
Diagnostic Approach and Pathophysiological Mechanisms of Anemia in Chronic Liver Disease—An Overview

 ,
,  , ,
, ,  , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
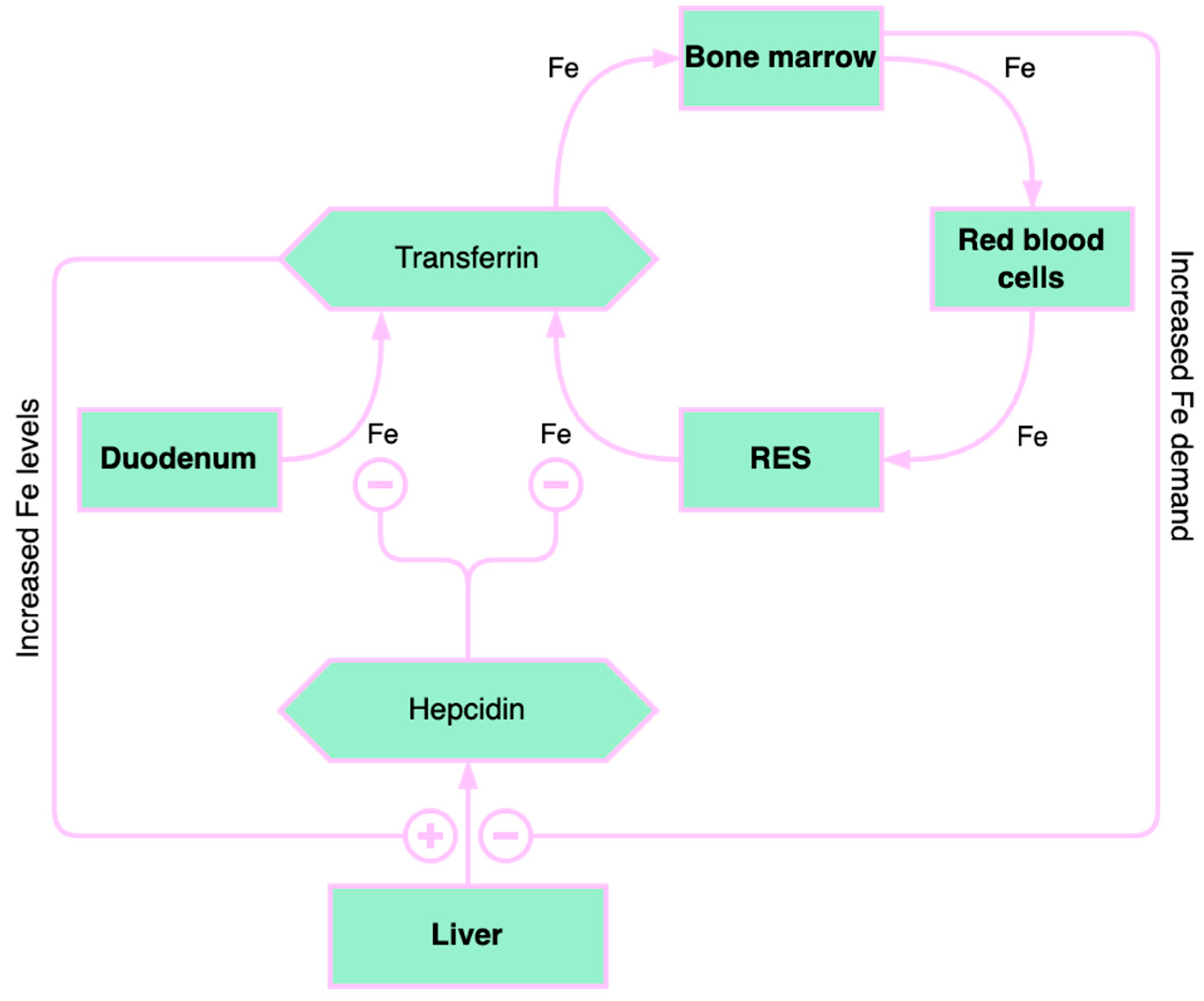
3. Role of Iron Deficiency in Chronic Liver Diseases
3.1. Liver Iron Storage and Chronic Liver Disease
3.2. The Causes of Iron Deficiency in Chronic Liver Diseases
4. Pathophysiological Mechanisms of Anemia in Alcoholic Liver Disease
5. Portal Hypertension
6. Hematological Hypersplenism
7. Abnormalities of Hemostasis in Liver Diseases
7.1. Primary Hemostasis—Platelet Count and Function
7.2. Secondary Hemostasis—Procoagulant and Anticoagulant Factors
7.3. The Fibrinolytic System
8. Hemodilution Anemia
9. Aplastic Anemia in Chronic Liver Diseases
10. Treatment-Associated Anemia in Chronic Viral Hepatitis
11. Anemia in Rare Liver Diseases
11.1. Zieve Syndrome
11.2. Wilson Disease
11.3. Hereditary Hemochromatosis
11.4. Primary Biliary Cholangitis
11.5. Alpha 1 Antitrypsin Deficiency
12. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Maruyama, S.; Hirayama, C.; Yamamoto, S.; Koda, M.; Udagawa, A.; Kadowaki, Y.; Inoue, M.; Sagayama, A.; Umeki, K. Red blood cell status in alcoholic and non-alcoholic liver disease. J. Lab. Clin. Med. 2001, 138, 332–337. [Google Scholar] [CrossRef]
- Mathurin, S.A.; Agüero, A.P.; Dascani, N.A.; Prestera, J.A.; Gianserra, C.; Londero, E.; Chiorra, C. Anemia in hospitalized patients with cirrhosis: Prevalence, clinical relevance, and predictive factors. Acta Gastroenterol. Latinoam. 2009, 39, 103–111. [Google Scholar]
- Qamar, A.A.; Grace, N.D.; Groszmann, R.J.; Garcia–Tsao, G.; Bosch, J.; Burroughs, A.K.; Ripoll, C.; Maurer, R.; Planas, R.; Escorsell, A.; et al. Incidence, prevalence, and clinical significance of abnormal hematologic indices in compensated cirrhosis. Clin. Gastroenterol. Hepatol. 2009, 7, 689–695. [Google Scholar] [CrossRef]
- Kalaitzakis, E.; Josefsson, A.; Castedal, M.; Henfridsson, P.; Bengtsson, M.; Andersson, B.; Björnsson, E. Hepatic encephalopathy is related to anemia and fat-free mass depletion in liver transplant candidates with cirrhosis. Scand. J. Gastroenterol. 2013, 48, 577–584. [Google Scholar] [CrossRef]
- Reiberger, T.; Püspök, A.; Schoder, M.; Baumann-Durchschein, F.; Bucsics, T.; Datz, C.; Dolak, W.; Ferlitsch, A.; Finkenstedt, A.; Graziadei, I.; et al. Austrian consensus guidelines on the management and treatment of portal hypertension (Billroth III). Wien. Klin. Wochenschr. 2017, 129 (Suppl. 3), 135–158. [Google Scholar] [CrossRef]
- Ripoll, C.; Garcia-Tsao, G. The management of portal hypertensive gastropathy and gastric antral vascular ectasia. Dig. Liver Dis. 2011, 43, 345–351. [Google Scholar] [CrossRef]
- Luo, J.-C.; Leu, H.-B.; Hou, M.-C.; Huang, C.-C.; Lin, H.-C.; Lee, F.-Y.; Chang, F.-Y.; Chan, W.-L.; Lin, S.-J.; Chen, J.-W. Cirrhotic patients at increased risk of peptic ulcer bleeding: A nationwide population-based cohort study. Aliment. Pharmacol. Ther. 2012, 36, 542–550. [Google Scholar] [CrossRef]
- Gado, A.; Ebeid, B.; Axon, A. Prevalence and outcome of peptic ulcer bleeding in patients with liver cirrhosis. Alex. J. Med. 2014, 50, 143–148. [Google Scholar] [CrossRef]
