
Prediction of Monoclonal Antibody Pharmacokinetics in Pediatric Populations Using PBPK Modeling and Simulation



Abstract
1. Introduction
2. Methods
2.1. Case Studies
2.2. Software and Model Structure
2.3. Pediatric Translation
2.4. PBPK Parameters
3. Results
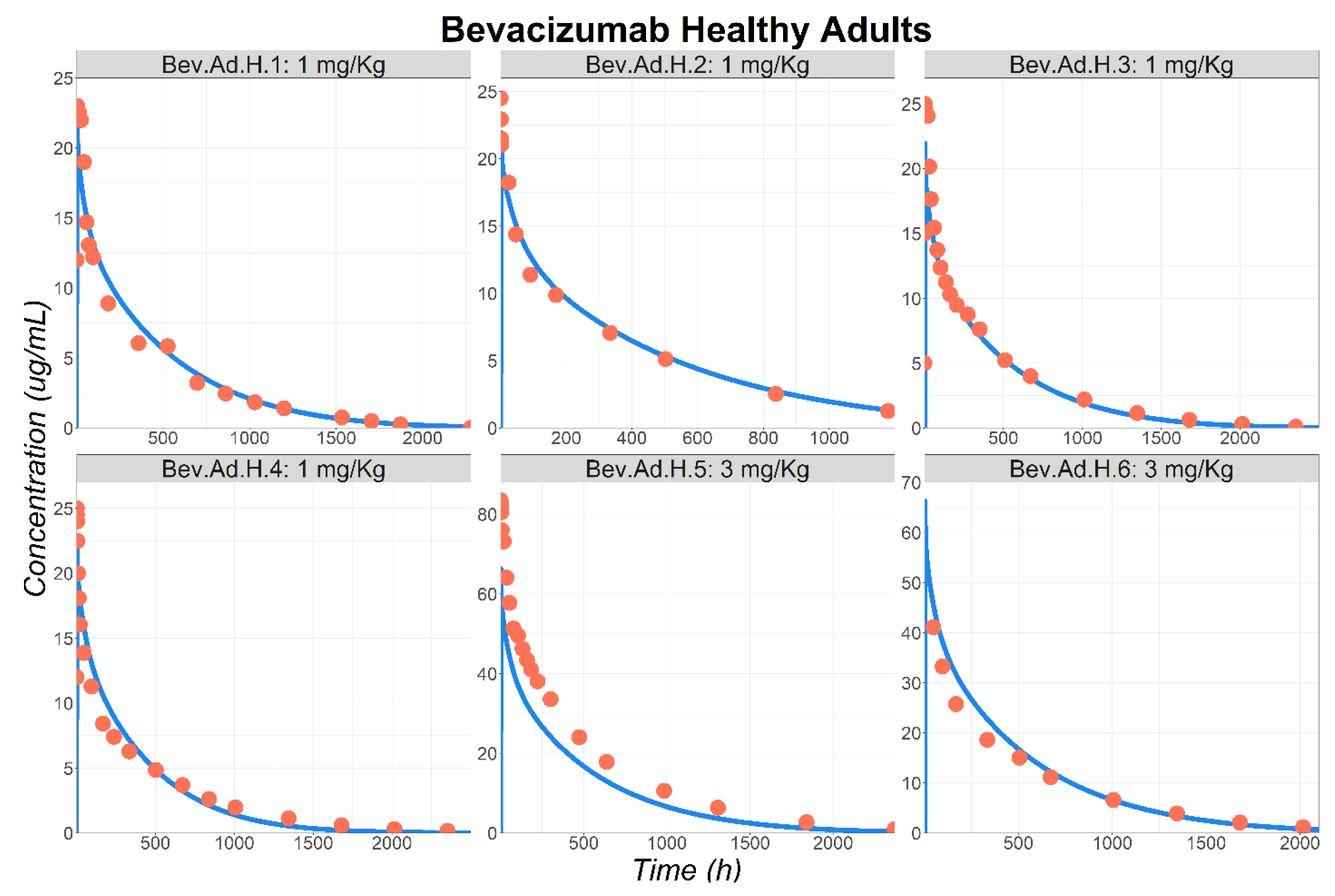
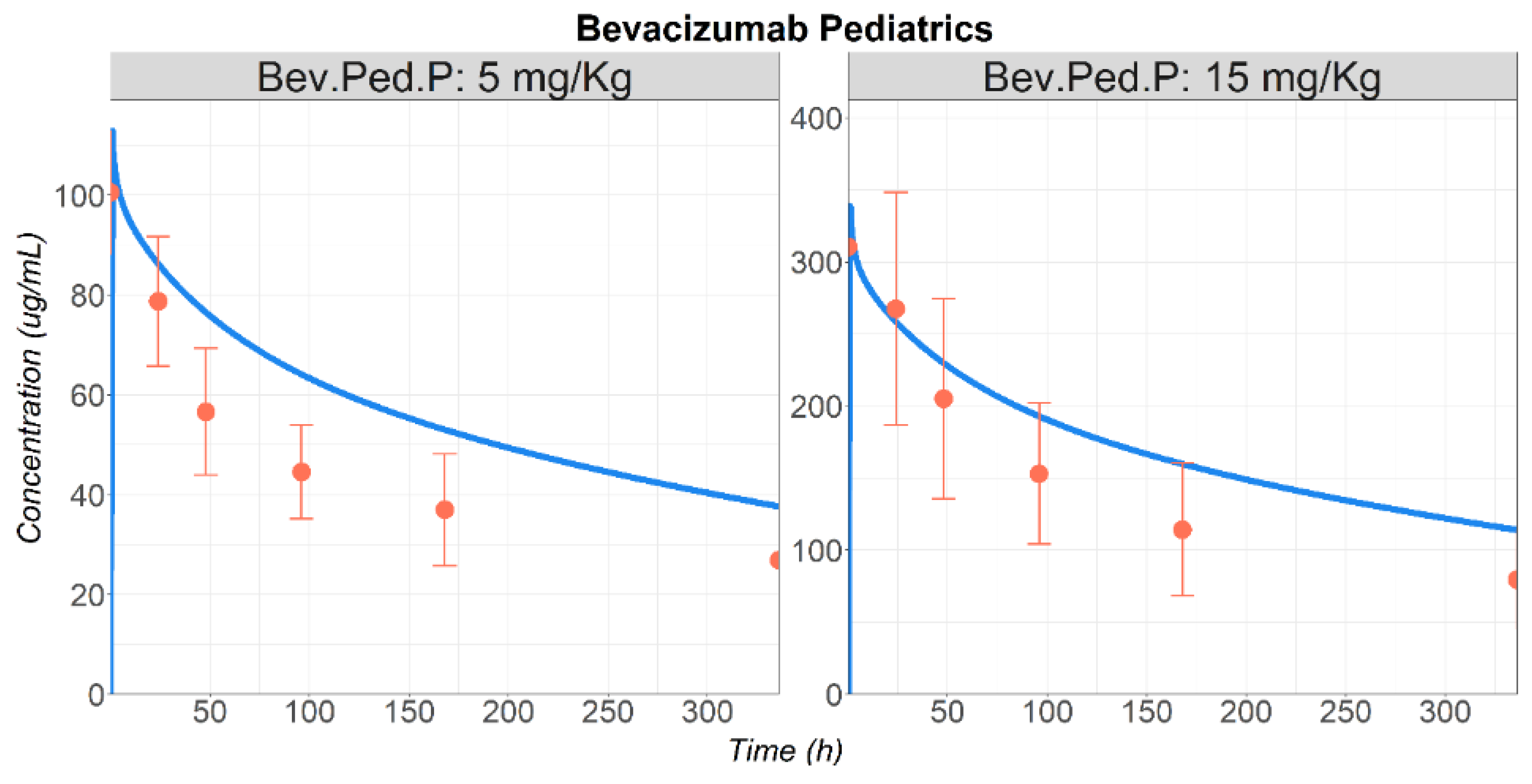
3.1. Bevacizumab
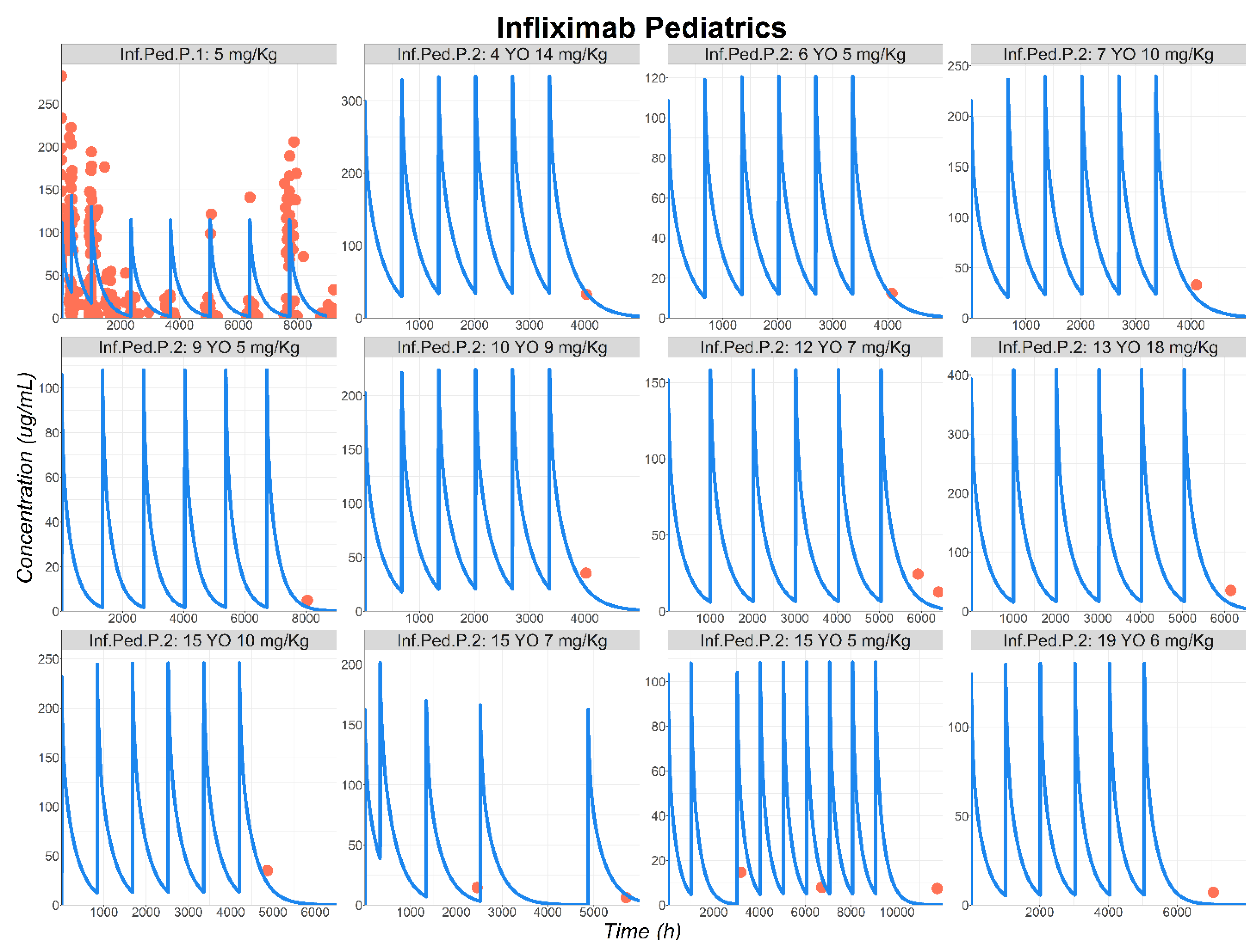
3.2. Infliximab
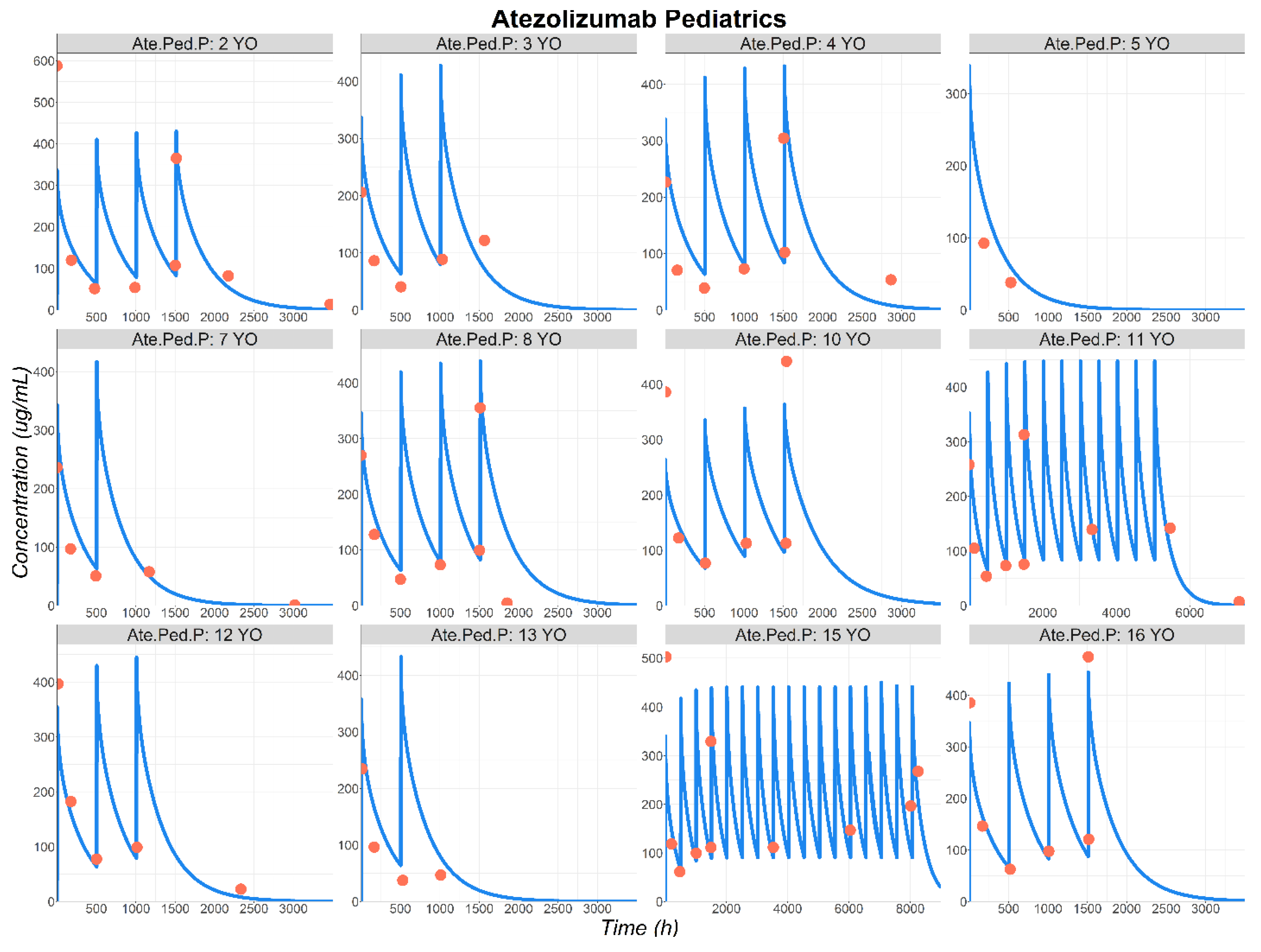
3.3. Atezolizumab
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Haraya, K.; Tsutsui, H.; Komori, Y.; Tachibana, T. Recent Advances in Translational Pharmacokinetics and Pharmacodynamics Prediction of Therapeutic Antibodies Using Modeling and Simulation. Pharmaceuticals 2022, 15, 508. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.N.; Ke, A.B. Physiologically Based Pharmacokinetic Modeling and Allometric Scaling in Pediatric Drug Development: Where Do We Draw the Line? J. Clin. Pharmacol. 2021, 61, S83–S93. [Google Scholar] [CrossRef]
- Zhang, X.; Yang, Y.; Grimstein, M.; Fan, J.; Grillo, J.A.; Huang, S.-M.; Zhu, H.; Wang, Y. Application of PBPK Modeling and Simulation for Regulatory Decision Making and Its Impact on US Prescribing Information: An Update on the 2018-2019 Submissions to the US FDA’s Office of Clinical Pharmacology. J. Clin. Pharmacol. 2020, 60, S160–S178. [Google Scholar] [CrossRef] [PubMed]
- Glassman, P.M.; Balthasar, J.P. Physiologically-based pharmacokinetic modeling to predict the clinical pharmacokinetics of monoclonal antibodies. J. Pharmacokinet. Pharmacodyn. 2016, 43, 427–446. [Google Scholar] [CrossRef] [PubMed]
- Liu, L. Pharmacokinetics of monoclonal antibodies and Fc-fusion proteins. Protein Cell 2018, 9, 15–32. [Google Scholar] [CrossRef]
- Pyzik, M.; Sand, K.M.K.; Hubbard, J.J.; Andersen, J.T.; Sandlie, I.; Blumberg, R.S. The Neonatal Fc Receptor (FcRn): A Misnomer? Front. Immunol. 2019, 10, 1540. [Google Scholar] [CrossRef]
- Niederalt, C.; Kuepfer, L.; Solodenko, J.; Eissing, T.; Siegmund, H.-U.; Block, M.; Willmann, S.; Lippert, J. A generic whole body physiologically based pharmacokinetic model for therapeutic proteins in PK-Sim. J. Pharmacokinet. Pharmacodyn. 2018, 45, 235–257. [Google Scholar] [CrossRef]
