
mPEG-PCL Nanoparticles to Improve Oral Bioavailability of Acalabrutinib: Effect of Polymer Lipophilicity and Hydrophilicity on Physicochemical Properties and In Vivo Performance in Rats



Abstract
1. Introduction
2. Materials and Methods
2.1. Materials and Animal Models
2.2. Instruments and Software Systems
2.3. Methods
2.3.1. Preparation and Characterization of Conjugated Polymer(s)
Preparation of mPEG-Polycaprolactone (mPEG-PCL) Co-Polymer(s)
Characterization of mPEG-PCL Co-Polymer(s)
2.3.2. Preparation of Polymer Lipid Hybrid Nanoparticles
Preformulation Studies and Preliminary Trials
Implementation of DoE
Method of Preparation
2.3.3. Method of Analysis Using HPLC-UV
2.3.4. Characterization of Optimized ACP-PLHNs
Determination of PS, PDI, and Surface Charge
Morphological Evaluation
Loading Efficiency (LE) and Entrapment Efficiency (EE)
Thermal Analysis
Powder X-Ray Diffraction (P-XRD)
Estimation of Residual Solvents
In Vitro Drug Release Studies
Stability Studies
Preventive Evaluations
Colloidal Stability
Hemolytic Potential
Effect on RBC Morphology
In Vivo Oral Pharmacokinetics (PK) and Tissue Distribution Study
Single Oral PK Studies
Drug Distribution Studies on Spleen
3. Results
3.1. Preparation and Characterization of Different Grades of mPEG-PCL Co-Polymer(s)
3.2. Preformulation Studies
3.2.1. Selection of Formulation Components
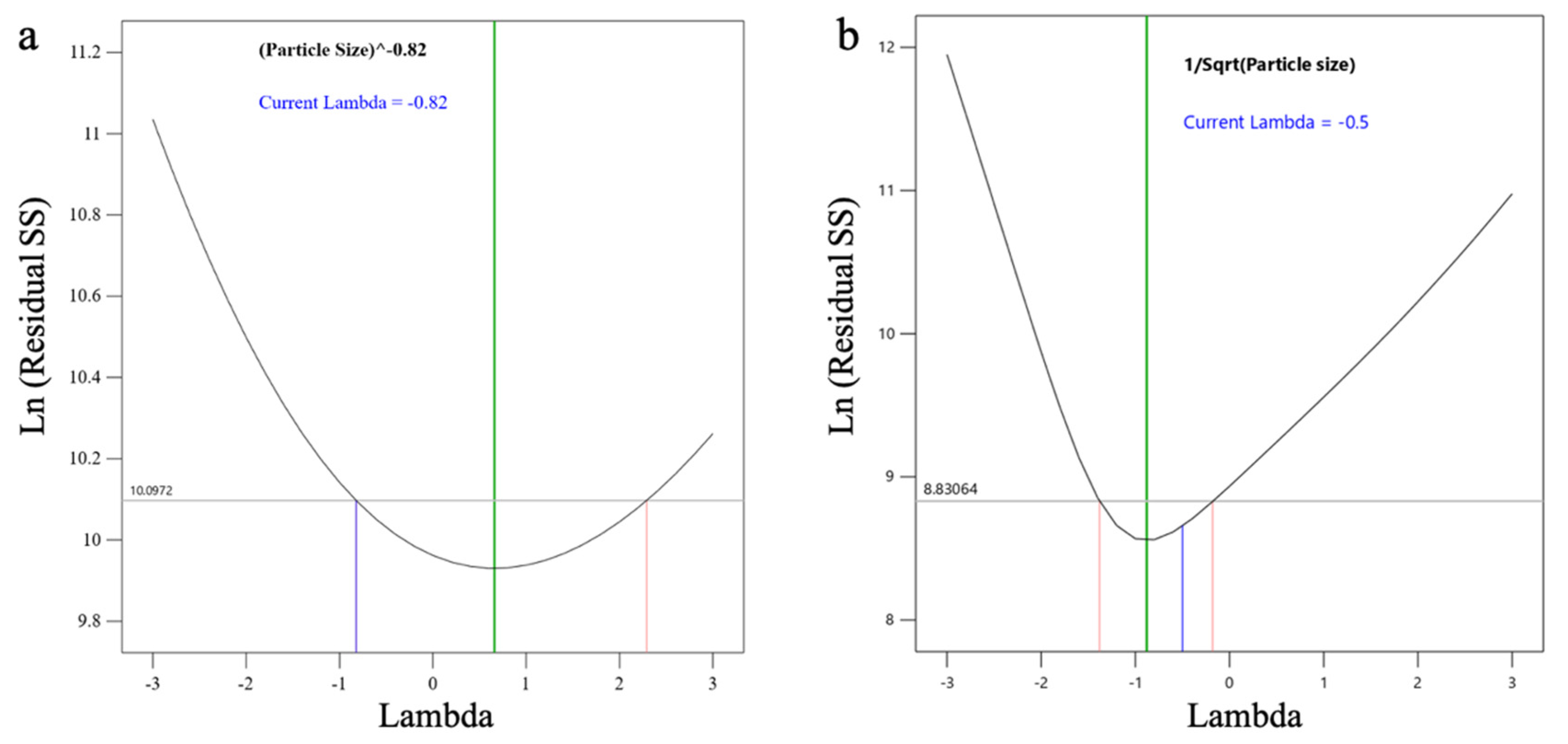
3.2.2. Selection of Critical Response Variables, Critical Factors, and Factorial Levels
3.3. Implementation of DoE
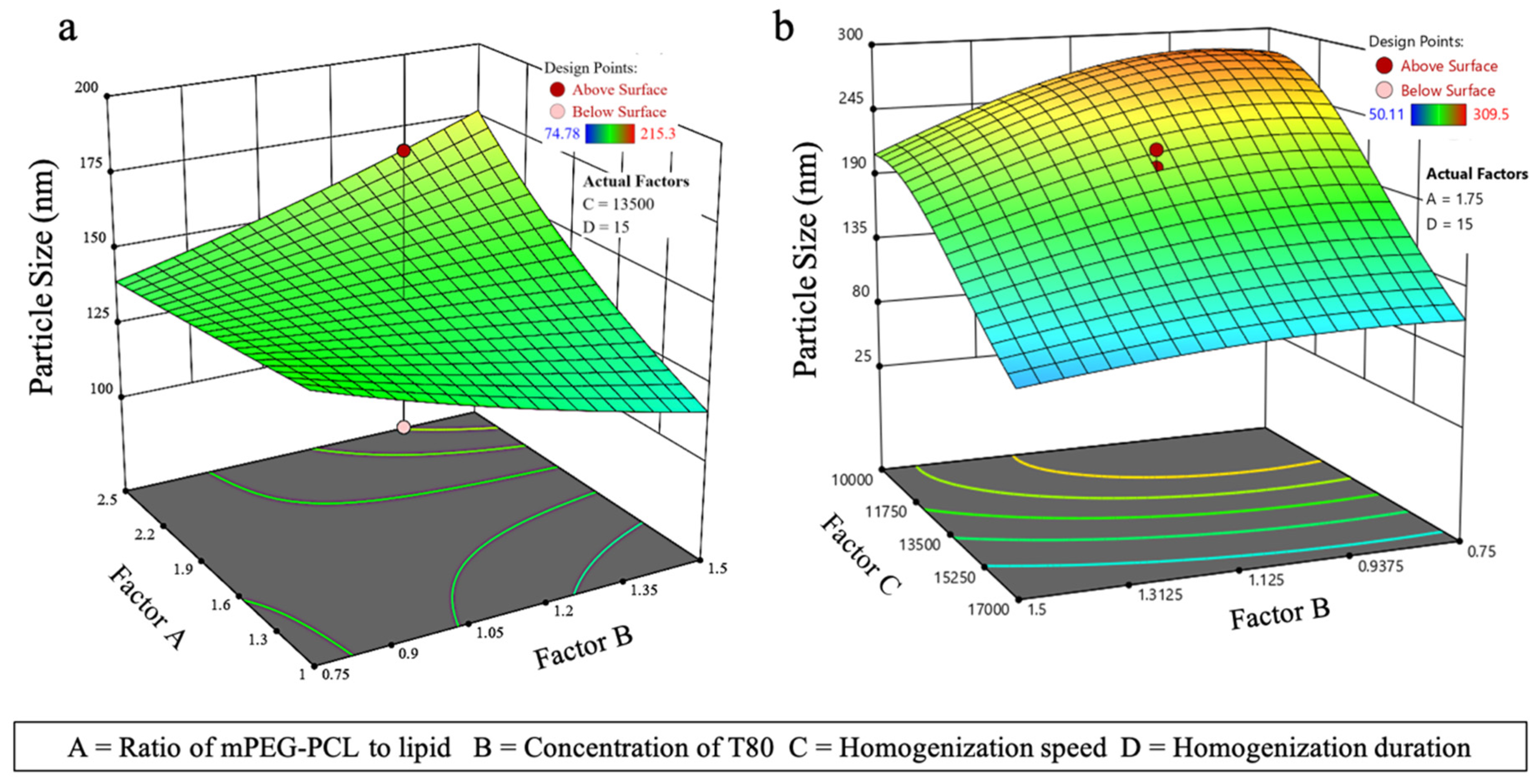
3.3.1. Effect on PS (Y1)
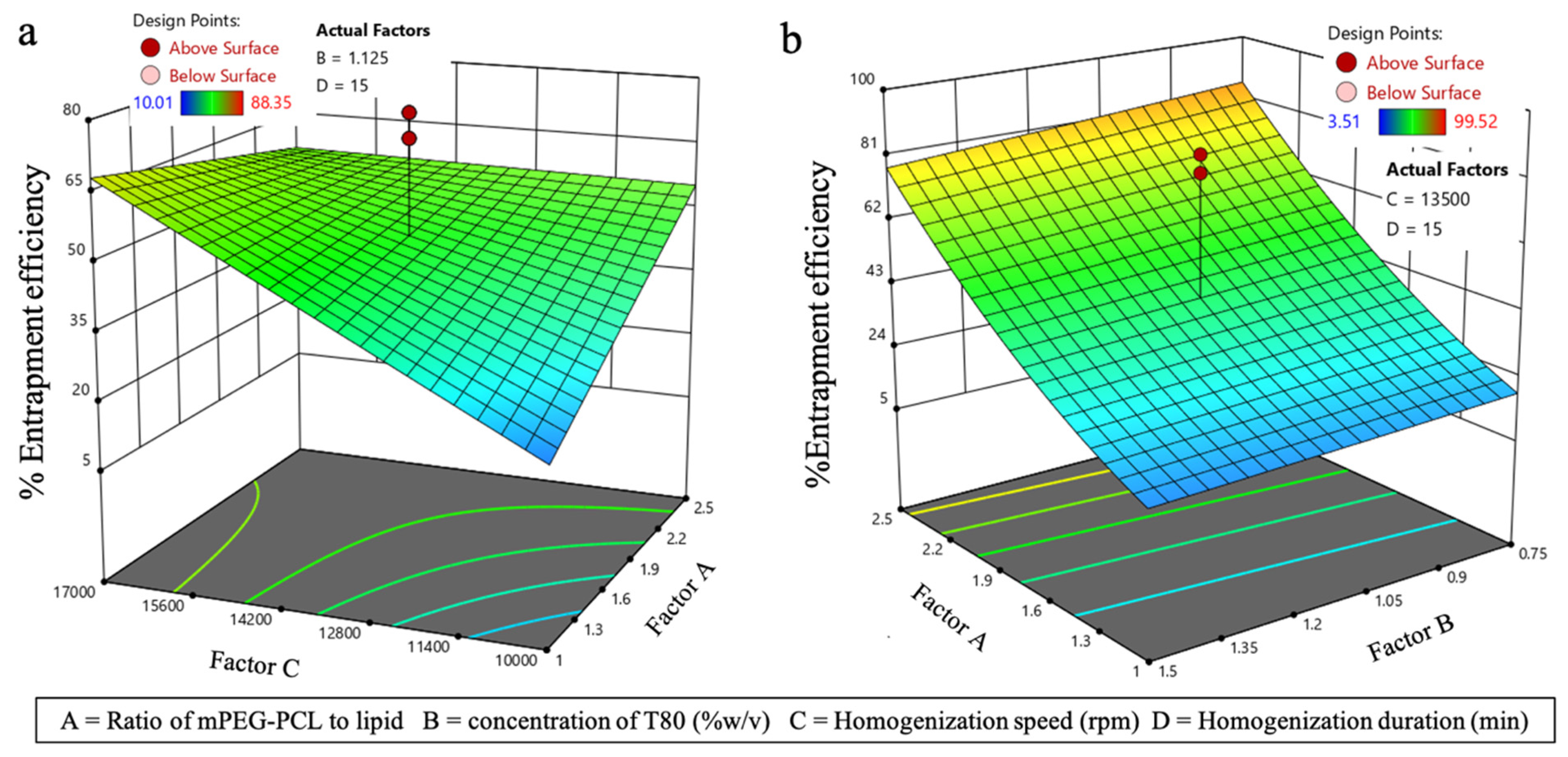
3.3.2. Effect on EE (Y2)
3.4. Physical Characterization of ACP-PLHNs
3.5. Loading Efficiency and Entrapment Efficiency
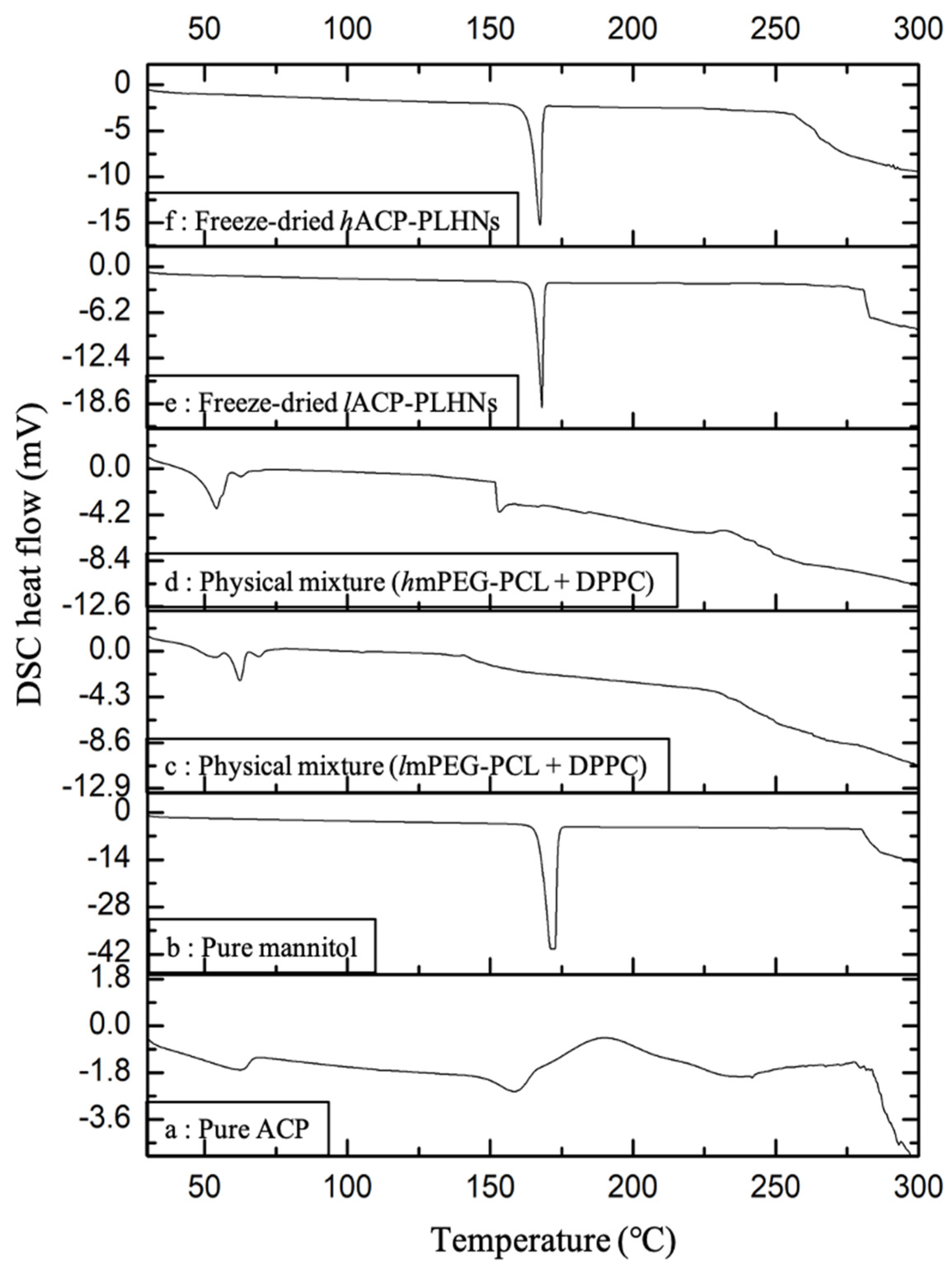
3.6. Thermal Analysis
3.7. P-XRD Analysis
3.8. Estimation of Residual Solvents
3.9. In Vitro Dissolution Study
3.10. Stability Studies
3.11. Preventive Evaluations
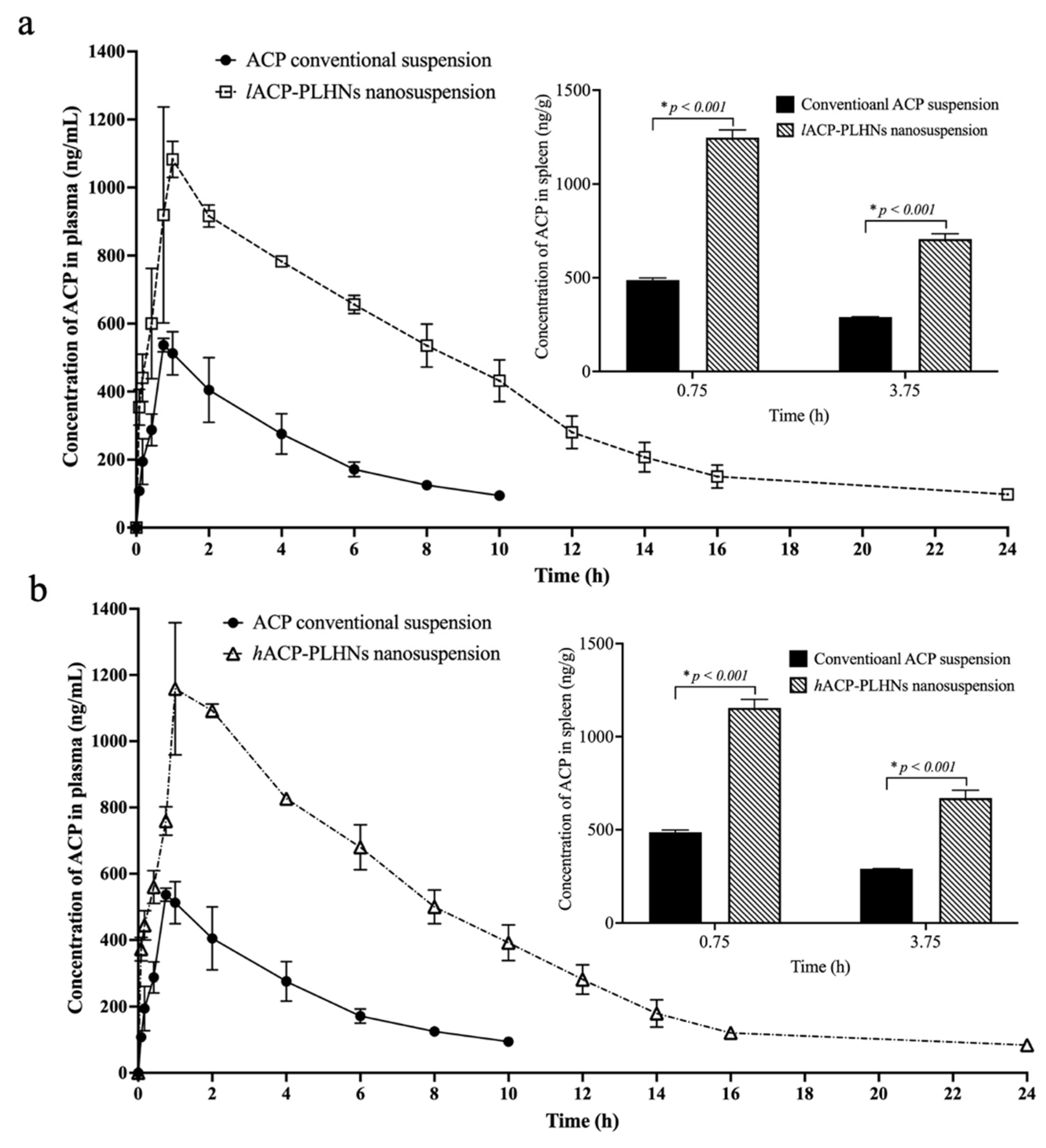
3.12. In Vivo PK and Tissue Distribution Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gajbhiye, K.R.; Salve, R.; Narwade, M.; Sheikh, A.; Kesharwani, P.; Gajbhiye, V. Lipid polymer hybrid nanoparticles: A custom-tailored next-generation approach for cancer therapeutics. Mol. Cancer 2023, 22, 160. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Kumar, M.; Kumar, P.; Verma, J.; Rosenholm, J.M.; Bansal, K.K.; Vaidya, A. Lipid–Polymer Hybrid Nanosystems: A Rational Fusion for Advanced Therapeutic Delivery. J. Funct. Biomater. 2023, 14, 437. [Google Scholar] [CrossRef] [PubMed]
