
Transthyretin Gene Variants and Associated Phenotypes in Danish Patients with Amyloid Cardiomyopathy

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
Statistics
3. Results
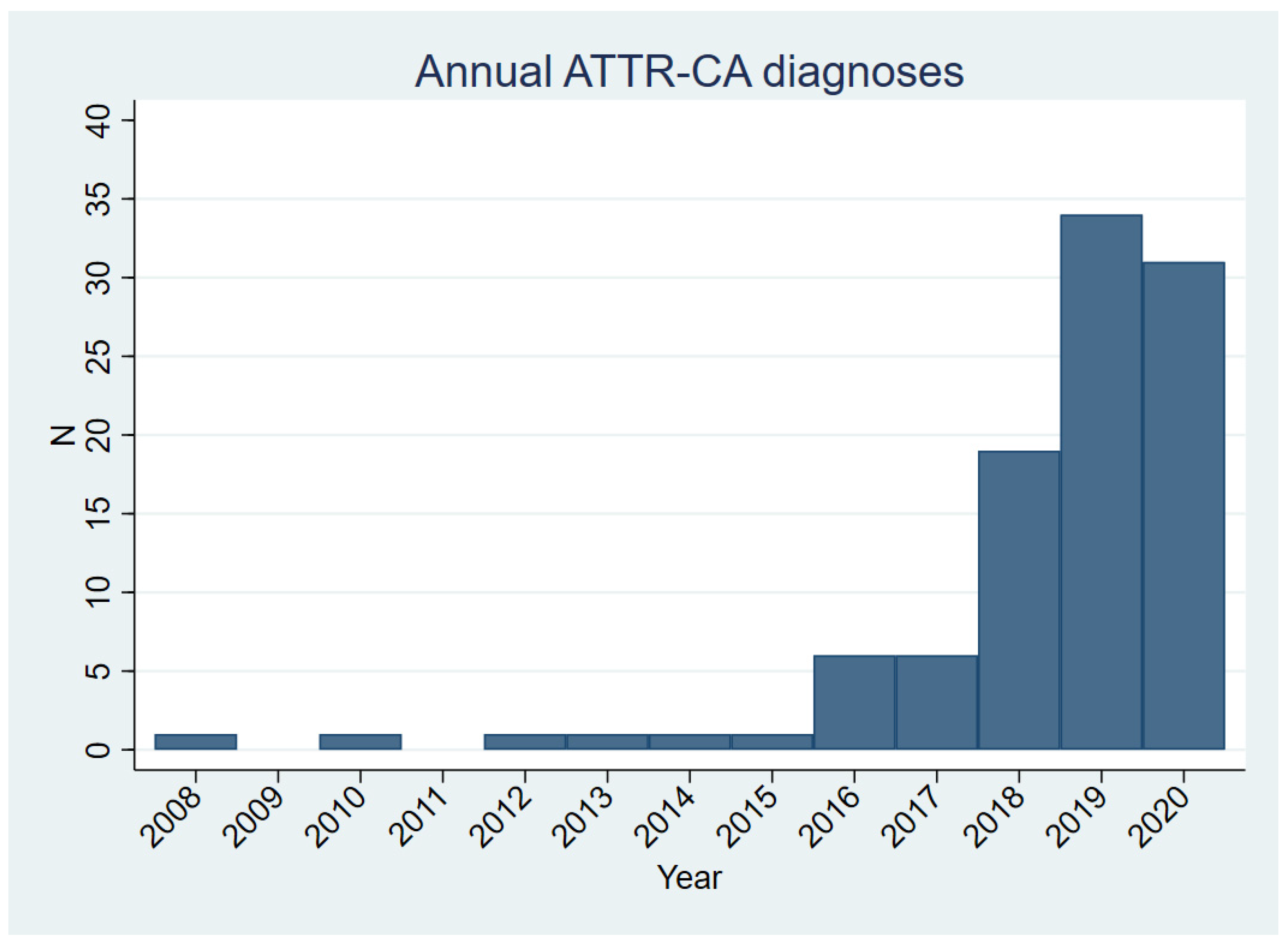
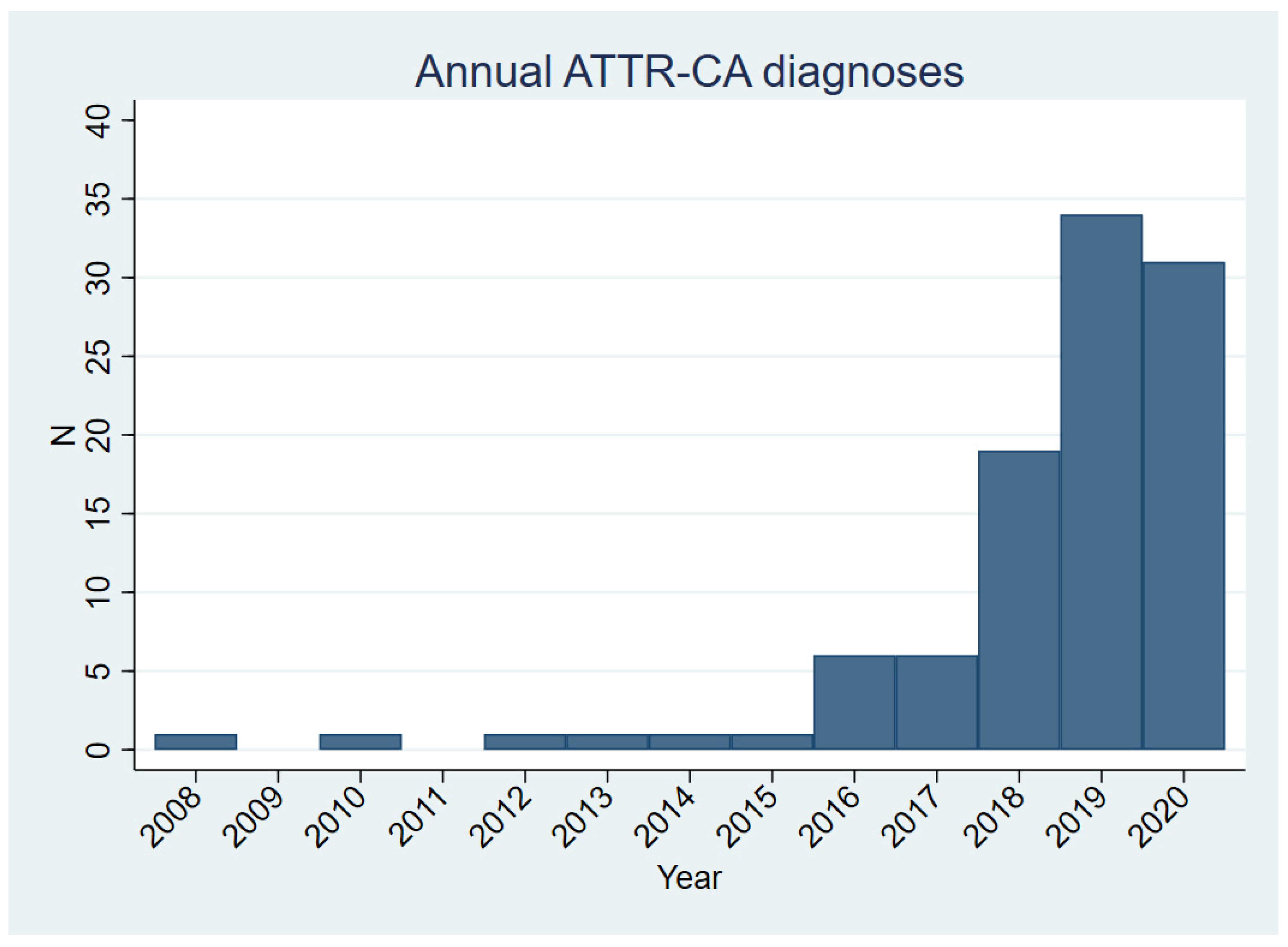
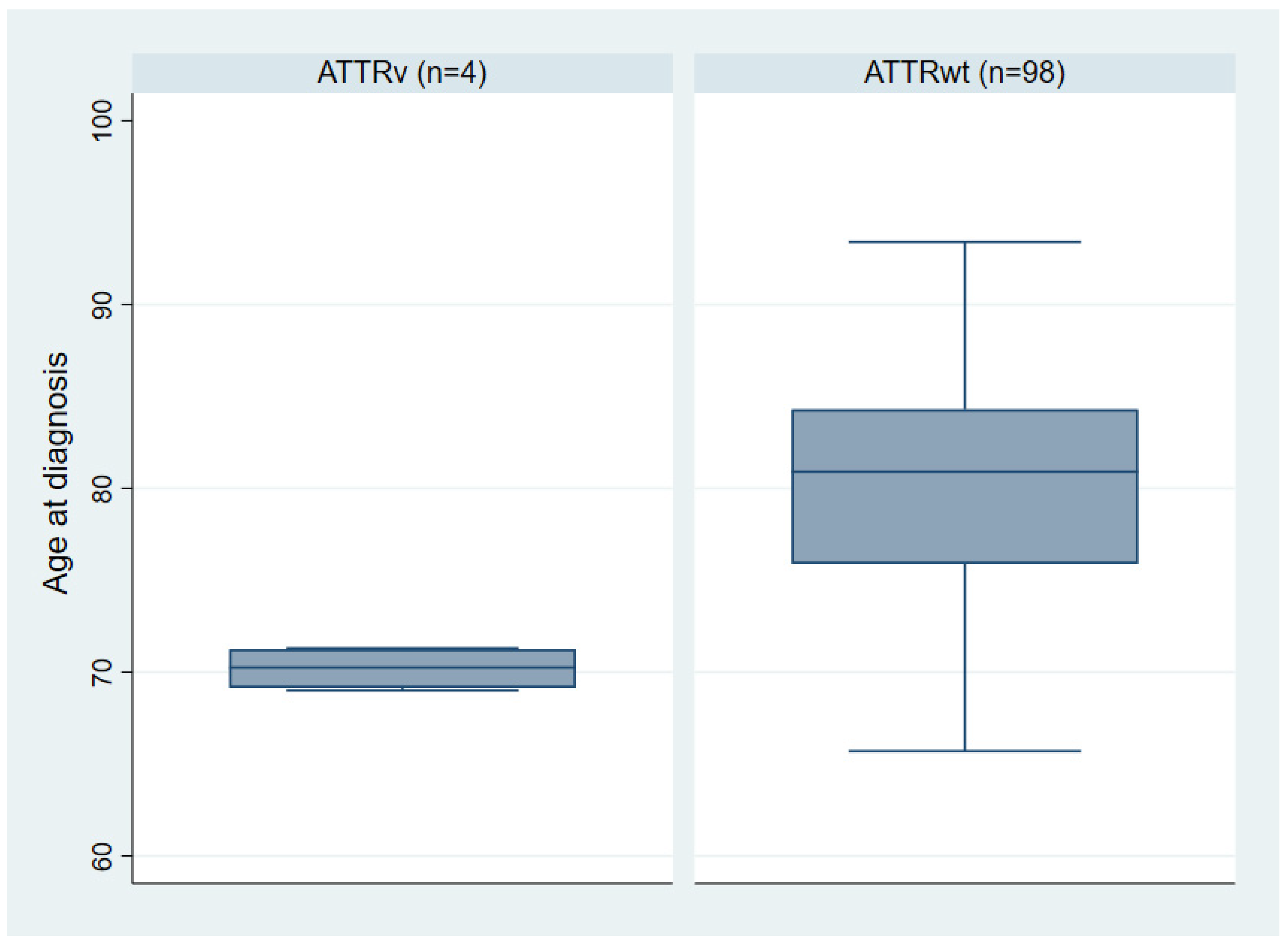
3.1. Clinical Characteristics of the ATTR-CA Cohort
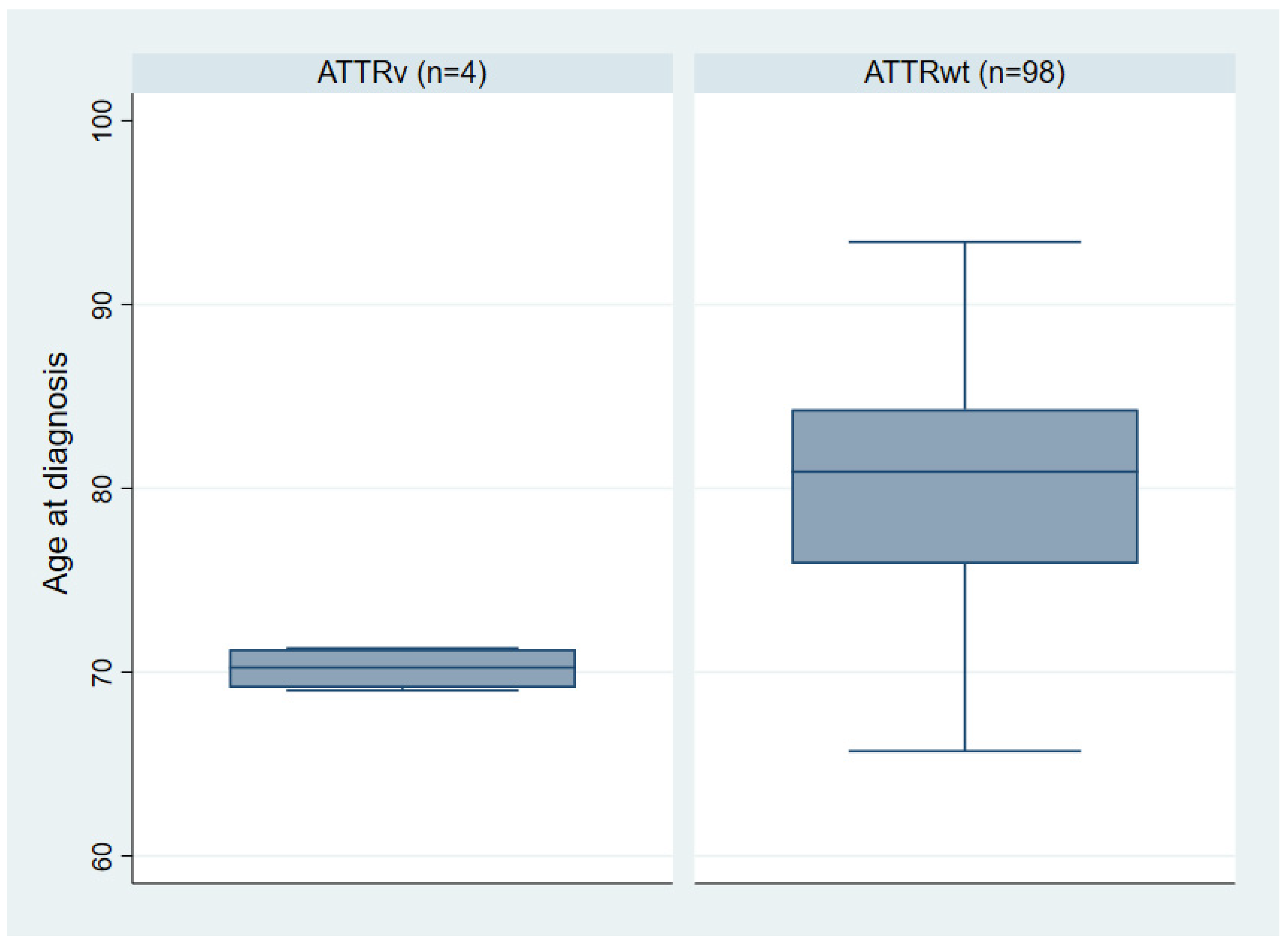
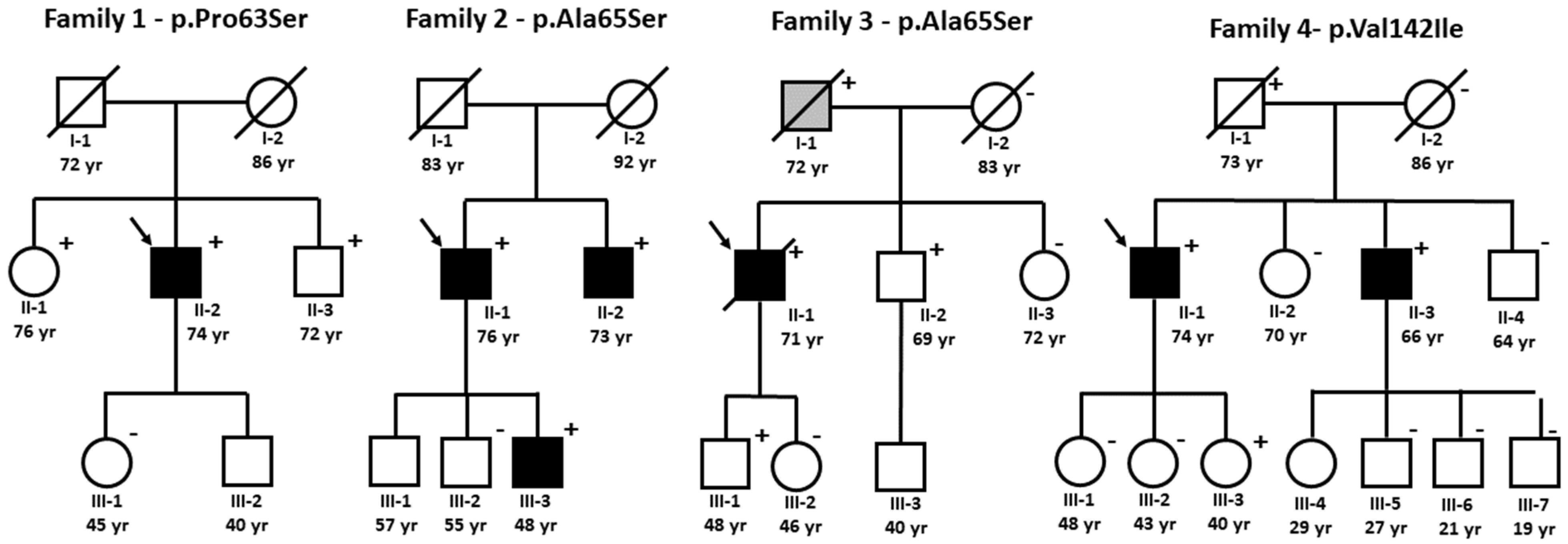
3.2. ATTRv Index Patients
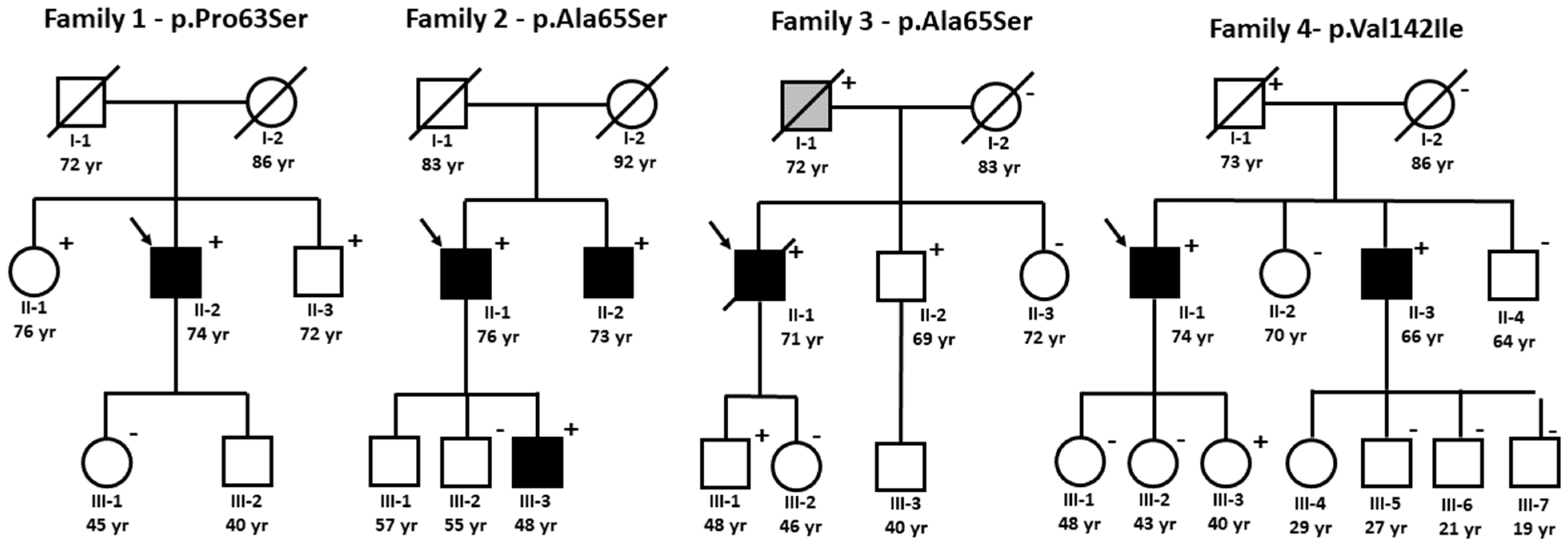
3.3. Family Studies of ATTRv
3.3.1. Family 1—p.Pro63Ser
3.3.2. Family 2 and 3—p.Ala65Ser
3.3.3. Family 4—p.Val142Ile
4. Discussion
5. Limitations
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Maurer, M.S.; Bokhari, S.; Damy, T.; Dorbala, S.; Drachman, B.M.; Fontana, M.; Grogan, M.; Kristen, A.V.; Lousada, I.; Nativi-Nicolau, J.; et al. Expert Consensus Recommendations for the Suspicion and Diagnosis of Transthyretin Cardiac Amyloidosis. Circ. Heart Fail. 2019, 12, e006075. [Google Scholar] [CrossRef]
- Gonzalez-Lopez, E.; Gallego-Delgado, M.; Guzzo-Merello, G.; Javier de Haro-del Moral, F.; Cobo-Marcos, M.; Robles, C.; Bornstein, B.; Salas, C.; Lara-Pezzi, E.; Alonso-Pulpon, L.; et al. Wild-type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur. Heart. J. 2015, 36, 2585–2594. [Google Scholar] [CrossRef] [Green Version]
- Clemmensen, T.S.; Molgaard, H.; Sorensen, J.; Eiskjaer, H.; Andersen, N.F.; Mellemkjaer, S.; Andersen, M.J.; Tolbod, L.P.; Harms, H.J.; Poulsen, S.H. Inotropic myocardial reserve deficiency is the predominant feature of exercise haemodynamics in cardiac amyloidosis. Eur. J. Heart Fail. 2017, 19, 1457–1465. [Google Scholar] [CrossRef] [Green Version]
- Clemmensen, T.S.; Soerensen, J.; Hansson, N.H.; Tolbod, L.P.; Harms, H.J.; Eiskjaer, H.; Mikkelsen, F.; Wiggers, H.; Andersen, N.F.; Poulsen, S.H. Myocardial Oxygen Consumption and Efficiency in Patients with Cardiac Amyloidosis. J. Am. Heart Assoc. 2018, 7, e009974. [Google Scholar] [CrossRef] [Green Version]
