
Kinetic and Methodological Insights into Hydrophilic Drug Release from Mesoporous Silica Nanocarriers

 , , and
, , and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of Mesoporous Silica Nanoparticles (MSNs)
2.3. Characterization
2.4. Drug Loading
2.5. In Vitro Drug Release
2.5.1. Sample and Separate with Shaking/Stirring
2.5.2. Sample and Separate Using Independent Batch Shaking/Stirring
2.5.3. Dialysis Bag with Shaking/Stirring
2.6. Kinetics Analysis
2.7. Statistical Analysis
3. Results and Discussion
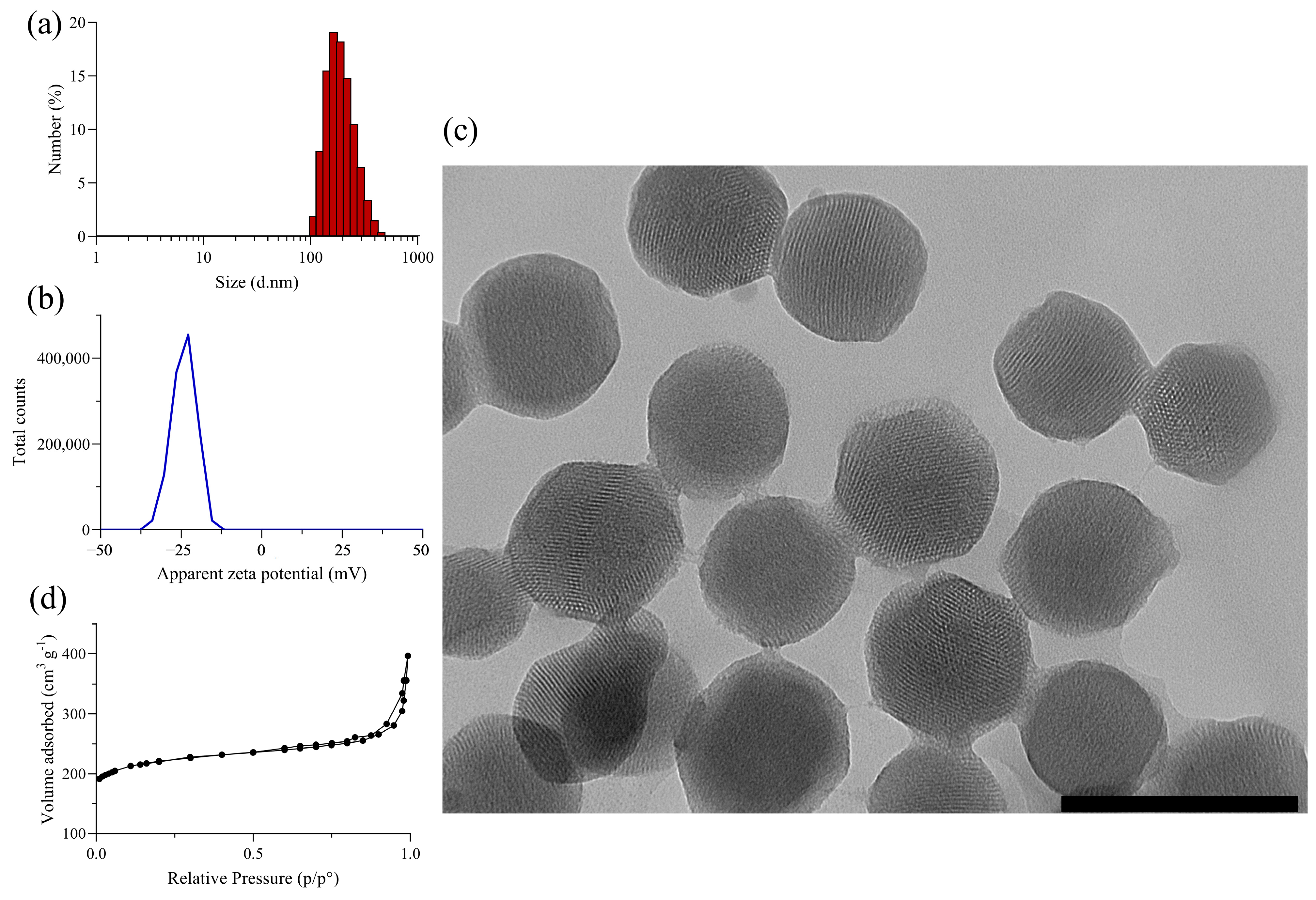
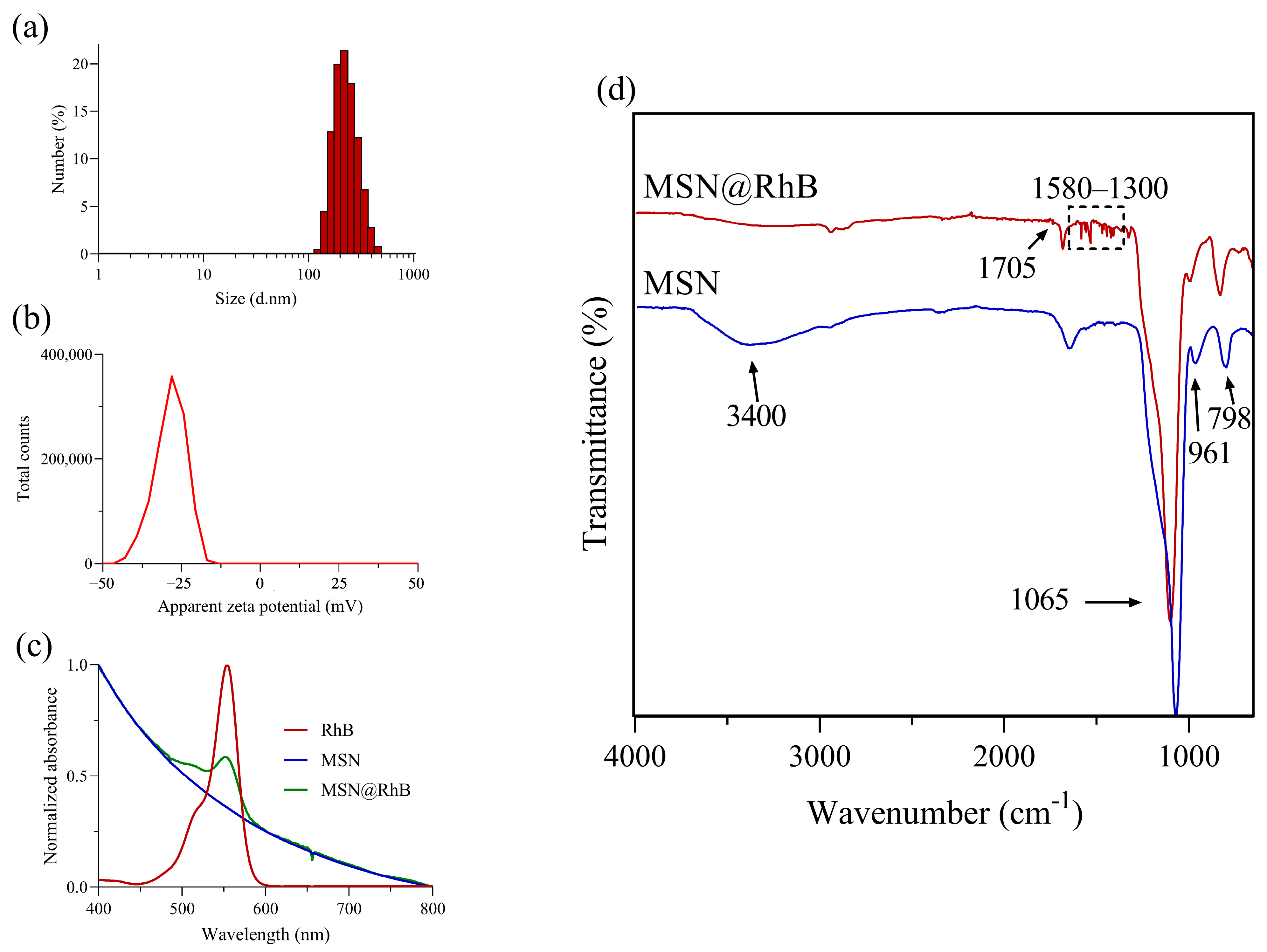
3.1. Synthesis and Characterization of Silica Nanomaterials
3.2. Release Kinetics of RhB from Mesoporous Silica Nanoparticles
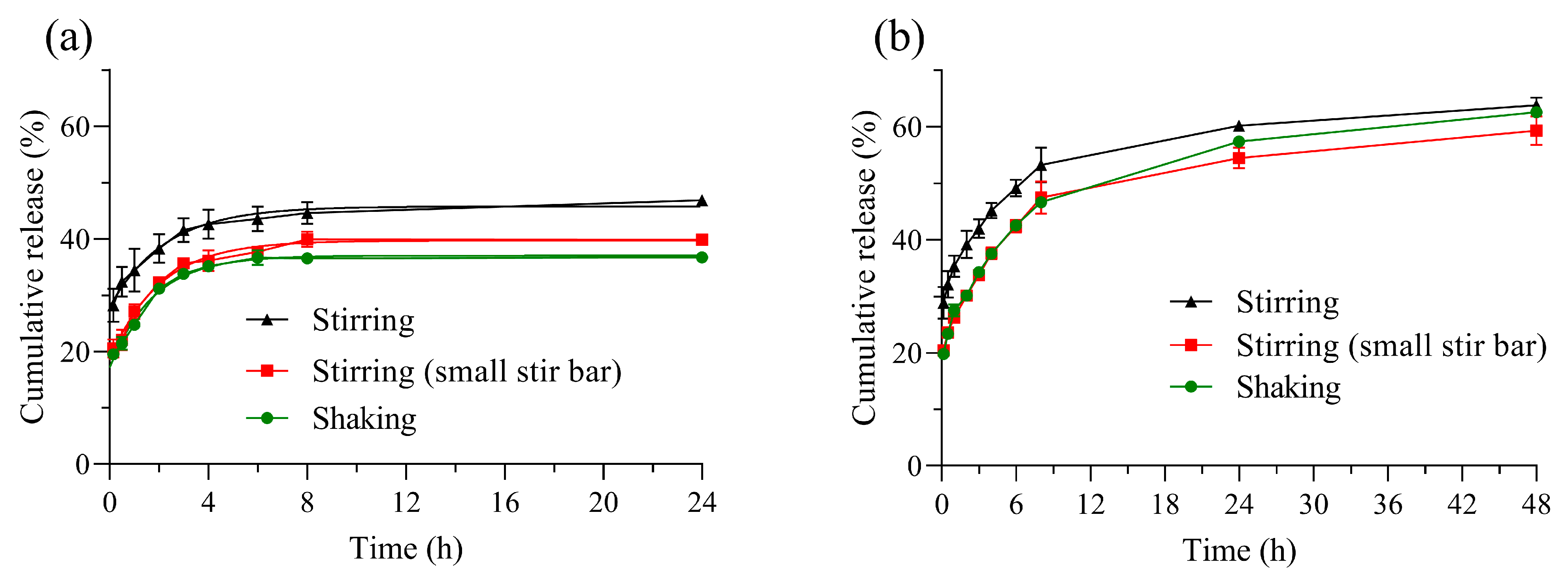
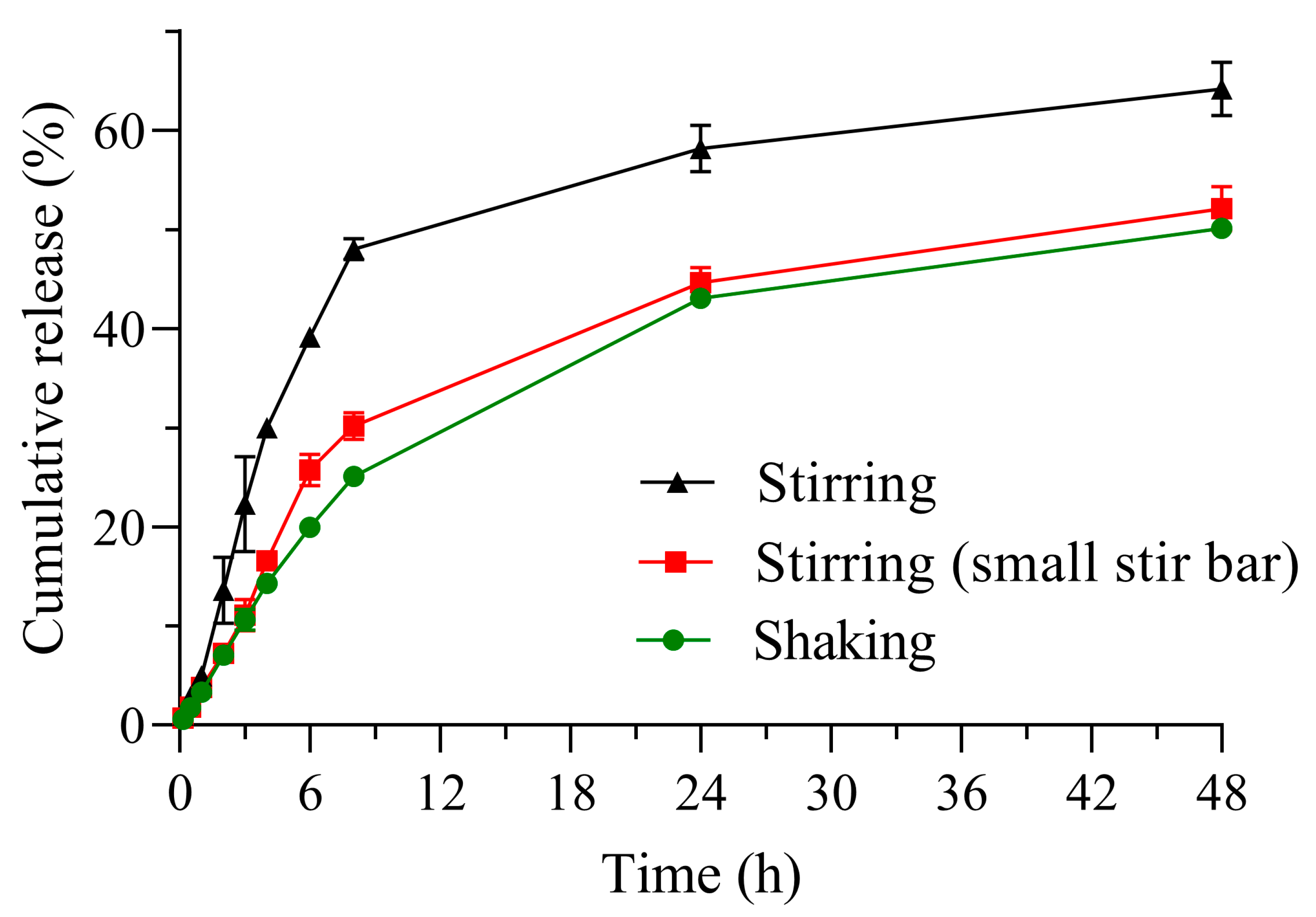
3.2.1. Comparison of Release Profiles Obtained by Different Methods
3.2.2. Kinetic Analysis of RhB Release
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Darrow, J.J.; Avorn, J.; Kesselheim, A.S. FDA Approval and Regulation of Pharmaceuticals, 1983–2018. JAMA 2020, 323, 164–176. [Google Scholar] [CrossRef] [PubMed]
