
Benign Recurrent Intrahepatic Cholestasis: Where Are We Now?


 , ,
, ,  and
and
Abstract
1. Introduction
2. Literature Search
3. History and Epidemiology of Cholestatic Syndromes: BRIC and PFIC
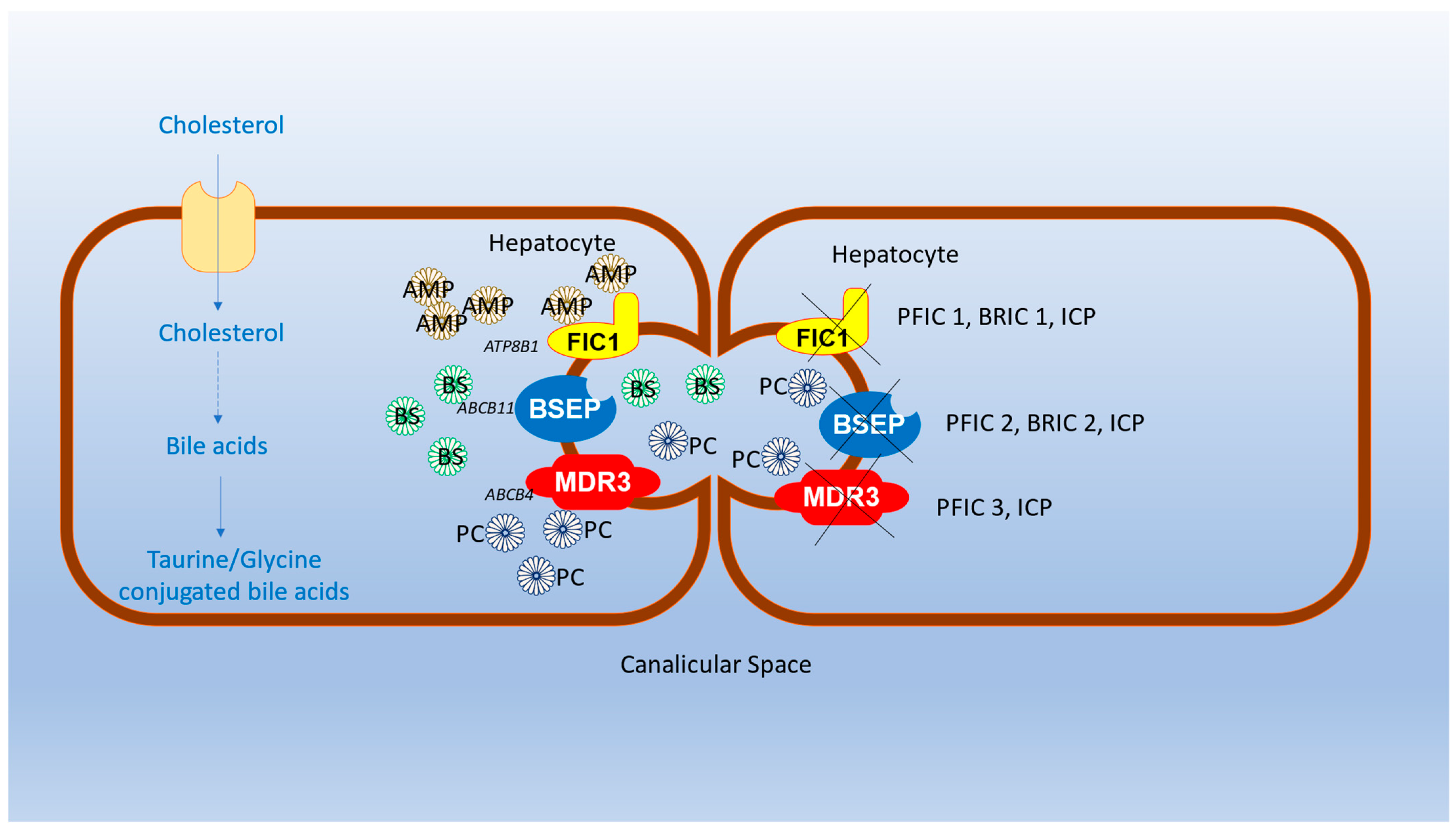
4. Genetic Aspects of BRIC and PFIC
5. Pathophysiology of BRIC
6. Triggering Factors for BRIC
7. Clinical Presentation of BRIC
8. Diagnosis of BRIC
9. Treatment Options and Future Challenges in BRIC

{kind=link}
| Author/Year | Age y.o. | Number of Attacks | Sex | BRIC-1/ BRIC-2 | Triggering Factor | Treatment | Remarks |
|---|---|---|---|---|---|---|---|
| Moghadamrad et al., 2013 [74] | 44 | Unknown | F | BRIC-2 | Gastroenteritis | None | |
| Oldakowska Jedynak et al., 2014 [71] | 14 | Unknown | M | BRIC-1 | Skin infection or tetracycline | None | |
| Kumar et al., 2016 [34] | 27 | >10 | M | BRIC 1 | Unknown | UDCA | |
| Schreiner et al., 2019 [75] | 16 | Unknown | M | BRIC-2 | Pharyngitis | UDCA | |
| Dold et al., 2019 [67] | 51 | 1 | F | BRIC 2 | Unknown | Nasobillary Drainage + Percutaneous Transgastral Biliodigestive Diversion | |
| Sohn et al., 2019 [26] | 6 | 3 | F | BRIC 1 | Unknown | Phenylbutyrate and Rifampicin | |
| Fotoulaki et al., 2019 [30] | 27 months | 1 | F | BRIC 2+ PFIC 2 | Cystitis | UDCA + Liver Transplantation after 5 years | |
| Halawi et al., 2020 [43] | 37 | 7 | F | BRIC-1 | Hyperthyroidism | UDCA + Thyroxine | |
| Arthur et al., 2020 [76] | 27 | 2 | F | BRIC-2 | Pregnancy | None | |
| Piazzolla et al., 2020 [19] | 29 | Unknown | M | BRIC-1 | Unknown | UDCA 15 mg/Kg + Cholestyramine 8 g/d | |
| Salyani et al., 2020 [63] | 21 | 4 | M | Unnknown | Skin abscess in 2 prior attacks | Rifampicin 150 mg × 2/d | |
| Schoneich et al., 2020 [70] | 24 | Unknown | Unknown | BRIC-1 | No | Dialysis | Resistant to Cholestyramine + Steroids + Rifampicin |
| Ayyash et al., 2021 [37] | 29 | 2 related to ICP | F | Unknown | Pregnancy | UDCA 600 mg × 2/d | Fetal Death at 36 weeks of gestation |
| Akbulut et al., 2021 [15] | 16 | >10 | M | BRIC-2 | Unknown | UDCA 20 mg/Kg/d + Cholestyramine 4 g/d | |
| Provenzano et al., 2021 [47] | 35 | 1 | F | BRIC 1 | Pregnancy | Childbirth | |
| Kornitzer et al., 2021 [21] | 15 | >10 | F | BRIC 1 | Unknown | UDCA | |
| Kalaranjini et al., 2021 [25] | 12 | >10 | M | BRIC-2 | Fever | UDCA | |
| Gupta et al., 2021 [2] | 26 | 3 | M | Unknown | Methamphetamine use | UDCA | |
| Chen et al., 2021 [27] | 34 | >10 | M | BRIC-1 | Pharyngitis | UDCA | |
| Koukoulioti et al., 2021 [61] | 47 | >10 | M | BRIC-1 | Unknown | Dialysis | Resistant to drugs on dialysis for many years |
| Bing et al., 2022 [20] | 17 | 1 | M | BRIC-1 | Unknown | UDCA Glycyrrhizin + Glucocorticoids + Dialysis | |
| Calhan et al., 2022 [44] | 59 | Unknown | M | Unknown | COVID-19 | UDCA + Cholestyramine + Rifampicin | |
| Miura et al., 2022 [24] | 16 | 1 | F | Unknown | Unknown | UDCA 300 mg/d + Cholestyramine 4 g/d | |
