
Pharmacokinetic Interaction Between Olaparib and Regorafenib in an Animal Model

 , , , ,
, , , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Animals
2.3. Study Protocol
2.4. UPLC-MS/MS Analysis of OLA, REG, M-2, and M-5
2.5. Pharmacokinetic Assay
2.6. Statistical Analysis
3. Results
3.1. Analytical Methods
3.1.1. OLA Analytical Method Validation
3.1.2. REG, M-2, and M-5 Analytical Method Validation
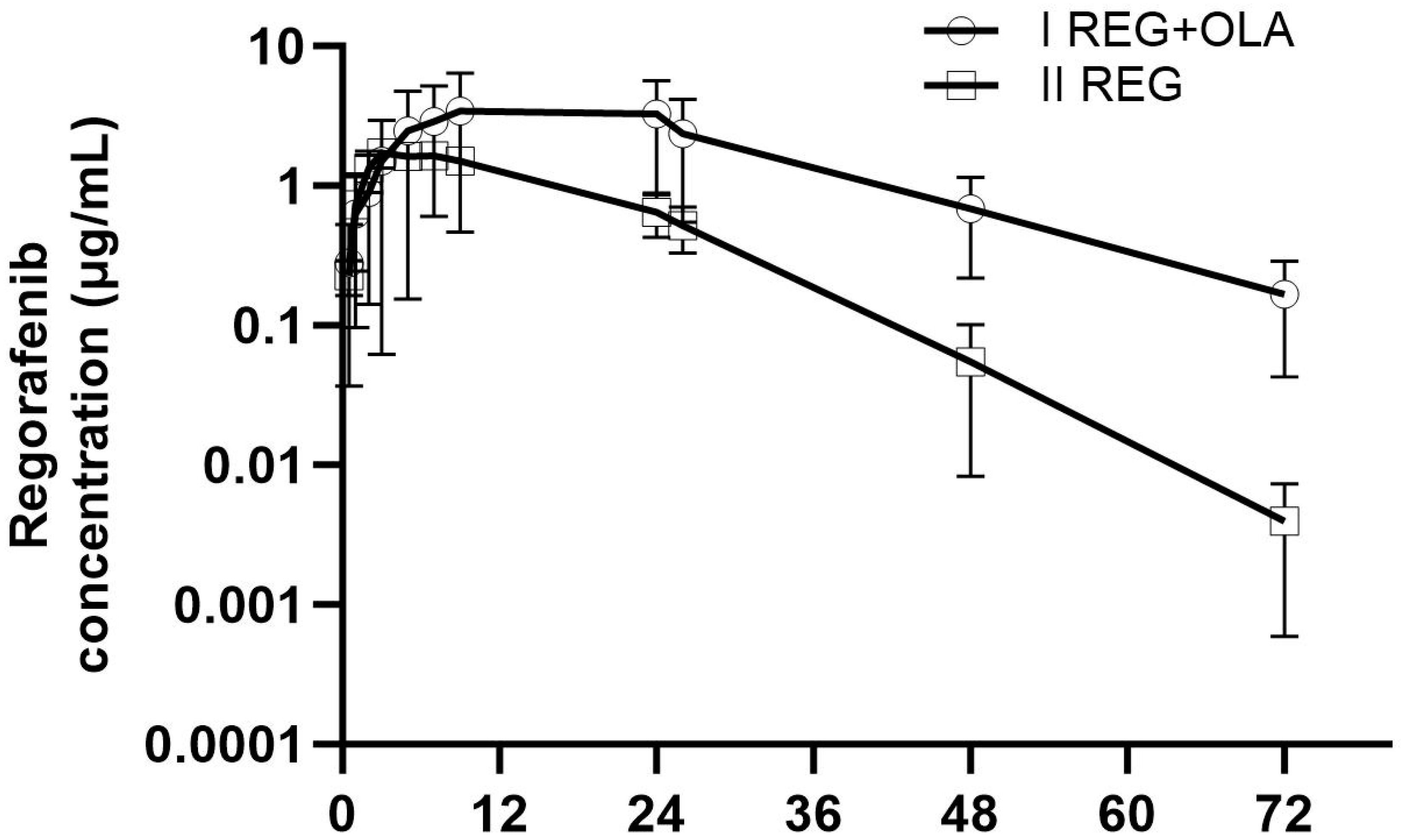
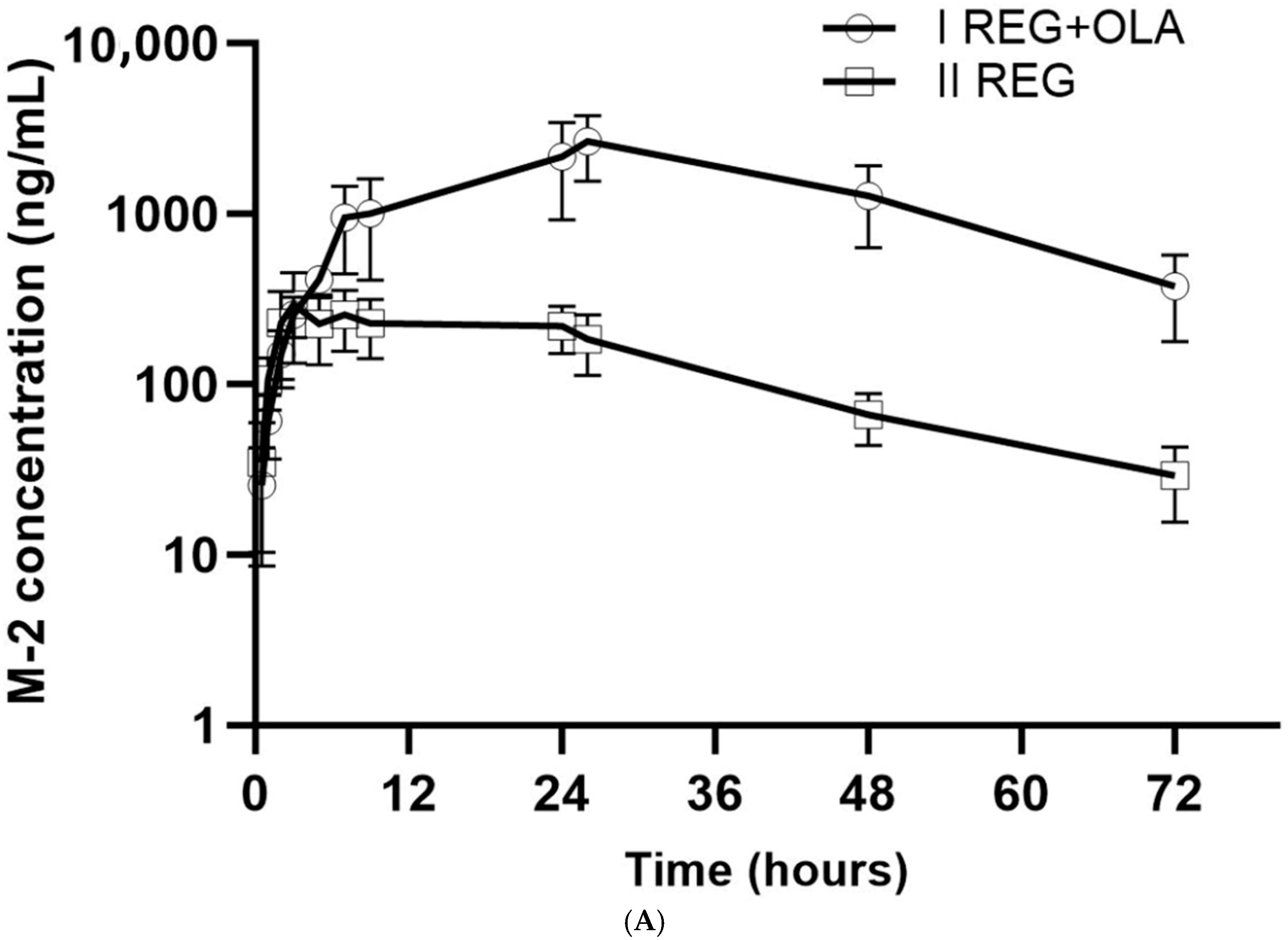
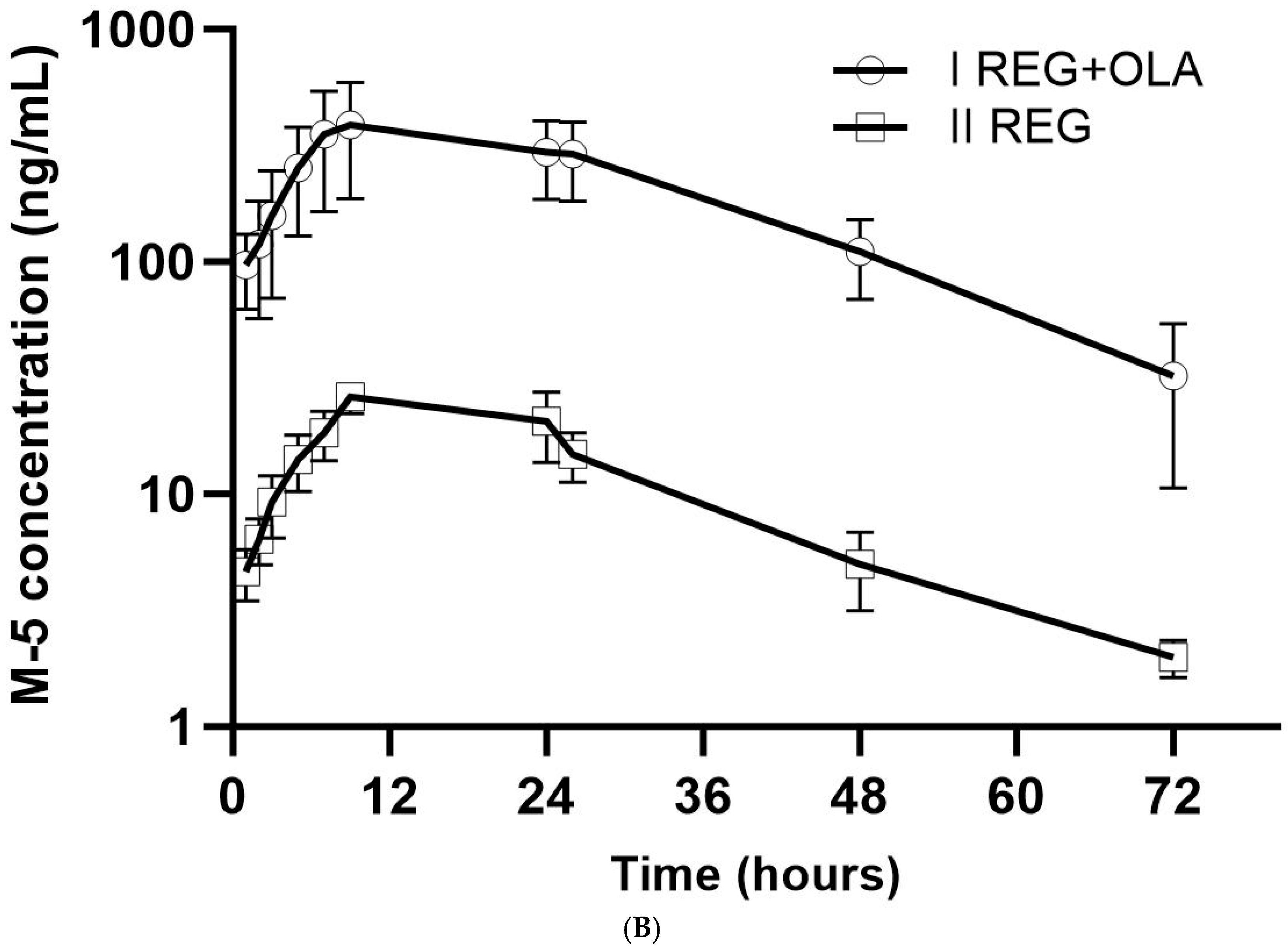
3.2. The Influence of OLA on the Pharmacokinetics of REG, M-2, and M-5
3.3. The Influence of REG on the Pharmacokinetics of OLA
4. Discussion
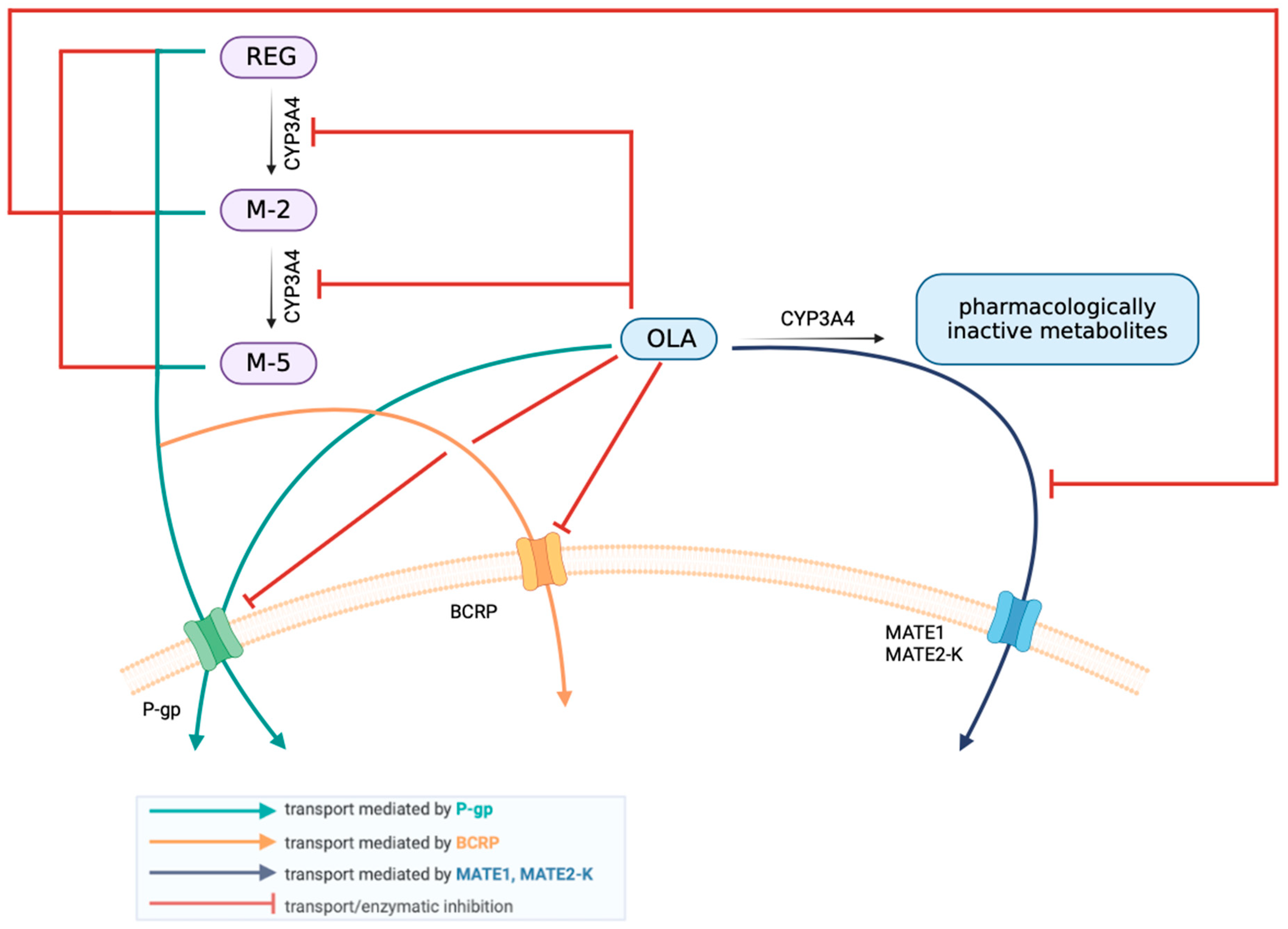
4.1. The Effect of OLA on the Pharmacokinetics of REG, M-2, and M-5
4.2. The Effect of REG on the Pharmacokinetics of OLA
4.3. Summary Including Interspecies Differences
4.4. Limitations of the Study