- Alexopoulou, A.; Vasilieva, L.; Kanellopoulou, T.; Pouriki, S.; Soultati, A.; Dourakis, S.P. Presence of spur cells as a highly predictive factor of mortality in patients with cirrhosis. J. Gastroenterol. Hepatol. 2014, 29, 830–834. [Google Scholar] [CrossRef]
- Vassiliadis, T.; Mpoumponaris, A.; Vakalopoulou, S.; Giouleme, O.; Gkissakis, D.; Grammatikos, N.; Soufleris, K.; Kakafika, A.; Tziomalos, K.; Patsiaoura, K.; et al. Spur cells and spur cell anemia in hospitalized patients with advanced liver disease: Incidence and correlation with disease severity and survival. Hepatol. Res. 2010, 40, 161–170. [Google Scholar] [CrossRef]
- Gonzalez-Casas, R.; Jones, E.A.; Moreno-Otero, R. Spectrum of anemia associated with chronic liver disease. World J. Gastroenterol. 2009, 15, 4653–4658. [Google Scholar] [CrossRef]
- Gkamprela, E.; Deutsch, M.; Pectasides, D. Iron deficiency anemia in chronic liver disease: Etiopathogenesis, diagnosis and treatment. Ann. Gastroenterol. 2017, 30, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Casas, R.; Garcia-Buey, L.; Jones, E.A.; Gisbert, J.P.; Moreno-Otero, R. Systematic review: Hepatitis-associated aplastic anaemia–a syndrome associated with abnormal immunological function. Aliment. Pharmacol. Ther. 2009, 30, 436–443. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.Q.; Zhan, Y.Q.; Hu, X.; Zhuang, Y.P.; Liu, H.Q.; Hong, M.F.; Zhong, H.J. Anemia Is Associated with Disease Severity, Hepatic Complications, and Progression of Wilson Disease: A Retrospective Cohort Study. Dig Dis. 2023, 41, 632–640. [Google Scholar] [CrossRef]
- Les, I.; Doval, E.; Flavià, M.; Jacas, C.; Cárdenas, G.; Esteban, R.; Guardia, J.; Córdoba, J. Quality of life in cirrhosis is related to potentially treatable factors. Eur. J. Gastroenterol. Hepatol. 2010, 22, 221–227. [Google Scholar] [CrossRef]
- Güngör, G.; Akyıldız, M.; Keskin, M.; Solak, Y.; Gaipov, A.; Bıyık, M.; Çifçi, S.; Ataseven, H.; Polat, H.; Demir, A. Is there any potential or additive effect of anemia on hepatorenal syndrome? Turk. J. Gastroenterol. 2016, 27, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Milward, E.; Johnstone, D.; Trinder, D.; Ramm, G.; Olynyk, J. The nexus of iron and inflammation in hepcidin regulation: SMADs, STATs, and ECSIT. Hepatology 2007, 45, 253–256. [Google Scholar] [CrossRef]
- Wang, C.-Y.; Babitt, J.L. Liver iron sensing and body iron homeostasis. Blood 2019, 133, 18–29. [Google Scholar] [CrossRef]
- Tsochatzis, E.; Papatheodoridis, G.V.; Koliaraki, V.; Hadziyannis, E.; Kafiri, G.; Manesis, E.K.; Mamalaki, A.; Archimandritis, A.J. Serum hepcidin levels are related to the severity of liver histological lesions in chronic hepatitis C. J. Viral Hepat. 2010, 17, 800–806. [Google Scholar] [CrossRef]
- Lyberopoulou, A.; Chachami, G.; Gatselis, N.K.; Kyratzopoulou, E.; Saitis, A.; Gabeta, S.; Eliades, P.; Paraskeva, E.; Zachou, K.; Koukoulis, G.K.; et al. Low serum hepcidin in patients with autoimmune liver diseases. PLoS ONE 2015, 10, e0135486. [Google Scholar] [CrossRef]
- Wang, X.H.; Cheng, P.P.; Jiang, F.; Jiao, X.Y. The effect of hepatitis B virus infection on hepcidin expression in hepatitis B patients. Ann. Clin. Lab. Sci. 2013, 43, 126–134. [Google Scholar]
- Lunova, M.; Trautwein, C.; Strnad, P.; Nahon, P. Reply to: “Hepatic hepcidin expression is decreased in cirrhosis and HCC”. J. Hepatol. 2015, 62, 979–980. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ganz, T. Hepcidin. Rinsho Ketsueki 2016, 57, 1913–1917. [Google Scholar] [PubMed]
