



Screening for Polymorphism, Cyclodextrin Complexation, and Co-Crystallization of the Non-Steroidal Anti-Inflammatory Drug Fenbufen: Isolation and Characterization of a Co-Crystal and an Ionic Co-Crystal of the API with a Common Coformer
 ,
,  ,
,  ,
,  and
and
Abstract

1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Polymorph Screening
2.2.2. Complexation with CDs
2.2.3. Co-Crystal Screening
2.2.4. Co-Crystal/Ionic Co-Crystal Analysis
Powder X-Ray Diffraction (PXRD)
Single-Crystal X-Ray Diffraction (SCXRD)
Hot Stage Microscopy (HSM)
Thermogravimetric Analysis (TGA)
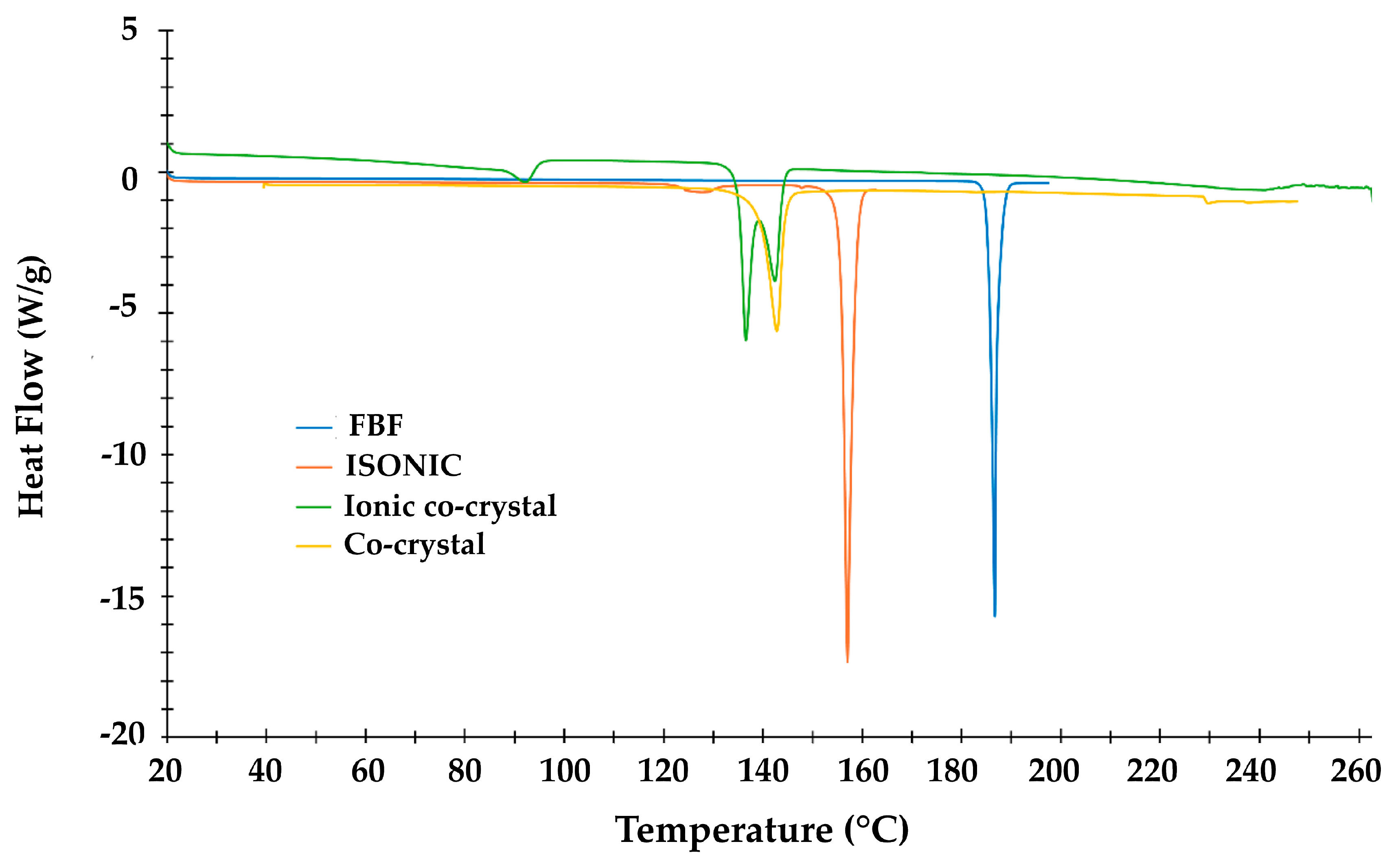
Differential Scanning Calorimetry (DSC)
Fourier-Transform Infrared (FT-IR) Spectroscopy
Elemental Analysis (EA)
Solubility Studies
3. Results and Discussion
3.1. Polymorph Screening
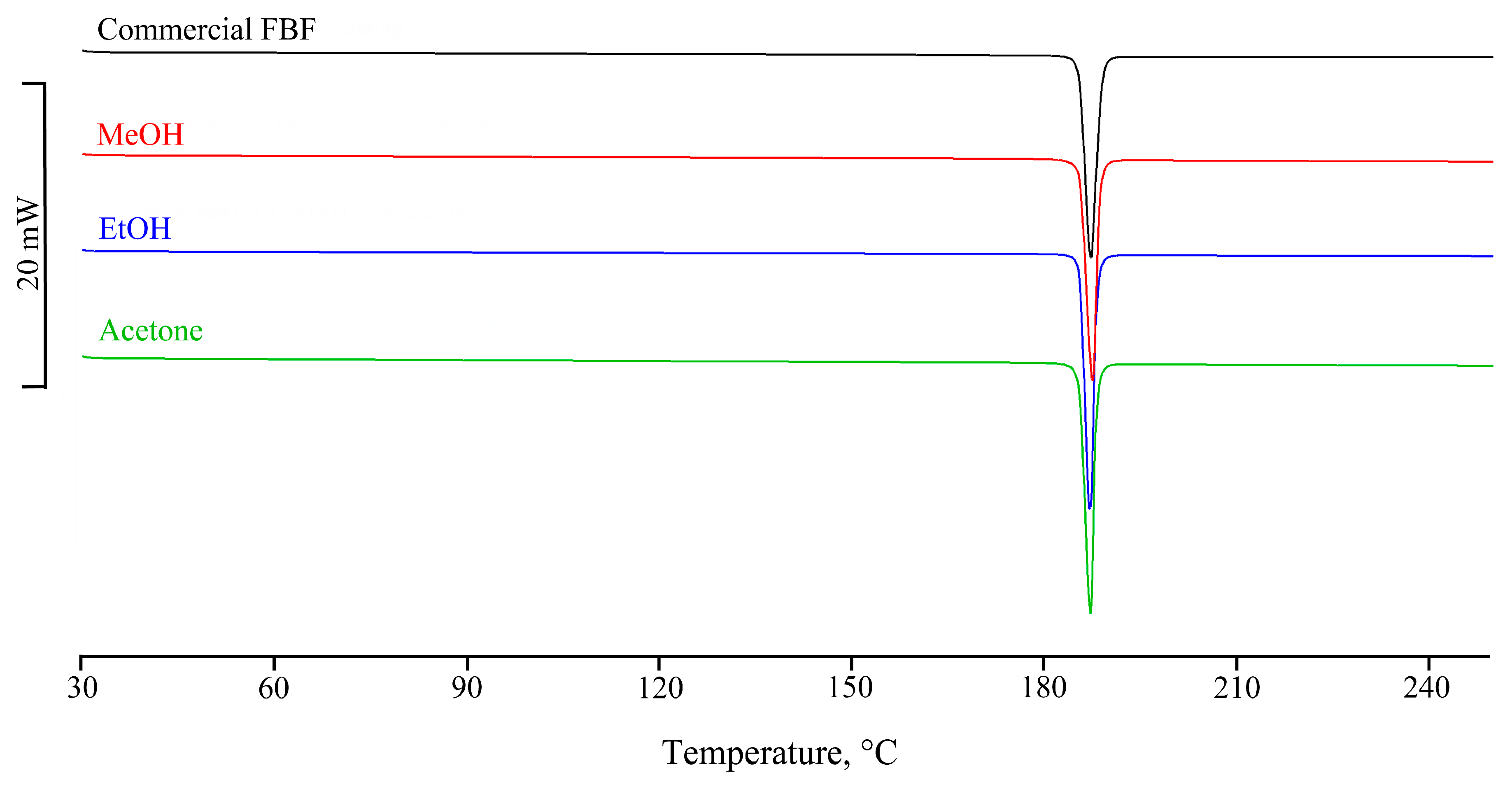
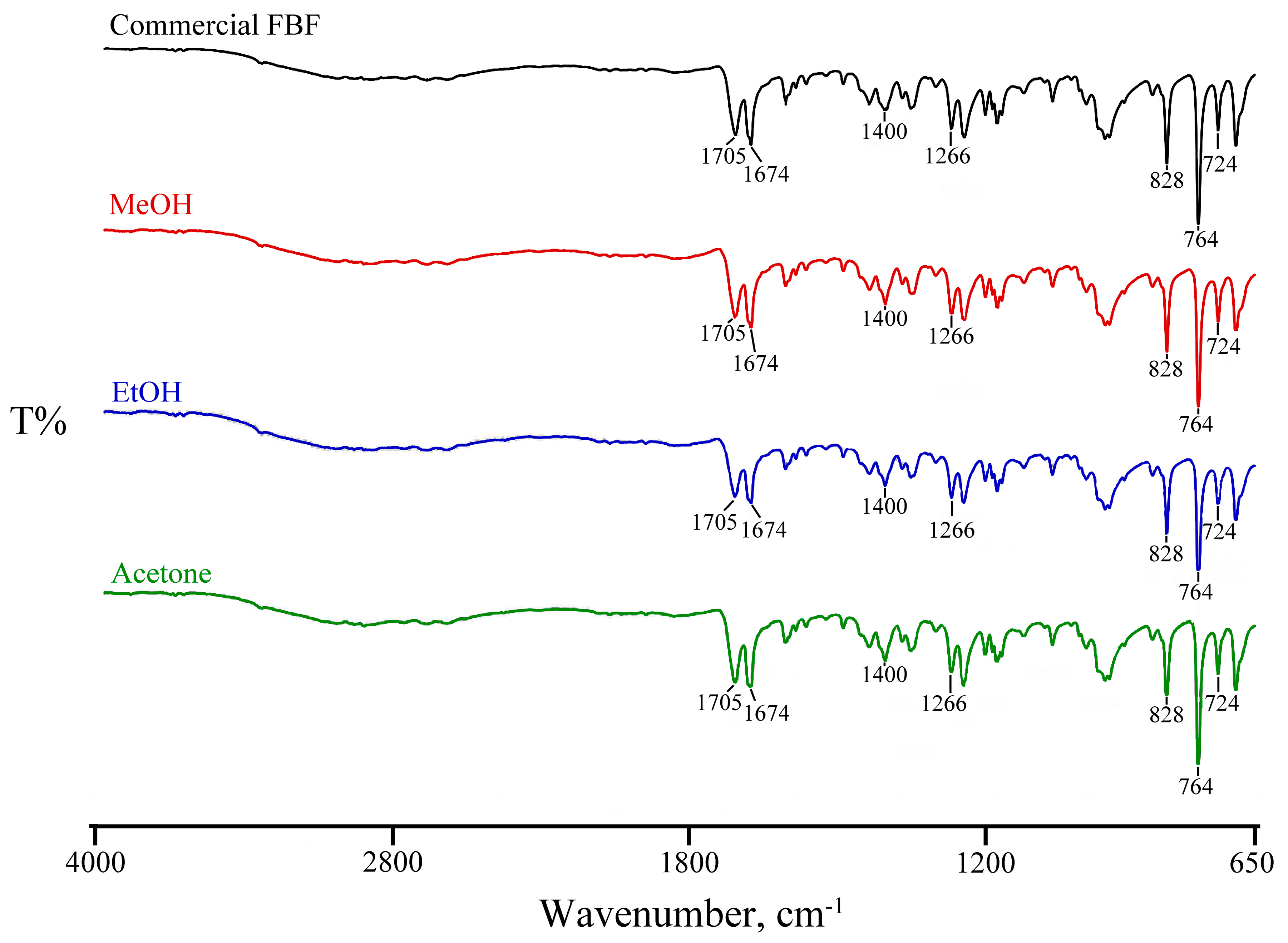
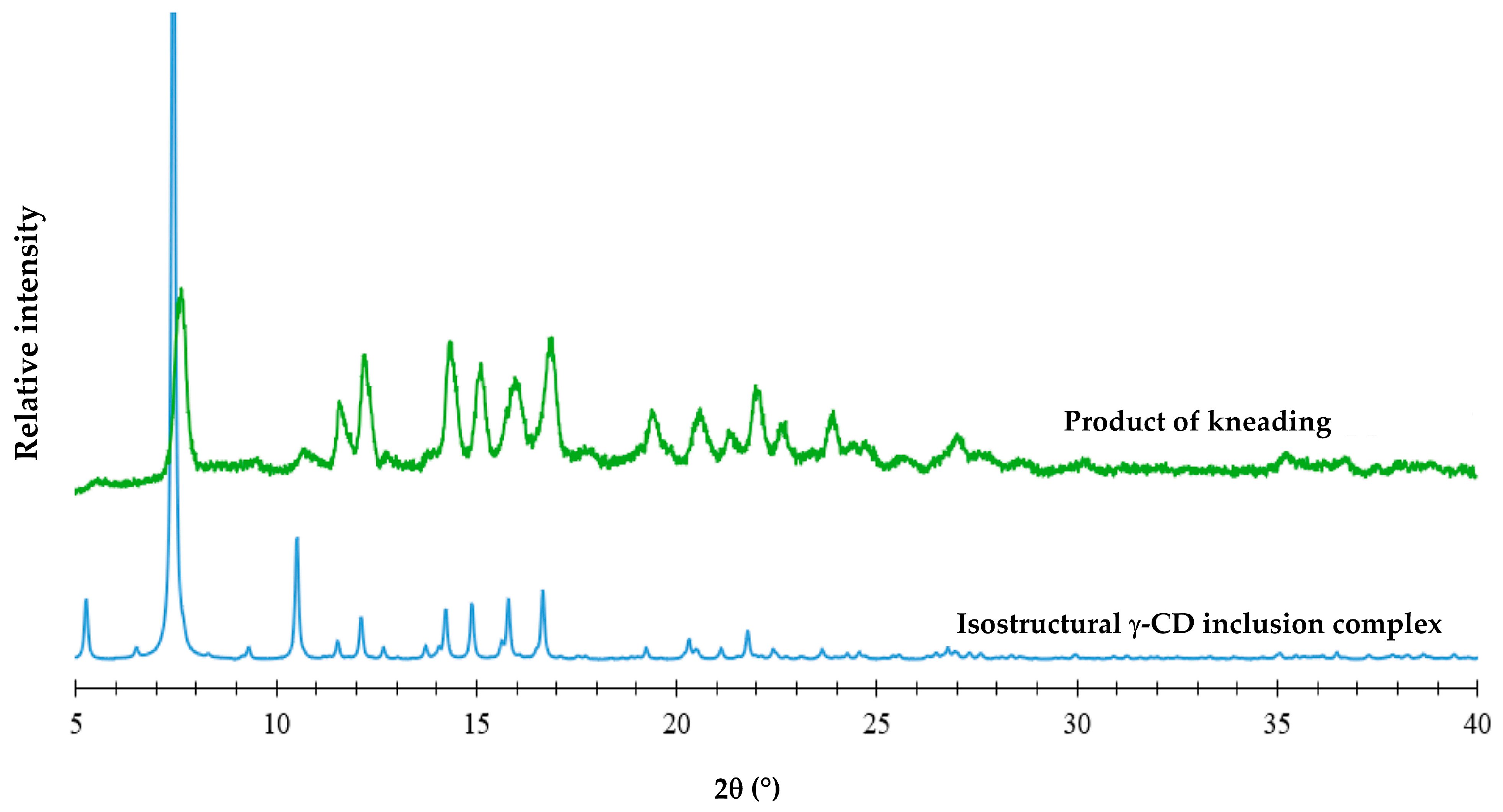
3.2. Attempted Complexation of FBF by Native Cyclodextrins
3.3. Co-Crystal and Ionic Co-Crystal Forms Containing FBF
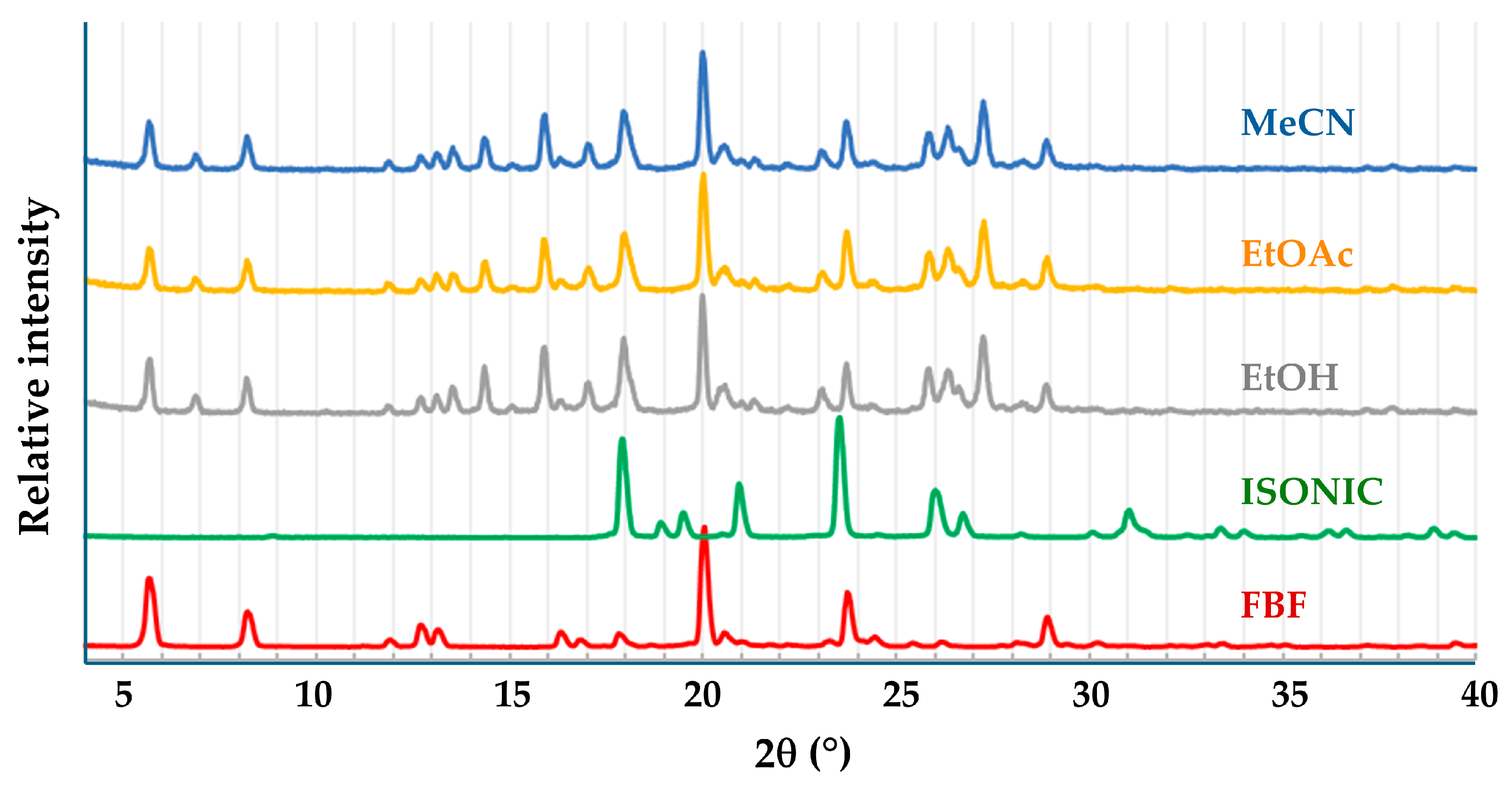
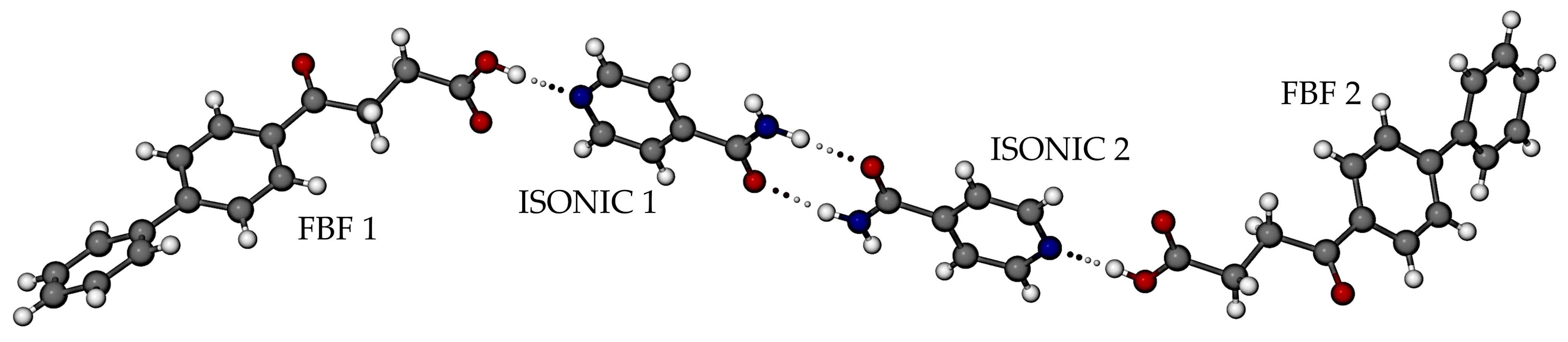
3.3.1. Co-Crystal FBF·ISONIC: Synthesis and Analysis
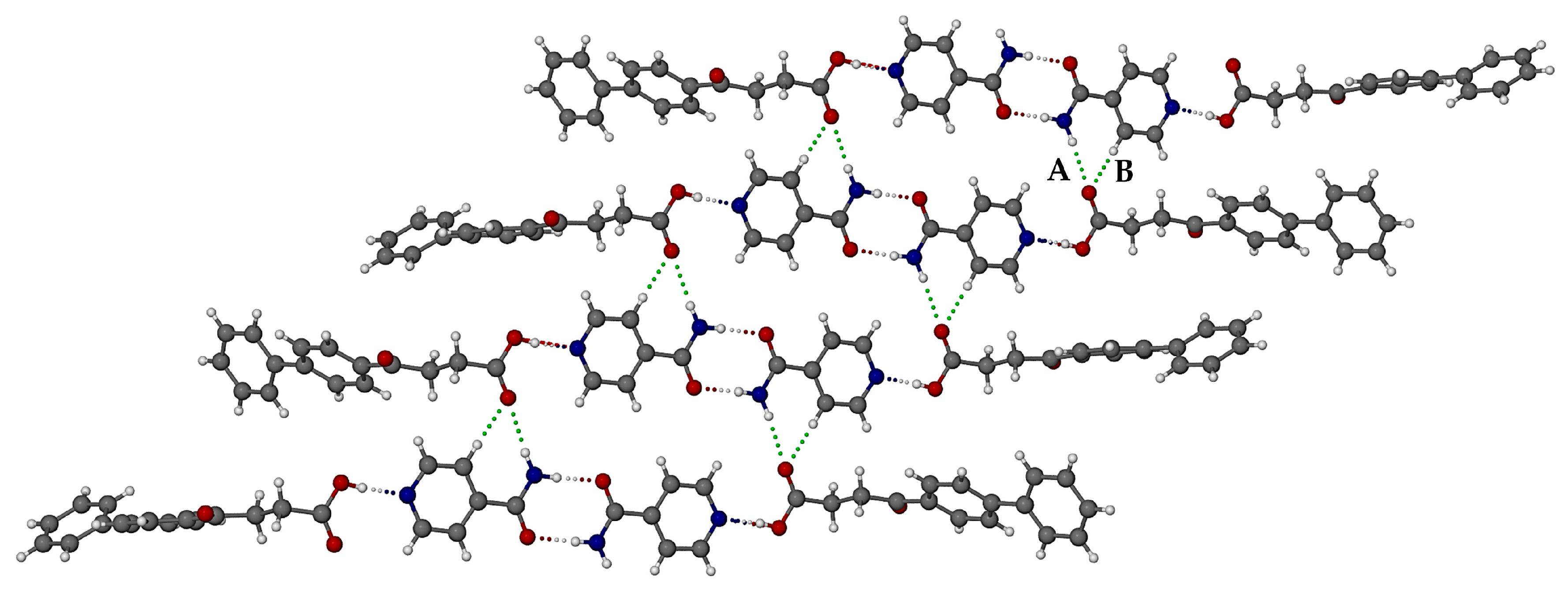