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Oliveira, R.F.; Oliveira, A.I.; Cruz, A.S.; Ribeiro, O.; Afreixo, V.; Pimentel, F. Polypharmacy and drug interactions in older patients with cancer receiving chemotherapy: Associated factors. BMC Geriatr. 2024, 24, 557. [Google Scholar] [CrossRef] [PubMed]
- Gholipourshahraki, T.; Aria, A.; Sharifi, M.; Moghadas, A.; Moghaddas, A. Potential Drug Interactions in Hospitalized Hematologic Cancer Patients: New Update with New Chemotherapy Regimens. J. Res. Pharm. Pract. 2024, 12, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Lam, C.S.; Hua, R.; Au-Doung, P.L.W.; Wu, Y.K.; Koon, H.K.; Zhou, K.R.; Loong, H.H.; Chung, V.C.; Cheung, Y.T. Association between potential supplement-drug interactions and liver diseases in patients with cancer: A large prospective cohort study. Clin. Nutr. ESPEN 2023, 58, 152–159. [Google Scholar] [CrossRef]
- Xi, Y.; Xu, P. Global colorectal cancer burden in 2020 and projections to 2040. Transl. Oncol. 2021, 14, 101174. [Google Scholar] [CrossRef]
- Klimeck, L.; Heisser, T.; Hoffmeister, M.; Brenner, H. Colorectal cancer: A health and economic problem. Best Pract. Res. Clin. Gastroenterol. 2023, 66, 101839. [Google Scholar] [CrossRef]
- Xu, D.; Liu, Y.; Tang, W.; Xu, L.; Liu, T.; Jiang, Y.; Zhou, S.; Qin, X.; Li, J.; Zhao, J.; et al. Regorafenib in Refractory Metastatic Colorectal Cancer: A Multi-Center Retrospective Study. Front. Oncol. 2022, 12, 838870. [Google Scholar] [CrossRef]
- Xu, X.; Yu, Y.; Liu, M.; Liang, L.; Liu, T. Efficacy and safety of regorafenib and fruquintinib as third-line treatment for colorectal cancer: A narrative review. Transl. Cancer Res. 2022, 11, 276–287. [Google Scholar] [CrossRef] [PubMed]
- Australian Public Assessment Report for Regorafenib, April 2019. Available online: https://www.tga.gov.au/sites/default/files/auspar-regorafenib-190404.pdf (accessed on 1 September 2024).