- Olefir, Y.V.; Sakanyan, E.I.; Luttseva, A.I.; Babeshina, L.G.; Shemeryankina, T.B. Validation of Methods and Procedures in Pharmacopoeial Monographs in the Framework of a Drug Standardization Program. Pharm. Chem. J. 2019, 52, 873–877. [Google Scholar] [CrossRef]
- Elder, D. ICH Q6A Specifications. In ICH Quality Guidelines; John Wiley & Sons: Hoboken, NJ, USA, 2017; pp. 433–466. ISBN 978-1-118-97114-7. [Google Scholar]
- Alshawwa, S.Z.; Kassem, A.A.; Farid, R.M.; Mostafa, S.K.; Labib, G.S. Nanocarrier Drug Delivery Systems: Characterization, Limitations, Future Perspectives and Implementation of Artificial Intelligence. Pharmaceutics 2022, 14, 883. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Lázaro, L.; Martín-Sabroso, C.; Aparicio-Blanco, J.; Torres-Suárez, A.I. Assessment of In Vitro Release Testing Methods for Colloidal Drug Carriers: The Lack of Standardized Protocols. Pharmaceutics 2024, 16, 103. [Google Scholar] [CrossRef]
- Weng, J.; Tong, H.H.Y.; Chow, S.F. In Vitro Release Study of the Polymeric Drug Nanoparticles: Development and Validation of a Novel Method. Pharmaceutics 2020, 12, 732. [Google Scholar] [CrossRef]