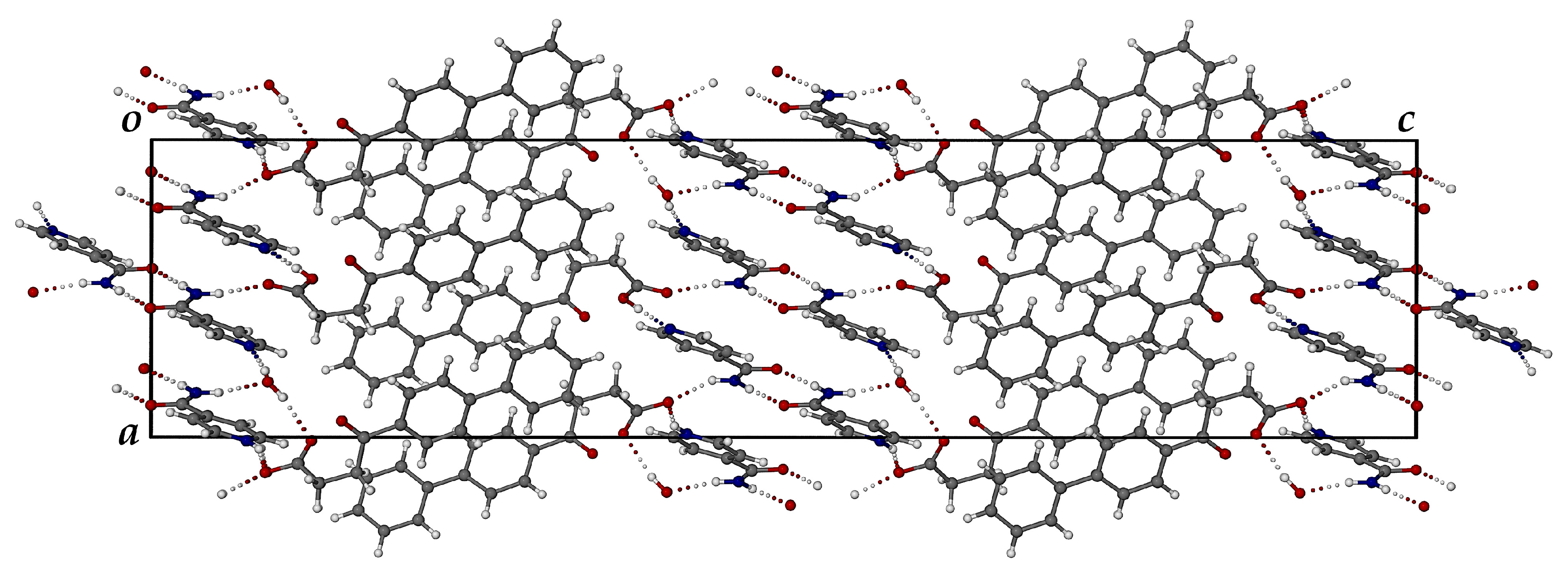
3.3.2. Ionic Co-Crystal Form: Synthesis and Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bernstein, J. Polymorphism of pharmaceuticals. In Polymorphism in Molecular Crystals; Oxford University Press: Oxford, UK, 2020; pp. 342–375. [Google Scholar] [CrossRef]
- Sarabia-Vallejo, Á.; del Mar Caja, M.; Olives, A.I.; Martín, M.A.; Menéndez, J.C. Cyclodextrin Inclusion Complexes for Improved Drug Bioavailability and Activity: Synthetic and Analytical Aspects. Pharmaceutics 2023, 15, 2345. [Google Scholar] [CrossRef] [PubMed]
- Poulson, B.J.; Alsulami, Q.A.; Sharfalddin, A.; El Agammy, E.F.; Mouffouk, F.; Emwas, A.-H.; Jaremko, L.; Jaremko, M. Cyclodextrins: Structural, Chemical, and Physical Properties, and Applications. Polysaccharides 2022, 3, 1. [Google Scholar] [CrossRef]
- Crini, G. A History of Cyclodextrins. Chem. Rev. 2014, 114, 10940–10975. [Google Scholar] [CrossRef] [PubMed]
- Bolla, G.; Sarma, B.; Nangia, A.K. Crystal Engineering of Pharmaceutical Cocrystals in the Discovery and Development of Improved Drugs. Chem. Rev. 2022, 122, 11514–11603. [Google Scholar] [CrossRef]