- Peyrin-Biroulet, L.; Williet, N.; Cacoub, P. Guidelines on the diagnosis and treatment of iron deficiency across indications: A systematic review. Am. J. Clin. Nutr. 2015, 102, 1585–1594. [Google Scholar] [CrossRef] [PubMed]
- Nahon, P.; Sutton, A.; Rufat, P.; Ziol, M.; Thabut, G.; Schischmanoff, P.; Vidaud, D.; Charnaux, N.; Couvert, P.; Ganne–Carrie, N.; et al. Liver iron, HFE gene mutations, and hepatocellular carcinoma occurrence in patients with cirrhosis. Gastroenterology 2008, 134, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Drakesmith, H.; Prentice, A.M. Hepcidin and the iron-infection axis. Science 2012, 338, 768–772. [Google Scholar] [CrossRef]
- Biciuşcă, V.; Popescu, M.; Petrescu, I.O.; Stan, I.S.; Durand, P.; Petrescu, M.; Velea, R.; Traşcă, D.M.; Popescu, I.A.S.; Udriştoiu, I.; et al. Hepatic pathological features in naïve patients with chronic hepatitis C who have developed thyroid disorder. Rom. J. Morphol. Embryol. 2021, 61, 1085–1097. [Google Scholar] [CrossRef]
- Stan, I.S.; Biciuşcă, V.; Durand, P.; Petrescu, A.-M.; Oancea, D.M.; Ciuciulete, A.R.; Petrescu, M.; Udriştoiu, I.; Camen, G.-C.; Bălteanu, M.A.; et al. Diagnostic and prognostic significance of hepatic steatosis in patients with chronic hepatitis C. Rom. J. Morphol. Embryol. 2021, 62, 765–775. [Google Scholar] [CrossRef]
- Handa, P.; Maliken, B.D.; Nelson, J.E.; Hennessey, K.A.; Vemulakonda, L.A.; Morgan-Stevenson, V.; Dhillon, B.K.; Gupta, R.; Yeh, M.M.; Kowdley, K.V. Differencesin hepatic expression of iron, inflammation and stress related genes in patients with nonalcoholic steatohepatitis. Ann. Hepatol. 2017, 16, 77–85. [Google Scholar] [CrossRef]
- Scheiner, B.; Semmler, G.; Maurer, F.; Schwabl, P.; Bucsics, T.A.; Paternostro, R.; Bauer, D.; Simbrunner, B.; Trauner, M.; Mandorfer, M.; et al. Prevalence of and risk factors for anaemia in patients with advanced chronic liver disease. Liver Int. 2020, 40, 194–204. [Google Scholar] [CrossRef]
- Mallet, M.; Rudler, M.; Thabut, D. Variceal bleeding in cirrhotic patients. Gastroenterol. Rep. 2017, 5, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Kate, V.; Sureshkumar, S.; Gurushankari, B.; Kalayarasan, R. Acute Upper Non-variceal and Lower Gastrointestinal Bleeding. J. Gastrointest. Surg. 2022, 26, 932–949. [Google Scholar] [CrossRef] [PubMed]
- Stein, J.; Connor, S.; Virgin, G.; Ong, D.E.; Pereyra, L. Anemia and iron deficiency in gastrointestinal and liver conditions. World J. Gastroenterol. 2016, 22, 7908–7925. [Google Scholar] [CrossRef] [PubMed]
- Kalafateli, M.; Triantos, C.K.; Nikolopoulou, V.; Burroughs, A. Non-variceal Gastrointestinal Bleeding in Patients with Liver Cirrhosis: A Review. Dig. Dis. Sci. 2012, 57, 2743–2754. [Google Scholar] [CrossRef]
- Kim, M.Y.; Choi, H.; Baik, S.K.; Yea, C.J.; Won, C.S.; Byun, J.W.; Park, S.Y.; Kwon, Y.H.; Kim, J.W.; Kim, H.S.; et al. Portal hypertensive gastropathy: Correlation with portal hypertension and prognosis in cirrhosis. Dig. Dis. Sci. 2010, 55, 3561–3567. [Google Scholar] [CrossRef]
- Selinger, C.P.; Ang, Y.S. Gastric antral vascular ectasia (GAVE): An update on clinical presentation, pathophysiology and treatment. Digestion 2008, 77, 131–137. [Google Scholar] [CrossRef]