- Date, T.; Nimbalkar, V.; Kamat, J.; Mittal, A.; Mahato, R.I.; Chitkara, D. Lipid-polymer hybrid nanocarriers for delivering cancer therapeutics. J. Control. Release 2018, 271, 60–73. [Google Scholar] [CrossRef]
- Persano, F.; Gigli, G.; Leporatti, S. Lipid-polymer hybrid nanoparticles in cancer therapy: Current overview and future directions. Nano Express 2021, 2, 012006. [Google Scholar] [CrossRef]
- Khalili, L.; Dehghan, G.; Sheibani, N.; Khataee, A. Smart active-targeting of lipid-polymer hybrid nanoparticles for therapeutic applications: Recent advances and challenges. Int. J. Biol. Macromol. 2022, 213, 166–194. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zang, A.; Wei, Y.; An, L.; Hong, D.; Shi, Y.; Zhang, J.; Su, S.; Fang, G. Hyaluronic acid capped, irinotecan and gene co-loaded lipid-polymer hybrid nanocarrier-based combination therapy platform for colorectal cancer. Drug Des. Dev. Ther. 2020, 14, 1095–1105. [Google Scholar] [CrossRef]
- Liu, Y.; Li, K.; Pan, J.; Liu, B.; Feng, S.-S. Folic acid conjugated nanoparticles of mixed lipid monolayer shell and biodegradable polymer core for targeted delivery of Docetaxel. Biomaterials 2010, 31, 330–338. [Google Scholar] [CrossRef]
- Kurd, M.; Malvajerd, S.S.; Rezaee, S.; Hamidi, M.; Derakhshandeh, K. Oral delivery of indinavir using mPEG-PCL nanoparticles: Preparation, optimization, cellular uptake, transport and pharmacokinetic evaluation. Artif. Cells Nanomed. Biotechnol. 2019, 47, 2123–2133. [Google Scholar] [CrossRef]
- Danafar, H. MPEG–PCL copolymeric nanoparticles in drug delivery systems. Cogent Med. 2016, 3, 1142411. [Google Scholar] [CrossRef]
- Khodaei, M.; Rostamizadeh, K.; Taromchi, A.H.; Monirinasab, H.; Fathi, M. DDAB cationic lipid-mPEG, PCL copolymer hybrid nano-carrier synthesis and application for delivery of siRNA targeting IGF-1R into breast cancer cells. Clin. Transl. Oncol. 2021, 23, 1167–1178. [Google Scholar] [CrossRef]
- Chen, Y.; Lu, Y.; Hu, D.; Peng, J.; Xiao, Y.; Hao, Y.; Pan, M.; Yuan, L.; Qian, Z. Cabazitaxel-loaded MPEG-PCL copolymeric nanoparticles for enhanced colorectal cancer therapy. Appl. Mater. Today 2021, 25, 101210. [Google Scholar] [CrossRef]
- Piazza, R.D.; Brandt, J.V.; Gobo, G.G.; Tedesco, A.C.; Primo, F.L.; Marques, R.F.C.; Junior, M.J. mPEG-co-PCL nanoparticles: The influence of hydrophobic segment on methotrexate drug delivery. Colloids Surfaces A Physicochem. Eng. Asp. 2018, 555, 142–149. [Google Scholar] [CrossRef]