- Clemmensen, T.S.; Eiskjaer, H.; Molgaard, H.; Larsen, A.H.; Soerensen, J.; Andersen, N.F.; Tolbod, L.P.; Harms, H.J.; Poulsen, S.H. Abnormal Coronary Flow Velocity Reserve and Decreased Myocardial Contractile Reserve Are Main Factors in Relation to Physical Exercise Capacity in Cardiac Amyloidosis. J. Am. Soc. Echocardiogr. 2018, 31, 71–78. [Google Scholar] [CrossRef]
- Sekijima, Y.; Uchiyama, S.; Tojo, K.; Sano, K.; Shimizu, Y.; Imaeda, T.; Hoshii, Y.; Kato, H.; Ikeda, S. High prevalence of wild-type transthyretin deposition in patients with idiopathic carpal tunnel syndrome: A common cause of carpal tunnel syndrome in the elderly. Hum. Pathol. 2011, 42, 1785–1791. [Google Scholar] [CrossRef] [Green Version]
- Gillmore, J.D.; Damy, T.; Fontana, M.; Hutchinson, M.; Lachmann, H.J.; Martinez-Naharro, A.; Quarta, C.C.; Rezk, T.; Whelan, C.J.; Gonzalez-Lopez, E.; et al. A new staging system for cardiac transthyretin amyloidosis. Eur. Heart J. 2018, 39, 2799–2806. [Google Scholar] [CrossRef]
- Law, S.; Petrie, A.; Chacko, L.; Cohen, O.C.; Ravichandran, S.; Gilbertson, J.A.; Rowczenio, D.; Wechalekar, A.; Martinez-Naharro, A.; Lachmann, H.J.; et al. Disease progression in cardiac transthyretin amyloidosis is indicated by serial calculation of National Amyloidosis Centre transthyretin amyloidosis stage. ESC Heart Fail. 2020, 7, 3942–3949. [Google Scholar] [CrossRef]
- Damy, T.; Kristen, A.V.; Suhr, O.B.; Maurer, M.S.; Plante-Bordeneuve, V.; Yu, C.R.; Ong, M.L.; Coelho, T.; Rapezzi, C.; Thaos Investigators. Transthyretin cardiac amyloidosis in continental Western Europe: An insight through the Transthyretin Amyloidosis Outcomes Survey (THAOS). Eur. Heart J. 2019, ehz173. [Google Scholar] [CrossRef] [Green Version]