- Granito, A.; Marinelli, S.; Forgione, A.; Renzulli, M.; Benevento, F.; Piscaglia, F.; Tovoli, F. Regorafenib Combined with Other Systemic Therapies: Exploring Promising Therapeutic Combinations in HCC. J. Hepatocell. Carcinoma 2021, 8, 477–492. [Google Scholar] [CrossRef]
- Rey, J.B.; Launay-Vacher, V.; Tournigand, C. Regorafenib as a single-agent in the treatment of patients with gastrointestinal tumors: An overview for pharmacists. Target. Oncol. 2015, 10, 199–213. [Google Scholar] [CrossRef]
- Papageorgiou, G.I.; Fergadis, E.; Skouteris, N.; Christakos, E.; Tsakatikas, S.A.; Lianos, E.; Kosmas, C. Case Report: Combination of Olaparib with Chemotherapy in a Patient with ATM-Deficient Colorectal Cancer. Front. Oncol. 2021, 11, 788809. [Google Scholar] [CrossRef]
- Wang, C.; Jette, N.; Moussienko, D.; Bebb, D.G.; Lees-Miller, S.P. ATM-Deficient Colorectal Cancer Cells Are Sensitive to the PARP Inhibitor Olaparib. Transl. Oncol. 2017, 10, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Jette, N.R.; Kumar, M.; Radhamani, S.; Arthur, G.; Goutam, S.; Yip, S.; Kolinsky, M.; Williams, G.J.; Bose, P.; Lees-Miller, S.P. ATM-Deficient Cancers Provide New Opportunities for Precision Oncology. Cancers 2020, 12, 687. [Google Scholar] [CrossRef] [PubMed]
- Gong, H.; Su, Y.; Zhao, L.; Ma, L.; Zhang, L.; Hou, L.; Li, T.; Niu, S.; Zhang, H.; Li, C.; et al. Efficacy and safety of targeted drugs in advanced or metastatic gastric and gastroesophageal junction cancer: A network meta-analysis. J. Clin. Pharm. Ther. 2022, 47, 493–506. [Google Scholar] [CrossRef]
- Australian Public Assessment Report for Olaparib, February 2018. Available online: https://www.tga.gov.au/sites/default/files/auspar-olaparib-210415.pdf (accessed on 1 September 2024).
- EMA Recommends Extension of Therapeutic Indications for Olaparib to Patients with Endometrial Cancer. 17 July 2024. Available online: https://www.esmo.org/oncology-news/ema-recommends-extension-of-therapeutic-indications-for-olaparib-to-patients-with-endometrial-cancer (accessed on 20 August 2024).
- Yang, X.D.; Kong, F.E.; Qi, L.; Lin, J.X.; Yan, Q.; Loong, J.H.C.; Xi, S.Y.; Zhao, Y.; Zhang, Y.; Yuan, Y.F.; et al. PARP inhibitor Olaparib overcomes Sorafenib resistance through reshaping the pluripotent transcriptome in hepatocellular carcinoma. Mol. Cancer 2021, 20, 20. [Google Scholar] [CrossRef]
- Lai, Y.H.; Tung, K.C.; Chen, S.C. Durable response to olaparib combined low-dose cisplatin in advanced hepatocellular carcinoma with FANCA mutation: A case report. Medicine 2022, 101, e30719. [Google Scholar] [CrossRef]
- Sun, G.; Miao, G.; Li, Z.; Zheng, W.; Zhou, C.; Sun, G.; Cao, H.; Li, Z.; Tang, W. Inhibition of PARP Potentiates Immune Checkpoint Therapy through miR-513/PD-L1 Pathway in Hepatocellular Carcinoma. J. Oncol. 2022, 2022, 6988923. [Google Scholar] [CrossRef]
- Xu, H.; Ma, Z.; Mo, X.; Chen, X.; Xu, F.; Wu, F.; Chen, H.; Zhou, G.; Xia, H.; Zhang, C. Inducing Synergistic DNA Damage by TRIP13 and PARP1 Inhibitors Provides a Potential Treatment for Hepatocellular Carcinoma. J. Cancer 2022, 13, 2226–2237. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Sun, Y.; Lin, H.; Chou, F.; Xiao, Y.; Jin, R.; Cai, X.; Chang, C. Olaparib and enzalutamide synergistically suppress HCC progression via the AR-mediated miR-146a-5p/BRCA1 signaling. FASEB J. 2020, 34, 5877–5891. [Google Scholar] [CrossRef]
- Zeng, Y.; Arisa, O.; Peer, C.J.; Fojo, A.; Figg, W.D. PARP inhibitors: A review of the pharmacology, pharmacokinetics, and pharmacogenetics. Semin. Oncol. 2024, 51, 19–24. [Google Scholar] [CrossRef]
- Dopazo, C.; Søreide, K.; Rangelova, E.; Mieog, S.; Carrion-Alvarez, L.; Diaz-Nieto, R.; Primavesi, F.; Stättner, S. Hepatocellular carcinoma. Eur. J. Surg. Oncol. 2024, 50, 107313. [Google Scholar] [CrossRef]
- Do, K.T.; Kochupurakkal, B.; Kelland, S.; de Jonge, A.; Hedglin, J.; Powers, A.; Quinn, N.; Gannon, C.; Vuong, L.; Parmar, K.; et al. Phase 1 Combination Study of the CHK1 Inhibitor Prexasertib and the PARP Inhibitor Olaparib in High-grade Serous Ovarian Cancer and Other Solid Tumors. Clin. Cancer Res. 2021, 27, 4710–4716. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.D.; Wethington, S.L.; Pagan, C.; Latif, N.; Tanyi, J.; Martin, L.P.; Morgan, M.; Burger, R.A.; Haggerty, A.; Zarrin, H.; et al. Combination ATR and PARP Inhibitor (CAPRI): A phase 2 study of ceralasertib plus olaparib in patients with recurrent, platinum-resistant epithelial ovarian cancer. Gynecol. Oncol. 2021, 163, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Wethington, S.L.; Shah, P.D.; Martin, L.; Tanyi, J.L.; Latif, N.; Morgan, M.; Torigian, D.A.; Rodriguez, D.; Smith, S.A.; Dean, E.; et al. Combination ATR (ceralasertib) and PARP (olaparib) Inhibitor (CAPRI) Trial in Acquired PARP Inhibitor-Resistant Homologous Recombination-Deficient Ovarian Cancer. Clin. Cancer Res. 2023, 29, 2800–2807. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, R.L.; Wijnhoven, P.W.G.; Ramos-Montoya, A.; Wilson, Z.; Illuzzi, G.; Falenta, K.; Jones, G.N.; James, N.; Chabbert, C.D.; Stott, J.; et al. Combined PARP and ATR inhibition potentiates genome instability and cell death in ATM-deficient cancer cells. Oncogene 2020, 39, 4869–4883. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Barry, W.T.; Birrer, M.; Westin, S.N.; Cadoo, K.A.; Shapiro, G.I.; Mayer, E.L.; O’Cearbhaill, R.E.; Coleman, R.L.; Kochupurakkal, B.; et al. Olaparib and α-specific PI3K inhibitor alpelisib for patients with epithelial ovarian cancer: A dose-escalation and dose-expansion phase 1b trial. Lancet Oncol. 2019, 20, 570–580. [Google Scholar] [CrossRef]