| Suzuki et al., 2022 [18] | 17 | Unknown | M | BRIC 1 | Unknown | UDCA/Prednisolone/Rifampicin 450 mg/day |
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Henkel, S.A.; Squires, J.H.; Ayers, M.; Ganoza, A.; Mckiernan, P.; Squires, J.E. Expanding etiology of progressive familial intrahepatic cholestasis. World J. Hepatol. 2019, 11, 450–463. [Google Scholar] [CrossRef]
- Gupta, S.; Ali, I.A.; Abreo, E.; Gujju, V.; Hayat, M. The Mystery of Episodic Recurrent Jaundice in a Young Male: Cholestasis With a normal gamma-glutamyl Transferase. Cureus 2021, 13, e13834. [Google Scholar] [CrossRef]
- Summerskill, W.H.; Walshe, J.M. Benign recurrent intrahepatic “obstructive” jaundice. Lancet 1959, 2, 686–690. [Google Scholar] [CrossRef]
- Tygstrup, N. Intermittent possibly familial intrahepatic cholestatic jaundice. Lancet 1960, 275, 1171–1172. [Google Scholar] [CrossRef]
- Tygstrup, N.; Jensen, B. Intermittent intrahepatic cholestasis of unknown etiology in five young males from the Faroe Islands. Acta Med. Scand. 1969, 185, 523–530. [Google Scholar] [CrossRef]
- Tygstrup, N.; Steig, B.A.; Juijn, J.A.; Bull, L.N.; Houwen, R.H. Recurrent familial intrahepatic cholestasis in the Faeroe Islands. Phenotypic heterogeneity but genetic homogeneity. Hepatology 1999, 29, 506–508. [Google Scholar] [CrossRef]
- Srivastava, A. Progressive familial intrahepatic cholestasis. J. Clin. Exp. Hepatol. 2014, 4, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Siddiqi, I.; Tadi, P. Progressive Familial Intrahepatic Cholestasis. [Updated 3 July 2023]. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Luketic, V.A.; Shiffman, M.L. Bening Recurrent intrahepatic cholestasis. Clin. Liver Dis. 1999, 3, 509–528. [Google Scholar] [CrossRef] [PubMed]
- van Ooteghem, N.A.; Klomp, L.W.; van Berge-Henegouwen, G.P.; Houwen, R.H. Benign recurrent intrahepatic cholestasis progressing to progressive familial intrahepatic cholestasis: Low GGT cholestasis is a clinical continuum. J. Hepatol. 2002, 36, 439–443. [Google Scholar] [CrossRef] [PubMed]
- van Mil, S.W.; Klomp, L.W.; Bull, L.N.; Houwen, R.H. FIC1 disease: A spectrum of intrahepatic cholestatic disorders. Semin. Liver Dis. 2001, 21, 535–544. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, R.; Suresh, N.; Sathiyasekeran, M.; Venkatakrishnan, L. Benign recurrent intrahepatic cholestasis Unravelling the paradox. Indian Pediatr. 2021, 58, 486–487. [Google Scholar] [CrossRef] [PubMed]
- García-Romero, R.; Morlan-Herrador, L.; Ros-Arnal, I.; Miramar, M.D.; Molera-Busons, C. Intrahepatic cholestasis, sometimes benign recurrent. Gastroenterol. Hepatol. 2021, 44, 719–720. [Google Scholar] [CrossRef] [PubMed]
- Davit-Spraul, A.; Gonzales, E.; Baussan, C.; Jacquemin, E. Progressive familial intrahepatic cholestasis. Orphanet. J. Rare Dis. 2009, 3, 509–528. [Google Scholar] [CrossRef] [PubMed]