- Wang, B.L.; Shen, Y.M.; Zhang, Q.W.; Li, Y.L.; Luo, M.; Liu, Z.; Li, Y.; Qian, Z.Y.; Gao, X.; Shi, H.S. Codelivery of curcumin and doxorubicin by MPEG-PCL results in improved efficacy of systemically administered chemotherapy in mice with lung cancer. Int. J. Nanomed. 2013, 8, 3521–3531. [Google Scholar] [CrossRef]
- Peng, W.; Jiang, X.-Y.; Zhu, Y.; Omari-Siaw, E.; Deng, W.-W.; Yu, J.-N.; Xu, X.-M.; Zhang, W.-M. Oral delivery of capsaicin using MPEG-PCL nanoparticles. Acta Pharmacol. Sin. 2014, 36, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Danafar, H.; Jaberizadeh, H.; Andalib, S. In vitro and in vivo delivery of gliclazide loaded mPEG-PCL micelles and its kinetic release and solubility study. Artif. Cells Nanomed. Biotechnol. 2018, 46, 1625–1636. [Google Scholar] [CrossRef]
- Wilmington, D. CALQUENCE® (Acalabrutinib) Tablets [Prescribing Information], 2022. Available online: https://www.fda.gov/medwatch (accessed on 21 January 2025).
- Center for Drug Evaluation and Research, Acalabrutinib (Package Insert), 2017. Available online: https://www.fda.gov/medwatch (accessed on 21 January 2025).
- Sinha, S.; Ravi, P.R.; Somvanshi, M.; Rajesh, R.S. Development and Validation of a Simple HPLC–UV-Based Bioanalytical Method for Estimation of Acalabrutinib in Rat Plasma and Its Application in Evaluation of Drug-Loaded Nanocrystal Formulation. Sep. Sci. Plus 2024, 7, e202400110. [Google Scholar] [CrossRef]
- Shettiwar, A.; Gupta, U.; Chatterjee, E.; Patra, B.; Aalhate, M.; Mahajan, S.; Maji, I.; Mehra, N.K.; Guru, S.K.; Singh, P.K. Oral linalool-based nanoemulsion of acalabrutinib for ameliorating its oral bioavailability and in vitro anticancer potential in T lymphoblast cell lines. Colloid Polym. Sci. 2024, 302, 1491–1511. [Google Scholar] [CrossRef]
- Danafar, H.; Davaran, S.; Rostamizadeh, K.; Valizadeh, H.; Hamidi, M. Biodegradable m-PEG/PCL Core-Shell Micelles: Preparation and Characterization as a Sustained Release Formulation for Curcumin. Adv. Pharm. Bull. 2014, 4, 501. [Google Scholar] [CrossRef]
- Murthy, A.; Ravi, P.R.; Kathuria, H.; Vats, R. Self-assembled lecithin-chitosan nanoparticles improve the oral bioavailability and alter the pharmacokinetics of raloxifene. Int. J. Pharm. 2020, 588, 119731. [Google Scholar] [CrossRef]
