
Evaluation of Antitumoral Activity in a 3D Cell Model of a Src Inhibitor Prodrug for Glioblastoma Treatment

 ,
,  ,
,  , , ,
, , ,  , ,
, ,  ,
,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals and HPLC-UV/MS Method
2.2. Cell Culture Medium, Fetal Bovine Serum, and Plasma Stability Assay
2.3. Parallel Artificial Membrane Permeability Assay (PAMPA)
2.4. Cell Culture
2.5. 3D-Bioprinted Cell Model
2.6. Cell Viability Assay
2.7. Western Blot
2.8. Data Analysis and Statistics
3. Results
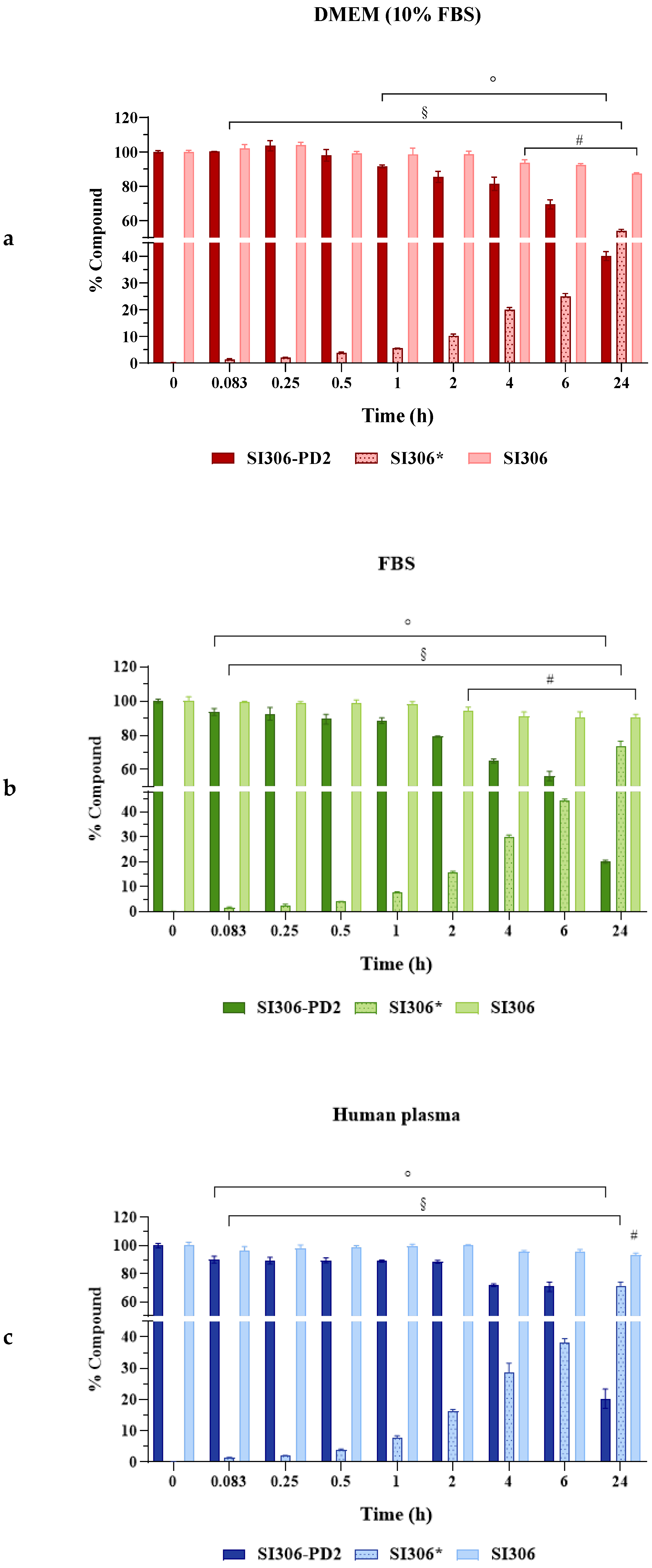
3.1. Evaluation of SI306 and SI306-PD2 Stability and SI306 Release from Prodrug
3.2. Permeability Through Biological Membranes
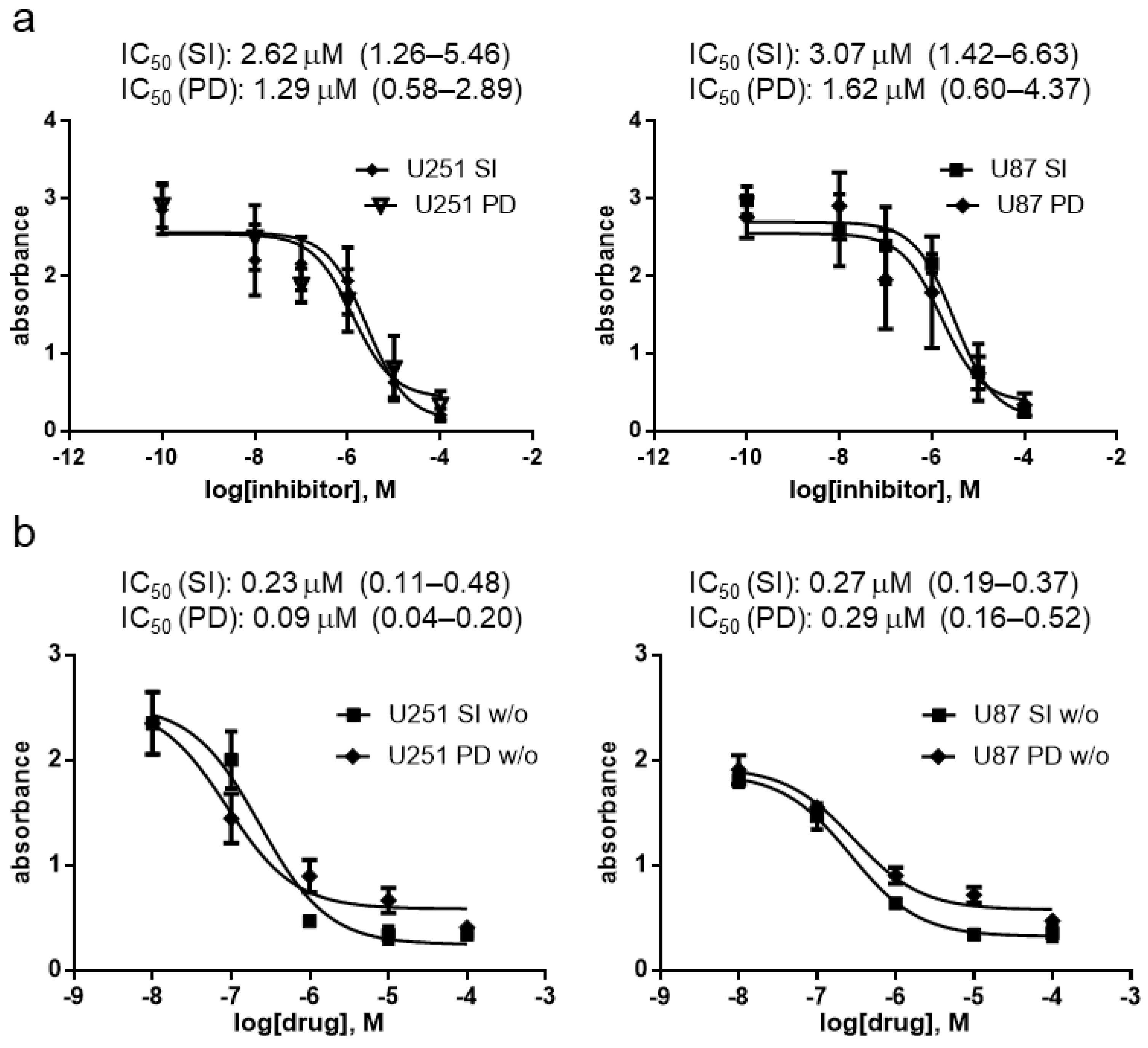
3.3. Antiproliferative Effect of SI306 and SI306-PD2 in 2D Cell Models
3.4. Antiproliferative Effect of SI306 and SI306-PD2 in 3D Cell Models
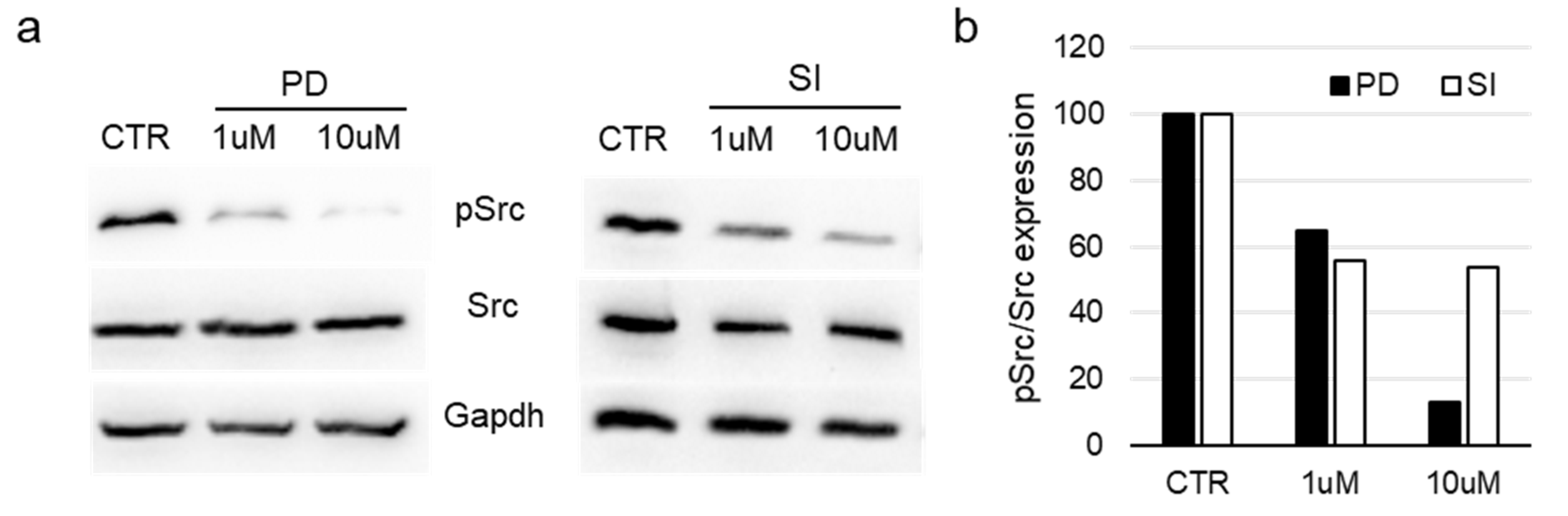
3.5. Evaluation of Src Activation in Bioprinted Cells
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A
Appendix A.1. Chemistry

Appendix A.2. Synthesis of 2-(2-Hydroxyethoxy)ethyl (3-Bromophenyl)(1-(2-chloro-2-phenylethyl)-6-((2-morpholinoethyl)thio)-1H-pyrazolo[3,4-d]pyrimidin-4-yl)carbamate (SI306-PD2)
References
- Sun, G.; Ayrapetov, M.K. Dissection of the catalytic and regulatory structure-function relationships of Csk protein tyrosine kinase. Front. Cell Dev. Biol. 2023, 11, 1148352. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. Combination immune checkpoint and targeted protein kinase inhibitors for the treatment of renal cell carcinomas. Pharmacol. Res. 2024, 203, 107181. [Google Scholar] [CrossRef] [PubMed]