- Guo, M.; Sun, X.; Chen, J.; Cai, T. Pharmaceutical cocrystals: A review of preparations, physicochemical properties and applications. Acta Pharm. Sin. B 2021, 11, 2537–2564. [Google Scholar] [CrossRef]
- Thakuria, R.; Sarma, B. Drug-Drug and Drug-Nutraceutical Cocrystal/Salt as Alternative Medicine for Combination Therapy: A Crystal Engineering Approach. Crystals 2018, 8, 101. [Google Scholar] [CrossRef]
- Zhou, Y.; Lv, C.; Gao, X.L.J.; Gao, Y.; Wang, T.; Huang, X. An Overview on Polymorph Preparation Methods of Active Pharmaceutical Ingredients. Cryst. Growth Des. 2024, 24, 584–600. [Google Scholar] [CrossRef]
- Yao, C.; Zhang, S.; Wang, L.; Tao, X. Recent Advances in Polymorph Discovery Methods of Organic Crystals. Cryst. Growth Des. 2023, 23, 637–654. [Google Scholar] [CrossRef]
- Chistyakov, D.; Sergeev, G. The Polymorphism of Drugs: New Approaches to the Synthesis of Nanostructured Polymorphs. Pharmaceutics 2020, 12, 34. [Google Scholar] [CrossRef]
- Cruz-Cabeza, A.J.; Feeder, N.; Davey, R.J. Open questions in organic crystal polymorphism. Commun. Chem. 2020, 3, 142. [Google Scholar] [CrossRef]
- Hilfiker, R. (Ed.) Polymorphism: In the Pharmaceutical Industry; Wiley-VCH: Weinheim, Germany, 2006; ISBN 978-3-527-31146-0. [Google Scholar] [CrossRef]