- Mager, D.E.; Jusko, W.J. General Pharmacokinetic Model for Drugs Exhibiting Target-Mediated Drug Disposition. J. Pharmacokinet. Pharmacodyn. 2001, 28, 507–532. [Google Scholar] [CrossRef]
- Yellepeddi, V.; Rower, J.; Liu, X.; Kumar, S.; Rashid, J.; Sherwin, C.M.T. State-of-the-Art Review on Physiologically Based Pharmacokinetic Modeling in Pediatric Drug Development. Clin. Pharmacokinet. 2019, 58, 1–13. [Google Scholar] [CrossRef]
- Basu, S.; Lien, Y.T.K.; Vozmediano, V.; Schlender, J.-F.; Eissing, T.; Schmidt, S.; Niederalt, C. Physiologically Based Pharmacokinetic Modeling of Monoclonal Antibodies in Pediatric Populations Using PK-Sim. Front. Pharmacol. 2020, 11, 868. [Google Scholar] [CrossRef]
- Chang, H.P.; Shakhnovich, V.; Frymoyer, A.; Funk, R.S.; Becker, M.L.; Park, K.T.; Shah, D.K. A population physiologically-based pharmacokinetic model to characterize antibody disposition in pediatrics and evaluation of the model using infliximab. Br. J. Clin. Pharmacol. 2022, 88, 290–302. [Google Scholar] [CrossRef] [PubMed]
- Malik, P.R.V.; Edginton, A.N. Physiologically-Based Pharmacokinetic Modeling vs. Allometric Scaling for the Prediction of Infliximab Pharmacokinetics in Pediatric Patients. CPT Pharmacomet. Syst. Pharmacol. 2019, 8, 835–844. [Google Scholar] [CrossRef]
- Huang, W.; Stader, F.; Chan, P.; Shemesh, C.S.; Chen, Y.; Gill, K.L.; Jones, H.M.; Li, L.; Rossato, G.; Wu, B.; et al. Development of a Pediatric Physiologically-Based Pharmacokinetic Model to Support Recommended Dosing of Atezolizumab in Children with Solid Tumors. Front. Pharmacol. 2022, 13, 974423. [Google Scholar] [CrossRef]
- Lim, A.; Sharma, P.; Stepanov, O.; Reddy, V.P. Application of Modelling and Simulation Approaches to Predict Pharmacokinetics of Therapeutic Monoclonal Antibodies in Pediatric Population. Pharmaceutics 2023, 15, 1552. [Google Scholar] [CrossRef]
- US FDA. FDA Label Bevacizumab [Internet]. 2004. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/125085s332lbl.pdf (accessed on 28 May 2025).