- Abou-Alfa, G.K.; Schwartz, L.; Ricci, S.; Amadori, D.; Santoro, A.; Figer, A.; De Greve, J.; Douillard, J.Y.; Lathia, C.; Schwartz, B.; et al. Phase II study of sorafenib in patients with advanced hepatocellular carcinoma. J. Clin. Oncol. 2006, 24, 4293–4300. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, M.; Porta, C. Sunitinib in the treatment of renal cell carcinoma: An update on recent evidence. Ther. Adv. Urol. 2017, 9, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Bernasconi, P.; Clauser, K.R.; Mani, D.R.; Finn, S.P.; Beroukhim, R.; Burns, M.; Julian, B.; Peng, X.P.; Hieronymus, H.; et al. Bead-based profiling of tyrosine kinase phosphorylation identifies SRC as a potential target for glioblastoma therapy. Nat. Biotechnol. 2009, 27, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, D.L.; Iida, M.; Dunn, E.F. The role of Src in solid tumors. Oncologist 2009, 14, 667–678. [Google Scholar] [CrossRef] [PubMed]
- Stettner, M.R.; Wang, W.; Nabors, L.B.; Bharara, S.; Flynn, D.C.; Grammer, J.R.; Gillespie, G.Y.; Gladson, C.L. Lyn kinase activity is the predominant cellular SRC kinase activity in glioblastoma tumor cells. Cancer Res. 2005, 65, 5535–5543. [Google Scholar] [CrossRef] [PubMed]
- Puls, L.N.; Eadens, M.; Messersmith, W. Current status of SRC inhibitors in solid tumor malignancies. Oncologist 2011, 16, 566–578. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Gong, C.; Zhou, H.; Liu, J.; Xia, X.; Ha, W.; Jiang, Y.; Liu, Q.; Xiong, H. Kinase Inhibitors and Kinase-Targeted Cancer Therapies: Recent Advances and Future Perspectives. Int. J. Mol. Sci. 2024, 25, 5489. [Google Scholar] [CrossRef] [PubMed]