- Censi, R.; Di Martino, P. Polymorph Impact on the Bioavailability and Stability of Poorly Soluble Drugs. Molecules 2015, 20, 18759–18776. [Google Scholar] [CrossRef]
- Dodziuk, H. (Ed.) Cyclodextrins and Their Complexes: Chemistry, Analytical Methods, Applications; Wiley-VCH Verlag GmbH & Co., KGaA: Weinheim, Germany, 2006. [Google Scholar] [CrossRef]
- Nicolaescu, O.E.; Belu, I.; Mocanu, A.G.; Manda, V.C.; Rău, G.; Pîrvu, A.S.; Ionescu, C.; Ciulu-Costinescu, F.; Popescu, M.; Ciocîlteu, M.V. Cyclodextrins: Enhancing Drug Delivery, Solubility and Bioavailability for Modern Therapeutics. Pharmaceutics 2025, 17, 288. [Google Scholar] [CrossRef] [PubMed]
- Musuc, A.M. Cyclodextrins: Advances in Chemistry, Toxicology, and Multifaceted Applications. Molecules. 2024, 29, 5319. [Google Scholar] [CrossRef]
- Mura, P. Analytical techniques for characterization of cyclodextrin complexes in the solid state: A review. J. Pharm. Biomed. Anal. 2015, 113, 226–238. [Google Scholar] [CrossRef] [PubMed]
- Friščić, T.; Jones, W. Cocrystal architecture and properties: Design and building of chiral and racemic structures by solid–solid reactions. Faraday Discuss 2007, 136, 167–229. [Google Scholar] [CrossRef]