- US FDA. FDA Label Infliximab [Internet]. 1998. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/103772s5401lbl.pdf (accessed on 28 May 2025).
- US FDA. FDA Label Atezolizumab [Internet]. 2016. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/761034s025lbl.pdf (accessed on 28 May 2025).
- Kut, C.; Mac Gabhann, F.; Popel, A.S. Where is VEGF in the body? A meta-analysis of VEGF distribution in cancer. Br. J. Cancer 2007, 97, 978–985. [Google Scholar] [CrossRef] [PubMed]
- Raimondo, F.; Azzaro, M.P.; Palumbo, G.A.; Bagnato, S.; Stagno, F.; Giustolisi, G.M.; Cacciola, E.; Sortino, G.; Guglielmo, P.; Giustolisi, R. Elevated vascular endothelial growth factor (VEGF) serum levels in idiopathic myelofibrosis. Leukemia 2001, 15, 976–980. [Google Scholar] [CrossRef]
- Stefanini, M.O.; Wu, F.T.; Mac Gabhann, F.; Popel, A.S. A compartment model of VEGF distribution in blood, healthy and diseased tissues. BMC Syst. Biol. 2008, 2, 77. [Google Scholar] [CrossRef]
- Ternant, D.; Ducourau, E.; Perdriger, A.; Corondan, A.; Le Goff, B.; Devauchelle-Pensec, V.; Solau-Gervais, E.; Watier, H.; Goupille, P.; Paintaud, G.; et al. Relationship between inflammation and infliximab pharmacokinetics in rheumatoid arthritis. Br. J. Clin. Pharmacol. 2014, 78, 118–128. [Google Scholar] [CrossRef]
- Demarchi, M.; Coliat, P.; Barthelemy, P.; Schott, R.; BenAbdelghani, M.; Kim, M.; Hii, J.C.S.; Feyaerts, P.; Ang, F.R.X.; Derde, M.P.; et al. A randomized phase I study comparing the pharmacokinetics of a bevacizumab (HD204) biosimilar to European Union- and United States of America-sourced bevacizumab. PLoS ONE 2021, 16, e0248222. [Google Scholar] [CrossRef]
- Hettema, W.; Wynne, C.; Lang, B.; Altendorfer, M.; Czeloth, N.; Lohmann, R.; Athalye, S.; Schliephake, D. A randomized, single-blind, Phase I trial (INVICTAN-1) assessing the bioequivalence and safety of BI 695502, a bevacizumab biosimilar candidate, in healthy subjects. Expert Opin. Investig. Drugs 2017, 26, 889–896. [Google Scholar] [CrossRef]
