
Genetics of Heritable Thoracic Aortic Disease



Abstract
:1. Introduction
1.1. Classification
1.2. Diagnostic Workup
2. Syndromic HTAD
2.1. Marfan Syndrome
2.2. Loeys–Dietz Syndrome
2.2.1. LDS Caused by Pathogenic Variants in TGFBR1 (OMIM #609192) and in TGFBR2 (OMIM #610168)
2.2.2. LDS Caused by Pathogenic Variants in SMAD3 (OMIM #613795)
2.2.3. LDS Caused by Pathogenic Variants in TGFB2 (OMIM #614816) and SMAD2 or TGFB3 (OMIM #615582)
2.3. Rare Syndromic HTAD
2.3.1. Vascular Ehlers–Danlos Syndrome (vEDS; OMIM #130050)
2.3.2. Meester–Loeys Syndrome (MRLS; OMIM #300989)
2.3.3. Filamin A-Related HTAD
2.3.4. LOX-Related HTAD
2.3.5. Gorlin–Chaudhry–Moss Syndrome (GCMS; OMIM #612289, ORPHA:2095)
2.3.6. Shprintzen–Goldberg Craniosynostosis Syndrome (SGS; OMIM #182212, ORPHA:2462)
2.3.7. Arterial Tortuosity Syndrome (ATORS; OMIM #208050, ORPHA:3342)
2.3.8. LTBP3-Related HTAD
2.3.9. ACTA2-Related sHTAD
2.3.10. Importin-Related HTAD-VISS Syndrome (OMIM #619472)
3. Thoracic Aortic Disease in Metabolic Storage Disease
4. Nonsyndromic HTAD
4.1. Bicuspid Aortic Valve Related HTAD
4.2. Familial and Sporadic Nonsyndromic HTAD
5. Genetic Testing and Surgical Intervention
6. Conclusions
Author Contributions
Funding
Informed Consent Statement
Conflicts of Interest
References
- Loeys, B.L.; Schwarze, U.; Holm, T.; Callewaert, B.L.; Thomas, G.H.; Pannu, H.; De Backer, J.F.; Oswald, G.L.; Symoens, S.; Manouvrier, S.; et al. Aneurysm Syndromes Caused by Mutations in the TGF-β Receptor. N. Engl. J. Med. 2006, 355, 788–798. [Google Scholar] [CrossRef] [PubMed]
- Verstraeten, A.; Luyckx, I.; Loeys, B. Aetiology and management of hereditary aortopathy. Nat. Rev. Cardiol. 2017, 14, 197–208. [Google Scholar] [CrossRef]
- Hostetler, E.M.; Regalado, E.S.; Guo, D.-C.; Hanna, N.; Arnaud, P.; Muiño-Mosquera, L.; Callewaert, B.L.; Lee, K.; Leal, S.M.; Wallace, S.E.; et al. SMAD3 pathogenic variants: Risk for thoracic aortic disease and associated complications from the Montalcino Aortic Consortium. J. Med. Genet. 2019, 56, 252–260. [Google Scholar] [CrossRef]
- Jondeau, G.; Ropers, J.; Regalado, E.; Braverman, A.; Evangelista, A.; Teixedo, G.; De Backer, J.; Muiño-Mosquera, L.; Naudion, S.; Zordan, C.; et al. International Registry of Patients Carrying TGFBR1 or TGFBR2 Mutations. Circ. Cardiovasc. Genet. 2016, 9, 548–558. [Google Scholar] [CrossRef] [Green Version]
- Franken, R.; Teixido-Tura, G.; Brion, M.; Forteza, A.; Palomares, J.F.R.; Gutierrez, L.; Dorado, D.G.; Pals, G.; Mulder, B.J.; Evangelista, A. Relationship between fibrillin-1 genotype and severity of cardiovascular involvement in Marfan syndrome. Heart 2017, 103, 1795–1799. [Google Scholar] [CrossRef]
- Arnaud, P.; Hanna, N.; Benarroch, L.; Aubart, M.; Bal, L.; Bouvagnet, P.; Busa, T.; Dulac, Y.; Dupuis-Girod, S.; Edouard, T.; et al. Genetic diversity and pathogenic variants as possible predictors of severity in a French sample of nonsyndromic heritable thoracic aortic aneurysms and dissections (nshTAAD). Genet. Med. 2019, 21, 2015–2024. [Google Scholar] [CrossRef]
- Elefteriades, J.A.; Farkas, E.A. Thoracic Aortic Aneurysm. Clinically Pertinent Controversies and Uncertainties. J. Am. Coll. Cardiol. 2010, 55, 841–857. [Google Scholar] [CrossRef] [PubMed]
- Loeys, B.L.; Dietz, H.C.; Braverman, A.C.; Callewaert, B.L.; De Backer, J.; Devereux, R.B.; Hilhorst-Hofstee, Y.; Jondeau, G.; Faivre, L.; Milewicz, D.M.; et al. The revised Ghent nosology for the Marfan syndrome. J. Med. Genet. 2010, 47, 476–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devereux, R.B.; De Simone, G.; Arnett, D.K.; Best, L.G.; Boerwinkle, E.; Howard, B.V.; Kitzman, D.; Lee, E.T.; Mosley, T.H. Weder, A. et al. Normal limits in relation to age, body size and gender of two-dimensional echocardiographic aortic root dimensions in persons ≥ 15 years of age. Am. J. Cardiol. 2012, 110, 1189–1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groth, K.A.; Hove, H.; Kyhl, K.; Folkestad, L.; Gaustadnes, M.; Vejlstrup, N.; Stochholm, K.; Østergaard, J.R.; Andersen, N.H.; Gravholt, C.H. Prevalence, incidence, and age at diagnosis in Marfan Syndrome. Orphanet J. Rare Dis. 2015, 10, 153. [Google Scholar] [CrossRef] [Green Version]
- Treasure, T.; Takkenberg, J.J.M.; Pepper, J. Surgical management of aortic root disease in Marfan syndrome and other congenital disorders associated with aortic root aneurysms. Heart 2014, 100, 1571–1576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Beaufort, H.W.L.; Trimarchi, S.; Korach, A.; Di Eusanio, M.; Gilon, D.; Montgomery, D.G.; Evangelista, A.; Braverman, A.C.; Chen, E.P.; Isselbacher, E.M.; et al. Aortic dissection in patients with Marfan syndrome based on the IRAD data. Ann. Cardiothorac. Surg. 2017, 6, 633–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gott, V.L.; Greene, P.S.; Alejo, D.E.; Cameron, D.E.; Naftel, D.C.; Miller, D.C.; Gillinov, A.M.; Laschinger, J.C.; Pyeritz, R.E.; Borst, H.G.; et al. Replacement of the aortic root in patients with Marfan’s syndrome. N. Engl. J. Med. 1999, 340, 1307–1313. [Google Scholar] [CrossRef]