- Zimmer, A.S.; Nichols, E.; Cimino-Mathews, A.; Peer, C.; Cao, L.; Lee, M.J.; Kohn, E.C.; Annunziata, C.M.; Lipkowitz, S.; Trepel, J.B.; et al. A phase I study of the PD-L1 inhibitor, durvalumab, in combination with a PARP inhibitor, olaparib, and a VEGFR1-3 inhibitor, cediranib, in recurrent women’s cancers with biomarker analyses. J. Immunother. Cancer 2019, 7, 197. [Google Scholar] [CrossRef]
- Fok, J.H.L.; Ramos-Montoya, A.; Vazquez-Chantada, M.; Wijnhoven, P.W.G.; Follia, V.; James, N.; Farrington, P.M.; Karmokar, A.; Willis, S.E.; Cairns, J.; et al. AZD7648 is a potent and selective DNA-PK inhibitor that enhances radiation, chemotherapy and olaparib activity. Nat. Commun. 2019, 10, 5065. [Google Scholar] [CrossRef]
- Li, J.; Wang, R.; Kong, Y.; Broman, M.M.; Carlock, C.; Chen, L.; Li, Z.; Farah, E.; Ratliff, T.L.; Liu, X. Targeting Plk1 to Enhance Efficacy of Olaparib in Castration-Resistant Prostate Cancer. Mol. Cancer Ther. 2017, 16, 469–479. [Google Scholar] [CrossRef]
- McCormick, A.; Swaisland, H.; Reddy, V.P.; Learoyd, M.; Scarfe, G. In vitro evaluation of the inhibition and induction potential of olaparib; a potent poly(ADP-ribose) polymerase inhibitor; on cytochrome P450. Xenobiotica 2018, 48, 555–564. [Google Scholar] [CrossRef]
- McCormick, A.; Swaisland, H. In vitro assessment of the roles of drug transporters in the disposition and drug-drug interaction potential of olaparib. Xenobiotica 2017, 47, 903–915. [Google Scholar] [CrossRef]
- Kobayashi, K.; Sugiyama, E.; Shinozaki, E.; Wakatsuki, T.; Tajima, M.; Kidokoro, H.; Aoyama, T.; Nakano, Y.; Kawakami, K.; Hashimoto, K.; et al. Associations among plasma concentrations of regorafenib and its metabolites; adverse events; and ABCG2 polymorphisms in patients with metastatic colorectal cancers. Cancer Chemother. Pharmacol. 2021, 87, 767–777. [Google Scholar] [CrossRef] [PubMed]
- Steinbronn, C.; Yang, X.; Yu, J.; Dimova, H.; Huang, S.M.; Ragueneau-Majlessi, I.; Isoherranen, N. Do Inhibitory Metabolites Impact DDI Risk Assessment? Analysis of in vitro and in vivo Data from NDA Reviews Between 2013 and 2018. Clin. Pharmacol. Ther. 2021, 110, 452–463. [Google Scholar] [CrossRef]
- Deng, F.; Sistonen, J.; Neuvonen, M.; Niemi, M. Inhibition of efflux transporters by poly ADP-ribose polymerase inhibitors. Basic Clin. Pharmacol. Toxicol. 2023, 133, 428–436. [Google Scholar] [CrossRef]
- Jin, S.; Paludetto, M.N.; Kurkela, M.; Kahma, H.; Neuvonen, M.; Xiang, X.; Cai, W.; Backman, J.T. In vitro assessment of inhibitory effects of kinase inhibitors on CYP2C9; 3A and 1A2: Prediction of drug-drug interaction risk with warfarin and direct oral anticoagulants. Eur. J. Pharm. Sci. 2024, 203, 106884. [Google Scholar] [CrossRef]
- Directive 2010/63/EU of the European Parliament and of the Council of 22 September 2010 on the Protection of Animals Used for Scientific Purposes. Available online: https://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2010:276:0033:0079:EN:PDF (accessed on 6 October 2023).
- Szkutnik-Fiedler, D.; Szałek, E.; Otto, F.; Czyrski, A.; Karaźniewicz-Łada, M.; Wolc, A.; Grześkowiak, E.; Lewandowski, K.; Karbownik, A. Pharmacokinetic interaction between regorafenib and atorvastatin in rats. Pharmacol. Rep. 2024, 76, 1184–1195. [Google Scholar] [CrossRef]
- Stanisławiak-Rudowicz, J.; Karbownik, A.; Szkutnik-Fiedler, D.; Otto, F.; Grabowski, T.; Wolc, A.; Grześkowiak, E.; Szałek, E. Bidirectional pharmacokinetic drug interactions between olaparib and metformin. Cancer Chemother. Pharmacol. 2024, 93, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Burden, N.; Chapman, K.; Sewell, F.; Robinson, V. Pioneering better science through the 3Rs: An introduction to the national centre for the replacement; refinement; and reduction of animals in research (NC3Rs). J. Am. Assoc. Lab. Anim. Sci. 2015, 54, 198–208. [Google Scholar] [PubMed]
- Su, G.; Qin, L.; Su, X.; Tao, C.; Wei, Y. Gender-dependent pharmacokinetics of olaparib in rats determined by ultra-high performance liquid chromatography/electrospray ionization tandem mass spectrometry. Biomed. Chromatogr. 2020, 34, e4791. [Google Scholar] [CrossRef]
- Gu, E.M.; Liu, Y.N.; Pan, L.; Hu, Y.; Ye, X.; Luo, P. A high throughput method for Monitoring of Sorafenib; regorafenib; cabozantinib and their metabolites with UPLC-MS/MS in rat plasma. Front. Pharmacol. 2022, 13, 955263. [Google Scholar] [CrossRef]
- ICH Guideline M10 on Bioanalytical Method Validation and Study Sample Analysis. 25 July 2022. Available online: https://www.ema.europa.eu/en/ich-m10-bioanalytical-method-validation-scientific-guideline (accessed on 15 November 2023).