- Hummel, M.; Bosje, T.; Shaw, A.; Liu, M.S.; Barve, A.; Kothekar, M.; Socinski, M.A.; Waller, C.F. A pharmacokinetics study of proposed bevacizumab biosimilar MYL-1402O vs EU-bevacizumab and US-bevacizumab. J. Cancer Res. Clin. Oncol. 2022, 148, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Wynne, C.; Xu, C.; Gan, Y.; Wang, C.; Thomas, B.E.; Yu, J.-C.; Li, S.; Zhang, L. A Global Phase I Clinical Study Comparing the Safety and Pharmacokinetics of Proposed Biosimilar BAT1706 and Bevacizumab (Avastin®) in Healthy Male Subjects. BioDrugs 2019, 33, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.; Lee, Y.J.; Choi, J.; Lee, D.; Park, M.; Petkova, M. A phase I, randomized, single-dose pharmacokinetic study comparing sb8 (bevacizumab biosimilar) with reference bevacizumab in healthy volunteers. Cancer Chemother. Pharmacol. 2020, 86, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Sinn, A.; García-Alvarado, F.; Gonzalez, V.; Huerga, C.; Bullo, F. A randomized, double blind, single dose, comparative study of the pharmacokinetics, safety and immunogenicity of MB02 (bevacizumab biosimilar) and reference bevacizumab in healthy male volunteers. Br. J. Clin. Pharmacol. 2022, 88, 1063–1073. [Google Scholar] [CrossRef]
- An, G. Concept of Pharmacologic Target-Mediated Drug Disposition in Large-Molecule and Small-Molecule Compounds. J. Clin. Pharmacol. 2020, 60, 149–163. [Google Scholar] [CrossRef]
- Romera, A. Bevacizumab biosimilar BEVZ92 versus reference bevacizumab in combination with FOLFOX or FOLFIRI as first-line treatment for metastatic colorectal cancer: A multicentre, open-label, randomised controlled trial. Lancet Gastroenterol. Hepatol. 2018, 3, 845–855. [Google Scholar] [CrossRef]
- Gordon, M.S.; Margolin, K.; Talpaz, M.; Sledge, G.W.; Holmgren, E.; Benjamin, R.; Stalter, S.; Shak, S.; Adelman, D. Phase I safety and pharmacokinetic study of recombinant human anti-vascular endothelial growth factor in patients with advanced cancer. J. Clin. Oncol. 2001, 19, 843–850. [Google Scholar] [CrossRef]
- Bender, J.L.G.; Adamson, P.C.; Reid, J.M.; Xu, L.; Baruchel, S.; Shaked, Y.; Kerbel, R.S.; Cooney-Qualter, E.M.; Stempak, D.; Chen, H.X.; et al. Phase I Trial and Pharmacokinetic Study of Bevacizumab in Pediatric Patients with Refractory Solid Tumors: A Children’s Oncology Group Study. J. Clin. Oncol. 2008, 26, 399–405. [Google Scholar] [CrossRef]