- Dash, S.G.; Thakur, T.S. Computational Screening of Multicomponent Solid Forms of 2-Aryl Propionate Class of NSAID, Zaltoprofen, and Their Experimental Variation. Cryst. Growth Des. 2021, 21, 449–461. [Google Scholar] [CrossRef]
- Wathoni, N.; Sari, W.A.; Elamin, K.M.; Mohammed, A.F.A.; Suharyani, I. A Review of Coformer Utilization in Multicomponent Crystal Formation. Molecules 2022, 27, 8693. [Google Scholar] [CrossRef] [PubMed]
- Cerreia Vioglio, P.; Chierotti, M.R.; Gobetto, R. Pharmaceutical aspects of salt and cocrystal forms of APIs and characterization challenges. Adv. Drug Deliv. Rev. 2017, 117, 86–110. [Google Scholar] [CrossRef]
- Wang, X.; Du, S.; Zhang, R.; Jia, X.; Yang, T.; Zhang, X. Drug-drug cocrystals: Opportunities and challenges. Asian J. Pharm. Sci. 2021, 16, 307–317. [Google Scholar] [CrossRef]
- Chemical Book. Available online: https://www.chemicalbook.com/ProductChemicalPropertiesCB2239125_EN.htm (accessed on 29 April 2025).
- Moore, R.A.; Derry, S.; McQuay, H.J. Single dose oral fenbufen for acute postoperative pain in adults. Cochrane Database Syst. Rev. 2009, 2009, CD007547. [Google Scholar] [CrossRef]
- DrugBank Online. Available online: https://go.drugbank.com/drugs/DB08981 (accessed on 29 April 2025).