- Vignaroli, G.; Iovenitti, G.; Zamperini, C.; Coniglio, F.; Calandro, P.; Molinari, A.; Fallacara, A.L.; Sartucci, A.; Calgani, A.; Colecchia, D.; et al. Prodrugs of Pyrazolo[3,4-d]pyrimidines: From Library Synthesis to Evaluation as Potential Anticancer Agents in an Orthotopic Glioblastoma Model. J. Med. Chem. 2017, 60, 6305–6320. [Google Scholar] [CrossRef] [PubMed]
- Greenwald, R.B.; Choe, Y.H.; McGuire, J.; Conover, C.D. Effective drug delivery by PEGylated drug conjugates. Adv. Drug Deliv. Rev. 2003, 55, 217–250. [Google Scholar] [CrossRef] [PubMed]
- Monteleone, L.; Marengo, B.; Musumeci, F.; Grossi, G.; Carbone, A.; Valenti, G.E.; Domenicotti, C.; Schenone, S. Anti-Survival Effect of SI306 and Its Derivatives on Human Glioblastoma Cells. Pharmaceutics 2022, 14, 1399. [Google Scholar] [CrossRef] [PubMed]
- Naghieh, S.; Sarker, M.; Sharma, N.K.; Barhoumi, Z.; Chen, X. Printability of 3D printed hydrogel scaffolds: Influence of hydrogel composition and printing parameters. Appl. Sci. 2019, 10, 292. [Google Scholar] [CrossRef]
- Poggialini, F.; Vagaggini, C.; Brai, A.; Pasqualini, C.; Crespan, E.; Maga, G.; Perini, C.; Cabella, N.; Botta, L.; Musumeci, F.; et al. Biological Evaluation and In Vitro Characterization of ADME Profile of In-House Pyrazolo[3,4-d]pyrimidines as Dual Tyrosine Kinase Inhibitors Active against Glioblastoma Multiforme. Pharmaceutics 2023, 15, 453. [Google Scholar] [CrossRef] [PubMed]