- Cole, S.; Kerwash, E.; Andersson, A. A summary of the current drug interaction guidance from the European Medicines Agency and considerations of future updates. Drug Metab. Pharmacokinet. 2020, 35, 2–11. [Google Scholar] [CrossRef]
- Choi, Y.H. Interpretation of Drug Interaction Using Systemic and Local Tissue Exposure Changes. Pharmaceutics 2020, 12, 417. [Google Scholar] [CrossRef]
- In Vitro Drug Interaction Studies—Cytochrome P450 Enzyme- and Transporter-Mediated Drug Interactions. Guidance for Industry. U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER). January 2020. Clinical Pharmacology. Available online: https://www.xenotech.com/access-adme-research-resources/resources/2020-fda-guidance-for-industry-2/ (accessed on 11 December 2023).
- Rousseau, B.; Boukerma, A.K.; Henriques, J.; Cohen, R.; Lucidarme, O.; Borg, C.; Tournigand, C.; Kim, S.; Bachet, J.B.; Mazard, T.; et al. Impact of trough concentrations of regorafenib and its major metabolites M-2 and M-5 on overall survival of chemorefractory metastatic colorectal cancer patients: Results from a multicentre GERCOR TEXCAN phase II study. Eur. J. Cancer 2022, 168, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Zhou, Z.; Tay-Sontheimer, J.; Levy, R.H.; Ragueneau-Majlessi, I. Risk of clinically relevant pharmacokinetic-based drug-drug interactions with drugs approved by the U.S. Food and Drug Administration between 2013 and 2016. Drug Metab. Dispos. 2018, 46, 835–845. [Google Scholar] [CrossRef] [PubMed]
- Molenaar-Kuijsten, L.; Van Balen, D.E.M.; Beijnen, J.H.; Steeghs, N.; Huitema, A.D.R. A review of CYP3A drug–drug interaction studies: Practical guidelines for patients using targeted oral anticancer drugs. Front. Pharmacol. 2021, 12, 670862. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Chen, M.; Anderson, J.T.; Sun, X.; Hu, S.; Sparreboom, A.; Baker, S.D. Interaction Between Sex and Organic Anion-Transporting Polypeptide 1b2 on the Pharmacokinetics of Regorafenib and Its Metabolites Regorafenib-N-Oxide and Regorafenib-Glucuronide in Mice. Clin. Transl. Sci. 2019, 12, 400–407. [Google Scholar] [CrossRef]
- Khachatryan, V.; Muazzam, A.; Hamal, C.; Velugoti, L.S.D.R.; Tabowei, G.; Gaddipati, G.N.; Mukhtar, M.; Alzubaidee, M.J.; Dwarampudi, R.S.; Mathew, S.; et al. The Role of Regorafenib in the Management of Advanced Gastrointestinal Stromal Tumors: A Systematic Review. Cureus 2022, 14, e28665. [Google Scholar] [CrossRef]
- Fukudo, M.; Asai, K.; Tani, C.; Miyamoto, M.; Ando, K.; Ueno, N. Pharmacokinetics of the oral multikinase inhibitor regorafenib and its association with real-world treatment outcomes. Investig. New Drugs 2021, 39, 1422–1431. [Google Scholar] [CrossRef]
- Zhao, D.; Long, X.; Wang, J. Transporter-mediated drug-drug interactions involving poly (ADP-ribose) polymerase inhibitors (Review). Oncol. Lett. 2023, 25, 161. [Google Scholar] [CrossRef]
- Sun, Z.; Zhang, Z.; Ji, M.; Yang, H.; Cromie, M.; Gu, J.; Wang, C.; Yang, L.; Yu, Y.; Gao, W.; et al. BDE47 induces rat CYP3A1 by targeting the transcriptional regulation of miR-23b. Sci. Rep. 2016, 6, 31958. [Google Scholar] [CrossRef]
- Bogaards, J.J.; Bertrand, M.; Jackson, P.; Oudshoorn, M.J.; Weaver, R.J.; van Bladeren, P.J.; Walther, B. Determining the best animal model for human cytochrome P450 activities: A comparison of mouse, rat, rabbit, dog, micropig, monkey and man. Xenobiotica 2000, 30, 1131–1152. [Google Scholar] [CrossRef]
- Zuber, R.; Anzenbacherová, E.; Anzenbacher, P. Cytochromes P450 and experimental models of drug metabolism. J. Cell. Mol. Med. 2002, 6, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Stivarga (regorafenib) Tablets. Drug Approval Package. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2012/203085Orig1s000TOC.cfm (accessed on 10 November 2024).