- Mukherjee, A.; Waters, A.K.; Kalyan, P.; Achrol, A.S.; Kesari, S.; Yenugonda, V.M. Lipid–polymer hybrid nanoparticles as a next-generation drug delivery platform: State of the art, emerging technologies, and perspectives. Int. J. Nanomed. 2019, 14, 1937–1952. [Google Scholar] [CrossRef]
- Mohanty, A.K.; Jana, U.; Manna, P.K.; Mohanta, G.P. Synthesis and evaluation of MePEG-PCL diblock copolymers: Surface properties and controlled release behavior. Prog. Biomater. 2015, 4, 89. [Google Scholar] [CrossRef] [PubMed]
- Hemmati, K.; Alizadeh, R.; Ghaemy, M. Synthesis and characterization of controlled drug release carriers based on functionalized amphiphilic block copolymers and super-paramagnetic iron oxide nanoparticles. Polym. Adv. Technol. 2015, 27, 504–514. [Google Scholar] [CrossRef]
- Khodaverdi, E.; Tayarani-Najaran, Z.; Minbashi, E.; Alibolandi, M.; Hossein, J.; Sepahi, S.; Kamali, H.; Hadizadeh, F. Docetaxel-Loaded Mixed Micelles and Polymersomes Composed of Poly (caprolactone)-Poly (ethylene glycol) (PEG-PCL) and Poly (lactic acid)-Poly (ethylene glycol) (PEG-PLA): Preparation and In-vitro Characterization. Iran. J. Pharm. Res. 2019, 18, 142. [Google Scholar] [PubMed]
- Khanmohammadi, M.R.; Azqhandi, M.H.A.; Farahani, B.V.; Najafipour, R. Methoxy poly(ethylene glycol)-block-polycaprolactone copolymer: Formulation and optimization by experimental design; determination of diblock molar mass by multivariate regression analysis of 1H NMR spectra. Anal. Methods 2013, 5, 2840–2847. [Google Scholar] [CrossRef]
- Zhong, Y.; Tong, R. Living Ring-Opening Polymerization of O-Carboxyanhydrides: The Search for Catalysts. Front. Chem. 2018, 6, 641. [Google Scholar] [CrossRef]
- Chen, T.; Qin, Z.; Qi, Y.; Deng, T.; Ge, X.; Wang, J.; Hou, X. Degradable polymers from ring-opening polymerization of α-angelica lactone, a five-membered unsaturated lactone. Polym. Chem. 2011, 2, 1190–1194. [Google Scholar] [CrossRef]
- Sinha, S.; Ravi, P.R.; Somvanshi, M.; Rashmi, S.R. Solid lipid nanoparticles for increased oral bioavailability of acalabrutinib in chronic lymphocytic leukaemia. Nanoscale Res. Lett. 2024, 19, 218. [Google Scholar] [CrossRef]
- Zein, R.; Sharrouf, W.; Selting, K. Physical Properties of Nanoparticles That Result in Improved Cancer Targeting. J. Oncol. 2020, 2020, 5194780. [Google Scholar] [CrossRef]
- Albanese, A.; Tang, P.S.; Chan, W.C.W. The effect of nanoparticle size, shape, and surface chemistry on biological systems. Annu. Rev. Biomed. Eng. 2012, 14, 1–16. [Google Scholar] [CrossRef]