- Brown, E.E.; Lee, Y.Z.J.; Halushka, M.K.; Steenbergen, C.; Johnson, N.M.; Almansa, J.; Tedford, R.J.; Cingolani, O.; Russell, S.D.; Sharma, K.; et al. Genetic testing improves identification of transthyretin amyloid (ATTR) subtype in cardiac amyloidosis. Amyloid 2017, 24, 92–95. [Google Scholar] [CrossRef]
- Svendsen, I.H.; Steensgaard-Hansen, F.; Nordvag, B.Y. A clinical, echocardiographic and genetic characterization of a Danish kindred with familial amyloid transthyretin methionine 111 linked cardiomyopathy. Eur. Heart J. 1998, 19, 782–789. [Google Scholar] [CrossRef]
- Frederiksen, T.; Gotzsche, H.; Harboe, N.; Kiaer, W.; Mellemgaard, K. Familial primary amyloidosis with severe amyloid heart disease. Am. J. Med. 1962, 33, 328–348. [Google Scholar] [CrossRef]
- Phelan, D.; Collier, P.; Thavendiranathan, P.; Popovic, Z.B.; Hanna, M.; Plana, J.C.; Marwick, T.H.; Thomas, J.D. Relative apical sparing of longitudinal strain using two-dimensional speckle-tracking echocardiography is both sensitive and specific for the diagnosis of cardiac amyloidosis. Heart 2012, 98, 1442–1448. [Google Scholar] [CrossRef]
- Perugini, E.; Guidalotti, P.L.; Salvi, F.; Cooke, R.M.; Pettinato, C.; Riva, L.; Leone, O.; Farsad, M.; Ciliberti, P.; Bacchi-Reggiani, L.; et al. Noninvasive etiologic diagnosis of cardiac amyloidosis using 99mTc-3,3-diphosphono-1,2-propanodicarboxylic acid scintigraphy. J. Am. Coll. Cardiol. 2005, 46, 1076–1084. [Google Scholar] [CrossRef] [Green Version]
- Galderisi, M.; Cosyns, B.; Edvardsen, T.; Cardim, N.; Delgado, V.; Di Salvo, G.; Donal, E.; Sade, L.E.; Ernande, L.; Garbi, M.; et al. Standardization of adult transthoracic echocardiography reporting in agreement with recent chamber quantification, diastolic function, and heart valve disease recommendations: An expert consensus document of the European Association of Cardiovascular Imaging. Eur. Heart. J. Cardiovasc. Imaging 2017, 18, 1301–1310. [Google Scholar]