- Akbulut, U.E.; Randa, N.C.; Işık, İ.A.; Atalay, A. Beinign recurrent intrahepatic cholestasis type 2 in a child. A case report and novel mutation. Turk. Arch. Pediatr. 2021, 1, 72–74. [Google Scholar] [CrossRef]
- Lu, L.; Chinese Society of Hepatology and Chinese Medical Association. Guidelines for the Management of Cholestatic Liver Diseases (2021). J. Clin. Transl. Hepatol. 2022, 10, 757–769. [Google Scholar] [CrossRef] [PubMed]
- Kato, K.; Umetsu, S.; Togawa, T.; Ito, K.; Kawabata, T.; Arinaga-Hino, T.; Tsumura, N.; Yasuda, R.; Mihara, Y.; Kusano, H.; et al. Clinicopathologic Features, Genetics, Treatment, and Long-Term Outcomes in Japanese Children and Young Adults with Benign Recurrent Intrahepatic Cholestasis: A Multicenter Study. J. Clin. Med. 2023, 12, 5979. [Google Scholar] [CrossRef]
- Suzuki, H.; Arinaga-Hino, T.; Sano, T.; Mihara, Y.; Kusano, H.; Mizuochi, T.; Togawa, T.; Ito, S.; Ide, T.; Kuwahara, R.; et al. Case Report: A Rare Case of Benign Recurrent Intrahepatic Cholestasis Type 1 with a Novel Heterozygous Pathogenic Variant of ATP8B1. Front. Med. 2022, 9, 891659. [Google Scholar] [CrossRef]
- Piazzolla, M.; Castellaneta, N.; Novelli, A.; Agolini, E.; Cocciadiferro, D.; Resta, L.; Duda, L.; Barone, M.; Ierardi, E.; Di Leo, A. Nonsense variant of ATP8B1 gene in heterozygosis and benign recurrent intrahepatic cholestasis: A case report and review of the literature. World J. Hepatol. 2020, 12, 64–71. [Google Scholar] [CrossRef]
- Bing, H.; Li, Y.L.; Li, D.; Zhang, C.; Chang, B. Case Report: A Rare Heterozygous ATP8B1 Mutation in a BRIC1 Patient: Haploinsufficiency? Front. Med. 2022, 9, 897108. [Google Scholar] [CrossRef]
- Kornitzer, G.A.; Alvarez, F. Case Report: A Novel Single Variant TJP2 Mutation in a Case of Benign Recurrent Intrahepatic Cholestasis. JPGN Rep. 2021, 2, e087. [Google Scholar] [CrossRef]
- Karolczak, S.; Deshwar, A.R.; Aristegui, E.; Kamath, B.M.; Lawlor, M.W.; Andreoletti, G.; Volpatti, J.; Ellis, J.L.; Yin, C.; Dowling, J.J.; et al. Loss of Mtm1 causes cholestatic liver disease in a model of X-linked myotubular myopathy. J. Clin. Investig. 2023, 133, e166275. [Google Scholar] [CrossRef]
- Wu, H.; Wu, L.; Zhang, Q.; Zhang, B.F. Case report: A rare case of pyruvate kinase deficiency and Crigler-Najjar syndrome type II with a novel pathogenic variant of PKLR and UGT1A1 mutation. Front Genet. 2023, 14, 1229271. [Google Scholar] [CrossRef] [PubMed]
- Miura, R.; Kawaoka, T.; Imamura, M.; Kosaka, M.; Johira, Y.; Shirane, Y.; Murakami, S.; Yano, S.; Amioka, K.; Naruto, K.; et al. Benign Recurrent Intrahepatic Cholestasis Type 1 with Novel Nonsense Mutations in the ATP8B1 Gene. Case Rep. Gastroenterol. 2022, 16, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Kalaranjini, K.V.; Glaxon, J.A.; Vasudevan, S.; Arunkumar, M.L. Benign recurrent intrahepatic cholestasis 2 deficiency in a child. Report from a tertiary care center in South India. Indian J. Pathol. Microbiol. 2021, 64 (Suppl. S1), S146–S148. [Google Scholar] [CrossRef] [PubMed]