- LiverTox: Clinical and Research Information on Drug-Induced Liver Injury. In National Institute of Diabetes and Digestive and Kidney Diseases; Nonsteroidal Antiinflammatory Drugs (NSAIDs): Bethesda, MD, USA, 2012. Available online: https://www.ncbi.nlm.nih.gov/books/ (accessed on 25 April 2025).
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Cryst. 2016, B72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.-L.; Bu, F.-Z.; Yu, Y.-M.; Meng, S.-S.; Wu, Z.-Y.; Yan, C.W.; Li, Y.-T. The molecular salt of pyrimethamine and fenbufen for enhancing dissolubility via an assisted efficacy-increasing approach of dual-drug salt formation: A combined study including theory analysis and experiment validation. J. Mol. Struct. 2023, 1284, 135455. [Google Scholar] [CrossRef]
- Miranda, G.M.; Ramos, E.; Santos, V.O.; Bessa, J.R.; Teles, Y.C.F.; Yahouédéhou, S.C.M.A.; Goncalves, M.S.; Ribeiro-Filho, J. Inclusion Complexes of Non-Steroidal Anti-Inflammatory Drugs with Cyclodextrins: A Systematic Review. Biomolecules 2021, 11, 361. [Google Scholar] [CrossRef] [PubMed]
- Kraszni, M.; Ágh, F.; Horváth, D.; Mirzahosseini, A.; Horváth, P. Effect of Substitution Degree and Homogeneity on Cyclodextrin-Ligand Complex Stability: Comparison of Fenbufen and Fenoprofen Using CD and NMR Spectroscopy. Int. J. Mol. Sci. 2023, 24, 7544. [Google Scholar] [CrossRef]