- Rasmussen, T.B.; Vase, H.; Ladefoged, B.; Dybro, A.M.; Clemmensen, T.; Terkelsen, A.J.; Molgaard, H.; Petersen, L.N.; Poulsen, S.H. Prevalence of hereditary transthyretin cardiac amyloidosis (mATTR) in Western Denmark. Eur. Heart J. 2020, 41 (Suppl. S2), ehaa946.2120. [Google Scholar] [CrossRef]
- Mullertz, K.M.; Baerentzen, S.; Jensen, H.K.; Molgaard, H. Phenotypic characterization of late onset cardiac amyloidosis caused by the transthyretin mutation TTRA45S, p.(Ala65Ser). Amyloid 2017, 24, 70–71. [Google Scholar] [CrossRef]
- Treibel, T.A.; Fontana, M.; Gilbertson, J.A.; Castelletti, S.; White, S.K.; Scully, P.R.; Roberts, N.; Hutt, D.F.; Rowczenio, D.M.; Whelan, C.J.; et al. Occult Transthyretin Cardiac Amyloid in Severe Calcific Aortic Stenosis: Prevalence and Prognosis in Patients Undergoing Surgical Aortic Valve Replacement. Circ. Cardiovasc. Imaging 2016, 9, e005066. [Google Scholar] [CrossRef] [Green Version]
- Castano, A.; Narotsky, D.L.; Hamid, N.; Khalique, O.K.; Morgenstern, R.; DeLuca, A.; Rubin, J.; Chiuzan, C.; Nazif, T.; Vahl, T.; et al. Unveiling transthyretin cardiac amyloidosis and its predictors among elderly patients with severe aortic stenosis undergoing transcatheter aortic valve replacement. Eur. Heart J. 2017, 38, 2879–2887. [Google Scholar] [CrossRef] [Green Version]
- Hahn, V.S.; Yanek, L.R.; Vaishnav, J.; Ying, W.; Vaidya, D.; Lee, Y.Z.J.; Riley, S.J.; Subramanya, V.; Brown, E.E.; Hopkins, C.D.; et al. Endomyocardial Biopsy Characterization of Heart Failure with Preserved Ejection Fraction and Prevalence of Cardiac Amyloidosis. JACC Heart Fail. 2020, 8, 712–724. [Google Scholar] [CrossRef]
- Maurizi, N.; Rella, V.; Fumagalli, C.; Salerno, S.; Castelletti, S.; Dagradi, F.; Torchio, M.; Marceca, A.; Meda, M.; Gasparini, M.; et al. Prevalence of cardiac amyloidosis among adult patients referred to tertiary centres with an initial diagnosis of hypertrophic cardiomyopathy. Int. J. Cardiol. 2020, 300, 191–195. [Google Scholar] [CrossRef]