- Erbel, R.; Aboyans, V.; Boileau, C.; Bossone, E.; Di Bartolomeo, R.; Eggebrecht, H.; Evangelista, A.; Falk, V.; Frank, H.; Gaemperli, O.; et al. 2014 ESC guidelines on the diagnosis and treatment of aortic diseases. Eur. Heart J. 2014, 35, 2873–2926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumgartner, H.; De Backer, J.; Babu-Narayan, S.V.; Budts, W.; Chessa, M.; Diller, G.-P.; Lung, B.; Kluin, J.; Lang, I.M.; Meijboom, F.; et al. 2020 ESC Guidelines for the management of adult congenital heart disease. Eur. Heart J. 2020, 42, 563–645. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Isselbacher, E.M.; Nienaber, C.A.; Pyeritz, R.E.; Eagle, K.A.; Tsai, T.T.; Cooper, J.V.; Januzzi, J.L.; Braverman, A.C.; Montgomery, D.G.; et al. Type-selective benefits of medications in treatment of acute aortic dissection (from the International Registry of Acute Aortic Dissection [IRAD]). Am. J. Cardiol. 2012, 109, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Update on Clinical Trials of Losartan with and Without β-Blockers to Block Aneurysm Growth in Patients With Marfan Syndrome: A Review. JAMA Cardiol. 2019, 4, 702–707. [CrossRef]
- Franken, R.; den Hartog, A.W.; Radonic, T.; Micha, D.; Maugeri, A.; van Dijk, F.S.; Meijers-Heijboer, H.E.; Timmermans, J.; Scholte, A.J.; van den Berg, M.P.; et al. Beneficial Outcome of Losartan Therapy Depends on Type of FBN1 Mutation in Marfan Syndrome. Circ. Cardiovasc. Genet. 2015, 8, 383–388. [Google Scholar] [CrossRef] [Green Version]
- Takeda, N.; Inuzuka, R.; Maemura, S.; Morita, H.; Nawata, K.; Fujita, D.; Taniguchi, Y.; Yamauchi, H.; Yagi, H.; Kato, M.; et al. Impact of Pathogenic FBN1 Variant Types on the Progression of Aortic Disease in Patients With Marfan Syndrome. Circ. Genom. Precis. Med. 2018, 11, e002058. [Google Scholar] [CrossRef] [Green Version]
- De Backer, J.; Campens, L.; Muiño Mosquera, L. Looking for the Missing Links: Challenges in the Search for Genotype-Phenotype Correlation in Marfan Syndrome. Circ. Genom. Precis. Med. 2018, 11, e002185. [Google Scholar] [CrossRef]
- Baudhuin, L.M.; Kotzer, K.E.; Lagerstedt, S.A. Increased frequency of FBN1 truncating and splicing variants in Marfan syndrome patients with aortic events. Genet. Med. 2015, 17, 177–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnaud, P.; Milleron, O.; Hanna, N.; Ropers, J.; Ould Ouali, N.; Affoune, A.; Langeois, M.; Eliahou, L.; Arnoult, F.; Renard, P.; et al. Clinical relevance of genotype–phenotype correlations beyond vascular events in a cohort study of 1500 Marfan syndrome patients with FBN1 pathogenic variants. Genet. Med. 2021, 23, 1296. [Google Scholar] [CrossRef]
- Milleron, O.; Arnoult, F.; Delorme, G.; Detaint, D.; Pellenc, Q.; Raffoul, R.; Tchitchinadze, M.; Langeois, M.; Guien, C.; Beroud, C.; et al. Pathogenic FBN1 Genetic Variation and Aortic Dissection in Patients With Marfan Syndrome. J. Am. Coll. Cardiol. 2020, 75, 843–853. [Google Scholar] [CrossRef] [PubMed]
- Hiratzka, L.F.; Bakris, G.L.; Beckman, J.A.; Bersin, R.M.; Carr, V.F.; Casey, D.E.; Eagle, K.A.; Hermann, L.K.; Isselbacher, E.M.; Kazerooni, E.A.; et al. 2010 ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM guidelines for the diagnosis and management of patients with thoracic aortic disease: Executive summary: A report of the American college of cardiology foundation/american heart association task force on practice guidelines, American association for thoracic surgery, American college of radiology, American stroke association. Circulation 2010, 121, 1544–1579. [Google Scholar]
- Aubart, M.; Gobert, D.; Aubart-Cohen, F.; Detaint, D.; Hanna, N.; d’Indya, H.; Lequintrec, J.-S.; Renard, P.; Vigneron, A.-M.; Dieudé, P.; et al. Early-Onset Osteoarthritis, Charcot-Marie-Tooth Like Neuropathy, Autoimmune Features, Multiple Arterial Aneurysms and Dissections: An Unrecognized and Life Threatening Condition. PLoS ONE 2014, 9, e96387. [Google Scholar] [CrossRef] [Green Version]
- Van de Laar, I.M.B.H.; Oldenburg, R.A.; Pals, G.; Roos-Hesselink, J.W.; de Graaf, B.M.; Verhagen, J.M.A.; Hoedemaekers, Y.M.; Willemsen, R.; Severijnen, L.-A.; Venselaar, H.; et al. Mutations in SMAD3 cause a syndromic form of aortic aneurysms and dissections with early-onset osteoarthritis. Nat. Genet. 2011, 43, 121–126. [Google Scholar] [CrossRef]