- Tintori, C.; Fallacara, A.L.; Radi, M.; Zamperini, C.; Dreassi, E.; Crespan, E.; Maga, G.; Schenone, S.; Musumeci, F.; Brullo, C.; et al. Combining X-ray crystallography and molecular modeling toward the optimization of pyrazolo[3,4-d]pyrimidines as potent c-Src inhibitors active in vivo against neuroblastoma. J. Med. Chem. 2015, 58, 347–361. [Google Scholar] [CrossRef] [PubMed]
- LaBonia, G.J.; Lockwood, S.Y.; Heller, A.A.; Spence, D.M.; Hummon, A.B. Drug penetration and metabolism in 3D cell cultures treated in a 3D printed fluidic device: Assessment of irinotecan via MALDI imaging mass spectrometry. Proteomics 2016, 16, 1814–1821. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Hummon, A.B. Quantitative determination of irinotecan and the metabolite SN-38 by nanoflow liquid chromatography-tandem mass spectrometry in different regions of multicellular tumor spheroids. J. Am. Soc. Mass Spectrom. 2015, 26, 577–586. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time (min) | % Eluent A H2O (FA 0.1%v/v) | % Eluent B ACN (FA 0.1%v/v) | Flow (µL/min) | Injection Volume (µL) | Wavelength (nm) | Scan Range (m/z) a |
|---|---|---|---|---|---|---|
| 0–1 | 95 | 5 | 600 | 10 | 254 | 100–2000 |
| 15–19 | 5 | 95 | ||||
| 20 | 95 | 5 |
| Time (h) | Stability/Release (% ± SD) | |||||
|---|---|---|---|---|---|---|
| DMEM (10% FBS) | FBS | Plasma | ||||
| SI306-PD2 (SI306) b | SI306 | SI306-PD2 (SI306) b | SI306 | SI306-PD2 (SI306) b | SI306 | |
| 0 | 100.02 ± 0.80 (0.11 ± 0.02) | 100.11 ± 0.91 | 100.06 ± 1.13 (0.15 ± 0.03) | 100.24 ± 2.48 | 101.03 ± 1.58 (0.13 ± 0.04) | 100.50 ± 2.32 |