- Lopes, L.R.; Futema, M.; Akhtar, M.M.; Lorenzini, M.; Pittman, A.; Syrris, P.; Elliott, P.M. Prevalence of TTR variants detected by whole-exome sequencing in hypertrophic cardiomyopathy. Amyloid 2019, 26, 243–247. [Google Scholar] [CrossRef]
- Akinboboye, O.; Shah, K.; Warner, A.L.; Damy, T.; Taylor, H.A.; Gollob, J.; Powell, C.; Karsten, V.; Vest, J.; Maurer, M.S. DISCOVERY: Prevalence of transthyretin (TTR) mutations in a US-centric patient population suspected of having cardiac amyloidosis. Amyloid 2020, 27, 223–230. [Google Scholar] [CrossRef]
- Rowczenio, D.; Quarta, C.C.; Fontana, M.; Whelan, C.J.; Martinez-Naharro, A.; Trojer, H.; Baginska, A.; Ferguson, S.M.; Gilbertson, J.; Rezk, T.; et al. Analysis of the TTR gene in the investigation of amyloidosis: A 25-year single UK center experience. Hum. Mutat. 2019, 40, 90–96. [Google Scholar] [CrossRef] [Green Version]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Gillmore, J.D.; Booth, D.R.; Pepys, M.B.; Hawkins, P.N. Hereditary cardiac amyloidosis associated with the transthyretin Ile122 mutation in a white man. Heart 1999, 82, e2. [Google Scholar] [CrossRef]
- Hamidi Asl, K.; Nakamura, M.; Yamashita, T.; Benson, M.D. Cardiac amyloidosis associated with the transthyretin Ile122 mutation in a Caucasian family. Amyloid 2001, 8, 263–269. [Google Scholar]
- Rapezzi, C.; Quarta, C.C.; Obici, L.; Perfetto, F.; Longhi, S.; Salvi, F.; Biagini, E.; Lorenzini, M.; Grigioni, F.; Leone, O.; et al. Disease profile and differential diagnosis of hereditary transthyretin-related amyloidosis with exclusively cardiac phenotype: An Italian perspective. Eur. Heart J. 2013, 34, 520–528. [Google Scholar] [CrossRef] [Green Version]
- Dungu, J.N.; Papadopoulou, S.A.; Wykes, K.; Mahmood, I.; Marshall, J.; Valencia, O.; Fontana, M.; Whelan, C.J.; Gillmore, J.D.; Hawkins, P.N.; et al. Afro-Caribbean Heart Failure in the United Kingdom: Cause, Outcomes, and ATTR V122I Cardiac Amyloidosis. Circ. Heart Fail. 2016, 9, e003352. [Google Scholar] [CrossRef]