- Sohn, M.J.; Woo, M.H.; Seong, M.W.; Park, S.S.; Kang, G.H.; Moon, J.S.; Ko, J.S. Benign Recurrent Intrahepatic Cholestasis Type 2 in Siblings with Novel ABCB11 mutations. Pediatr. Gastroenterol. Hepatol. Nutr. 2019, 22, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Wu, D.; Jiang, W.; Lei, T.; Lu, C.; Zhou, T. Case report: A novel homozygous variant identified in a Chinese patient with benign recurrent intrahepatic cholestasis type 1. Front. Med. 2021, 8, 705489. [Google Scholar] [CrossRef] [PubMed]
- Strautnieks, S.S.; Byrne, J.A.; Pawlikowska, L.; Cebecauerová, D.; Rayner, A.; Dutton, L.; Meier, Y.; Antoniou, A.; Stieger, B.; Arnell, H.; et al. Severe bile salt export pump deficiency: 82 different ABCB11 mutations in 109 families. Gastroenterology 2008, 134, 1203–1214. [Google Scholar] [CrossRef] [PubMed]
- Khabou, B.; Kallabi, F.; Abdelaziz, R.B.; Maaloul, I.; Aloulou, H.; Chehida, A.B.; Kammoun, T.; Barbu, V.; Boudawara, T.S.; Fakhfakh, F.; et al. Molecular and computational characterization of ABCB11 and ABCG5 variants in Tunisian patients with neonatal/infantile low-GGT intrahepatic cholestasis: Genetic diagnosis and genotype-phenotype correlation assessment. Ann. Hum. Genet. 2023; early view. [Google Scholar] [CrossRef] [PubMed]
- Fotoulaki, M.; Giza, S.; Jirsa, M.; Grammatikopoulos, T.; Miquel, R.; Hytiroglou, P.; Tsitouridis, I.; Knisely, A.S. Beyond an Obvious Cause of Cholestasis in a Toddler: Compound Heterozygosity for ABCB11 Mutations. Pediatrics 2019, 143, e20182146. [Google Scholar] [CrossRef]
- Corpechot, C.; Barbu, V.; Chazouillères, O.; Broué, P.; Girard, M.; Roquelaure, B.; Chrétien, Y.; Dong, C.; Lascols, O.; Housset, C.; et al. Genetic contribution of ABCC2 to Dubin Johnson syndrome and inherited cholestatic disorders. Liver Int. 2020, 40, 163–174. [Google Scholar] [CrossRef]
- Schonfeld, E.A.; Brown, R.S., Jr. Genetic causes of liver disease: When to suspect a genetic etiology, initial lab testing and the basics of management. Med. Clin. N. Am. 2019, 103, 991–1003. [Google Scholar] [CrossRef]
- Halawi, A.; Ibrahim, N.; Bitar, R. Triggers of benign recurrent intrahepatic cholestasis and its pathophysiology: A review of literature. Acta Gastroenterol. Belg. 2021, 84, 477–486. [Google Scholar] [CrossRef]
- Kumar, P.; Charaniya, R.; Ahuja, A.; Mittal, S.; Sahoo, R. Benign Recurrent Intrahepatic Cholestasis in a Young Adult. J. Clin. Diagn. Res. 2016, 10, OD01-2. [Google Scholar] [CrossRef]
- Sohail, M.I.; Dönmez-Cakil, Y.; Szöllősi, D.; Stockner, T.; Chiba, P. The Bile Salt Export Pump: Molecular Structure, Study Models and Small-Molecule Drugs for the Treatment of Inherited BSEP Deficiencies. Int. J. Mol. Sci. 2021, 22, 784. [Google Scholar] [CrossRef]