- Dirix, L.; Swaisland, H.; Verheul, H.M.; Rottey, S.; Leunen, K.; Jerusalem, G.; Rolfo, C.; Nielsen, D.; Molife, L.R.; Kristeleit, R.; et al. Effect of Itraconazole and Rifampin on the Pharmacokinetics of Olaparib in Patients with Advanced Solid Tumors: Results of Two Phase I Open-label Studies. Clin. Ther. 2016, 38, 2286–2299. [Google Scholar] [CrossRef]
- Pasanen, M. Species differences in CYP enzymes. In Citocromo P40; Agnosto, M.C., Comez-Lechon, M.J., Eds.; Instituto de Espana, Real academia Nacional de Farmacia: Madrid, Spain, 2004; pp. 63–90. [Google Scholar]
- Kojima, A.; Sogabe, A.; Nadai, M.; Katoh, M. Species differences in oxidative metabolism of regorafenib. Xenobiotica 2021, 51, 1400–1407. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, T.; Naritomi, Y.; Tetsuka, K.; Nakamori, F.; Moriguchi, H.; Yamano, K.; Terashita, S.; Tabata, K.; Teramura, T. Species differences in intestinal glucuronidation activities between humans, rats, dogs and monkeys. Xenobiotica 2014, 44, 205–216. [Google Scholar] [CrossRef]
- Kazuki, Y.; Kobayashi, K.; Hirabayashi, M.; Abe, S.; Kajitani, N.; Kazuki, K.; Takehara, S.; Takiguchi, M.; Satoh, D.; Kuze, J.; et al. Humanized UGT2 and CYP3A transchromosomic rats for improved prediction of human drug metabolism. Proc. Natl. Acad. Sci. USA 2019, 116, 3072–3081. [Google Scholar] [CrossRef] [PubMed]
- Verscheijden, L.F.M.; Koenderink, J.B.; de Wildt, S.N.; Russel, F.G.M. Differences in P-glycoprotein activity in human and rodent blood-brain barrier assessed by mechanistic modelling. Arch. Toxicol. 2021, 95, 3015–3029. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Yuan, H.; Li, N.; Song, G.; Zheng, Y.; Baratta, M.; Hua, F.; Thurston, A.; Wang, J.; Lai, Y. Identification of interspecies difference in efflux transporters of hepatocytes from dog, rat, monkey and human. Eur. J. Pharm. Sci. 2008, 35, 114–126. [Google Scholar] [CrossRef]
- Sharma, S.; Singh, D.K.; Mettu, V.S.; Yue, G.; Ahire, D.; Basit, A.; Heyward, S.; Prasad, B. Quantitative Characterization of Clinically Relevant Drug-Metabolizing Enzymes and Transporters in Rat Liver and Intestinal Segments for Applications in PBPK Modeling. Mol. Pharm. 2023, 20, 1737–1749. [Google Scholar] [CrossRef]
- Kawahara, I.; Nishikawa, S.; Yamamoto, A.; Kono, Y.; Fujita, T. Assessment of contribution of BCRP to intestinal absorption of various drugs using portal-systemic blood concentration difference model in mice. Pharmacol. Res. Perspect. 2020, 8, e00544. [Google Scholar] [CrossRef]
- Sharma, S.; Mettu, V.S.; Prasad, B. Interplay of Breast Cancer Resistance Protein (Bcrp/Abcg2), Sex, and Fed State in Oral Pharmacokinetic Variability of Furosemide in Rats. Pharmaceutics 2023, 15, 542. [Google Scholar] [CrossRef]
- Nakanishi, T.; Tamai, I. Interaction of Drug or Food with Drug Transporters in Intestine and Liver. Curr. Drug Metab. 2015, 16, 753–764. [Google Scholar] [CrossRef]
- Li, N.; Palandra, J.; Nemirovskiy, O.V.; Lai, Y. LC-MS/MS mediated absolute quantification and comparison of bile salt export pump and breast cancer resistance protein in livers and hepatocytes across species. Anal. Chem. 2009, 81, 2251–2259. [Google Scholar] [CrossRef] [PubMed]
- Uchida, Y.; Yagi, Y.; Takao, M.; Tano, M.; Umetsu, M.; Hirano, S.; Usui, T.; Tachikawa, M.; Terasaki, T. Comparison of absolute protein abundances of transporters and receptors among blood-brain barriers at different cerebral regions and blood-spinal cord barrier in human and rat. Mol. Pharm. 2020, 17, 2006–2020. [Google Scholar] [CrossRef]
- Cao, X.; Gibbs, S.; Fang, L.; Miller, H.; Landowski, C.; Shin, H.; Lennernas, H.; Zhong, Y.; Amidon, G.; Yu, L.; et al. Why is it Challenging to Predict Intestinal Drug Absorption and Oral Bioavailability in Human Using Rat Model. Pharm. Res. 2006, 23, 1675–1686. [Google Scholar] [CrossRef] [PubMed]
- Hotta, K.; Ueyama, J.; Tatsumi, Y.; Tsukiyama, I.; Sugiura, Y.; Saito, H.; Matsuura, K.; Hasegawa, T. Lack of Contribution of Multidrug Resistance-associated Protein and Organic Anion-transporting Polypeptide to Pharmacokinetics of Regorafenib, a Novel Multi-Kinase Inhibitor, in Rats. Anticancer Res. 2015, 35, 4681–4689. [Google Scholar] [PubMed]
- Hryn, V.; Kostylenko, Y.; Yushchenko, Y.; Lavrenko, A.; Ryabushko, O. General comparative anatomy of human and white rat digestive systems: A bibliographic analysis. Wiad. Lek. 2018, 71, 1599–1602. [Google Scholar]
- Wu, T.C.; Ma, C.; Jeong, H.; Yao, H.M.; Chiou, W.L. Comparison of gastrointestinal absorption between human and rat: The role of intestinal absorptive surface area and unstirred aqueous layer. Food Chem. Toxicol. 2002, 40, 1727–1728. [Google Scholar] [CrossRef]