- Spahr, L.; Villeneuve, J.-P.; Dufresne, M.-P.; Tassé, D.; Bui, B.; Willems, B.; Fenyves, D.; Pomier-Layrargues, G. Gastric antral vascular ectasia in cirrhotic patients: Absence of relation with portal hypertension. Gut 1999, 44, 739–742. [Google Scholar] [CrossRef]
- Canlas, K.R.; Dobozi, B.M.; Lin, S.; Smith, A.D.; Rockey, D.C.; Muir, A.J.; Agrawal, N.M.; Poleski, M.H.; Patel, K.; McHutchison, J.G. Using Capsule Endoscopy toIdentify GI Tract Lesions in Cirrhotic Patients With Portal Hypertension and Chronic Anemia. J. Clin. Gastroenterol. 2008, 42, 844–848. [Google Scholar] [CrossRef]
- Halsted, C.H.; Villanueva, J.A.; Devlin, A.M.; Chandler, C.J. Metabolic interactions of alcohol and folate. J. Nutr. 2002, 132, 2367S–2372S. [Google Scholar] [CrossRef]
- Medici, V.; Halsted, C.H. Folate, alcohol, and liver disease. Mol. Nutr. Food Res. 2012, 57, 596–606. [Google Scholar] [CrossRef]
- Green, R.; Dwyre, D.M. Evaluation of macrocytic anemias. Semin. Hematol. 2015, 52, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Grattagliano, I.; Calamita, G.; Cocco, T.; Wang, D.Q.; Portincasa, P. Pathogenic role of oxidative and nitrosative stress in primary biliary cirrhosis. World J. Gastroenterol. 2014, 20, 5746–5759. [Google Scholar] [CrossRef] [PubMed]
- Marginean, C.M.; Pirscoveanu, D.; Popescu, M.; Vasile, C.M.; Docea, A.O.; Mitruț, R.; Mărginean, I.C.; Iacob, G.A.; Firu, D.M.; Mitruț, P. Challenges in Diagnosis and Therapeutic Approach of Acute on Chronic Liver Failure—A Review of Current Evidence. Biomedicines 2023, 11, 1840. [Google Scholar] [CrossRef] [PubMed]
- Wiley, J.S.; Moore, M.R. Heme Biosynthesis and Its Disorders: Porphyrias and Sideroblastic Anemias; Churchill Livingstone: London, UK, 2000. [Google Scholar]
- Holloway, R. Sideroblastic anaemia secondary to chronic alcoholism: A case study and review. N. Z. J. Med. Lab. Sci. 2007, 61, 69. [Google Scholar]
- Privitera, G.; Meli, G. An unusual cause of anemia in cirrhosis: Spur cell anemia, a case report withreview of literature. Gastroenterol. Hepatol. Bed Bench 2016, 9, 335. [Google Scholar]
- Roy, A.; Rodge, G.; Goenka, M.K. Spur Cell Anaemia in Cirrhosis: A Narrative Review. J. Clin. Exp. Hepatol. 2023, 13, 500–508. [Google Scholar] [CrossRef]
- Gerber, B.; Stussi, G. Reversibility of spur cell anemia. Blood 2011, 118, 4304. [Google Scholar] [CrossRef]
- Nakatsuka, H.; Sato, Y.; Oya, H.; Yamamoto, S.; Kurosaki, I.; Shirai, Y.; Hatakeyama, K. A case of spur cell anemia resolved by living donor liver transplantation. Kanzo 2007, 48, 181–186. [Google Scholar] [CrossRef]
- Iwakiri, Y. Pathophysiology of portal hypertension. Clin. Liver Dis. 2014, 18, 281–291. [Google Scholar] [CrossRef]
- Garcia-Pagan, J.C.; De Gottardi, A.; Bosch, J. Review article: The modern management of portal hypertension--primary and secondary prophylaxis of variceal bleeding in cirrhotic patients. Aliment. Pharmacol. Ther. 2008, 28, 178–186. [Google Scholar] [CrossRef]
- Albillos, A. Preventing first variceal hemorrhage in cirrhosis. J. Clin. Gastroenterol. 2007, 41 (Suppl. 3), S305–S311. [Google Scholar] [CrossRef]
- Abraldes, J.G.; Bosch, J. The treatment of acute variceal bleeding. J. Clin. Gastroenterol. 2007, 41 (Suppl. 3), S312–S317. [Google Scholar] [CrossRef] [PubMed]