- Han, K.; Peyret, T.; Quartino, A.; Gosselin, N.H.; Gururangan, S.; Casanova, M.; Merks, J.H.M.; Massimino, M.; Grill, J.; Daw, N.C.; et al. Bevacizumab dosing strategy in paediatric cancer patients based on population pharmacokinetic analysis with external validation. Br. J. Clin. Pharmacol. 2016, 81, 148–160. [Google Scholar] [CrossRef]
- Shin, D.; Kim, Y.; Kim, Y.S.; Körnicke, T.; Fuhr, R. A Randomized, Phase I Pharmacokinetic Study Comparing SB2 and Infliximab Reference Product (Remicade(®)) in Healthy Subjects. BioDrugs Clin. Immunother. Biopharm. Gene Ther. 2015, 29, 381–388. [Google Scholar] [CrossRef]
- Park, W.; Lee, S.J.; Yun, J.; Yoo, D.H. Comparison of the pharmacokinetics and safety of three formulations of infliximab (CT-P13, EU-approved reference infliximab and the US-licensed reference infliximab) in healthy subjects: A randomized, double-blind, three-arm, parallel-group, single-dose, Phase I study. Expert Rev. Clin. Immunol. 2015, 11 (Suppl. S1), 25–31. [Google Scholar] [CrossRef]
- Yuan, D.; Rode, F.; Cao, Y. A Minimal Physiologically Based Pharmacokinetic Model with a Nested Endosome Compartment for Novel Engineered Antibodies. AAPS J. 2018, 20, 48. [Google Scholar] [CrossRef] [PubMed]
- Maini, R.N.; Breedveld, F.C.; Kalden, J.R.; Smolen, J.S.; Davis, D.; Macfarlane, J.D.; Antoni, C.; Leeb, B.; Elliott, M.J.; Woody, J.N.; et al. Therapeutic efficacy of multiple intravenous infusions of anti-tumor necrosis factor alpha monoclonal antibody combined with low-dose weekly methotrexate in rheumatoid arthritis. Arthritis Rheumatol. 1998, 41, 1552–1563. [Google Scholar] [CrossRef]
- Gottlieb, A.B.; Masuda, S.; Ramamurthi, R.; Abdulghani, A.; Romano, P.; Chaudhari, U.; Dooley, L.T.; Fasanmade, A.A.; Wagner, C.L. Pharmacodynamic and pharmacokinetic response to anti-tumor necrosis factor-α monoclonal antibody (infliximab) treatment of moderate to severe psoriasis vulgaris. J. Am. Acad. Dermatol. 2003, 48, 68–75. [Google Scholar] [CrossRef]