- Schepers, D.; Tortora, G.; Morisaki, H.; MacCarrick, G.; Lindsay, M.; Liang, D.; Mehta, S.G.; Hague, J.; Verhagen, J.; van de Laar, I.; et al. A mutation update on the LDS-associated genes TGFB2/3 and SMAD2/3. Hum. Mutat. 2018, 39, 621–634. [Google Scholar] [CrossRef] [Green Version]
- Wischmeijer, A.; Van Laer, L.; Tortora, G.; Bolar, N.A.; Van Camp, G.; Fransen, E.; Peeters, N.; di Bartolomeo, R.; Pacini, D.; Gargiulo, G.; et al. Thoracic Aortic Aneurysm in Infancy in Aneurysms-Osteoarthritis Syndrome Due to a Novel SMAD 3 Mutation: Further Delineation of the Phenotype. Am. J. Med. Genet. Part A 2013, 161, 1028–1035. [Google Scholar] [CrossRef]
- Boileau, C.; Guo, D.-C.; Hanna, N.; Regalado, E.S.; Detaint, D.; Gong, L.; Varret, M.; Prakash, S.K.; Li, A.H.; d’Indy, H.; et al. TGFB2 mutations cause familial thoracic aortic aneurysms and dissections associated with mild systemic features of Marfan syndrome. Nat. Genet. 2012, 44, 916–921. [Google Scholar] [CrossRef] [Green Version]
- Renard, M.; Callewaert, B.; Malfait, F.; Campens, L.; Sharif, S.; del Campo, M.; Valenzuela, I.; Mcwilliam, C.; Coucke, P.; De Paepe, A.; et al. Thoracic aortic-aneurysm and dissection in association with significant mitral valve disease caused by mutations in TGFB2. Int. J. Cardiol. 2013, 165, 584–587. [Google Scholar] [CrossRef]
- Malfait, F.; Francomano, C.; Byers, P.; Belmont, J.; Berglund, B.; Black, J.; Bloom, L.; Bowen, J.M.; Brady, A.F.; Burrows, N.P.; et al. The 2017 international classification of the Ehlers-Danlos syndromes; The 2017 international classification of the Ehlers-Danlos syndromes. Am. J. Med. Genet. Part C Semin. Med. Genet. 2017, 175, 8–26. [Google Scholar] [CrossRef] [Green Version]
- Beighton, P.; De Paepe, A.; Steinmann, B.; Tsipouras, P.; Wenstrup, R.J. Ehlers-Danlos syndromes: Revised nosology, Villefranche, 1997. Ehlers-Danlos National Foundation (USA) and Ehlers-Danlos Support Group (UK). Am. J. Med. Genet. 1998, 77, 31–37. [Google Scholar] [CrossRef]
- Pepin, M.; Schwarze, U.; Superti-Furga, A.; Byers, P.H. Clinical and Genetic Features of Ehlers–Danlos Syndrome Type IV, the Vascular Type. N. Engl. J. Med. 2000, 342, 673–680. [Google Scholar] [CrossRef]
- Schwarze, U.; Schievink, W.I.; Petty, E.; Jaff, M.R.; Babovic-Vuksanovic, D.; Cherry, K.J.; Pepin, M.; Byers, P.H. Haploinsufficiency for One COL3A1 Allele of Type III Procollagen Results in a Phenotype Similar to the Vascular Form of Ehlers-Danlos Syndrome, Ehlers-Danlos Syndrome Type IV. Am. J. Hum. Genet. 2001, 69, 989–1001. [Google Scholar] [CrossRef] [Green Version]
- Monroe, G.R.; Harakalova, M.; van der Crabben, S.N.; Majoor-Krakauer, D.; Bertoli-Avella, A.M.; Moll, F.L.; Oranen, B.I.; Dooijes, D.; Vink, A.; Knoers, N.V.; et al. Familial Ehlers-Danlos syndrome with lethal arterial events caused by a mutation in COL5A1. Am. J. Med. Genet. Part A 2015, 167, 1196–1203. [Google Scholar] [CrossRef] [PubMed]
- Leistritz, D.F.; Pepin, M.G.; Schwarze, U.; Byers, P.H. COL3A1 haploinsufficiency results in a variety of Ehlers-Danlos syndrome type IV with delayed onset of complications and longer life expectancy. Genet. Med. 2011, 13, 717–722. [Google Scholar] [CrossRef] [PubMed]
- Pepin, M.G.; Schwarze, U.; Rice, K.M.; Liu, M.; Leistritz, D.; Byers, P.H. Survival is affected by mutation type and molecular mechanism in vascular Ehlers–Danlos syndrome (EDS type IV). Genet. Med. 2014, 16, 881–888. [Google Scholar] [CrossRef] [Green Version]
- Frank, M.; Adham, S.; Seigle, S.; Legrand, A.; Mirault, T.; Henneton, P.; Albuisson, J.; Denarié, N.; Mazzella, J.M.; Mousseaux, E.; et al. Vascular Ehlers-Danlos Syndrome: Long-Term Observational Study. J. Am. Coll. Cardiol. 2019, 73, 1948–1957. [Google Scholar] [CrossRef]
- Frank, M.; Albuisson, J.; Ranque, B.; Golmard, L.; Mazzella, J.-M.; Bal-Theoleyre, L.; Fauret, A.-L.; Mirault, T.; Denarié, N.; Mousseaux, E.; et al. The type of variants at the COL3A1 gene associates with the phenotype and severity of vascular Ehlers–Danlos syndrome. Eur. J. Hum. Genet. 2015, 23, 1657–1664. [Google Scholar] [CrossRef] [PubMed]
- Bergqvist, D.; Björck, M.; Wanhainen, A. Treatment of Vascular Ehlers-Danlos Syndrome. Ann. Surg. 2013, 258, 257–261. [Google Scholar] [CrossRef]
- Ong, K.-T.; Perdu, J.; De Backer, J.; Bozec, E.; Collignon, P.; Emmerich, J.; Fauret, A.-L.; Fiessinger, J.-N.; Germain, D.P.; Georgesco, G.; et al. Effect of celiprolol on prevention of cardiovascular events in vascular Ehlers-Danlos syndrome: A prospective randomised, open, blinded-endpoints trial. Lancet 2010, 376, 1476–1484. [Google Scholar] [CrossRef]