- Müllenbach, R.; Bennett, A.; Tetlow, N.; Patel, N.; Hamilton, G.; Cheng, F.; Chambers, J.; Howard, R.; Taylor-Robinson, S.D.; Williamson, C. ATP8B1 mutations in British cases with intrahepatic cholestasis of pregnancy. Gut 2005, 54, 829–834. [Google Scholar] [CrossRef]
- Ayyash, M.; Smith, N.; Keerthy, M.; Singh, A.; Shaman, M. Bening recurrent intrahepatic cholestasis in pregnancy. Fetal death at 36 weeks of gestation. Case Rep. Obstet. Gynecol. 2021, 6, 5086846. [Google Scholar] [CrossRef]
- Nguyen, K.D.; Sundaram, V.; Ayoub, W.S. Atypical causes of cholestasis. World J. Gastroenterol. 2014, 20, 9418–9426. [Google Scholar] [CrossRef] [PubMed]
- Sticova, E.; Jirsa, M.; Pawłowska, J. New insights in genetic cholestasis: From molecular mechanisms to clinical implications. Can. J. Gastroenterol. Hepatol. 2018, 2018, 2313675. [Google Scholar] [CrossRef]
- Nakanishi, Y.; Saxena, R. Pathophysiology and Diseases of the Proximal Pathways of the Biliary System. Arch. Pathol. Lab. Med. 2015, 139, 858–866. [Google Scholar] [CrossRef] [PubMed]
- van der Woerd, W.L.; van Mil, S.W.; Stapelbroek, J.M.; Klomp, L.W.; van de Graaf, S.F.; Houwen, R.H. Familial cholestasis: Progressive familial intrahepatic cholestasis, benign recurrent intrahepatic cholestasis and intrahepatic cholestasis of pregnancy. Best Pract. Res. Clin. Gastroenterol. 2010, 24, 541–553. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, C.G.; Geier, A. Effect of drug transporter pharmacogenetics on cholestasis. Expert. Opin. Drug. Metab. Toxicol. 2014, 10, 1533–1551. [Google Scholar] [CrossRef] [PubMed]
- Halawi, A.; Bitar, R.; Ibrahim, N. Hyperthyroidism as a potential trigger for benign recurrent intrahepatic cholestasis. ACG Case Rep. J. 2020, 7, e00423. [Google Scholar] [CrossRef]
- Calhan, T.; Yivli, E. Coronavirus disease 2019 (COVID-19) as a potential trigger for benign recurrent intrahepatic cholestasis. Clin. Case Rep. 2022, 10, e05557. [Google Scholar] [CrossRef]
- Soroka, C.J.; Boyer, J.L. Biosynthesis and trafficking of the bile salt export pump, BSEP: Therapeutic implications of BSEP mutations. Mol. Aspects Med. 2014, 37, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Huynh, M.T.; Nguyen, T.T.; Grison, S.; Lascols, O.; Fernandez, E.; Barbu, V. Clinical characteristics and genetic profiles of young and adult patients with cholestatic liver disease. Rev. Esp. Enferm. Dig. 2019, 111, 775–788. [Google Scholar] [CrossRef]
- Provenzano, A.; Farina, A.; Seidenari, A.; Azzaroli, F.; Serra, C.; Della Gatta, A.; Zuffardi, O.; Giglio, S.R. Prenatal Noninvasive Trio-WES in a Case of Pregnancy Related Liver Disorder. Diagnostics 2021, 11, 1904. [Google Scholar] [CrossRef] [PubMed]
- Giovannoni, I.; Callea, F.; Bellacchio, E.; Torre, G.; De Ville De Goyet, J.; Francalanci, P. Genetics and Molecular Modeling of New Mutations of Familial Intrahepatic Cholestasis in a Single Italian Center. PLoS ONE 2015, 10, e0145021. [Google Scholar] [CrossRef]
