

Mechanistic Insights into Cytokine Antagonist-Drug Interactions: A Physiologically Based Pharmacokinetic Modelling Approach with Tocilizumab as a Case Study


Abstract
1. Introduction
2. Materials and Methods
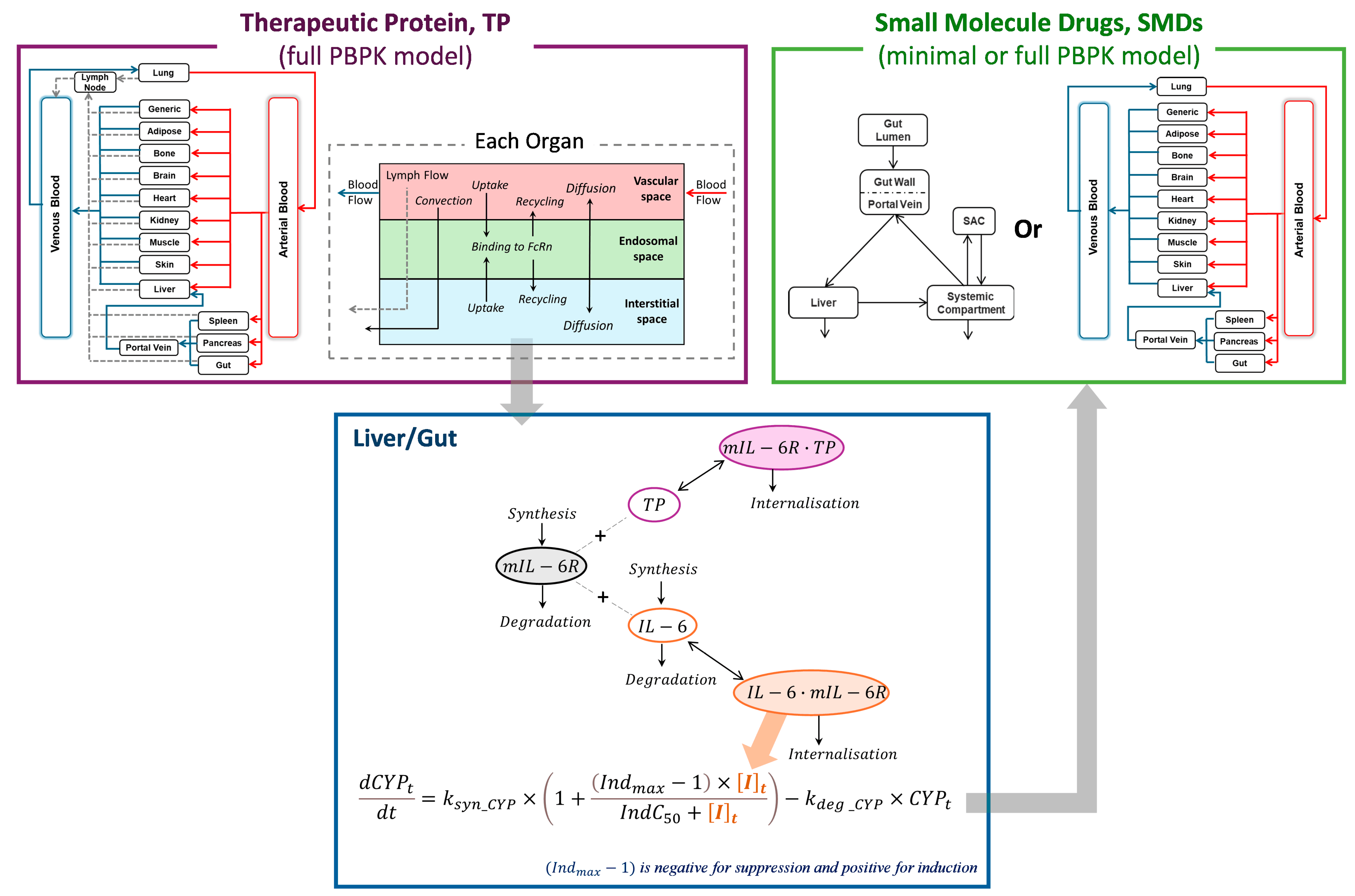
2.1. Integration of PBPK Models for Simulating TP-DIs
2.1.1. Modelling Tocilizumab Profiles Following IV and SC Administration
2.1.2. Modelling of Interactions Between Tocilizumab, IL-6, and mIL-6R
2.1.3. Modelling of IL-6/mIL-6R Complex-Driven Suppression of CYP Enzymes