- Dubacher, N.; Münger, J.; Gorosabel, M.C.; Crabb, J.; Ksiazek, A.A.; Caspar, S.M.; Bakker, E.N.T.P.; Van Bavel, E.; Ziegler, U.; Carrel, T.; et al. Celiprolol but not losartan improves the biomechanical integrity of the aorta in a mouse model of vascular Ehlers-Danlos syndrome. Cardiovasc. Res. 2020, 116, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Meester, J.A.N.; Vandeweyer, G.; Pintelon, I.; Lammens, M.; Van Hoorick, L.; De Belder, S.; Waitzman, K.; Young, L.; Markham, L.W.; Vogt, J.; et al. Loss-of-function mutations in the X-linked biglycan gene cause a severe syndromic form of thoracic aortic aneurysms and dissections. Genet. Med. 2017, 19, 386–395. [Google Scholar] [CrossRef] [Green Version]
- Sheen, V.L.; Jansen, A.; Chen, M.H.; Parrini, E.; Morgan, T.; Ravenscroft, R.; Ganesh, V.; Underwood, T.; Wiley, J.; Leventer, R.; et al. Filamin A mutations cause periventricular heterotopia with Ehlers-Danlos syndrome. Neurology 2005, 64, 254–262. [Google Scholar] [CrossRef]
- Chen, M.H.; Choudhury, S.; Hirata, M.; Khalsa, S.; Chang, B.; Walsh, C.A. Thoracic aortic aneurysm in patients with loss of function Filamin A mutations: Clinical characterization, genetics, and recommendations; Thoracic aortic aneurysm in patients with loss of function Filamin A mutations: Clinical characterization, genetics, and recommendations. Am. J. Med. Genet. A 2018, 176, 337–350. [Google Scholar] [PubMed]
- Guo, D.C.; Regalado, E.S.; Gong, L.; Duan, X.; Santos-Cortez, R.L.P.; Arnaud, P.; Ren, Z.; Cai, B.; Hostetler, E.M.; Moran, R.; et al. LOX mutations predispose to thoracic aortic aneurysms and dissections. Circ. Res. 2016, 118, 928–934. [Google Scholar] [CrossRef] [Green Version]
- Wolford, B.N.; Hornsby, W.E.; Guo, D.; Zhou, W.; Lin, M.; Farhat, L.; McNamara, J.; Driscoll, A.; Wu, X.; Schmidt, E.M.; et al. Clinical Implications of Identifying Pathogenic Variants in Individuals With Thoracic Aortic Dissection. Circ. Genom. Precis. Med. 2019, 12, e002476. [Google Scholar] [CrossRef] [Green Version]
- García, M.E.R.; Vinagre, F.J.C.; Cruz-Rojo, J.; Lorenzo, L.G.; Carnicero-Rodríguez, P.; Pozo, J.S.-D.; Martínez-Azorín, F. A rare male patient with Fontaine progeroid syndrome caused by p.R217H de novo mutation in SLC25A24. Am. J. Med. Genet. Part A 2018, 176, 2479–2486. [Google Scholar] [CrossRef] [PubMed]
- Ehmke, N.; Graul-Neumann, L.; Smorag, L.; Koenig, R.; Segebrecht, L.; Magoulas, P.; Scaglia, F.; Kilic, E.; Hennig, A.F.; Adolphs, N.; et al. De Novo Mutations in SLC25A24 Cause a Craniosynostosis Syndrome with Hypertrichosis, Progeroid Appearance, and Mitochondrial Dysfunction. Am. J. Hum. Genet. 2017, 101, 833–843. [Google Scholar] [CrossRef] [Green Version]
- Legué, J.; François, J.H.M.; van Rijswijk, C.S.P.; van Brakel, T.J. Is Gorlin–Chaudhry–Moss syndrome associated with aortopathy? Eur. J. Cardio-Thorac. Surg. 2020, 58, 654–655. [Google Scholar] [CrossRef]
- Schepers, D.; Doyle, A.J.; Oswald, G.; Sparks, E.; Myers, L.; Willems, P.J.; Mansour, S.; Simpson, M.A.; Frysira, H.; Maat-Kievit, A.; et al. The SMAD-binding domain of SKI: A hotspot for de novo mutations causing Shprintzen-Goldberg syndrome. Eur. J. Hum. Genet. 2015, 23, 224–228. [Google Scholar] [CrossRef] [PubMed]
- Doyle, A.J.; Doyle, J.J.; Bessling, S.L.; Maragh, S.; Lindsay, M.E.; Schepers, D.; Gillis, E.; Mortier, G.; Homfray, T.; Sauls, K.; et al. Mutations in the TGF-β repressor SKI cause Shprintzen-Goldberg syndrome with aortic aneurysm. Nat. Genet. 2012, 44, 1249–1254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callewaert, B.L.; Willaert, A.; Kerstjens-Frederikse, W.S.; De Backer, J.; Devriendt, K.; Albrecht, B.; Ramos-Arroyo, M.A.; Doco-Fenzy, M.; Hennekam, R.C.M.; Pyeritz, R.E.; et al. Arterial tortuosity syndrome: Clinical and molecular findings in 12 newly identified families. Hum. Mutat. 2008, 29, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Beyens, A.; Albuisson, J.; Boel, A.; Al-Essa, M.; Al-Manea, W.; Bonnet, D.; Bostan, O.; Boute, O.; Busa, T.; Canham, N.; et al. Arterial tortuosity syndrome: 40 new families and literature review. Genet. Med. 2018, 20, 1236–1245. [Google Scholar] [CrossRef] [Green Version]
- Guo, D.-C.; Regalado, E.S.; Pinard, A.; Chen, J.; Lee, K.; Rigelsky, C.; Zilberberg, L.; Hostetler, E.M.; Aldred, M.; Wallace, S.E.; et al. LTBP3 Pathogenic Variants Predispose Individuals to Thoracic Aortic Aneurysms and Dissections. Am. J. Hum. Genet. 2018, 102, 706–712. [Google Scholar] [CrossRef] [Green Version]
- Zhu, G.; Luo, M.; Chen, Q.; Zhang, Y.; Zhao, K.; Zhang, Y.; Shu, C.; Yang, H.; Zhou, Z. Novel LTBP3 mutations associated with thoracic aortic aneurysms and dissections. Orphanet J. Rare Dis. 2021, 16, 513. [Google Scholar] [CrossRef]