- Lv, Y.; Gong, X.; Xie, X.; Wang, B.; Yang, Y.; Li, Y. Clinical Study on the Relationship between Hematocytopenia and Splenomegaly Caused by Cirrhotic Portal Hypertension. Cell Biochem. Biophys. 2014, 70, 355–360. [Google Scholar] [CrossRef]
- Shah, S.H.; Hayes, P.C.; Allan, P.L.; Nicoll, J.; Finlayson, N.D. Measurement of spleen size and its relation to hypersplenism and portal hemodynamics in portal hypertension due to hepatic cirrhosis. Am J Gastroenterol. 1996, 91, 2580–2583. [Google Scholar]
- Tan, C.H.; Hall, J.A.; Hammonds, K.; Dodlapati, J.; Linz, W.J.; Henderson, S.M. Relationship betweensplenomegaly and transfusion requirements in patients with cirrhosis. In Baylor University Medical Center Proceedings; Taylor & Francis: London, UK, 2020; Volume 34, pp. 44–48. [Google Scholar]
- Laffi, G.; Marra, F.; Tarquini, R.; Abbate, R. Coagulation defects in cirrhosis—Old dogmas not yet ready for burial. J. Thromb. Haemost. 2006, 4, 2068–2069. [Google Scholar] [CrossRef]
- Greer, J.P.; Rodgers, G.M.; Glader, B.; Arber, D.A.; Appelbaum, F.R.; Dispenzieri, A.; Fehniger, T.A.; Michaelis, L.; Leonard, J.P. Wintrobe’s Clinical Hematology, 14th ed.; Wolters Kluwer: Philadelphia, PA, USA, 2019; pp. 1212–1214. [Google Scholar]
- Giannini, E.G.; Peck-Radosavljevic, M. Platelet Dysfunction: Status of Thrombopoietin in Thrombocytopenia Associated with Chronic Liver Failure. Semin. Thromb. Hemost. 2015, 41, 455–461. [Google Scholar] [PubMed]
- Martin, T.G.; Somberg, K.A.; Meng, Y.G.; Cohen, R.L.; Heid, C.A.; de Sauvage, F.J.; Shuman, M.A. Thrombopoietin levels in patients with cirrhosis before and after orthotopic liver transplantation. Ann. Intern. Med. 1997, 127, 285–288. [Google Scholar] [CrossRef]
- Eissa, L.A.; Gad, L.S.; Rabie, A.M.; El-Gayar, A.M. Thrombopoietin level in patients with chronic liver disease. Ann. Hepatol. 2008, 7, 235–244. [Google Scholar] [CrossRef]
- European Association for the Study of the Liver. EASL Clinical Practice Guidelines for the management of patients with decompensated cirrhosis. J. Hepatol. 2018, 69, 406–460, Erratum in J. Hepatol. 2018, 69, 1207. [Google Scholar] [CrossRef]
- Kim, K.; Ong, F.; Varadi, G.; Gupta, S.; Jorge, V.M. A Systematic Review and Meta-Analysis of Safety and Efficacy for Pre-Procedural Use of Thrombopoietin Receptor Agonists in Hepatic Cirrhosis Patients. Blood 2019, 134, 1094. [Google Scholar] [CrossRef]
- Mitchell, O.; Feldman, D.M.; Diakow, M.; Sigal, S.H. The pathophysiology of thrombocytopenia in chronic liver disease. Hepatic Med. Évid. Res. 2016, 8, 39–50. [Google Scholar] [CrossRef]
- Christodoulou, D.; Katsanos, K.; Zervou, E.; Theopistos, V.; Papathanasopoulos, A.; Christou, L.; Tsianos, E.V. Platelet IgG antibodies are significantly increased in chronic liver disease. Ann. Gastroenterol. 2011, 24, 47–52. [Google Scholar]
- Ikura, Y.; Ohsawa, M.; Okada, M.; Iwai, Y.; Wakasa, K. The significance of platelet consumption in the development of thrombocytopenia in patients with cirrhosis. Am. J. Med. Sci. 2013, 346, 199–203. [Google Scholar] [CrossRef] [PubMed]
- Voican, I.; Onisâi, M.; Nicolescu, A.; Vlădăreanu, A.M.; Vlădăreanu, R. Heparin induced thrombocytopenia: A review. Farmacia 2012, 60, 773–784. [Google Scholar]