- Smith, C.E.; Soti, S.; Jones, T.A.; Nakagawa, A.; Xue, D.; Yin, H. Non-steroidal Anti-inflammatory Drugs Are Caspase Inhibitors. Cell Chem. Biol. 2017, 16, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Xiao, W.; Jiang, W.; Li, K.; Guo, X.; Zong, G.; Wang, C.; Bao, J.; Chen, J.; Cheng, Z.; et al. Fenbufen Alleviates Severe Acute Pancreatitis by Suppressing Caspase-1/Caspase-11-mediated Pyroptosis in Mice. Curr. Mol. Pharmacol. 2024, 17, e110523216783. [Google Scholar] [CrossRef]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler Wood, P.A. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Crystallogr. 2020, 53 Pt 1, 226–235. [Google Scholar] [CrossRef]
- Program Analysis. Version 3.1 for Windows, Soft Imaging System GmbH, Digital Solutions for Imaging and Microscopy; Program Analysis: Münster, Germany, 2013. [Google Scholar]
- TA Instruments, Universal Analysis 2000 for Windows. Version 4.7A, TA Instruments—Waters LLC, New Castle, Delaware, © 1998–2009. Available online: https://www.pubcompare.ai/product/wSPhCZIBPBHhf-iFqyCY/ (accessed on 5 May 2025).