2.2. Virtual Populations
2.3. Clinical Data and Simulations
2.3.1. Clinical Study Simulations
2.3.2. Prospective Application Simulations
2.3.3. Sensitivity Analysis
3. Results
3.1. Prediction of Tocilizumab Concentration–Time Profiles Across IV and SC Dose Regimens
3.2. Prediction of the Time Course of TP-DIs Between Tocilizumab and Simvastatin
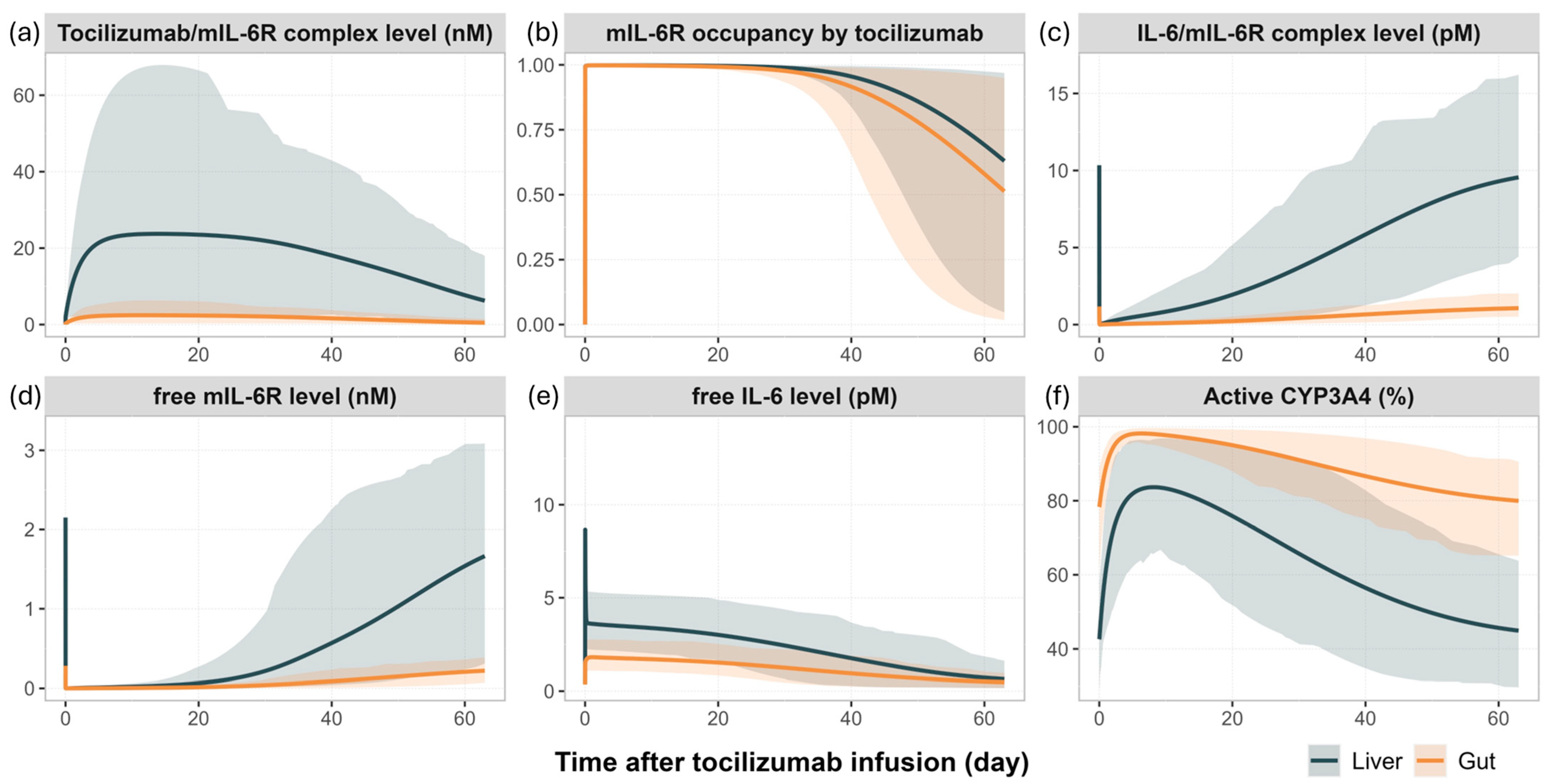
3.3. Mechanistic Insights into IL-6 Signalling and CYP3A4 Modulation Following Tocilizumab Administration