- Desai, N. Challenges in Development of Nanoparticle-Based Therapeutics. AAPS J. 2012, 14, 282–295. [Google Scholar] [CrossRef]
- Joseph, T.M.; Kar Mahapatra, D.; Esmaeili, A.; Piszczyk, Ł.; Hasanin, M.S.; Kattali, M.; Haponiuk, J.; Thomas, S. Nanoparticles: Taking a Unique Position in Medicine. Nanomaterials 2023, 13, 574. [Google Scholar] [CrossRef]
- Narayan, R.; Nayak, U.Y.; Raichur, A.M.; Garg, S. Mesoporous Silica Nanoparticles: A Comprehensive Review on Synthesis and Recent Advances. Pharmaceutics 2018, 10, 118. [Google Scholar] [CrossRef]
- D’Souza, S. A Review of In Vitro Drug Release Test Methods for Nano-Sized Dosage Forms. Adv. Pharm. 2014, 2014, 304757. [Google Scholar] [CrossRef]
- Wacker, M.G.; Lu, X.; Burke, M.; Nir, I.; Fahmy, R. Testing the In-Vitro Product Performance of Nanomaterial-Based Drug Products: View of the USP Expert Panel. Dissolution Technol. 2022, 29, 6–20. [Google Scholar] [CrossRef]
