. Clin. Microbiol. 2002, 40, 1475–1480. [Google Scholar] [CrossRef]
- Cloeckaert, A.; Verger, J.M.; Grayon, M.; Grépinet, O. Restriction site polymorphism of the genes encoding the major 25 kDa and 36 kDa outer-membrane proteins of Brucella. Microbiology 1995, 141 Pt 9, 2111–2121. [Google Scholar] [CrossRef]
- Georgi, E.; Walter, M.C.; Pfalzgraf, M.T.; Northoff, B.H.; Holdt, L.M.; Scholz, H.C.; Zoeller, L.; Zange, S.; Antwerpen, M.H. Whole genome sequencing of Brucella melitensis isolated from 57 patients in Germany reveals high diversity in strains from Middle East. PLoS ONE 2017, 12, e0175425. [Google Scholar] [CrossRef]
- Lucero, N.E.; Ayala, S.M.; Escobar, G.I.; Jacob, N.R. Brucella isolated in humans and animals in Latin America from 1968 to 2006. Epidemiol. Infect. 2008, 136, 496–503. [Google Scholar] [CrossRef]
- Janowicz, A.; De Massis, F.; Ancora, M.; Cammà, C.; Patavino, C.; Battisti, A.; Prior, K.; Harmsen, D.; Scholz, H.; Zilli, K.; et al. Core genome multilocus sequence typing and single nucleotide polymorphism analysis in the epidemiology of Brucella melitensis Infections. J. Clin. Microbiol. 2018, 56, e00517-18. [Google Scholar] [CrossRef]
- Le Flèche, P.; Jacques, I.; Grayon, M.; Al Dahouk, S.; Bouchon, P.; Denoeud, F.; Nöckler, K.; Neubauer, H.; Guilloteau, L.A.; Vergnaud, G. Evaluation and selection of tandem repeat loci for a Brucella MLVA typing assay. BMC Microbiol. 2006, 6, 9. [Google Scholar] [CrossRef]
- Babetsa, M.; Boukouvala, E.; Gelasakis, A.I.; Papadopoulos, A.I.; Zdragas, A.; Ekaterinadou, L.V. Detection of a Brucella melitensis Rev. 1 vaccine strain and Rev. 1-like strains in unvaccinated small ruminants and aborted fetuses. Open Access J. Vet. Sci. Med. 2019, 1, 103. [Google Scholar]
- Banai, M. Control of small ruminant brucellosis by use of Brucella melitensis Rev.1 vaccine: Laboratory aspects and field observations. Vet. Microbiol. 2002, 90, 497–519. [Google Scholar] [CrossRef]
- Salmon-Divon, M.; Banai, M.; Bardenstein, S.; Blum, S.E.; Kornspan, D. Complete Genome Sequence of the live attenuated vaccine strain Brucella melitensis Rev.1. Genome Announc. 2018, 6, e00175-18. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Gene | Intact Gene | Partial Sequence within the Gene | ||
|---|---|---|---|---|
| Forward (P1) | Reverse (P2) | Forward (P3) | Reverse (P4) | |
| omp2a BMEI1306 | GGCTATTCAAAATTCTGGCG | ATCGATTCTCACGCTTTCGT | TGGAGGTCAGAAATGAAC | GAGTGCGAAACGAGCGC |
| tRNA (uracil-5)-methyl transferase BMEI1496 | ACACTGGCAGGATGAACCTT | GCGTTATAGGGCAGAAGCTC | CGGAAATCATCGCGCGTCT | AGGCACCTTGCCGAAATG |
| Serial No. | Ear Tag No. | File No. | Amplified Sequences | ||
|---|---|---|---|---|---|
| omp2a | tRNA (uracil-5-)-methyltransferase Sequence | ||||
| Rev. 1 Sequence | Field Strain Sequence | ||||
| 201 | 4898849 R | E238264 | + | Negative | |
| 202 | 3673916 Y 2014 | E238264 | + | + | |
| 203 | 4505960 R | E238264 | Negative | Negative | Negative |
| 204 | 4505924 R | E238264 | + | Negative | |
| 205 | 4813646 Y 2020 | E238264 | Negative | Negative | + |
| 206 | 3991347 Y 2016 | E238264 | Negative | Negative | + |
| 207 | 4002118 Y 2017 | E238264 | + | + | |
| 208 | 4898827 R | E238264 | + | Negative | |
| 209 | 4898865 R | E238264 | + | Negative | |
| 210 | 3651960 R | E238264 | Negative | Negative | Negative |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mathur, S.; Bardenstein, S.; Cohen, D.; Banai, M. Serum PCR Diagnosis of Brucella melitensis Infection in Rev. 1 Vaccinated Sheep. Microbiol. Res. 2023, 14, 21-33. https://doi.org/10.3390/microbiolres14010002
Mathur S, Bardenstein S, Cohen D, Banai M. Serum PCR Diagnosis of Brucella melitensis Infection in Rev. 1 Vaccinated Sheep. Microbiology Research. 2023; 14(1):21-33. https://doi.org/10.3390/microbiolres14010002
Chicago/Turabian StyleMathur, Shubham, Svetlana Bardenstein, Daniel Cohen, and Menachem Banai. 2023. "Serum PCR Diagnosis of Brucella melitensis Infection in Rev. 1 Vaccinated Sheep" Microbiology Research 14, no. 1: 21-33. https://doi.org/10.3390/microbiolres14010002
APA StyleMathur, S., Bardenstein, S., Cohen, D., & Banai, M. (2023). Serum PCR Diagnosis of Brucella melitensis Infection in Rev. 1 Vaccinated Sheep. Microbiology Research, 14(1), 21-33. https://doi.org/10.3390/microbiolres14010002