- Morisaki, H.; Akutsu, K.; Ogino, H.; Kondo, N.; Yamanaka, I.; Tsutsumi, Y.; Yoshimuta, T.; Okajima, T.; Matsuda, H.; Minatoya, K.; et al. Human Mutation RESEARCH ARTICLE Mutation of ACTA2 Gene as an Important Cause of Familial and Nonfamilial Nonsyndromatic Thoracic Aortic Aneurysm and/or Dissection (TAAD). Hum. Mutat. 2009, 30, 1406–1411. [Google Scholar] [CrossRef]
- Guo, D.-C.; Pannu, H.; Tran-Fadulu, V.; Papke, C.L.; Yu, R.K.; Avidan, N.; Bourgeois, S.; Estrera, A.L.; Safi, H.J.; Sparks, E.; et al. Mutations in smooth muscle α-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat. Genet. 2007, 39, 1488–1493. [Google Scholar] [CrossRef]
- Guo, D.C.; Papke, C.L.; Tran-Fadulu, V.; Regalado, E.S.; Avidan, N.; Johnson, R.J.; Kim, D.H.; Pannu, H.; Willing, M.C.; Sparks, E.; et al. Mutations in smooth muscle alpha-actin (ACTA2) cause coronary artery disease, stroke, and Moyamoya disease, along with thoracic aortic disease. Am. J. Hum. Genet. 2009, 84, 617–627. [Google Scholar] [CrossRef] [Green Version]
- Regalado, E.S.; Guo, D.; Prakash, S.; Bensend, T.A.; Flynn, K.; Estrera, A.; Safi, H.; Liang, D.; Hyland, J.; Child, A.; et al. Aortic Disease Presentation and Outcome Associated With ACTA2 Mutations. Circ. Cardiovasc. Genet. 2015, 8, 457–464. [Google Scholar] [CrossRef] [Green Version]
- Meuwissen, M.E.C.; Lequin, M.H.; Bindels-de Heus, K.; Bruggenwirth, H.T.; Knapen, M.F.C.M.; Dalinghaus, M.; de Coo, R.; van Bever, Y.; Winkelman, B.H.J.; Mancini, G.M.S. ACTA2 mutation with childhood cardiovascular, autonomic and brain anomalies and severe outcome. Am. J. Med. Genet. A 2013, 161A, 1376–1380. [Google Scholar] [CrossRef]
- Milewicz, D.M.; Østergaard, J.R.; Ala-Kokko, L.M.; Khan, N.; Grange, D.K.; Mendoza-Londono, R.; Bradley, T.J.; Olney, A.H.; Adès, L.; Maher, J.F.; et al. De novo ACTA2 mutation causes a novel syndrome of multisystemic smooth muscle dysfunction. Am. J. Med. Genet. A 2010, 152A, 2437–2443. [Google Scholar] [CrossRef] [Green Version]
- Munot, P.; Saunders, D.E.; Milewicz, D.M.; Regalado, E.S.; Ostergaard, J.R.; Braun, K.P.; Kerr, T.; Lichtenbelt, K.D.; Philip, S.; Rittey, C.; et al. A novel distinctive cerebrovascular phenotype is associated with heterozygous Arg179 ACTA2 mutations. Brain 2012, 135, 2506–2514. [Google Scholar] [CrossRef] [PubMed]
- Regalado, E.S.; Mellor-Crummey, L.; De Backer, J.; Braverman, A.C.; Ades, L.; Benedict, S.; Bradley, T.J.; Brickner, M.E.; Chatfield, K.C.; Child, A.; et al. Clinical History and Management Recommendations of the Smooth Muscle Dysfunction Syndrome Due to ACTA2 Arginine 179 Alterations. Genet. Med. 2018, 20, 1206–1215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yetman, A.T.; Starr, L.J.; Bleyl, S.B.; Meyers, L.; Delaney, J.W. Progressive Aortic Dilation Associated With ACTA2 Mutations Presenting in Infancy. Pediatrics 2015, 136, e262–e266. [Google Scholar] [CrossRef] [Green Version]
- Disabella, E.; Grasso, M.; Gambarin, F.I.; Narula, N.; Dore, R.; Favalli, V.; Serio, A.; Antoniazzi, E.; Mosconi, M.; Pasotti, M.; et al. Risk of dissection in thoracic aneurysms associated with mutations of smooth muscle alpha-actin 2 (ACTA2). Heart 2011, 97, 321–326. [Google Scholar] [CrossRef]
- Bertoli-Avella, A.M.; Kandaswamy, K.K.; Khan, S.; Ordonez-Herrera, N.; Tripolszki, K.; Beetz, C.; Rocha, M.E.; Urzi, A.; Hotakainen, R.; Leubauer, A.; et al. Combining exome/genome sequencing with data repository analysis reveals novel gene–disease associations for a wide range of genetic disorders. Genet. Med. 2021, 23, 1551–1568. [Google Scholar] [CrossRef] [PubMed]
- Van Gucht, I.; Meester, J.A.N.; Bento, J.R.; Bastiaansen, M.; Bastianen, J.; Luyckx, I.; Van Den Heuvel, L.; Neutel, C.H.G.; Guns, P.J.; Vermont, M.; et al. A human importin-β-related disorder: Syndromic thoracic aortic aneurysm caused by bi-allelic loss-of-function variants in IPO8. Am. J. Hum. Genet. 2021, 108, 1115–1125. [Google Scholar] [CrossRef]
- Ziegler, A.; Duclaux-Loras, R.; Revenu, C.; Charbit-Henrion, F.; Begue, B.; Duroure, K.; Grimaud, L.; Guihot, A.L.; Desquiret-Dumas, V.; Zarhrate, M.; et al. Bi-allelic variants in IPO8 cause a connective tissue disorder associated with cardiovascular defects, skeletal abnormalities, and immune dysregulation. Am. J. Hum. Genet. 2021, 108, 1126–1137. [Google Scholar] [CrossRef]
- Fuchs, M.M.; Attenhofer Jost, C.; Babovic-Vuksanovic, D.; Connolly, H.M.; Egbe, A. Long-Term Outcomes in Patients With Turner Syndrome: A 68-Year Follow-Up. J. Am. Heart Assoc. 2019, 8, e011501. [Google Scholar] [CrossRef] [Green Version]
- Thunström, S.; Krantz, E.; Thunström, E.; Hanson, C.; Bryman, I.; Landin-Wilhelmsen, K. Incidence of Aortic Dissection in Turner Syndrome: A 23-Year Prospective Cohort Study. Circulation 2019, 139, 2802–2804. [Google Scholar] [CrossRef] [PubMed]