- TA Instruments, TRIOS. Version 4.1.0.31739, TA Instruments—Waters LLC, New Castle, Delaware, © 2016. Available online: https://www.tainstruments.com/support/software-downloads-support/downloads/ (accessed on 5 May 2025).
- Caira, M.R. On the isostructurality of cyclodextrin inclusion complexes and its practical utility. Rev. Roum. Chim. 2001, 46, 371–386. [Google Scholar]
- Pezzilli, R.; Morselli-Labate, A.M.; Corinaldesi, R. NSAIDs and Acute Pancreatitis: A Systematic Review. Pharmaceuticals 2010, 3, 558–571. [Google Scholar] [CrossRef]
- Vishweshwar, P.; Nangia, A.; Lynch, V.M. Recurrence of Carboxylic Acid−Pyridine Supramolecular Synthon in the Crystal Structures of Some Pyrazinecarboxylic Acids. J. Org. Chem 2002, 67, 556–565. [Google Scholar] [CrossRef]
- Good Laboratory Practice Bioscience. Available online: https://www.glpbio.com/fenbufen.html (accessed on 25 April 2025).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Title 1 | Ton (°C) | Tpeak (°C) | ΔH (Jg−1) |
|---|---|---|---|
| FBF | 186.0 | 187.0 | 159.1 |
| ISONIC | 155.8 | 157.4 | 209.2 |
| IONIC CO-CRYSTAL | 86.9 | 92.1 | 25.4 |
| 135.3 | 136.4 | 121.5 | |
| 139.0 | 142.3 | 44.7 | |
| CO-CRYSTAL | 128.5 | 143.3 | 118.8 |
| Solid Phase | Water (mg/mL) | FaSSIF, pH 6.5 (mg/mL) |
|---|---|---|
| FBF | 0.0022 | 6.92 ± 0.8 |
| FBF in ionic crystal | 0.68 | 8.65 ± 0.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frösler, H.M.; Mancapa, N.R.; Catenacci, L.; Sorrenti, M.; Bonferoni, M.C.; Caira, M.R. Screening for Polymorphism, Cyclodextrin Complexation, and Co-Crystallization of the Non-Steroidal Anti-Inflammatory Drug Fenbufen: Isolation and Characterization of a Co-Crystal and an Ionic Co-Crystal of the API with a Common Coformer. Pharmaceutics 2025, 17, 842. https://doi.org/10.3390/pharmaceutics17070842
Frösler HM, Mancapa NR, Catenacci L, Sorrenti M, Bonferoni MC, Caira MR. Screening for Polymorphism, Cyclodextrin Complexation, and Co-Crystallization of the Non-Steroidal Anti-Inflammatory Drug Fenbufen: Isolation and Characterization of a Co-Crystal and an Ionic Co-Crystal of the API with a Common Coformer. Pharmaceutics. 2025; 17(7):842. https://doi.org/10.3390/pharmaceutics17070842
Chicago/Turabian StyleFrösler, Hannah M., Neo Refiloe Mancapa, Laura Catenacci, Milena Sorrenti, Maria Cristina Bonferoni, and Mino R. Caira. 2025. "Screening for Polymorphism, Cyclodextrin Complexation, and Co-Crystallization of the Non-Steroidal Anti-Inflammatory Drug Fenbufen: Isolation and Characterization of a Co-Crystal and an Ionic Co-Crystal of the API with a Common Coformer" Pharmaceutics 17, no. 7: 842. https://doi.org/10.3390/pharmaceutics17070842
APA StyleFrösler, H. M., Mancapa, N. R., Catenacci, L., Sorrenti, M., Bonferoni, M. C., & Caira, M. R. (2025). Screening for Polymorphism, Cyclodextrin Complexation, and Co-Crystallization of the Non-Steroidal Anti-Inflammatory Drug Fenbufen: Isolation and Characterization of a Co-Crystal and an Ionic Co-Crystal of the API with a Common Coformer. Pharmaceutics, 17(7), 842. https://doi.org/10.3390/pharmaceutics17070842