- Koganov, E.S.; Carmichael, S.L.; Forde, E.E.; Freliger, A.L.; Michelson, A.D. Platelet Function in Thrombocytopenic Patients with Chronic Liver Disease. Blood 2017, 130, 2314. [Google Scholar] [CrossRef]
- Tischendorf, M.; Miesbach, W.; Chattah, U.; Chattah, Z.; Maier, S.; Welsch, C.; Zeuzem, S.; Lange, C.M. Differential Kinetics of Coagulation Factors and Natural Anticoagulants in Patients with Liver Cirrhosis: Potential Clinical Implications. PLoS ONE 2016, 11, e0155337. [Google Scholar] [CrossRef] [PubMed]
- Lisman, T.; Leebeek, F.W.G.; de Groot, P.G. Haemostatic abnormalities in patients with liver disease. J. Hepatol. 2002, 37, 280–287. [Google Scholar] [CrossRef] [PubMed]
- Girolami, A.; Ferrari, S.; Cosi, E.; Santarossa, C.; Randi, M.L. Vitamin K-dependent coagulation factors that may be responsible for both bleeding and thrombosis (FII, FVII, and FIX). Clin. Appl. Thromb. 2018, 24, 42S–47S. [Google Scholar] [CrossRef]
- Singhal, A.; Karachristos, A.; Bromberg, M.; Daly, E.; Maloo, M.; Jain, A.K. Hypercoagulability in end-stage liver disease: Prevalence and its correlation with severity of liver disease and portal vein thrombosis. Clin. Appl. Thromb. 2012, 18, 594–598. [Google Scholar] [CrossRef]
- Fisher, C.; Patel, V.C.; Stoy, S.H.; Singanayagam, A.; Adelmeijer, J.; Wendon, J.; Shawcross, D.; Lisman, T.; Bernal, W. Balanced haemostasis with both hypo- and hyper-coagulable features in critically ill patients with acute-on-chronic-liver failure. J. Crit. Care 2018, 43, 54–60. [Google Scholar] [CrossRef]
- Hartman, M.; Szalai, C.; Saner, F.H. Hemostasis in liver transplantation: Pathophysiology, monitoring, and treatment. World J. Gastroenterol. 2016, 22, 1541–1550. [Google Scholar] [CrossRef] [PubMed]
- Ferro, D.; Celestini, A.; Violi, F. Hyperfibrinogenolysis in liver disease. Clin. Liver Dis. 2009, 13, 21–31. [Google Scholar] [CrossRef]
- Drevon, L.; Maslah, N.; Soret-Dulphy, J.; Dosquet, C.; Ravdan, O.; Vercellino, L.; Belkhodja, C.; Parquet, N.; Brignier, A.C.; Schlageter, M.H.; et al. Anemia andhemodilution: Analysis of a single center cohort based on 2858 red cell mass measurements. Haematologica 2021, 106, 1167–1171. [Google Scholar] [CrossRef] [PubMed]
- Singer, C.E.; Vasile, C.M.; Popescu, M.; Popescu, A.I.S.; Marginean, I.C.; Iacob, G.A.; Popescu, M.D.; Marginean, C.M. Role of Iron Deficiency in Heart Failure—Clinical and Treatment Approach: An Overview. Diagnostics 2023, 13, 304. [Google Scholar] [CrossRef]
- Alshaibani, A.; Dufour, C.; Risitano, A.; de Latour, R.; Aljurf, M. Hepatitis-associated aplastic anemia. Hematol./Oncol. Stem Cell Ther. 2020, in press. [Google Scholar] [CrossRef] [PubMed]
- Rauff, B.; Idrees, M.; Shah, S.A.R.; Butt, S.; Butt, A.M.; Ali, L.; Hussain, A.; Ali, M. Hepatitis Associated Aplastic Anemia: A review. Virol. J. 2011, 8, 87. [Google Scholar] [CrossRef]
- Ikawa, Y.; Nishimura, R.; Kuroda, R.; Mase, S.; Araki, R.; Maeba, H.; Wada, T.; Toma, T.; Koizumi, S.; Yachie, A. Expansion of a liver-infiltratingcytotoxic T-lymphocyte clone in concert with the development of hepatitis-associated aplastic anaemia. Br. J. Haematol. 2013, 161, 599–602. [Google Scholar] [CrossRef] [PubMed]
- Young, N.S.; Calado, R.T.; Scheinberg, P. Current concepts in the pathophysiology and treatment of aplastic anemia. Blood 2006, 108, 2509–2519. [Google Scholar] [CrossRef]