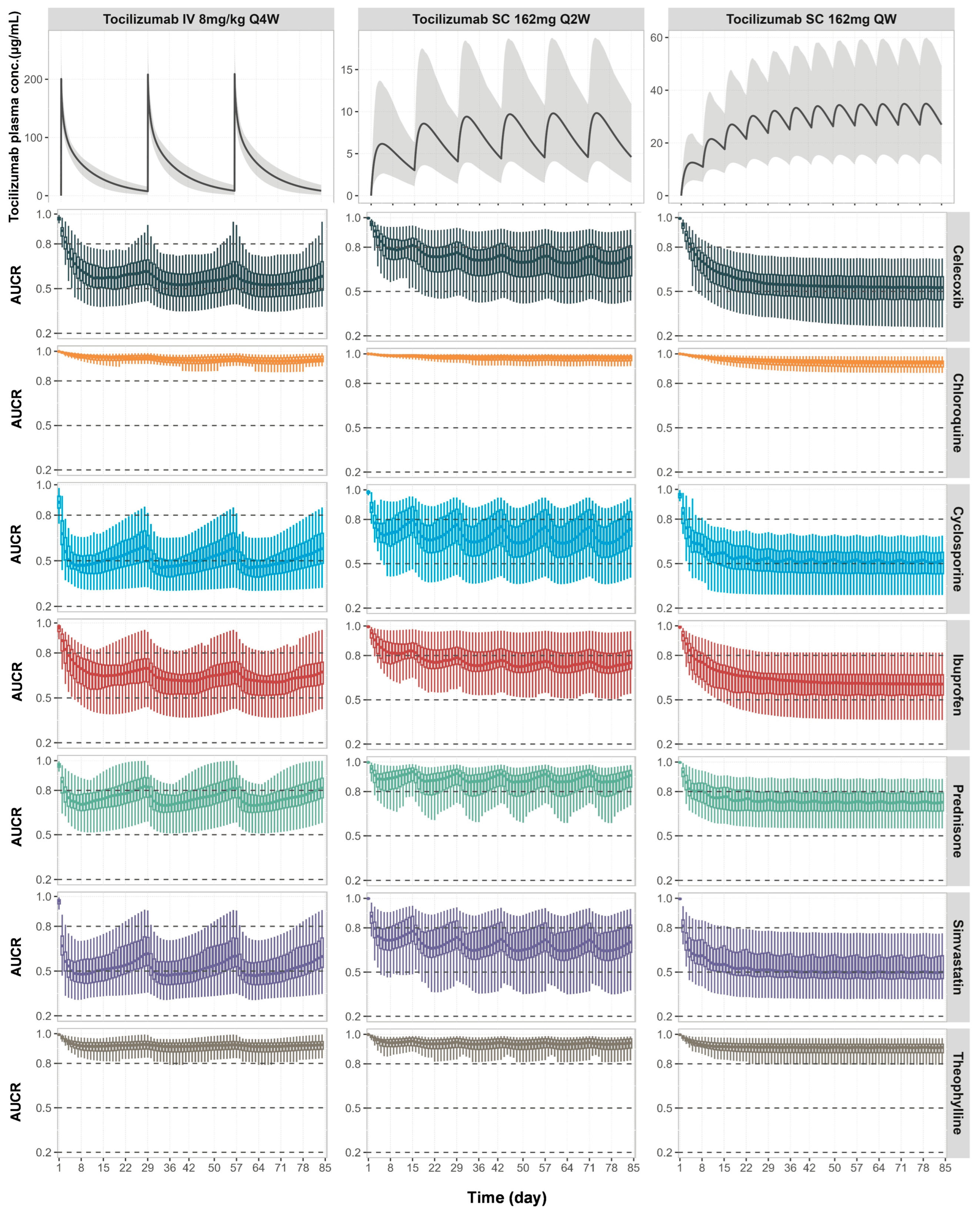
3.4. Prospective Predictions of TP-DIs in RA Patients with Comedications of Tocilizumab and CYP Substrate Drugs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- U.S. Food & Drug Administration, Drug-Drug Interaction Assessment for Therapeutic Proteins, Guidance for Industry. Available online: www.fda.gov/regulatory-information/search-fda-guidance-documents/drug-drug-interaction-assessment-therapeutic-proteins-guidance-industry (accessed on 6 June 2023).