- Singh, R.; Lillard, J.W. Nanoparticle-based targeted drug delivery. Exp. Mol. Pathol. 2009, 86, 215. [Google Scholar] [CrossRef]
- Cheow, W.S.; Hadinoto, K. Factors affecting drug encapsulation and stability of lipid–polymer hybrid nanoparticles. Colloids Surfaces B Biointerfaces 2011, 85, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Matta, H.B.; Tripathi, R.M.; Kazi, M.A.; Khanna, M.S.; Prasad, M.; Shaikh, S.S. Crystalline Polymorphic Forms of Acalabrutinib. WIPO2019/159097A1, 2019. [Google Scholar]
- Impurities: Guideline for Residual Solvents Q3C(R8), Rockville, MD, USA, 2021. Available online: https://database.ich.org/sites/default/files/ICH_Q3C-R8_Guideline_Step4_2021_0422_1.pdf (accessed on 22 October 2024).
- Toxicological Profile for Chloroform- Draft for Public Comment, Atlanta, Georgia, 2024. Available online: https://www.atsdr.cdc.gov/toxprofiles/tp6.pdf (accessed on 22 October 2024).
- Pepin, X.J.; Sanderson, N.J.; Blanazs, A.; Grover, S.; Ingallinera, T.G.; Mann, J.C. Bridging in vitro dissolution and in vivo exposure for acalabrutinib. Part I. Mechanistic modelling of drug product dissolution to derive a P-PSD for PBPK model input. Eur. J. Pharm. Biopharm. 2019, 142, 421–434. [Google Scholar] [CrossRef] [PubMed]
- Sisson, A.L.; Ekinci, D.; Lendlein, A. The contemporary role of ε-caprolactone chemistry to create advanced polymer architectures. Polymer 2013, 54, 4333–4350. [Google Scholar] [CrossRef]
- Leonenko, Z.; Finot, E.; Ma, H.; Dahms, T.S.; Cramb, D. Investigation of Temperature-Induced Phase Transitions in DOPC and DPPC Phospholipid Bilayers Using Temperature-Controlled Scanning Force Microscopy. Biophys. J. 2004, 86, 3783–3793. [Google Scholar] [CrossRef]
- Lazzari, S.; Moscatelli, D.; Codari, F.; Salmona, M.; Morbidelli, M.; Diomede, L. Colloidal stability of polymeric nanoparticles in biological fluids. J. Nanopart. Res. 2012, 14, 920. [Google Scholar] [CrossRef]
- Lampert, A.I.; Wotherspoon, A.; Van Noorden, S.; Hasserjian, R.P. High expression of CD23 in the proliferation centers of chronic lymphocytic leukemia in lymph nodes and spleen. Hum. Pathol. 1999, 30, 648–654. [Google Scholar] [CrossRef]
- Feng, G.; Wang, X. Role of spleen tyrosine kinase in the pathogenesis of chronic lymphocytic leukemia. Leuk. Lymphoma 2014, 55, 2699–2705. [Google Scholar] [CrossRef]