- Cariani, E.; Pelizzari, A.M.; Rodella, A.; Gargiulo, F.; Imberti, L.; Manca, N.; Rossi, G. Immune-mediated hepatitis-associated aplastic anemia caused by the emergence of a mutant hepatitis B virus undetectable by standard assays. J. Hepatol. 2007, 46, 743–747. [Google Scholar] [CrossRef]
- De Franceschi, L.; Fattovich, G.; Turrini, F.; Ayi, K.; Brugnara, C.; Manzato, F.; Noventa, F.; Stanzial, A.M.; Solero, P.; Corrocher, R. Hemolytic anemia induced by ribavirin therapy in patients with chronic hepatitis C virus infection: Role of membrane oxidativedamage. Hepatology 2000, 31, 997–1004. [Google Scholar] [CrossRef]
- Van Vlierbergh, H.; Delanghe, J.R.; De Vos, M.; Leroux-Roel, G. Factors influencing ribavirin-induced hemolysis. J. Hepatol. 2001, 34, 911–916. [Google Scholar] [CrossRef] [PubMed]
- Lakshmipathy, A.; Krishnamani, R.; Felix, J. Unusual lipid and metabolic abnormalities secondary to alcohol abuse. Hosp. Physician 2004, 40, 43–46. [Google Scholar]
- Ampuero, J.; Romero-Gómez, M. Pharmacogenetics of ribavirin-induced anemia in hepatitis C. Pharmacogenomics 2016, 17, 1587–1594. [Google Scholar] [CrossRef] [PubMed]
- Senatore, F.J.; McDonald, K. Pitfalls of treating alcoholic hepatitis: Recognizing hemolytic anemia in Zieve’s syndrome. Am. J. Gastroenterol. 2016, 111, 577–579. [Google Scholar] [CrossRef]
- Grudeva-Popova, J.G.; Spasova, M.I.; Chepileva, K.G.; Zaprianov, Z.H. Acute hemolytic anemia as an initial clinical manifesta-tion of Wilson’s disease. Folia Med. 2000, 42, 42–46. [Google Scholar]
- Brake, S. Hemolytic Anemia Associated with Fulminant Hepatitis in Wilson’s Disease: Diagnosis and Management. Blood 2009, 114, 5088. [Google Scholar] [CrossRef]
- Sood, R.; Bakashi, R.; Hegade, V.S.; Kelly, S.M. Diagnosis and management of hereditary haemochromatosis. Br. J. Gen. Pr. 2013, 63, 331–332. [Google Scholar] [CrossRef]
- Khan, A.A.; Hadi, Y.; Hassan, A.; Kupec, J. Polycythemia and anemia in hereditary hemochromatosis. Cureus 2020, 12, e7607. [Google Scholar] [CrossRef]
- Carey, E.J.; Ali, A.H.; Lindor, K.D. Primary biliary cirrhosis. Lancet 2015, 386, 1565–1575, Erratum in Lancet 2015, 386, 1536. [Google Scholar] [CrossRef]
- Hirschfield, G.M.; Gershwin, M.E. The immunobiology and pathophysiology of primary biliary cirrhosis. Annu. Rev. Pathol. Mech. Dis. 2013, 8, 303–330. [Google Scholar] [CrossRef]
- Chung, C.S.; Hsu, Y.C.; Huang, S.Y.; Jeng, Y.M.; Chen, C.H. Primary biliary cirrhosis associated with pernicious anemia. Can. Fam. Physician 2010, 56, 889–891. [Google Scholar] [PubMed]
- Gonzalez-Moreno, E.I.; Martinez-Cabriales, S.A.; Cruz-Moreno, M.A.; Borjas-Almaguer, O.D.; Cortez-Hernandez, C.A.; Bosques-Padilla, F.J.; Garza, A.A.; Gonzalez-Gonzalez, J.A.; Garcia-Compean, D.; Ocampo-Candiani, J.; et al. Primary biliary cholangitis associated with warm autoimmune hemolytic anemia. J. Dig. Dis. 2016, 17, 128–131. [Google Scholar] [CrossRef]
- Ekeowa, U.I.; Marciniak, S.J.; Lomas, D.A. A(1)-Antitrypsin Deficiency and Inflammation. Expert Rev. Clin. Immunol. 2011, 7, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Carroll, T.P.; O’Connor, C.A.; Floyd, O.; McPartlin, J.; Kelleher, D.P.; O’Brien, G.; Dimitrov, B.D.; Morris, V.B.; Taggart, C.C.; McElvaney, N.G. The prevalence of alpha-1 antitrypsin deficiency in Ireland. Respir. Res. 2011, 12, 91. [Google Scholar] [CrossRef] [PubMed]