| 0.083 | 100.22 ± 0.10 (1.37 ± 0.23) § | 101.97 ± 2.32 | 93.64 ± 2.06 ° (1.55 ± 0.29) § | 99.67 ± 0.21 | 90.12 ± 2.45 ° (1.37 ± 0.23) § | 96.64 ± 2.74 |
| 0.25 | 103.59 ± 2.97 (2.06 ± 0.09) § | 103.96 ± 1.68 | 92.56 ± 3.74 ° (2.24 ± 0.73) § | 98.42 ± 1.19 | 89.41 ± 2.37 ° (2.06 ± 0.09) § | 97.99 ± 2.48 |
| 0.5 | 98.01 ± 3.40 (3.79 ± 0.31) § | 99.17 ± 1.10 | 89.39 ± 2.87 ° (4.00 ± 0.14) § | 98.49 ± 2.18 | 89.57 ± 1.82 ° (3.88 ± 0.34) § | 98.95 ± 1.18 |
| 1 | 91.48 ± 0.86 ° (5.55 ± 0.09) § | 98.52 ± 3.69 | 88.58 ± 1.66 ° (7.94 ± 0.14) § | 97.91 ± 1.79 | 89.05 ± 0.75 ° (7.67 ± 0.76) § | 99.48 ± 1.70 |
| 2 | 85.45 ± 3.20 ° (10.21 ± 0.63) § | 98.64 ± 1.79 | 79.26 ± 0.34 ° (15.82 ± 0.36) § | 93.95 ± 2.42 # | 88.55 ± 1.18 ° (16.29 ± 0.54) § | 99.92 ± 0.61 |
| 4 | 81.41 ± 3.94 ° (19.99 ± 0.81) § | 93.56 ± 1.79 # | 64.90 ± 1.21 ° (30.02 ± 0.66) § | 90.92 ± 2.68 # | 71.86 ± 1.03 ° (28.80 ± 2.84) § | 95.72 ± 0.76 |
| 6 | 69.38 ± 2.73 ° (24.95 ± 1.09) § | 92.30 ± 0.84 # | 56.13 ± 2.72 ° (44.32 ± 0.78) § | 90.51 ± 3.24 # | 70.80 ± 3.25 ° (38.02 ± 1.46) § | 95.54 ± 1.74 |
| 24 | 40.07 ± 1.70 ° (53.92 ± 0.90) § | 87.39 ± 0.41 # | 20.06 ± 0.66 ° (73.80 ± 2.65) § | 90.02 ± 2.28 # | 20.29 ± 3.07 ° (71.59 ± 2.45) § | 93.12 ± 1.70 # |
| t1/2 a | 18.09 * | >100 | 10.65 * | >100 | 10.88 * | >100 |
| Cpd | PAMPA Assay | |||
|---|---|---|---|---|
| GI | BBB | |||
| Papp cm/s × 10−6 a | MR (% ± SD) b | Papp cm/s × 10−6 a | MR (% ± SD) b | |
| SI306 | 5.93 ± 0.38 | 60.09 ± 3.38 | 8.44 ± 1.03 § | 39.95 ± 0.97 § |
| SI306-PD2 | 18.45 ± 2.32 * | 5.26 ± 2.70 * | 19.51 ± 0.46 *§ | 3.60 ± 1.15 * |