- Reichert, M.C.; Hall, R.A.; Krawczyk, M.; Lammert, F. Genetic determinants of cholangiopathies: Molecular and systems genetics. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1484–1490. [Google Scholar] [CrossRef] [PubMed]
- Togawa, T.; Sugiura, T.; Ito, K.; Endo, T.; Aoyama, K.; Ohashi, K.; Negishi, Y.; Kudo, T.; Ito, R.; Kikuchi, A.; et al. Molecular Genetic Dissection and Neonatal/Infantile Intrahepatic Cholestasis Using Targeted Next Generation Sequencing. J. Pediatr. 2016, 171, 171–177.e1-4. [Google Scholar] [CrossRef] [PubMed]
- Bull, L.N.; Ellmers, R.; Foskett, P.; Strautnieks, S.; Sambrotta, M.; Czubkowski, P.; Jankowska, I.; Wagner, B.; Deheragoda, M.; Thompson, R.J. Cholestasis due to USP53 Deficiency. J. Pediatr. Gastroenterol. Nutr. 2021, 72, 667–673. [Google Scholar] [CrossRef]
- Schonfeld, E.A.; Brown, R.S., Jr. Genetic Testing in Liver Disease: What to order, in Whom, and When. Clin. Liver Dis. 2017, 21, 673–686. [Google Scholar] [CrossRef]
- Lee, S.J.; Kim, J.E.; Choe, B.H.; Seo, A.N.; Bae, H.I.; Hwang, S.K. Early Diagnosis of ABCB11 Spectrum Liver Disorders by Next Generation Sequencing. Pediatr. Gastroenterol. Hepatol. Nutr. 2017, 20, 114–123. [Google Scholar] [CrossRef]
- Naik, J.; de Waart, D.R.; Utsunomiya, K.; Duijst, S.; Mok, K.H.; Oude Elferink, R.P.; Bosma, P.J.; Paulusma, C.C. ATP8B1 and ATP11C: Two Lipid Flippases Important for Hepatocyte Function. Dig. Dis. 2015, 33, 314–318. [Google Scholar] [CrossRef]
- Zheng, Y.; Guo, H.; Chen, L.; Cheng, W.; Yan, K.; Zhang, Z.; Li, M.; Jin, Y.; Hu, G.; Wang, C.; et al. Diagnostic yield and novel candidate genes by next generation sequencing in 166 children with intrahepatic cholestasis. Hepatol. Int. 2023. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, Y.; Gong, J.Y.; Li, L.T.; Li, J.Q.; Zhang, M.H.; Lu, Y.; Xie, X.B.; Hong, Y.R.; Yu, Z.; et al. Low-GGT intrahepatic cholestasis associated with biallelic USP53 variants: Clinical, histological and ultrastructural characterization. Liver Int. 2020, 40, 1142–1150. [Google Scholar] [CrossRef]
- Vij, M.; Sankaranarayanan, S. Biallelic Mutations in Ubiquitin-Specific Peptidase 53 (USP53) Causing Progressive Intrahepatic Cholestasis. Report of a Case With Review of Literature. Pediatr. Dev. Pathol. Off. J. Soc. Pediatr. Pathol. Paediatr. Pathol. Soc. 2022, 25, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Folmer, D.E.; van der Mark, V.A.; Ho-Mok, K.S.; Oude Elferink, R.P.; Paulusma, C.C. Differential effects of progressive familial intrahepatic cholestasis type 1 and benign recurrent intrahepatic cholestasis type 1 mutations on canalicular localization of ATP8B1. Hepatology 2009, 50, 1597–1605. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, A.; Sabu, Y.; Naoi, S.; Ito, S.; Nakano, S.; Minowa, K.; Mizuochi, T.; Ito, K.; Abukawa, D.; Kaji, S.; et al. Assessment of Adenosine Triphosphatase Phospholipid Transporting 8B1 (ATP8B1) Function in Patients With Cholestasis With ATP8B1 Deficiency by Using Peripheral Blood Monocyte-Derived Macrophages. Hepatol. Commun. 2020, 5, 52–62. [Google Scholar] [CrossRef] [PubMed]