- Lopez, L.; Arheart, K.L.; Colan, S.D.; Stein, N.S.; Lopez-Mitnik, G.; Lin, A.E.; Reller, M.D.; Ventura, R.; Silberbach, M. Turner syndrome is an independent risk factor for aortic dilation in the young. Pediatrics 2008, 121, e1622–e1627. [Google Scholar] [CrossRef] [PubMed]
- Carlson, M.; Silberbach, M. Dissection of the aorta in Turner syndrome: Two cases and review of 85 cases in the literature. Case Rep. 2009, 2009, bcr0620091998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Gharbawy, A.H.; Bhat, G.; Murillo, J.E.; Thurberg, B.L.; Kampmann, C.; Mengel, K.E.; Kishnani, P.S. Expanding the clinical spectrum of late-onset Pompe disease: Dilated arteriopathy involving the thoracic aorta, a novel vascular phenotype uncovered. Mol. Genet. Metab. 2011, 103, 362–366. [Google Scholar] [CrossRef]
- Barbey, F.; Qanadli, S.D.; Juli, C.; Brakch, N.; Palaek, T.; Rizzo, E.; Jeanrenaud, X.; Eckhardt, B.; Linhart, A. Aortic remodelling in Fabry disease. Eur. Heart J. 2010, 31, 347–353. [Google Scholar] [CrossRef] [Green Version]
- Anastasakis, A.; Papatheodorou, E.; Steriotis, A.K. Fabry disease and cardiovascular involvement. Curr. Pharm. Des. 2013, 19, 5997–6008. [Google Scholar] [CrossRef] [PubMed]
- Anastasakis, A.; Sevdalis, E.; Papatheodorou, E.; Stefanadis, C. Anderson-Fabry disease: A cardiomyopathy that can be cured. Hell. J. Cardiol. 2011, 52, 316–326. [Google Scholar]
- De Oliviera Poswar, F.; de Souza, C.F.M.; Giugliani, R.; Baldo, G. Aortic root dilatation in patients with mucopolysaccharidoses and the impact of enzyme replacement therapy. Heart Vessel. 2019, 34, 290–295. [Google Scholar] [CrossRef]
- Bolourchi, M.; Renella, P.; Wang, R. Aortic Root Dilatation in Mucopolysaccharidosis I–VII. Int. J. Mol. Sci. 2016, 17, 2004. [Google Scholar] [CrossRef] [Green Version]
- Belfiore, M.P.; Iacobellis, F.; Acampora, E.; Caiazza, M.; Rubino, M.; Monda, E.; Magaldi, M.R.; Tarallo, A.; Sasso, M.; De Pasquale, V.; et al. Aortopathies in mouse models of Pompe, Fabry and Mucopolysaccharidosis IIIB lysosomal storage diseases. PLoS ONE 2020, 15, e0233050. [Google Scholar] [CrossRef]
- Coffey, S.; Cairns, B.J.; Iung, B. The modern epidemiology of heart valve disease. Heart 2016, 102, 75–85. [Google Scholar] [CrossRef]
- Freeze, S.L.; Landis, B.J.; Ware, S.M.; Helm, B.M. Bicuspid Aortic Valve: A Review with Recommendations for Genetic Counseling. J. Genet. Couns. 2016, 25, 1171–1178. [Google Scholar] [CrossRef] [Green Version]
- Galian-Gay, L.; Carro Hevia, A.; Teixido-Turà, G.; Rodríguez Palomares, J.; Gutiérrez-Moreno, L.; Maldonado, G.; Gonzàlez-Alujas, M.T.; Sao-Aviles, A.; Gallego, P.; Calvo-Iglesias, F.; et al. Familial clustering of bicuspid aortic valve and its relationship with aortic dilation in first-degree relatives. Heart 2019, 105, 603–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huntington, K.; Hunter, A.G.; Chan, K.L. A prospective study to assess the frequency of familial clustering of congenital bicuspid aortic valve. J. Am. Coll. Cardiol. 1997, 30, 1809–1812. [Google Scholar] [CrossRef]
- Tadros, T.M.; Klein, M.D.; Shapira, O.M. Ascending Aortic Dilatation Associated With Bicuspid Aortic Valve. Circulation 2009, 119, 880–890. [Google Scholar] [CrossRef] [PubMed]
- Davies, R.R.; Kaple, R.K.; Mandapati, D.; Gallo, A.; Botta, D.M.; Elefteriades, J.A.; Coady, M.A. Natural History of Ascending Aortic Aneurysms in the Setting of an Unreplaced Bicuspid Aortic Valve. Ann. Thorac. Surg. 2007, 83, 1338–1344. [Google Scholar] [CrossRef] [PubMed]
- Prakash, S.K.; Bossé, Y.; Muehlschlegel, J.D.; Michelena, H.I.; Limongelli, G.; Della Corte, A.; Pluchinotta, F.R.; Russo, M.G.; Evangelista, A.; Benson, D.W.; et al. A roadmap to investigate the genetic basis of bicuspid aortic valve and its complications: Insights from the International BAVCon (Bicuspid Aortic Valve Consortium). J. Am. Coll. Cardiol. 2014, 64, 832–839. [Google Scholar] [CrossRef] [Green Version]
- Pileggi, S.; De Chiara, B.; Magnoli, M.; Franzosi, M.G.; Merlanti, B.; Bianchini, F.; Moreo, A.; Romeo, G.; Russo, C.F.; Rizzo, S.; et al. Sequencing of NOTCH1 gene in an Italian population with bicuspid aortic valve: Preliminary results from the GISSI OUTLIERS VAR study. Gene 2019, 715, 143970. [Google Scholar] [CrossRef]
- Kent, K.C.; Crenshaw, M.L.; Goh, D.L.; Dietz, H.C. Genotype–phenotype correlation in patients with bicuspid aortic valve and aneurysm. J. Thorac. Cardiovasc. Surg. 2013, 146, 158–165.e1. [Google Scholar] [CrossRef] [Green Version]
- Luyckx, I.; Maccarrick, G.; Kempers, M.; Meester, J.; Geryl, C.; Rombouts, O.; Peeters, N.; Claes, C.; Boeckx, N.; Sakalihasan, N.; et al. European Journal of Human Genetics Confirmation of the role of pathogenic SMAD6 variants in bicuspid aortic valve-related aortopathy. Eur. J. Hum. Genet. 2019, 5, 3552. [Google Scholar]