| Atenolol | 0.73 ± 0.06 | 53.58 ± 1.96 | 1.01 ± 0.20 | 21.03 ± 1.14 |
| Propranolol | 19.16 ± 0.37 | 8.89 ± 1.12 | 17.16 ± 0.31 | 10.73 ± 1.33 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Clementi, L.; Poggialini, F.; Musumeci, F.; Taglienti, J.; Cornacchia, E.; Vagaggini, C.; Carbone, A.; Grossi, G.; Dreassi, E.; Angelucci, A.; et al. Evaluation of Antitumoral Activity in a 3D Cell Model of a Src Inhibitor Prodrug for Glioblastoma Treatment. Pharmaceutics 2025, 17, 704. https://doi.org/10.3390/pharmaceutics17060704
Clementi L, Poggialini F, Musumeci F, Taglienti J, Cornacchia E, Vagaggini C, Carbone A, Grossi G, Dreassi E, Angelucci A, et al. Evaluation of Antitumoral Activity in a 3D Cell Model of a Src Inhibitor Prodrug for Glioblastoma Treatment. Pharmaceutics. 2025; 17(6):704. https://doi.org/10.3390/pharmaceutics17060704
Chicago/Turabian StyleClementi, Letizia, Federica Poggialini, Francesca Musumeci, Julia Taglienti, Emanuele Cornacchia, Chiara Vagaggini, Anna Carbone, Giancarlo Grossi, Elena Dreassi, Adriano Angelucci, and et al. 2025. "Evaluation of Antitumoral Activity in a 3D Cell Model of a Src Inhibitor Prodrug for Glioblastoma Treatment" Pharmaceutics 17, no. 6: 704. https://doi.org/10.3390/pharmaceutics17060704
APA StyleClementi, L., Poggialini, F., Musumeci, F., Taglienti, J., Cornacchia, E., Vagaggini, C., Carbone, A., Grossi, G., Dreassi, E., Angelucci, A., & Schenone, S. (2025). Evaluation of Antitumoral Activity in a 3D Cell Model of a Src Inhibitor Prodrug for Glioblastoma Treatment. Pharmaceutics, 17(6), 704. https://doi.org/10.3390/pharmaceutics17060704