- DeSesso, J.M.; Jacobson, C.F. Anatomical and physiological parameters affecting gastrointestinal absorption in humans and rats. Food Chem. Toxicol. 2001, 39, 209–228. [Google Scholar] [CrossRef]
- Colclough, N.; Ruston, L.; Wood, J.M.; MacFaul, P.A. Species differences in drug plasma protein binding. Med. Chem. Commun. 2014, 5, 963–967. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pharmacokinetic Parameters | IREG+OLA (n = 8) | IIREG (n = 8) | p-Value IREG+OLA vs. IIREG | Gmean Ratio ** (90% CI) IREG+OLA vs. IIREG |
|---|---|---|---|---|
| REG | ||||
| Cmax (µg/mL) | 3.69 ± 2.97 (80.5) | 1.87 ± 0.31 (16.5) | 0.5054 | 1.37 (0.73; 2.58) |
| AUC0–t (ng × h/mL) | 106.92 ± 84.63 (79.2) | 36.76 ± 5.84 (15.9) | 0.0518 | 1.71 (0.73; 4.04) |
| AUC0–∞ (ng × h/mL) | 124.47 ± 79.63 (64.0) | 36.85 ± 5.83 (15.8) | 0.0289 | 2.54 (1.38; 4.67) |
| tmax (h) | 14.1 ± 8.2 (58.2) | 5.8 ± 2.1 (36.9) | 0.0069 | 2.27 (1.47; 3.51) |
| kel (h−1) | 0.06 ± 0.02 (26.1) | 0.11 ± 0.03 (22.8) | 0.0015 * | 0.56 (0.44; 0.71) |
| t0.5 (h) | 11.65 ± 3.70 (31.7) | 6.37 ± 1.33 (20.8) | 0.0003 | 1.79 (1.41; 2.26) |
| Cl/F (mL/h × kg) | 129.86 ± 160.06 (123.1) | 220.83 ± 29.74 (13.5) | 0.0205 | 0.37 (0.20; 0.68) |
| Vd/F (mL) | 2025.57 ± 2097.73 (103.5) | 2062.53 ± 615.85 (29.7) | 0.3357 | 0.66 (0.34; 1.27) |
| M-2 | ||||
| Cmax (ng/mL) | 2469.92 ± 1257.44 (50.9) | 342.10 ± 132.87 (38.8) | 0.0007 | 6.83 (4.52; 10.33) |
| AUC0–t (ng × h/mL) | 85,176.80 ± 35,788.00 (42.0) | 9615.36 ± 2275.41 (23.7) | 0.0007 | 8.38 (5.92; 11.87) |
| AUC0–∞ (ng × h/mL) | 2469.92 ± 1257.44 (50.9) | 11,469.93 ± 2133.05 (18.6) | 0.0007 | 6.83 (4.52; 10.33) |
| Metabolite M-2/Parent ratio | ||||
| Cmax | 1.24 ± 0.89 (71.9) | 0.44 ± 0.73 (39.76) | 0.0007 | 5.73 (3.34; 9.83) |
| AUC0–t | 1.44 ± 1.09 (75.9) | 0.27 ± 0.08 (29.77) | 0.0007 | 6.39 (3.07; 13.29) |
| M-5 | ||||
| Cmax (ng/mL) | 428.18 ± 194.48 (45.4) | 26.23 ± 3.98 (15.2) | 0.0007 | 14.23 (10.39; 19.63) |
| AUC0–t (ng × h/mL) | 14,324.20 ± 5574.43 (38.9) | 803.19 ± 177.02 (22.0) | 0.0007 | 16.13 (12.03; 21.62) |
| AUC0–∞ (ng × h/mL) | 15,959.41 ± 4837.53 (30.31) | 847.88 ± 178.97 (21.1) | 0.0016 | 18.43 (14.19; 23.93) |
| Metabolite M-5/Parent ratio | ||||
| Cmax | 0.28 ± 0.22 (78.1) | 0.03 ± 0.06 (170.1) | 0.0007 | 11.93 (5.47; 26.01) |
| AUC0–t | 0.26 ± 0.17 (66.2) | 0.02 ± 0.01 (23.33) | 0.0007 | 12.29 (4.22; 35.74) |
| AUC0–∞ | 0.37 ± 0.19 (50.9) | 0.02 ± 0.01 (22.82) | 0.0040 | 9.61 (4.41; 20.93) |
| Pharmacokinetic Parameters | IREG+OLA (n = 8) | IIIOLA (n = 8) | IREG+OLA vs. IIIOLA p-Value | Gmean Ratio ** (90% CI) IREG+OLA vs. IIIOLA |
|---|---|---|---|---|
| Cmax (µg/mL) | 17.87 ± 7.66 (42.9) | 8.72 ± 6.80 (78.0) | 0.0241 | 2.86 (1.36; 6.03) |
| AUC0–t (µg × h/mL) | 96.53 ± 33.23 (34.4) | 28.28 ± 28.16 (99.6) | 0.0006 | 5.98 (2.42; 14.77) |
| AUC0–∞ (µg × h/mL) | 97.14 ± 33.26 (34.2) | 28.58 ± 28.31 (99.1) | 0.0006 | 5.93 (2.41; 14.62) |
| kel (1/h) | 0.25 ± 0.06 (23.2) | 0.44 ± 0.34 (77.5) | 0.0583 * | 0.59 (0.30; 1.13) |
| tmax (h) | 1.28 ± 1.11 (86.3) | 1.16 ± 0.92 (78.9) | 0.9120 * | 1.32 (0.62; 2.80) |
| t0.5 (h) | 3.01 ± 0.86 (28.6) | 4.79 ± 6.06 (126.5) | 0.0650 * | 1.71 (0.88; 3.30) |
| Cl/F (L/h) | 0.43 ± 0.16 (38.3) | 7.84 ± 15.21 (193.9) | 0.0006 * | 0.15 (0.06; 0.37) |
| Vd/F (L) | 1.88 ± 1.05 (55.9) | 22.49 ± 24.81 (110.3) | 0.0281 * | 0.26 (0.10; 0.65) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szkutnik-Fiedler, D.; Karbownik, A.; Otto, F.; Maciejewska, J.; Kuźnik, A.; Grabowski, T.; Wolc, A.; Grześkowiak, E.; Stanisławiak-Rudowicz, J.; Szałek, E. Pharmacokinetic Interaction Between Olaparib and Regorafenib in an Animal Model. Pharmaceutics 2024, 16, 1575. https://doi.org/10.3390/pharmaceutics16121575
Szkutnik-Fiedler D, Karbownik A, Otto F, Maciejewska J, Kuźnik A, Grabowski T, Wolc A, Grześkowiak E, Stanisławiak-Rudowicz J, Szałek E. Pharmacokinetic Interaction Between Olaparib and Regorafenib in an Animal Model. Pharmaceutics. 2024; 16(12):1575. https://doi.org/10.3390/pharmaceutics16121575
Chicago/Turabian StyleSzkutnik-Fiedler, Danuta, Agnieszka Karbownik, Filip Otto, Julia Maciejewska, Alicja Kuźnik, Tomasz Grabowski, Anna Wolc, Edmund Grześkowiak, Joanna Stanisławiak-Rudowicz, and Edyta Szałek. 2024. "Pharmacokinetic Interaction Between Olaparib and Regorafenib in an Animal Model" Pharmaceutics 16, no. 12: 1575. https://doi.org/10.3390/pharmaceutics16121575
APA StyleSzkutnik-Fiedler, D., Karbownik, A., Otto, F., Maciejewska, J., Kuźnik, A., Grabowski, T., Wolc, A., Grześkowiak, E., Stanisławiak-Rudowicz, J., & Szałek, E. (2024). Pharmacokinetic Interaction Between Olaparib and Regorafenib in an Animal Model. Pharmaceutics, 16(12), 1575. https://doi.org/10.3390/pharmaceutics16121575