- Gillis, E.; Kumar, A.A.; Luyckx, I.; Preuss, C.; Cannaerts, E.; van de Beek, G.; Wieschendorf, B.; Alaerts, M.; Bolar, N.; Vandeweyer, G.; et al. Candidate Gene Resequencing in a Large Bicuspid Aortic Valve-Associated Thoracic Aortic Aneurysm Cohort: SMAD6 as an Important Contributor. Front. Physiol. 2017, 8, 400. [Google Scholar] [CrossRef] [PubMed]
- Gould, R.A.; Aziz, H.; Woods, C.E.; Seman-Senderos, M.A.; Sparks, E.; Preuss, C.; Wünnemann, F.; Bedja, D.; Moats, C.R.; McClymont, S.A.; et al. ROBO4 variants predispose individuals to bicuspid aortic valve and thoracic aortic aneurysm. Nat. Genet. 2019, 51, 42–50. [Google Scholar] [CrossRef]
- Luyckx, I.; Kumar, A.A.; Reyniers, E.; Dekeyser, E.; Vanderstraeten, K.; Vandeweyer, G.; Wünnemann, F.; Preuss, C.; Mazzella, J.-M.; Goudot, G.; et al. Copy number variation analysis in bicuspid aortic valve-related aortopathy identifies TBX20 as a contributing gene. Eur. J. Hum. Genet. 2019, 27, 1033–1043. [Google Scholar] [CrossRef] [PubMed]
- Coady, M.A.; Davies, R.R.; Roberts, M.; Goldstein, L.J.; Rogalski, M.J.; Rizzo, J.A.; Hammond, G.L.; Kopf, G.S.; Elefteriades, J.A. Familial Patterns of Thoracic Aortic Aneurysms. Arch. Surg. 1999, 134, 361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LeMaire, S.A.; McDonald, M.-L.N.; Guo, D.; Russell, L.; Miller, C.C.; Johnson, R.J.; Bekheirnia, M.R.; Franco, L.M.; Nguyen, M.; Pyeritz, R.E.; et al. Genome-wide association study identifies a susceptibility locus for thoracic aortic aneurysms and aortic dissections spanning FBN1 at 15q21.1. Nat. Genet. 2011, 43, 996–1000. [Google Scholar] [CrossRef]
- Weerakkody, R.; Ross, D.; Parry, D.A.; Ziganshin, B.; Vandrovcova, J.; Gampawar, P.; Abdullah, A.; Biggs, J.; Dumfarth, J.; Ibrahim, Y.; et al. Targeted genetic analysis in a large cohort of familial and sporadic cases of aneurysm or dissection of the thoracic aorta. Genet. Med. 2018, 20, 1414–1422. [Google Scholar] [CrossRef]
- Ke, T.; Han, M.; Zhao, M.; Wang, Q.K.; Zhang, H.; Zhao, Y.; Ruan, X.; Li, H.; Xu, C.; Sun, T. Alpha-actin-2 mutations in Chinese patients with a non-syndromatic thoracic aortic aneurysm. BMC Med. Genet. 2016, 17, 45. [Google Scholar] [CrossRef] [Green Version]
- Van De Laar, I.M.B.H.; Arbustini, E.; Loeys, B.; Björck, E.; Murphy, L.; Groenink, M.; Kempers, M.; Timmermans, J.; Roos-Hesselink, J.; Benke, K.; et al. European reference network for rare vascular diseases (VASCERN) consensus statement for the screening and management of patients with pathogenic ACTA2 variants. Orphanet J. Rare Dis. 2019, 14, 264. [Google Scholar] [CrossRef]
- Hannuksela, M.; Stattin, E.-L.; Klar, J.; Ameur, A.; Johansson, B.; Sörensen, K.; Carlberg, B. A novel variant in MYLK causes thoracic aortic dissections: Genotypic and phenotypic description. BMC Med. Genet. 2016, 17, 61. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Guo, D.-C.; Cao, J.; Gong, L.; Kamm, K.E.; Regalado, E.; Li, L.; Shete, S.; He, W.-Q.; Zhu, M.-S.; et al. Mutations in myosin light chain kinase cause familial aortic dissections. Am. J. Hum. Genet. 2010, 87, 701–707. [Google Scholar] [CrossRef] [Green Version]
- Wallace, S.E.; Regalado, E.S.; Gong, L.; Janda, A.L.; Guo, D.-C.; Russo, C.F.; Kulmacz, R.J.; Hanna, N.; Jondeau, G.; Boileau, C.; et al. MYLK pathogenic variants aortic disease presentation, pregnancy risk, and characterization of pathogenic missense variants. Genet. Med. 2019, 21, 144–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, L.; Vranckx, R.; Van Kien, P.K.; Lalande, A.; Boisset, N.; Mathieu, F.; Wegman, M.; Glancy, L.; Gasc, J.-M.; Brunotte, F.; et al. Mutations in myosin heavy chain 11 cause a syndrome associating thoracic aortic aneurysm/aortic dissection and patent ductus arteriosus. Nat. Genet. 2006, 38, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Ziganshin, B.A.; Bailey, A.E.; Coons, C.; Dykas, D.; Charilaou, P.; Tanriverdi, L.H.; Liu, L.; Tranquilli, M.; Bale, A.E.; Elefteriades, J.A. Routine Genetic Testing for Thoracic Aortic Aneurysm and Dissection in a Clinical Setting. Ann. Thorac. Surg. 2015, 100, 1604–1611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renard, M.; Francis, C.; Ghosh, R.; Scott, A.F.; Witmer, P.D.; Adès, L.C.; Andelfinger, G.U.; Arnaud, P.; Boileau, C.; Callewaert, B.L.; et al. Clinical Validity of Genes for Heritable Thoracic Aortic Aneurysm and Dissection. J. Am. Coll. Cardiol. 2018, 72, 605–615. [Google Scholar] [CrossRef]