- Quarta, C.C.; Buxbaum, J.N.; Shah, A.M.; Falk, R.H.; Claggett, B.; Kitzman, D.W.; Mosley, T.H.; Butler, K.R.; Boerwinkle, E.; Solomon, S.D. The amyloidogenic V122I transthyretin variant in elderly black Americans. N. Engl. J. Med. 2015, 372, 21–29. [Google Scholar] [CrossRef] [Green Version]
- Janunger, T.; Anan, I.; Holmgren, G.; Lovheim, O.; Ohlsson, P.I.; Suhr, O.B.; Tashima, K. Heart failure caused by a novel amyloidogenic mutation of the transthyretin gene: ATTR Ala45Ser. Amyloid 2000, 7, 137–140. [Google Scholar] [CrossRef]
- Suhr, O.B.; Andersen, O.; Aronsson, T.; Jonasson, J.; Kalimo, H.; Lundahl, C.; Lundgren, H.; Melberg, A.; Nyberg, J.; Olsson, M.; et al. Report of five rare or previously unknown amyloidogenic transthyretin mutations disclosed in Sweden. Amyloid 2009, 16, 208–214. [Google Scholar] [CrossRef]
- Jacobson, D.R.; Pastore, R.D.; Yaghoubian, R.; Kane, I.; Gallo, G.; Buck, F.S.; Buxbaum, J.N. Variant-sequence transthyretin (isoleucine 122) in late-onset cardiac amyloidosis in black Americans. N. Engl. J. Med. 1997, 336, 466–473. [Google Scholar] [CrossRef]
- Maurer, M.S.; Schwartz, J.H.; Gundapaneni, B.; Elliott, P.M.; Merlini, G.; Waddington-Cruz, M.; Kristen, A.V.; Grogan, M.; Witteles, R.; Damy, T.; et al. Tafamidis Treatment for Patients with Transthyretin Amyloid Cardiomyopathy. N. Engl. J. Med. 2018, 379, 1007–1016. [Google Scholar] [CrossRef]
- Benson, M.D.; Waddington-Cruz, M.; Berk, J.L.; Polydefkis, M.; Dyck, P.J.; Wang, A.K.; Plante-Bordeneuve, V.; Barroso, F.A.; Merlini, G.; Obici, L.; et al. Inotersen Treatment for Patients with Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 2018, 379, 22–31. [Google Scholar] [CrossRef]
- Adams, D.; Gonzalez-Duarte, A.; O’Riordan, W.D.; Yang, C.C.; Ueda, M.; Kristen, A.V.; Tournev, I.; Schmidt, H.H.; Coelho, T.; Berk, J.L.; et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 2018, 379, 11–21. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| No TTR Variant (n = 98) | TTR Variant (n = 4) | p-Value | |