- ICH Guideline M12 on Drug Interaction Studies. Available online: https://database.ich.org/sites/default/files/ICH_M12_Step4_Guideline_2024_0521_0.pdf (accessed on 10 June 2024).
- Morgan, E.T.; Goralski, K.B.; Piquette-Miller, M.; Renton, K.W.; Robertson, G.R.; Chaluvadi, M.R.; Charles, K.A.; Clarke, S.J.; Kacevska, M.; Liddle, C.; et al. Regulation of drug-metabolizing enzymes and transporters in infection, inflammation, and cancer. Drug Metab. Dispos. 2008, 36, 205–216. [Google Scholar] [CrossRef] [PubMed]
- Harvey, R.D.; Morgan, E.T. Cancer, inflammation, and therapy: Effects on cytochrome p450-mediated drug metabolism and implications for novel immunotherapeutic agents. Clin. Pharmacol. Ther. 2014, 96, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.F.; Jones, H.M.; Gill, K.L. Physiologically Based Pharmacokinetic Modeling To Predict Drug-Biologic Interactions with Cytokine Modulators: Are These Relevant and Is Interleukin-6 Enough? Drug Metab. Dispos. 2022, 50, 1322–1331. [Google Scholar] [CrossRef]
- Coutant, D.E.; Hall, S.D. Disease-Drug Interactions in Inflammatory States via Effects on CYP-Mediated Drug Clearance. J. Clin. Pharmacol. 2018, 58, 849–863. [Google Scholar] [CrossRef]
- Rose-John, S. IL-6 trans-signaling via the soluble IL-6 receptor: Importance for the pro-inflammatory activities of IL-6. Int. J. Biol. Sci. 2012, 8, 1237–1247. [Google Scholar] [CrossRef] [PubMed]
- Machavaram, K.K.; Almond, L.M.; Rostami-Hodjegan, A.; Gardner, I.; Jamei, M.; Tay, S.; Wong, S.; Joshi, A.; Kenny, J.R. A physiologically based pharmacokinetic modeling approach to predict disease-drug interactions: Suppression of CYP3A by IL-6. Clin. Pharmacol. Ther. 2013, 94, 260–268. [Google Scholar] [CrossRef]
- Mihara, M.; Kasutani, K.; Okazaki, M.; Nakamura, A.; Kawai, S.; Sugimoto, M.; Matsumoto, Y.; Ohsugi, Y. Tocilizumab inhibits signal transduction mediated by both mIL-6R and sIL-6R, but not by the receptors of other members of IL-6 cytokine family. Int. Immunopharmacol. 2005, 5, 1731–1740. [Google Scholar] [CrossRef]
- Nishimoto, N.; Terao, K.; Mima, T.; Nakahara, H.; Takagi, N.; Kakehi, T. Mechanisms and pathologic significances in increase in serum interleukin-6 (IL-6) and soluble IL-6 receptor after administration of an anti-IL-6 receptor antibody, tocilizumab, in patients with rheumatoid arthritis and Castleman disease. Blood 2008, 112, 3959–3964. [Google Scholar] [CrossRef]
- Nishimoto, N.; Yoshizaki, K.; Maeda, K.; Kuritani, T.; Deguchi, H.; Sato, B.; Imai, N.; Suemura, M.; Kakehi, T.; Takagi, N.; et al. Toxicity, pharmacokinetics, and dose-finding study of repetitive treatment with the humanized anti-interleukin 6 receptor antibody MRA in rheumatoid arthritis. Phase I/II clinical study. J. Rheumatol. 2003, 30, 1426–1435. [Google Scholar]