- Yu, M.; Yuan, W.; Li, D.; Schwendeman, A.; Schwendeman, S.P. Predicting Drug Release Kinetics from Nanocarriers inside Dialysis Bags. J. Control. Release 2019, 315, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Wallace, S.J.; Li, J.; Nation, R.L.; Boyd, B.J. Drug Release from Nanomedicines: Selection of Appropriate Encapsulation and Release Methodology. Drug Deliv. Transl. Res. 2012, 2, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Zhan, H.; Sun, X.; Zhang, Z.-R.; Deng, L. A Density-Changing Centrifugation Method for Efficient Separation of Free Drugs from Drug-Loaded Particulate Delivery Systems. AAPS J. 2019, 21, 33. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Lapitsky, Y. Pitfalls in Analyzing Release from Chitosan/Tripolyphosphate Micro- and Nanoparticles. Eur. J. Pharm. Biopharm. 2019, 142, 204–215. [Google Scholar] [CrossRef]
- van Leeuwen, T.; Kuchel, R.P.; Knothe Tate, M.L.; Zetterlund, P.B. Paclitaxel Release from Hollow PMMA Nanoparticles: Factors Affecting Release Rate as Quantified via Dialysis and Membrane Centrifugation. Colloids Surf. A Physicochem. Eng. Asp. 2023, 675, 131992. [Google Scholar] [CrossRef]
- Moreno-Bautista, G.; Tam, K.C. Evaluation of Dialysis Membrane Process for Quantifying the in Vitro Drug-Release from Colloidal Drug Carriers. Colloids Surf. A Physicochem. Eng. Asp. 2011, 389, 299–303. [Google Scholar] [CrossRef]
- Modi, S.; Anderson, B.D. Determination of Drug Release Kinetics from Nanoparticles: Overcoming Pitfalls of the Dynamic Dialysis Method. Mol. Pharm. 2013, 10, 3076–3089. [Google Scholar] [CrossRef]
- Abouelmagd, S.A.; Sun, B.; Chang, A.C.; Ku, Y.J.; Yeo, Y. Release Kinetics Study of Poorly Water-Soluble Drugs from Nanoparticles: Are We Doing It Right? Mol. Pharm. 2015, 12, 997–1003. [Google Scholar] [CrossRef]
- Ortiz, A.C.; Yañez, O.; Salas-Huenuleo, E.; Morales, J.O. Development of a Nanostructured Lipid Carrier (NLC) by a Low-Energy Method, Comparison of Release Kinetics and Molecular Dynamics Simulation. Pharmaceutics 2021, 13, 531. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration Liposome Drug Products: Chemistry, Manufacturing, and Controls; Human Pharmacokinetics and Bioavailability and Labeling Documentation. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/liposome-drug-products-chemistry-manufacturing-and-controls-human-pharmacokinetics-and (accessed on 22 April 2025).