|---|---|---|---|
| Mean age at diagnosis (years) [range] | 80.0 ± 6.2 [65.7–93.4] | 70.2 ± 1.2 [69–71.3] | 0.005 |
| Male sex | 93% | 100% | 0.582 |
| Diagnostic method | |||
| DPD-scintigraphy Endomyocardial biopsy Protein mass spectrometry | 47% 33% 20% | 50% 25% 25% | |
| LV end-diastolic diameter (mm) | 47.5 ± 5.6 | 49.5 ± 6.2 | 0.524 |
| LV ejection fraction (%) | 48.1 ± 10.7 | 39.8 ± 21.2 | 0.480 |
| Interventricular septum (mm) | 17 ± 3 | 20 ± 8 | 0.437 |
| LV global longitudinal strain (%) | −12.4 ± 3.7 | −9.5 ± 4.1 | 0.204 |
| NT-proBNP (ng/L) * | 1871 [889; 3706] | 2641 [1962; 3772] | 0.343 |
| eGFR (mL/min) | 63.1 ± 19.7 | 58.5 ± 27.7 | 0.737 |
| Mean NAC-stage | 1.5 ± 0.7 | 1.75 ± 1.0 | 0.534 |
| NAC-stage 1 NAC stage 2 NAC-stage 3 | 63% 24% 13% | 50% 25% 25% | |
| Atrial fibrillation or -flutter | 63% | 100% | 0.126 |
| Pacemaker | 24% | 75% | 0.026 |
| Implantable cardioverter defibrillator | 3% | 75% | 0.001 |
| Previous carpal tunnel surgery | 38% | 100% | 0.137 |
| Obstructive coronary artery disease | 27% | 25% | 0.946 |
| ID | Age * | ECG | TTE | DPD (PG) | NT-proBNP (ng/L) | eGFR (mL/min) | CTS | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IVS (mm) | LVEDD (mm) | LVEF (%) | GLS (%) | RALS | LAE | DD | |||||||
| Family 1—p.Pro63Ser | |||||||||||||
| II-1 | 76 | SR | 14 | 47 | 60 | n/a | n/a | + | 2 | 0 | 444 | 88 | - |
| II-2 | 71 | AF | 15 | 58 | 30 | −5.7 | 1.93 | + | 2 | 3 | 3091 | 75 | + |
| II-3 | 72 | SR, AVB | 10 | 49 | 52 | -21 | 0.81 | - | 1 | 0 | 81 | 90 | - |
| Family 2—p.Ala65Ser | |||||||||||||
| II-1 | 69 | AF, AVB, nsVT | 28 | 47 | 51 | −6.7 | 1.12 | + | 1 | n/a | 2191 | 46 | + |
| II-2 | 73 | SR | 11 | 49 | 62 | −21.1 | 0.69 | - | 1 | 3 | <50 | >90 | - |
| III-3 | 49 | SR | 10 | 48 | 69 | −21.3 | 0.69 | - | 1 | 2–3 | <50 | >90 | - |