- Rawstron, A.C.; Bennett, F.L.; O’COnnor, S.J.; Kwok, M.; Fenton, J.A.; Plummer, M.; de Tute, R.; Owen, R.G.; Richards, S.J.; Jack, A.S.; et al. Monoclonal B-cell lymphocytosis and chronic lymphocytic leukemia. N. Engl. J. Med. 2008, 359, 575–583. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Factor Code | Factors and Units | High Level (+1) | Low Level (−1) | Alpha High (+α) | Alpha Low (−α) | Center Point Level |
|---|---|---|---|---|---|---|
| A | Ratio of mPEG-PCL to lipid | 2.5 | 1 | 3.25 | 0.25 | 1.75 |
| B | Concentration of T80 (% w/v) | 1.5 | 0.75 | 1.875 | 0.375 | 1.125 |
| C | Homogenization speed (rpm) | 17,000 | 10,000 | 18,800 | 3800 | 11,300 |
| D | Homogenization duration (min) | 20 | 10 | 25 | 5 | 15 |
| The critical responses of both lACP-PLHNs and hACP-PLHNs. | ||||||
| Y1 | PS (nm) | Less than 250 nm is desirable | ||||
| Y2 | PDI | Less than 0.6 is desirable | ||||
| Y3 | EE | Higher than 50% is desirable | ||||
| Batch No. | Material Attributes | Process Parameters of High Shear Homogenization | CRVs | |||
|---|---|---|---|---|---|---|
| Ratio of mPEG-PCL to Lipid | Concentration of T80 (% w/v) | PS (nm) | PDI | EE (%) | ||
| Batches prepared using lipophilic grade of mPEG-PCL | ||||||
| 01-L | 2:1 | 0.25 | Speed = 10,000 rpm Duration = 20 min | 220.38 | 0.402 | 64.96 |
| 02-L | 1:1 | 0.5 | Speed = 10,000 rpm Duration = 15 min | 218.64 | 0.323 | 5.82 |
| 03-L | 2:1 | 1.0 | Speed = 15,000 rpm Duration = 10 min | 153.81 | 0.226 | 56.17 |
| 04-L | 2:1 | 1.0 | Speed = 20,000 rpm Duration = 15 min | 210.55 | 0.372 | 43.60 |
| Batches prepared using hydrophilic grade of mPEG-PCL | ||||||
| 01-H | 2:1 | 0.25 | Speed = 10,000 rpm Duration = 20 min | 280.19 | 0.439 | 80.84 |
| 02-H | 1:1 | 0.5 | Speed = 10,000 rpm Duration = 15 min | 312.24 | 0.401 | 38.47 |
| 03-H | 2:1 | 1.0 | Speed = 15,000 rpm Duration = 10 min | 204.75 | 0.211 | 62.32 |
| 04-H | 2:1 | 0.75 | Speed = 20,000 rpm Duration = 15 min | 134.17 | 0.238 | 60.42 |
| Std Run | Factor A | Factor B | Factor C | Factor D | PS (nm) | EE (%) |
|---|---|---|---|---|---|---|
| 9 | 1 | 0.75 | 10,000 | 20 | 147.8 | 10.01 |
| 10 | 2.5 | 0.75 | 10,000 | 20 | 114.8 | 70.4 |
| 21 | 1.75 | 1.125 | 6500 | 15 | 175.8 | 13.56 |
| 7 | 1 | 1.5 | 17,000 | 10 | 74.78 | 27.73 |
| 26 | 1.75 | 1.125 | 13,500 | 15 | 100.3 | 71.17 |
| 22 | 1.75 | 1.125 | 20,500 | 15 | 185.3 | 84.57 |
| 20 | 1.75 | 1.875 | 13,500 | 15 | 143.4 | 25.36 |
| 27 | 1.75 | 1.125 | 13,500 | 15 | 135.9 | 32.25 |
| 5 | 1 | 0.75 | 17,000 | 10 | 106.8 | 85.46 |
| 19 | 1.75 | 0.375 | 13,500 | 15 | 174.4 | 19.92 |