- van der Woerd, W.L.; Houwen, R.H.; van de Graaf, S.F. Current and future therapies for inherited cholestatic liver diseases. World J. Gastroenterol. 2017, 23, 763–775. [Google Scholar] [CrossRef] [PubMed]
- Koukoulioti, E.; Ziagaki, A.; Weber, S.N.; Lammert, F.; Berg, T. Long-Term Colestyramine Treatment Prevents Cholestatic Attacks in Refractory Benign Recurrent Intrahepatic Cholestasis Type 1 Disease. Hepatology 2021, 74, 522–524. [Google Scholar] [CrossRef] [PubMed]
- Alhebbi, H.; Peer-Zada, A.A.; Al-Hussaini, A.A.; Algubaisi, S.; Albassami, A.; AlMasri, N.; Alrusayni, Y.; Alruzug, I.M.; Alharby, E.; Samman, M.A.; et al. New paradigms of USP53 disease. Normal CG, cholestasis, BRIC, cholangiopathy and responsiveness to rifampicin. J. Hum. Genet. 2021, 66, 151–159. [Google Scholar] [CrossRef]
- Salyani, A.; Barasa, L.; Rajula, A.; Ali, S.K. Benign Recurrent Intrahepatic Cholestasis (BRIC): An African Case Report. Case Rep. Gastrointest. Med. 2020, 10, 894293. [Google Scholar] [CrossRef]
- Helgadottir, H.; Folvik, G.; Vesterhus, M. Improvement of cholestatic episodes in patients with benign recurrent intrahepatic cholestasis (BRIC) treated with rifampicin. A long-term follow-up. Scand. J. Gastroenterol. 2023, 58, 512–520. [Google Scholar] [CrossRef] [PubMed]
- Kagawa, T.; Orii, R.; Hirose, S.; Arase, Y.; Shiraishi, K.; Mizutani, A.; Tsukamoto, H.; Mine, T. Ursodeoxycholic acid stabilizes the bile salt export pump in the apical membranein MDCK II cells. J. Gastroenterol. 2014, 49, 890–899. [Google Scholar] [CrossRef]
- Choudhury, A.; Kulkarni, A.V.; Sahoo, B.; Bihari, C. Endoscopic nasobiliary drainage: An effective treatment option for benign recurrent intrahepatic cholestasis(BRIC). BMJ Case Rep. 2017, 2017, bcr2016218874. [Google Scholar] [CrossRef] [PubMed]
- Dold, L.; Tschada, A.; Strassburg, C.P.; Weismüller, T.J. Percutaneous transgastral biliodigestive diversion as treatment option for benign recurrent intrahepatic cholestasis. Liver Int. 2019, 39, 222. [Google Scholar] [CrossRef] [PubMed]
- Yakar, T.; Demir, M.; Gokturk, H.S.; Unler Kanat, A.G.; Parlakgumus, A.; Ozer, B.; Serin, E. Nasobiliary Drainage for Benign Recurrent Intrahepatic Cholestasis in Patients Refractory to Sandard Therapy. Clin. Investig. Med. 2016, 39, 27522. [Google Scholar]
- Appleby, V.J.; Hutchinson, J.M.; Davies, M.H. Safety and efficacy of long term nasobiliary drainage to treat intractable pruritus in cholestatic liver disease. Frontline Gastroenterol. 2015, 6, 252–254. [Google Scholar] [CrossRef]