- Smolen, J.S.; Choe, J.-Y.; Prodanovic, N.; Niebrzydowski, J.; Staykov, I.; Dokoupilova, E.; Baranauskaite, A.; Yatsyshyn, R.; Mekic, M.; Porawska, W.; et al. Safety, immunogenicity and efficacy after switching from reference infliximab to biosimilar SB2 compared with continuing reference infliximab and SB2 in patients with rheumatoid arthritis: Results of a randomised, double-blind, phase III transition study. Ann. Rheum. Dis. 2018, 77, 234–240. [Google Scholar] [CrossRef] [PubMed]
- Fasanmade, A.A.; Adedokun, O.J.; Ford, J.; Hernandez, D.; Johanns, J.; Hu, C.; Davis, H.M.; Zhou, H. Population pharmacokinetic analysis of infliximab in patients with ulcerative colitis. Eur. J. Clin. Pharmacol. 2009, 65, 1211–1228. [Google Scholar] [CrossRef]
- Fasanmade, A.A.; Adedokun, O.J.; Blank, M.; Zhou, H.; Davis, H.M. Pharmacokinetic Properties of Infliximab in Children and Adults with Crohn’s Disease: A Retrospective Analysis of Data from 2 Phase III Clinical Trials. Clin. Ther. 2011, 33, 946–964. [Google Scholar] [CrossRef]
- Hyams, J.; Crandall, W.; Kugathasan, S.; Griffiths, A.; Olson, A.; Johanns, J.; Liu, G.; Travers, S.; Heuschkel, R.; Markowitz, J.; et al. Induction and Maintenance Infliximab Therapy for the Treatment of Moderate-to-Severe Crohn’s Disease in Children. Gastroenterology 2007, 132, 863–873. [Google Scholar] [CrossRef]
- Funk, R.S.; Shakhnovich, V.; Cho, Y.K.; Polireddy, K.; Jausurawong, T.; Gress, K.; Becker, M.L. Factors associated with reduced infliximab exposure in the treatment of pediatric autoimmune disorders: A cross-sectional prospective convenience sampling study. Pediatr. Rheumatol. 2021, 19, 62. [Google Scholar] [CrossRef]
- Piester, T.; Frymoyer, A.; Christofferson, M.; Yu, H.; Bass, D.; Park, K.T. A Mobile Infliximab Dosing Calculator for Therapy Optimization in Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2018, 24, 227–234. [Google Scholar] [CrossRef]
- Herbst, R.S.; Soria, J.-C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef] [PubMed]