- Schmitt, C.; Kuhn, B.; Zhang, X.; Kivitz, A.J.; Grange, S. Disease-drug-drug interaction involving tocilizumab and simvastatin in patients with rheumatoid arthritis. Clin. Pharmacol. Ther. 2011, 89, 735–740. [Google Scholar] [CrossRef]
- Wang, L.; Chen, Y.; Zhou, W.; Miao, X.; Zhou, H. Utilization of physiologically-based pharmacokinetic model to assess disease-mediated therapeutic protein-disease-drug interaction in immune-mediated inflammatory diseases. Clin. Transl. Sci. 2022, 15, 464–476. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Zhuang, Y.; Xu, Z.; Wang, W.; Zhou, H. Development of a Physiologically Based Pharmacokinetic Model to Predict Disease-Mediated Therapeutic Protein-Drug Interactions: Modulation of Multiple Cytochrome P450 Enzymes by Interleukin-6. AAPS J. 2016, 18, 767–776. [Google Scholar] [CrossRef] [PubMed]
- Gill, K.L.; Gardner, I.; Li, L.; Jamei, M. A Bottom-Up Whole-Body Physiologically Based Pharmacokinetic Model to Mechanistically Predict Tissue Distribution and the Rate of Subcutaneous Absorption of Therapeutic Proteins. AAPS J. 2016, 18, 156–170. [Google Scholar] [CrossRef]
- Jamei, M.; Bajot, F.; Neuhoff, S.; Barter, Z.; Yang, J.; Rostami-Hodjegan, A.; Rowland-Yeo, K. A mechanistic framework for in vitro-in vivo extrapolation of liver membrane transporters: Prediction of drug-drug interaction between rosuvastatin and cyclosporine. Clin. Pharmacokinet. 2014, 53, 73–87. [Google Scholar] [CrossRef]
- Jamei, M.; Turner, D.; Yang, J.; Neuhoff, S.; Polak, S.; Rostami-Hodjegan, A.; Tucker, G. Population-based mechanistic prediction of oral drug absorption. AAPS J. 2009, 11, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Aukland, K.; Reed, R.K. Interstitial-lymphatic mechanisms in the control of extracellular fluid volume. Physiol. Rev. 1993, 73, 1–78. [Google Scholar] [CrossRef]
- Rippe, B.; Haraldsson, B. Fluid and protein fluxes across small and large pores in the microvasculature. Application of two-pore equations. Acta Physiol. Scand. 1987, 131, 411–428. [Google Scholar] [CrossRef]
- Li, L.; Gardner, I.; Dostalek, M.; Jamei, M. Simulation of monoclonal antibody pharmacokinetics in humans using a minimal physiologically based model. AAPS J. 2014, 16, 1097–1109. [Google Scholar] [CrossRef]
- Mager, D.E.; Krzyzanski, W. Quasi-equilibrium pharmacokinetic model for drugs exhibiting target-mediated drug disposition. Pharm. Res. 2005, 22, 1589–1596. [Google Scholar] [CrossRef]
- Schwabe, C.; Illes, A.; Ullmann, M.; Ghori, V.; Vincent, E.; Petit-Frere, C.; Monnet, J.; Racault, A.S.; Wynne, C. Pharmacokinetics and pharmacodynamics of a proposed tocilizumab biosimilar MSB11456 versus both the US-licensed and EU-approved products: A randomized, double-blind trial. Expert Rev. Clin. Immunol. 2022, 18, 533–543. [Google Scholar] [CrossRef]
- ACTEMRA® (Tocilizumab) Injection. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2024/125276s144,125472s056lbl.pdf (accessed on 24 March 2025).