| 28 | 1.75 | 1.125 | 13,500 | 15 | 190.1 | 31.83 |
| 16 | 2.5 | 1.5 | 17,000 | 20 | 127.9 | 52.54 |
| 14 | 2.5 | 0.75 | 17,000 | 20 | 106.6 | 86.27 |
| 13 | 1 | 0.75 | 17,000 | 20 | 160.1 | 88.35 |
| 3 | 1 | 1.5 | 10,000 | 10 | 172.9 | 12.46 |
| 2 | 2.5 | 0.75 | 10,000 | 10 | 162.8 | 58.97 |
| 15 | 1 | 1.5 | 17,000 | 20 | 114.2 | 77.91 |
| 25 | 1.75 | 1.125 | 13,500 | 15 | 117.4 | 76.8 |
| 23 | 1.75 | 1.125 | 13,500 | 5 | 165.5 | 46.85 |
| 17 | 0.25 | 1.125 | 13,500 | 15 | 96.18 | 14.83 |
| 12 | 2.5 | 1.5 | 10,000 | 20 | 215.3 | 71.84 |
| 18 | 3.25 | 1.125 | 13,500 | 15 | 167.2 | 55.15 |
| 11 | 1 | 1.5 | 10,000 | 20 | 132.6 | 30.1 |
| 24 | 1.75 | 1.125 | 13,500 | 25 | 137.1 | 82.88 |
| 4 | 2.5 | 1.5 | 10,000 | 10 | 149.7 | 32.8 |
| 1 | 1 | 0.75 | 10,000 | 10 | 165.1 | 50.18 |
| 6 | 2.5 | 0.75 | 17,000 | 10 | 121.4 | 68.84 |
| 8 | 2.5 | 1.5 | 17,000 | 10 | 154.4 | 12.23 |
| Std Run | Factor A | Factor B | Factor C | Factor D | PS (nm) | %EE |
|---|---|---|---|---|---|---|
| 13 | 1 | 0.75 | 17,000 | 20 | 304.9 | 15.5 |
| 7 | 1 | 1.5 | 17,000 | 10 | 158.8 | 7.51 |
| 12 | 2.5 | 1.5 | 10,000 | 20 | 130.9 | 63.2 |
| 14 | 2.5 | 0.75 | 17,000 | 20 | 163.2 | 81.63 |
| 9 | 1 | 0.75 | 10,000 | 20 | 186.8 | 7.68 |
| 15 | 1 | 1.5 | 17,000 | 20 | 169.6 | 6.43 |
| 19 | 1.75 | 0.375 | 13,500 | 15 | 165.4 | 71.08 |
| 24 | 1.75 | 1.125 | 13,500 | 25 | 309.5 | 61.66 |
| 28 | 1.75 | 1.125 | 13,500 | 15 | 186.9 | 79.27 |
| 26 | 1.75 | 1.125 | 13,500 | 15 | 212.3 | 25.24 |
| 18 | 3.25 | 1.125 | 13,500 | 15 | 188.2 | 77.3 |
| 5 | 1 | 0.75 | 17,000 | 10 | 135.1 | 6.57 |
| 16 | 2.5 | 1.5 | 17,000 | 20 | 97.37 | 57.49 |
| 6 | 2.5 | 0.75 | 17,000 | 10 | 112.5 | 93.67 |
| 27 | 1.75 | 1.125 | 13,500 | 15 | 226.9 | 5.75 |
| 23 | 1.75 | 1.125 | 13,500 | 5 | 261.9 | 44.15 |
| 1 | 1 | 0.75 | 10,000 | 10 | 159.7 | 8.7 |
| 20 | 1.75 | 1.875 | 13,500 | 15 | 128.4 | 50.42 |
| 4 | 2.5 | 1.5 | 10,000 | 10 | 114 | 92.95 |
| 21 | 1.75 | 1.125 | 6500 | 15 | 198.8 | 59.51 |
| 22 | 1.75 | 1.125 | 20,500 | 15 | 50.11 | 48.37 |
| 17 | 0.25 | 1.125 | 13,500 | 15 | 171.7 | 8.07 |
| 10 | 2.5 | 0.75 | 10,000 | 20 | 243.3 | 91.07 |
| 3 | 1 | 1.5 | 10,000 | 10 | 133.8 | 16.4 |
| 2 | 2.5 | 0.75 | 10,000 | 10 | 142.3 | 78.14 |
| 25 | 1.75 | 1.125 | 13,500 | 15 | 202.1 | 84.61 |
| 8 | 2.5 | 1.5 | 17,000 | 10 | 107.9 | 99.52 |
| 11 | 1 | 1.5 | 10,000 | 20 | 130.3 | 3.51 |
| Source | Y1 = PS | Y2 = EE | ||
|---|---|---|---|---|
| lACP-PLHNs | hACP-PLHNs | lACP-PLHNs | hACP-PLHNs | |