- Geoerger, B.; Zwaan, C.M.; Marshall, L.V.; Michon, J.; Bourdeaut, F.; Casanova, M.; Corradini, N.; Rossato, G.; Farid-Kapadia, M.; Shemesh, C.S.; et al. Atezolizumab for children and young adults with previously treated solid tumours, non-Hodgkin lymphoma, and Hodgkin lymphoma (iMATRIX): A multicentre phase 1–2 study. Lancet Oncol. 2020, 21, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Shemesh, C.S.; Chanu, P.; Jamsen, K.; Wada, R.; Rossato, G.; Donaldson, F.; Garg, A.; Winter, H.; Ruppel, J.; Wang, X.; et al. Population pharmacokinetics, exposure-safety, and immunogenicity of atezolizumab in pediatric and young adult patients with cancer. J. Immunother. Cancer 2019, 7, 314. [Google Scholar] [CrossRef] [PubMed]
- Barber, J.; Al-Majdoub, Z.M.; Couto, N.; Howard, M.; Elmorsi, Y.; Scotcher, D.; Alizai, N.; De Wildt, S.; Stader, F.; Sepp, A.; et al. Toward systems-informed models for biologics disposition: Covariates of the abundance of the neonatal Fc Receptor (FcRn) in human tissues and implications for pharmacokinetic modelling. Eur. J. Pharm. Sci. 2023, 182, 106375. [Google Scholar] [CrossRef]
- Hardiansyah, D.; Ng, C.M. Effects of the FcRn developmental pharmacology on the pharmacokinetics of therapeutic monoclonal IgG antibody in pediatric subjects using minimal physiologically-based pharmacokinetic modelling. mAbs 2018, 10, 1144–1156. [Google Scholar] [CrossRef]
- Levy, G. Pharmacologic target-mediated drug disposition. Clin. Pharmacol. Ther. 1994, 56, 248–252. [Google Scholar] [CrossRef]
- Ober, R.J.; Martinez, C.; Vaccaro, C.; Zhou, J.; Ward, E.S. Visualizing the Site and Dynamics of IgG Salvage by the MHC Class I-Related Receptor, FcRn. J. Immunol. 2004, 172, 2021–2029. [Google Scholar] [CrossRef]
- Kavanaugh, A.; St Clair, E.W.; McCune, W.J.; Braakman, T.; Lipsky, P. Chimeric anti-tumor necrosis factor-alpha monoclonal antibody treatment of patients with rheumatoid arthritis receiving methotrexate therapy. J. Rheumatol. 2000, 27, 841–850. [Google Scholar]
- Elliott, M.J.; Maini, R.N.; Feldmann, M.; Long-Fox, A.; Charles, P.; Bijl, J.A.; Woody, J.N. Repeated therapy with monoclonal antibody to tumour necrosis factor α (cA2) in patients with rheumatoid arthritis. Lancet 1994, 344, 1125–1127. [Google Scholar] [CrossRef]
- Chaudhari, U.; Romano, P.; Mulcahy, L.; Dooley, L.; Baker, D.; Gottlieb, A. Efficacy and safety of infliximab monotherapy for plaque-type psoriasis: A randomised trial. Lancet 2001, 357, 1842–1847. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Bevacizumab (Healthy/Patient) | Infliximab (Healthy/Patient) | Atezolizumab (Patient) | |||