- Haigler, H.T.; McKanna, J.A.; Cohen, S. Rapid stimulation of pinocytosis in human carcinoma cells A-431 by epidermal growth factor. J. Cell Biol. 1979, 83, 82–90. [Google Scholar] [CrossRef]
- Chung, S.; Nguyen, V.; Lin, Y.L.; Lafrance-Vanasse, J.; Scales, S.J.; Lin, K.; Deng, R.; Williams, K.; Sperinde, G.; Li, J.J.; et al. An in vitro FcRn- dependent transcytosis assay as a screening tool for predictive assessment of nonspecific clearance of antibody therapeutics in humans. MAbs 2019, 11, 942–955. [Google Scholar] [CrossRef]
- Frey, N.; Grange, S.; Woodworth, T. Population pharmacokinetic analysis of tocilizumab in patients with rheumatoid arthritis. J. Clin. Pharmacol. 2010, 50, 754–766. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Rafique, A.; Potocky, T.; Paccaly, A.; Nolain, P.; Lu, Q.; Iglesias-Rodriguez, M.; St John, G.; Nivens, M.C.; Kanamaluru, V.; et al. Differential Binding of Sarilumab and Tocilizumab to IL-6Ralpha and Effects of Receptor Occupancy on Clinical Parameters. J. Clin. Pharmacol. 2021, 61, 714–724. [Google Scholar] [CrossRef]
- Van Hall, G.; Steensberg, A.; Sacchetti, M.; Fischer, C.; Keller, C.; Schjerling, P.; Hiscock, N.; Moller, K.; Saltin, B.; Febbraio, M.A.; et al. Interleukin-6 stimulates lipolysis and fat oxidation in humans. J. Clin. Endocrinol. Metab. 2003, 88, 3005–3010. [Google Scholar] [CrossRef] [PubMed]
- Alfinito, E.; Beccaria, M.; Ciccarese, M. Biosensing Cytokine IL-6: A Comparative Analysis of Natural and Synthetic Receptors. Biosensors 2020, 10, 106. [Google Scholar] [CrossRef] [PubMed]
- Gerhartz, C.; Dittrich, E.; Stoyan, T.; Rose-John, S.; Yasukawa, K.; Heinrich, P.C.; Graeve, L. Biosynthesis and half-life of the interleukin-6 receptor and its signal transducer gp130. Eur. J. Biochem. 1994, 223, 265–274. [Google Scholar] [CrossRef]
- Dickmann, L.J.; Patel, S.K.; Rock, D.A.; Wienkers, L.C.; Slatter, J.G. Effects of interleukin-6 (IL-6) and an anti-IL-6 monoclonal antibody on drug-metabolizing enzymes in human hepatocyte culture. Drug Metab. Dispos. 2011, 39, 1415–1422. [Google Scholar] [CrossRef]
- Pan, X.; Gill, K.L.; Pansari, A.; Hatley, O.; Curry, L.; Jamei, M.; Gardner, I. Cytokine Dynamics in Action: A Mechanistic Approach to Assess Interleukin 6 Related Therapeutic Protein-Drug-Disease Interactions. Clin. Pharmacol. Ther. 2025, 117, 1369–1380. [Google Scholar] [CrossRef]
- Reeh, H.; Rudolph, N.; Billing, U.; Christen, H.; Streif, S.; Bullinger, E.; Schliemann-Bullinger, M.; Findeisen, R.; Schaper, F.; Huber, H.J.; et al. Response to IL-6 trans- and IL-6 classic signalling is determined by the ratio of the IL-6 receptor alpha to gp130 expression: Fusing experimental insights and dynamic modelling. Cell Commun. Signal. 2019, 17, 46. [Google Scholar] [CrossRef]
- Molmenti, E.P.; Ziambaras, T.; Perlmutter, D.H. Evidence for an acute phase response in human intestinal epithelial cells. J. Biol. Chem. 1993, 268, 14116–14124. [Google Scholar] [CrossRef] [PubMed]
- Machavaram, K.K.; Endo-Tsukude, C.; Terao, K.; Gill, K.L.; Hatley, O.J.; Gardner, I.; Parrott, N.; Ducray, P.S. Simulating the Impact of Elevated Levels of Interleukin-6 on the Pharmacokinetics of Various CYP450 Substrates in Patients with Neuromyelitis Optica or Neuromyelitis Optica Spectrum Disorders in Different Ethnic Populations. AAPS J. 2019, 21, 42. [Google Scholar] [CrossRef]
- Abduljalil, K.; Pan, X.; Pansari, A.; Jamei, M.; Johnson, T.N. Preterm Physiologically Based Pharmacokinetic Model. Part II: Applications of the Model to Predict Drug Pharmacokinetics in the Preterm Population. Clin. Pharmacokinet. 2020, 59, 501–518. [Google Scholar] [CrossRef] [PubMed]