- Schoeneich, K.; Frimmel, S.; Koball, S. Successful treatment of a patient with benign recurrent intrahepatic cholestasis type 1 with albumin dialysis. Artif. Organs 2020, 44, 341–342. [Google Scholar] [CrossRef]
- Ołdakowska-Jedynak, U.; Jankowska, I.; Hartleb, M.; Jirsa, M.; Pawłowska, J.; CzubChoudhury, A.; Kulkarni, A.V.; Sahoo, B.; Bihari Ckowski, P.; Krawczyk, M. Treatment of pruritus with Prometheus dialysis and absorption system in a patient with benign recurrent intrahepatic cholestasis. Hepatol. Res. 2014, 44, E304–E308. [Google Scholar] [CrossRef]
- Muntaha HS, T.; Munir, M.; Sajid, S.H.; Sarfraz, Z.; Sarfraz, A.; Robles-Velasco, K.; Sarfraz, M.; Felix, M.; Cherrez-Ojeda, I. Ileal Bile Acid Transporter Blockers for Cholestatic Liver Disease in Pediatric Patients with Alagille Syndrome: A Systematic Review and Meta-Analysis. J. Clin. Med. 2022, 11, 7526. [Google Scholar] [CrossRef] [PubMed]
- Kamath, B.M.; Stein, P.; Houwen, R.H.J.; Verkade, H.J. Potential of ileal bile acid transporter inhibition as a therapeutic target in Alagille syndrome and progressive familial intrahepatic cholestasis. Liver Int. 2020, 40, 1812–1822. [Google Scholar] [CrossRef] [PubMed]
- Moghadamrad, S.; Montani, M.; Weimann, R.; De Gottardi, A. Cholestasis in a patient with gallstones and a normal gamma-glutamyl transferase. Hepatology 2013, 57, 2539–2541. [Google Scholar] [CrossRef] [PubMed]
- Schreiner, P.; Stieger, B.; McLin, V.; Rougemont, A.L.; Keitel, V.; Dröge, C.; Müllhaupt, B. A rare cause of cholestatic jaundice in a North African teenager. Liver Int. 2019, 39, 2036–2041. [Google Scholar] [CrossRef]
- Arthur Lorio, E.; Valadez, D.; Alkhouri, N.; Loo, N. Cholestasis in Benign Recurrent Intrahepatic Cholestasis 2. ACG Case Rep. J. 2020, 7, e00412. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Geladari, E.V.; Vallianou, N.G.; Margellou, E.; Kounatidis, D.; Sevastianos, V.; Alexopoulou, A. Benign Recurrent Intrahepatic Cholestasis: Where Are We Now? Gastroenterol. Insights 2024, 15, 156-167. https://doi.org/10.3390/gastroent15010011
Geladari EV, Vallianou NG, Margellou E, Kounatidis D, Sevastianos V, Alexopoulou A. Benign Recurrent Intrahepatic Cholestasis: Where Are We Now? Gastroenterology Insights. 2024; 15(1):156-167. https://doi.org/10.3390/gastroent15010011
Chicago/Turabian StyleGeladari, Eleni V., Natalia G. Vallianou, Evangelia Margellou, Dimitris Kounatidis, Vassilios Sevastianos, and Alexandra Alexopoulou. 2024. "Benign Recurrent Intrahepatic Cholestasis: Where Are We Now?" Gastroenterology Insights 15, no. 1: 156-167. https://doi.org/10.3390/gastroent15010011
APA StyleGeladari, E. V., Vallianou, N. G., Margellou, E., Kounatidis, D., Sevastianos, V., & Alexopoulou, A. (2024). Benign Recurrent Intrahepatic Cholestasis: Where Are We Now? Gastroenterology Insights, 15(1), 156-167. https://doi.org/10.3390/gastroent15010011