|---|---|---|---|---|---|---|
| Molecular weight (kDa) | 149 | [15] | 149.1 | [16] | 145 | [17] |
| Vascular Reflection Coefficient | 0.99 | Fitted | 0.99 | Fitted | 0.99 | Fitted |
| Lymphatic Reflection Coefficient | 0.63 | Fitted | 0.60 | Fitted | 0.29 | Fitted |
| Kon FcRn pH 6 (1/μM/day) | 8000 | GP default | 8000 | GP default | 8000 | GP default |
| Koff FcRn pH 6 (1/day) | 500 | GP default | 500 | GP default | 500 | GP default |
| Endosomal clearance (1/day) | 1.24 × 104 | Fitted | 1.34 × 104/ 2.00 × 104 | Fitted | 1.8 × 104 | Fitted |
| TMDD integrated | Yes | No | No | |||
| Kon VEGF-A (1/μM/day) | 30 | [20] | NA | |||
| Koff VEGF-A (1/day) | 0.006 | [20] | ||||
| Kint (1/day) | 1 | |||||
| Expression of VEGF-A (μmol/mL-plasma) | 1.96 × 10−6/ 3.86 × 10−6 | [18,19,20] | ||||
| Kdeg complex (1/day) | 0.173 | |||||
| Ksyn VEGF-A (μmol/g-plasma/day) | 0.34 × 10−6/ 0.67 × 10−6 | [18,19,20] | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zunino, C.; Gualano, V.; Zhou, H.; Lukacova, V.; Le Merdy, M. Prediction of Monoclonal Antibody Pharmacokinetics in Pediatric Populations Using PBPK Modeling and Simulation. Pharmaceutics 2025, 17, 884. https://doi.org/10.3390/pharmaceutics17070884
Zunino C, Gualano V, Zhou H, Lukacova V, Le Merdy M. Prediction of Monoclonal Antibody Pharmacokinetics in Pediatric Populations Using PBPK Modeling and Simulation. Pharmaceutics. 2025; 17(7):884. https://doi.org/10.3390/pharmaceutics17070884
Chicago/Turabian StyleZunino, Chiara, Virginie Gualano, Haiying Zhou, Viera Lukacova, and Maxime Le Merdy. 2025. "Prediction of Monoclonal Antibody Pharmacokinetics in Pediatric Populations Using PBPK Modeling and Simulation" Pharmaceutics 17, no. 7: 884. https://doi.org/10.3390/pharmaceutics17070884
APA StyleZunino, C., Gualano, V., Zhou, H., Lukacova, V., & Le Merdy, M. (2025). Prediction of Monoclonal Antibody Pharmacokinetics in Pediatric Populations Using PBPK Modeling and Simulation. Pharmaceutics, 17(7), 884. https://doi.org/10.3390/pharmaceutics17070884