- Kilford, P.J.; Chen, K.F.; Crewe, K.; Gardner, I.; Hatley, O.; Ke, A.B.; Neuhoff, S.; Zhang, M.; Rowland Yeo, K. Prediction of CYP-mediated DDIs involving inhibition: Approaches to address the requirements for system qualification of the Simcyp Simulator. CPT Pharmacomet. Syst. Pharmacol. 2022, 11, 822–832. [Google Scholar] [CrossRef]
- Rowland Yeo, K.; Zhang, M.; Pan, X.; Ban Ke, A.; Jones, H.M.; Wesche, D.; Almond, L.M. Impact of Disease on Plasma and Lung Exposure of Chloroquine, Hydroxychloroquine and Azithromycin: Application of PBPK Modeling. Clin. Pharmacol. Ther. 2020, 108, 976–984. [Google Scholar] [CrossRef]
- Terrier, J.; Abouir, K.; Gaspar, F.; Daali, Y.; Samer, C.F. The Impact of Dexamethasone and Prednisone on Apixaban and Rivaroxaban Exposure in COVID-19 Patients: A Physiologically Based Pharmacokinetic Modeling Study. Clin. Pharmacol. Ther. 2025, 117, 554–560. [Google Scholar] [CrossRef] [PubMed]
- Abdallah, H.; Hsu, J.C.; Lu, P.; Fettner, S.; Zhang, X.; Douglass, W.; Bao, M.; Rowell, L.; Burmester, G.R.; Kivitz, A. Pharmacokinetic and Pharmacodynamic Analysis of Subcutaneous Tocilizumab in Patients With Rheumatoid Arthritis From 2 Randomized, Controlled Trials: SUMMACTA and BREVACTA. J. Clin. Pharmacol. 2017, 57, 459–468. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, Y.C.; Fettner, S.; Rowell, L.; Gott, T.; Grimsey, P.; Unsworth, A. Pharmacokinetics and pharmacodynamics of tocilizumab after subcutaneous administration in patients with rheumatoid arthritis. Int. J. Clin. Pharmacol. Ther. 2013, 51, 620–630. [Google Scholar] [CrossRef]
- Ferreira, R.C.; Freitag, D.F.; Cutler, A.J.; Howson, J.M.; Rainbow, D.B.; Smyth, D.J.; Kaptoge, S.; Clarke, P.; Boreham, C.; Coulson, R.M.; et al. Functional IL6R 358Ala allele impairs classical IL-6 receptor signaling and influences risk of diverse inflammatory diseases. PLoS Genet. 2013, 9, e1003444. [Google Scholar] [CrossRef]
- Ershad, A.; Taziki, S.; Ebrahimian, M.; Abadi, S.S.D. Acute cyclosporine overdose: A systematic review. Med. Clínica Práctica 2023, 6, 100358. [Google Scholar] [CrossRef]
- Ellis, E.F. Theophylline. In Allergic Diseases: Diagnosis and Treatment; Lieberman, P., Anderson, J.A., Eds.; Humana Press: Totowa, NJ, USA, 2007; pp. 343–359. [Google Scholar]
- Emery, P.; Keystone, E.; Tony, H.P.; Cantagrel, A.; Van Vollenhoven, R.; Sanchez, A.; Alecock, E.; Lee, J.; Kremer, J. IL-6 receptor inhibition with tocilizumab improves treatment outcomes in patients with rheumatoid arthritis refractory to anti-tumour necrosis factor biologicals: Results from a 24-week multicentre randomised placebo-controlled trial. Ann. Rheum. Dis. 2008, 67, 1516–1523. [Google Scholar] [CrossRef] [PubMed]
- Nawata, Y.; Eugui, E.M.; Lee, S.W.; Allison, A.C. IL-6 is the principal factor produced by synovia of patients with rheumatoid arthritis that induces B-lymphocytes to secrete immunoglobulins. Ann. N. Y. Acad. Sci. 1989, 557, 230–238, discussion 239. [Google Scholar] [CrossRef] [PubMed]
- Arvidson, N.G.; Gudbjornsson, B.; Elfman, L.; Ryden, A.C.; Totterman, T.H.; Hallgren, R. Circadian rhythm of serum interleukin-6 in rheumatoid arthritis. Ann. Rheum. Dis. 1994, 53, 521–524. [Google Scholar] [CrossRef]
- Dijkmans, B.A.; Van Rijthoven, A.W.; Goei The, H.S.; Boers, M.; Cats, A. Cyclosporine in rheumatoid arthritis. Semin. Arthritis Rheum. 1992, 22, 30–36. [Google Scholar] [CrossRef]
- Lee, J.I.; Zhang, L.; Men, A.Y.; Kenna, L.A.; Huang, S.M. CYP-mediated therapeutic protein-drug interactions: Clinical findings, proposed mechanisms and regulatory implications. Clin. Pharmacokinet. 2010, 49, 295–310. [Google Scholar] [CrossRef]