| Transformation | Power | Inverse square root | None | Square root |
| Model | 0.0496 | <0.0001 | 0.0041 | <0.0001 |
| A | 0.0966 | 0.6344 | 0.1409 | <0.0001 |
| B | 0.7748 | 0.0004 | 0.0701 | 0.4997 |
| C | 0.0572 | <0.0001 | 0.006 | 0.8956 |
| D | - | 0.0043 | 0.0463 | 0.7366 |
| AB | 0.0542 | 0.2561 | - | - |
| AC | - | 0.0042 | 0.0288 | - |
| BC | - | - | 0.1677 | - |
| BD | - | 0.0042 | - | - |
| A2 | - | 0.1406 | - | - |
| B2 | - | 0.0027 | - | - |
| C2 | - | <0.0001 | - | - |
| D2 | - | 0.0452 | - | - |
| A2C | - | <0.0001 | - | - |
| AB2 | - | 0.0243 | - | - |
| Lack of Fit | 0.7541 | 0.1593 | 0.7626 | 0.9971 |
| Parameters (Units) | Conventional ACP Suspension ^ | lACP-PLHN Nanosuspension | hACP-PLHN Nanosuspension | |
|---|---|---|---|---|
| ng/mL | 558.25 ± 22.45 | 1140.94 ± 133.72 | 1207.35 ± 116.64 | |
| h | 0.75 | 1 | 1 | |
| h * ng/mL | 2447.85 ± 269.42 | 9530.43 ± 634.51 | 9510.17 ± 281.71 | |
| Fabs # | - | 26.83 ± 1.35 | 83.69 ± 5.57 | 83.51 ± 2.48 |
| Frel * | - | - | 3.96 ± 0.73 | 3.93 ± 0.49 |
| h | 3.65 ± 0.15 | 7.23 ± 0.40 | 6.76 ± 0.22 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sinha, S.; Ravi, P.R.; Rashmi, S.R.; Szeleszczuk, Ł. mPEG-PCL Nanoparticles to Improve Oral Bioavailability of Acalabrutinib: Effect of Polymer Lipophilicity and Hydrophilicity on Physicochemical Properties and In Vivo Performance in Rats. Pharmaceutics 2025, 17, 774. https://doi.org/10.3390/pharmaceutics17060774
Sinha S, Ravi PR, Rashmi SR, Szeleszczuk Ł. mPEG-PCL Nanoparticles to Improve Oral Bioavailability of Acalabrutinib: Effect of Polymer Lipophilicity and Hydrophilicity on Physicochemical Properties and In Vivo Performance in Rats. Pharmaceutics. 2025; 17(6):774. https://doi.org/10.3390/pharmaceutics17060774
Chicago/Turabian StyleSinha, Swagata, Punna Rao Ravi, Sahadevan Rajesh Rashmi, and Łukasz Szeleszczuk. 2025. "mPEG-PCL Nanoparticles to Improve Oral Bioavailability of Acalabrutinib: Effect of Polymer Lipophilicity and Hydrophilicity on Physicochemical Properties and In Vivo Performance in Rats" Pharmaceutics 17, no. 6: 774. https://doi.org/10.3390/pharmaceutics17060774
APA StyleSinha, S., Ravi, P. R., Rashmi, S. R., & Szeleszczuk, Ł. (2025). mPEG-PCL Nanoparticles to Improve Oral Bioavailability of Acalabrutinib: Effect of Polymer Lipophilicity and Hydrophilicity on Physicochemical Properties and In Vivo Performance in Rats. Pharmaceutics, 17(6), 774. https://doi.org/10.3390/pharmaceutics17060774