- De Backer, J.; Bondue, A.; Budts, W.; Evangelista, A.; Gallego, P.; Jondeau, G.; Loeys, B.; Peña, M.L.; Teixido-Tura, G.; van de Laar, I.; et al. Genetic counselling and testing in adults with congenital heart disease: A consensus document of the ESC Working Group of Grown-Up Congenital Heart Disease, the ESC Working Group on Aorta and Peripheral Vascular Disease and the European Society of Human G. Eur. J. Prev. Cardiol. 2019, 27, 1423–1435. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Luo, M.; Fu, Y.; Cao, Y.; Yin, K.; Li, W.; Meng, C.; Ma, Y.; Zhang, J.; Fan, Y.; et al. Genetic testing of 248 Chinese aortopathy patients using a panel assay. Sci. Rep. 2016, 6, 33002. [Google Scholar] [CrossRef] [Green Version]
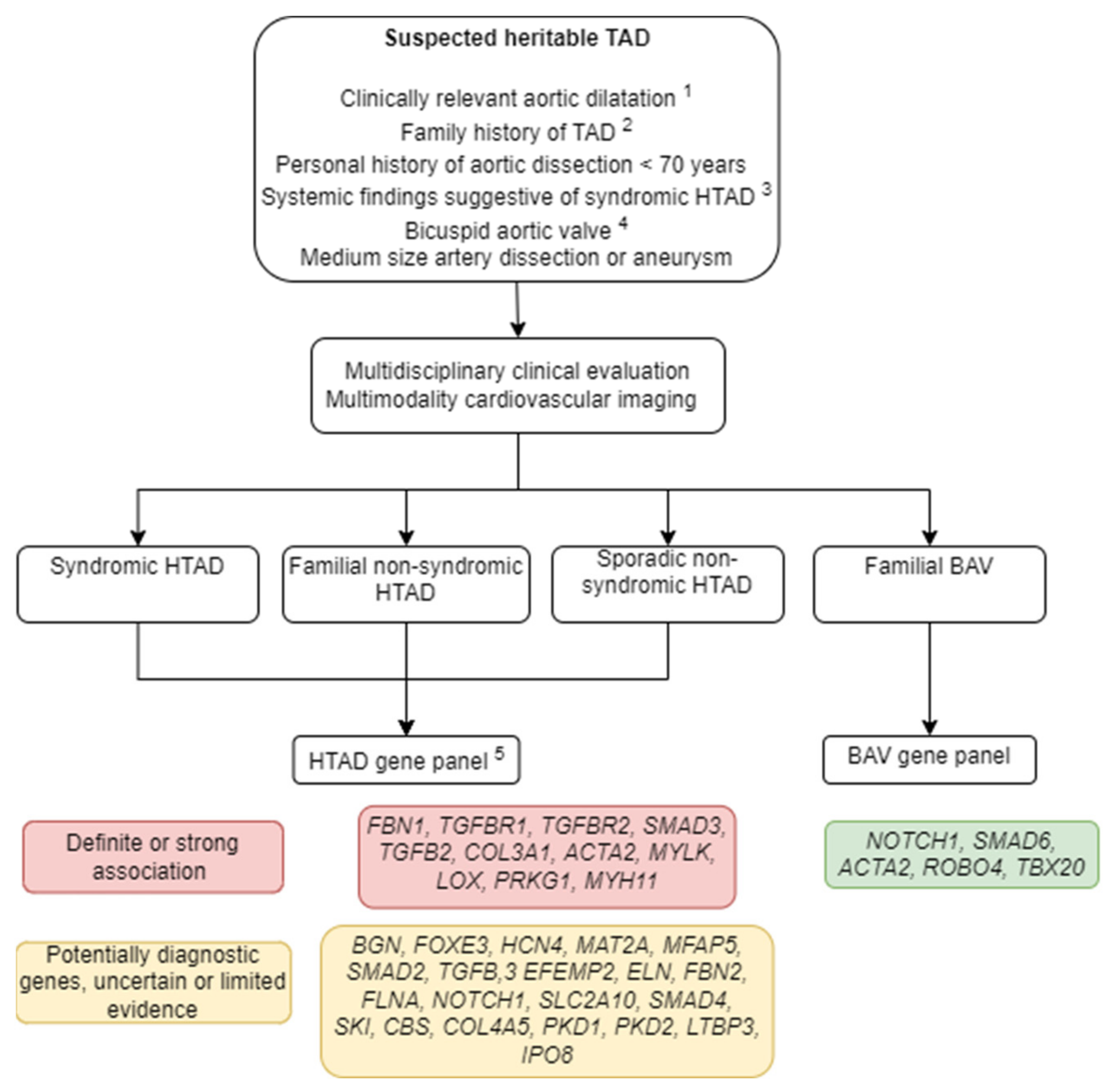

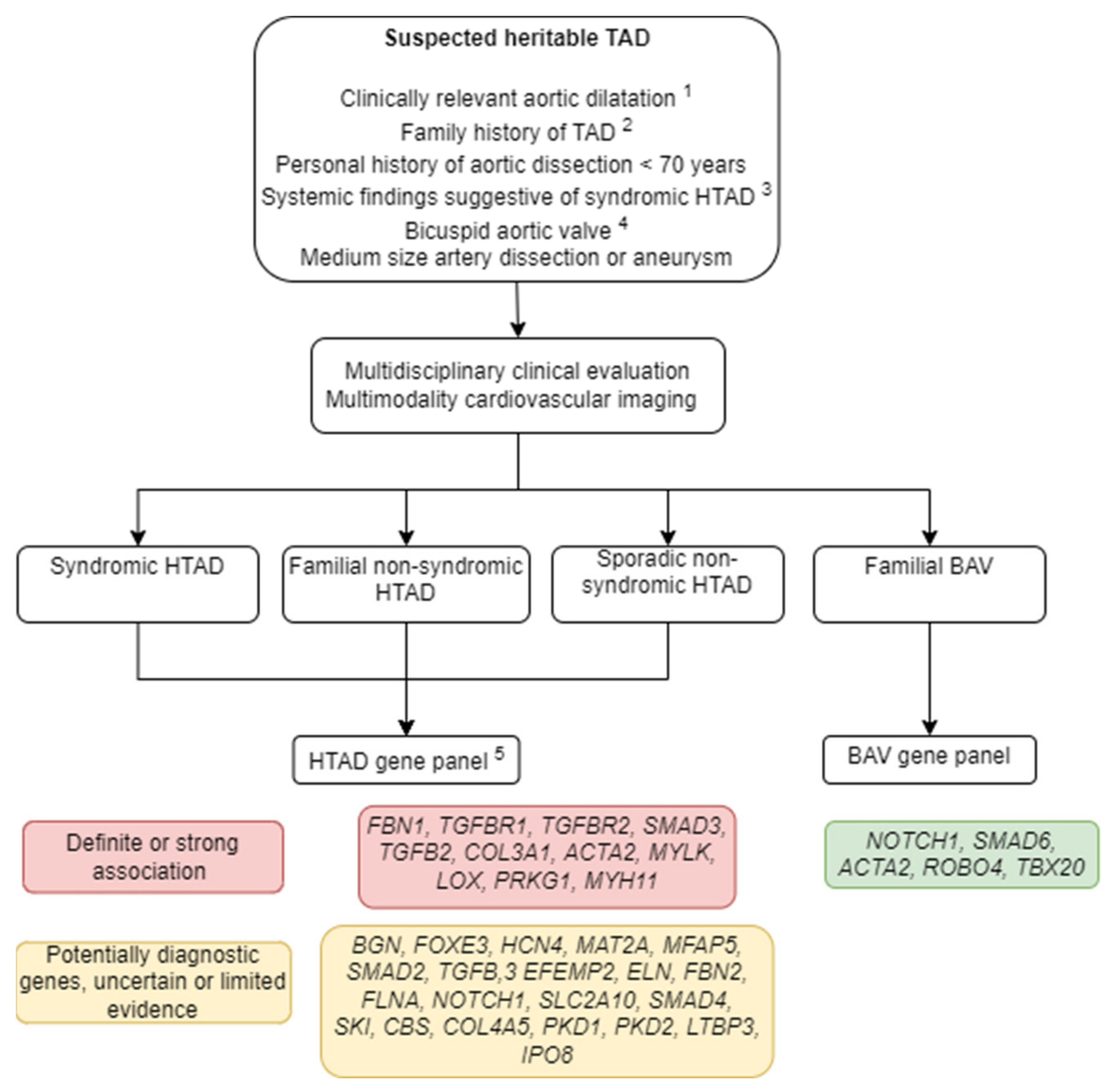{kind=link}
| Patient’s Age | Criteria for Genetic Testing a |
|---|---|
| All ages | Familial HTAD (≥2 relatives identified) |
| Personal history of aortic dissection | |
| Aortic root diameter Z-score >3.5 | |
| <18 years old | Aortic root diameter Z-score ≥3 |
| 18–60 years old | Aortic root diameter Z-score 2.5–3.5 |
| >60 | Aortic root diameter Z-score 2.5–3.5 without hypertension |
| Aortopathy | Genes | Aortic Size to Consider Surgical Operation a | |||
|---|---|---|---|---|---|
| ≥40–42 mm | ≥45 mm | ≥50 mm | ≥55 mm | ||
| Marfan syndrome | FBN1 | ≥1 high-risk factor b | No high-risk factors b | ||
| Loeys–Dietz syndrome | SMAD2, SMAD3, TGFB2, TGFB3, TGFBR1, TGFBR2 | TGFBR2 (females, severe phenotypic features, low body surface area) | SMAD3, TGFB2, TGFBR1, TGFBR2 | SMAD2, TGFB3 | |
| Vascular Ehlers–Danlos syndrome | COL3A1, COL1A1 | Surgical intervention not recommended due to high surgical risk; only as emergency treatment | |||
| Meester–Loeys syndrome | BGN (X--linked) | Aortic dissection in a patient with aortic size of 45 mm has been reported; individualized expert assessment is needed | |||
| Filamin-related HTAD | FLNA (X-linked) | Aortic rupture at aortic size of 42 mm has been reported; individualized expert assessment is needed | |||
| LOX-related HTAD | LOX | Aortic dissection at aortic size >50 mm has been reported; individualized expert assessment is needed | |||
| Gorlin–Chaudhry–Moss syndrome | SLC25A24 | Aortic dissection at aortic size of 51 mm has been reported; individualized expert assessment is needed | |||
| Shprintzen–Goldberg syndrome | SKI | Milder phenotype than Marfan syndrome or Loeys–Dietz syndrome; individualized expert assessment is needed | |||
| Importin-related HTAD | IPO8 (biallelic) | No aortic dissections have been reported | |||
| LTBP3-related HTAD | LTBP3 (biallelic) | Aortic dissection at aortic size >50 mm has been reported; individualized expert assessment is needed | |||
| ACTA2-related HTAD | ACTA2 (especially p.R179, p.R258) | Dissections have been reported at aortic aneurysm sizes of as low as 40 mm. More aggressive phenotype with p.R179 (multisystemic smooth muscle dysfunction syndrome) and p.R258 variants | |||
| Arterial tortuosity syndrome | SLC2A10 | No dissections reported; individualized expert assessment is needed | |||
| Turner syndrome | Complete or partial X chromosome monosomy | ASI >25 mm/m2, especially if ≥1 high-risk factor present c | |||
| Pompe disease | GAA (X-linked) | Aortic dissection at aortic size >50 mm has been reported; individualized expert assessment is needed | |||
| Fabry disease | GLA (X-linked) | No aortic dissections have been reported | |||
| Mucopolysaccharidoses | IDUA, IDS, SGSH, NAGLU, HGSNAT, GNS, GALNS, GLB1, ARSB, GUSB, HYAL1 | No aortic dissections have been reported | |||
| Bicuspid aortic valve-related aortopathy | NOTCH1, ACTA2, SMAD6, ROBO4, TBX20 | ≥1 high-risk factor present d | No high-risk factors present d | ||
| Nonsyndromic HTAD (familial or sporadic) | MYLK, MYH11, PRKG1, ACTA2, LOX, MFAP5, FOXE3, MAT2A, SMAD2, SMAD4, NOTCH1, PLOD1, TGFB2, TGFBR2, FBN1, FBN2, LTBP3 | MYLK, MYH11, PRKG1, ACTA2 (especially p.R179, p.R258) | All other genes; individualized expert assessment is needed | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papatheodorou, E.; Degiannis, D.; Anastasakis, A. Genetics of Heritable Thoracic Aortic Disease. Cardiogenetics 2022, 12, 63-79. https://doi.org/10.3390/cardiogenetics12010006
Papatheodorou E, Degiannis D, Anastasakis A. Genetics of Heritable Thoracic Aortic Disease. Cardiogenetics. 2022; 12(1):63-79. https://doi.org/10.3390/cardiogenetics12010006
Chicago/Turabian StylePapatheodorou, Efstathios, Dimitrios Degiannis, and Aris Anastasakis. 2022. "Genetics of Heritable Thoracic Aortic Disease" Cardiogenetics 12, no. 1: 63-79. https://doi.org/10.3390/cardiogenetics12010006
APA StylePapatheodorou, E., Degiannis, D., & Anastasakis, A. (2022). Genetics of Heritable Thoracic Aortic Disease. Cardiogenetics, 12(1), 63-79. https://doi.org/10.3390/cardiogenetics12010006