- Evers, R.; Dallas, S.; Dickmann, L.J.; Fahmi, O.A.; Kenny, J.R.; Kraynov, E.; Nguyen, T.; Patel, A.H.; Slatter, J.G.; Zhang, L. Critical review of preclinical approaches to investigate cytochrome p450-mediated therapeutic protein drug-drug interactions and recommendations for best practices: A white paper. Drug Metab. Dispos. 2013, 41, 1598–1609. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Sharma, A. Therapeutic protein-drug interactions: Plausible mechanisms and assessment strategies. Expert Opin. Drug Metab. Toxicol. 2016, 12, 1323–1331. [Google Scholar] [CrossRef] [PubMed]
- Diao, L.; Li, N.; Brayman, T.G.; Hotz, K.J.; Lai, Y. Regulation of MRP2/ABCC2 and BSEP/ABCB11 expression in sandwich cultured human and rat hepatocytes exposed to inflammatory cytokines TNF-alpha, IL-6, and IL-1beta. J. Biol. Chem. 2010, 285, 31185–31192. [Google Scholar] [CrossRef]
- Nguyen, T.V.; Ukairo, O.; Khetani, S.R.; McVay, M.; Kanchagar, C.; Seghezzi, W.; Ayanoglu, G.; Irrechukwu, O.; Evers, R. Establishment of a hepatocyte-kupffer cell coculture model for assessment of proinflammatory cytokine effects on metabolizing enzymes and drug transporters. Drug Metab. Dispos. 2015, 43, 774–785. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Value | Reference/Note |
|---|---|---|
| Molecular Weight (g/mol) | 148,000 | Drug label [23] |
| Subcutaneous Absorption | ||
| First-Order Absorption Model | ||
| ka (1/h) | 0.02 | Fitted to PK data after a single SC dose of 162 mg [22] |
| fa | 0.3 | |
| Distribution | ||
| Distribution Model | Full PBPK | |
| Kup (1/h) | 0.0298 | [24] |
| Krc (1/h) | 16.7 | Fitted based on endogenous IgG data [20] |
| FR | 1 | Assumed FR = 1 for all organs except adipose, muscle, and spleen, where FR = 0 |
| KD,Tocilizumab-FcRn (µM) | 0.46 | [25] |
| Elimination | ||
| CLcat (L/h) | 0.0175 | Fitted based on endogenous IgG data [20] |
| CLadd (L/h) | 0.009 | Optimised to recover the PK data after an IV dose of 4 mg/kg [26] |
| TMDD Parameters | ||
| TMDD Model | Quasi-equilibrium approximation | |
| Drug Target name | mIL-6R | |
| KD_Tocilizumab-mIL-6R (µM) | 1.96 × 10−4 | [27] |
| kint_Tocilizumab/mIL-6R complex (1/h) | 0.0186 | [27] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pan, X.; Liu, C.; Stader, F.; Derbalah, A.; Jamei, M.; Gardner, I. Mechanistic Insights into Cytokine Antagonist-Drug Interactions: A Physiologically Based Pharmacokinetic Modelling Approach with Tocilizumab as a Case Study. Pharmaceutics 2025, 17, 896. https://doi.org/10.3390/pharmaceutics17070896
Pan X, Liu C, Stader F, Derbalah A, Jamei M, Gardner I. Mechanistic Insights into Cytokine Antagonist-Drug Interactions: A Physiologically Based Pharmacokinetic Modelling Approach with Tocilizumab as a Case Study. Pharmaceutics. 2025; 17(7):896. https://doi.org/10.3390/pharmaceutics17070896
Chicago/Turabian StylePan, Xian, Cong Liu, Felix Stader, Abdallah Derbalah, Masoud Jamei, and Iain Gardner. 2025. "Mechanistic Insights into Cytokine Antagonist-Drug Interactions: A Physiologically Based Pharmacokinetic Modelling Approach with Tocilizumab as a Case Study" Pharmaceutics 17, no. 7: 896. https://doi.org/10.3390/pharmaceutics17070896
APA StylePan, X., Liu, C., Stader, F., Derbalah, A., Jamei, M., & Gardner, I. (2025). Mechanistic Insights into Cytokine Antagonist-Drug Interactions: A Physiologically Based Pharmacokinetic Modelling Approach with Tocilizumab as a Case Study. Pharmaceutics, 17(7), 896. https://doi.org/10.3390/pharmaceutics17070896