| Family 3—p.Ala65Ser | |||||||||||||
| II-1 | 69 | SR, AVB, nsVT | 18 | 50 | 45 | −7.7 | 1.58 | + | 3 | 3 | 4452 | 26 | + |
| II-2 | 69 | SR | 13 | 48 | 63 | −22.2 | 0.67 | + | 1 | 0 | <50 | 66 | - |
| III-1 | 48 | SR | 9 | 50 | 57 | −18.7 | 0.54 | - | - | 0 | <50 | >90 | - |
| Family 4: p.Val142Ile | |||||||||||||
| II-1 | 71 | SR | 15 | 41 | 52 | −14.6 | 1.18 | + | 2 | 3 | 1732 | 87 | + |
| II-3 | 64 | SR | 16 | 49 | 51 | −18.9 | 1.29 | + | 2 | 3 | 354 | 88 | + |
| III-3 | 40 | SR | 8 | 40 | 58 | −25.1 | 0.59 | - | - | - | <50 | >90 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rasmussen, T.B.; Ladefoged, B.T.; Dybro, A.M.; Clemmensen, T.S.; Sørensen, R.H.; Terkelsen, A.J.; Mølgaard, H.; Vase, H.; Poulsen, S.H. Transthyretin Gene Variants and Associated Phenotypes in Danish Patients with Amyloid Cardiomyopathy. Cardiogenetics 2022, 12, 1-11. https://doi.org/10.3390/cardiogenetics12010001
Rasmussen TB, Ladefoged BT, Dybro AM, Clemmensen TS, Sørensen RH, Terkelsen AJ, Mølgaard H, Vase H, Poulsen SH. Transthyretin Gene Variants and Associated Phenotypes in Danish Patients with Amyloid Cardiomyopathy. Cardiogenetics. 2022; 12(1):1-11. https://doi.org/10.3390/cardiogenetics12010001
Chicago/Turabian StyleRasmussen, Torsten B., Bertil T. Ladefoged, Anne M. Dybro, Tor S. Clemmensen, Rikke H. Sørensen, Astrid J. Terkelsen, Henning Mølgaard, Henrik Vase, and Steen H. Poulsen. 2022. "Transthyretin Gene Variants and Associated Phenotypes in Danish Patients with Amyloid Cardiomyopathy" Cardiogenetics 12, no. 1: 1-11. https://doi.org/10.3390/cardiogenetics12010001
APA StyleRasmussen, T. B., Ladefoged, B. T., Dybro, A. M., Clemmensen, T. S., Sørensen, R. H., Terkelsen, A. J., Mølgaard, H., Vase, H., & Poulsen, S. H. (2022). Transthyretin Gene Variants and Associated Phenotypes in Danish Patients with Amyloid Cardiomyopathy. Cardiogenetics, 12(1), 1-11. https://doi.org/10.3390/cardiogenetics12010001