- Blanco, I.; Lara, B.; de Serres, F. Efficacy of alpha1-antitrypsin augmentation therapy in conditions other than pulmonary emphysema. Orphanet J. Rare Dis. 2011, 6, 14. [Google Scholar] [CrossRef]
- Bals, R. Alpha-1-antitrypsin deficiency. Best Pract. Res. Clin. Gastroenterol. 2010, 24, 629–633. [Google Scholar] [CrossRef] [PubMed]
- Graziadei, I.; Gaggl, S.; Kaserbacher, R.; Braunsteiner, H.; Vogel, W. The acute-phase protein alpha 1-antitrypsin inhibits growth and proliferation of human early erythroid progenitor cells (burst-forming units-erythroid) and of human erythroleukemic cells (K562) in vitro by interfering with transferrin iron uptake. Blood 1994, 83, 260–268. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Anomalies | Mechanisms | |
|---|---|---|
| Primary hemostasis | Thrombocytopenia | immunological destruction; increased consumption of platelets; bacterial translocation; infection |
| Secondary hemostasis | Abnormalities in both pro- and anticoagulation factors | inefficient or inadequate synthesis; increased consumption (fibrinogen); malabsorption (vitamin K) |
| Fibrinolytic system | Impaired fibrinolysis | increased levels of tissue plasminogen activator; decreased levels of alpha-2 antiplasmin, XIII factors, and thrombin-activatable fibrinolysis inhibitor; increased fibrin degradation products |
| Liver Disease | Type of Anemia |
|---|---|
| Zieve syndrome | Coombs-negative hemolytic anemia |
| Wilson disease | Coombs-negative hemolytic anemia |
| Hereditary hemochromatosis | aplastic/megaloblastic anemia |
| Primary biliary cholangitis | megaloblastic/hemolytic/warm autoimmune hemolytic anemia |
| Alpha 1 antitrypsin deficiency | anemia of chronic disease |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marginean, C.M.; Pirscoveanu, D.; Popescu, M.; Docea, A.O.; Radu, A.; Popescu, A.I.S.; Vasile, C.M.; Mitrut, R.; Marginean, I.C.; Iacob, G.A.; et al. Diagnostic Approach and Pathophysiological Mechanisms of Anemia in Chronic Liver Disease—An Overview. Gastroenterol. Insights 2023, 14, 327-341. https://doi.org/10.3390/gastroent14030024
Marginean CM, Pirscoveanu D, Popescu M, Docea AO, Radu A, Popescu AIS, Vasile CM, Mitrut R, Marginean IC, Iacob GA, et al. Diagnostic Approach and Pathophysiological Mechanisms of Anemia in Chronic Liver Disease—An Overview. Gastroenterology Insights. 2023; 14(3):327-341. https://doi.org/10.3390/gastroent14030024
Chicago/Turabian StyleMarginean, Cristina Maria, Denisa Pirscoveanu, Mihaela Popescu, Anca Oana Docea, Antonia Radu, Alin Iulian Silviu Popescu, Corina Maria Vasile, Radu Mitrut, Iulia Cristina Marginean, George Alexandru Iacob, and et al. 2023. "Diagnostic Approach and Pathophysiological Mechanisms of Anemia in Chronic Liver Disease—An Overview" Gastroenterology Insights 14, no. 3: 327-341. https://doi.org/10.3390/gastroent14030024
APA StyleMarginean, C. M., Pirscoveanu, D., Popescu, M., Docea, A. O., Radu, A., Popescu, A. I. S., Vasile, C. M., Mitrut, R., Marginean, I. C., Iacob, G. A., Firu, D. M., & Mitrut, P. (2023). Diagnostic Approach and Pathophysiological Mechanisms of Anemia in Chronic Liver Disease—An Overview. Gastroenterology Insights, 14(3), 327-341. https://doi.org/10.3390/gastroent14030024