- Skoczen, S.L.; Stern, S.T. Improved Ultrafiltration Method to Measure Drug Release from Nanomedicines Utilizing a Stable Isotope Tracer. In Characterization of Nanoparticles Intended for Drug Delivery; McNeil, S.E., Ed.; Springer: New York, NY, USA, 2018; pp. 223–239. ISBN 978-1-4939-7352-1. [Google Scholar]
- Nagpal, S.; Png, J.; Kahouadji, L.; Wacker, M.G. A Bio-Predictive Release Assay for Liposomal Prednisolone Phosphate. J. Control. Release 2024, 374, 61–75. [Google Scholar] [CrossRef]
- Đorđević, S.; Gonzalez, M.M.; Conejos-Sánchez, I.; Carreira, B.; Pozzi, S.; Acúrcio, R.C.; Satchi-Fainaro, R.; Florindo, H.F.; Vicent, M.J. Current Hurdles to the Translation of Nanomedicines from Bench to the Clinic. Drug Deliv. Transl. Res. 2022, 12, 500–525. [Google Scholar] [CrossRef] [PubMed]
- Paswan, S.K.; Saini, T. Comparative Evaluation of in Vitro Drug Release Methods Employed for Nanoparticle Drug Release Studies. Clin. Trials 2021, 14, 17. [Google Scholar] [CrossRef]
- Wolska, E.; Szymańska, M. Comparison of the In Vitro Drug Release Methods for the Selection of Test Conditions to Characterize Solid Lipid Microparticles. Pharmaceutics 2023, 15, 511. [Google Scholar] [CrossRef] [PubMed]
- Slowing, I.; Trewyn, B.G.; Lin, V.S.-Y. Effect of Surface Functionalization of MCM-41-Type Mesoporous Silica Nanoparticles on the Endocytosis by Human Cancer Cells. J. Am. Chem. Soc. 2006, 128, 14792–14793. [Google Scholar] [CrossRef]
- Ortiz, A.C.; Jackson, N.; Buelvas, N.; Jerez, A.; López-Muñoz, R.A.; Morales, J.; Arriagada, F. Release Kinetics Approach of Stimuli-Responsive Mesoporous Silica Nanocarriers: pH-Sensitive Linker versus pH-Sensitive Framework. J. Drug Deliv. Sci. Technol. 2024, 91, 105212. [Google Scholar] [CrossRef]
- Ritger, P.L.; Peppas, N.A. A Simple Equation for Description of Solute Release II. Fickian and Anomalous Release from Swellable Devices. J. Control. Release 1987, 5, 37–42. [Google Scholar] [CrossRef]
- Shah, V.P.; Tsong, Y.; Sathe, P.; Liu, J.-P. In Vitro Dissolution Profile Comparison—Statistics and Analysis of the Similarity Factor, F2. Pharm. Res. 1998, 15, 889–896. [Google Scholar] [CrossRef]
- Xie, F.; Ji, S.; Cheng, Z. In Vitro Dissolution Similarity Factor (F2) and in Vivo Bioequivalence Criteria, How and When Do They Match? Using a BCS Class II Drug as a Simulation Example. Eur. J. Pharm. Sci. 2015, 66, 163–172. [Google Scholar] [CrossRef]
- Maurya, N.S.; Mittal, A.K.; Cornel, P.; Rother, E. Biosorption of Dyes Using Dead Macro Fungi: Effect of Dye Structure, Ionic Strength and pH. Bioresour. Technol. 2006, 97, 512–521. [Google Scholar] [CrossRef]
- Yu, Y.; Murthy, B.N.; Shapter, J.G.; Constantopoulos, K.T.; Voelcker, N.H.; Ellis, A.V. Benzene Carboxylic Acid Derivatized Graphene Oxide Nanosheets on Natural Zeolites as Effective Adsorbents for Cationic Dye Removal. J. Hazard. Mater. 2013, 260, 330–338. [Google Scholar] [CrossRef]
- Rasalingam, S.; Peng, R.; Koodali, R.T. An Insight into the Adsorption and Photocatalytic Degradation of Rhodamine B in Periodic Mesoporous Materials. Appl. Catal. B Environ. 2015, 174–175, 49–59. [Google Scholar] [CrossRef]
- Ouachtak, H.; El Haouti, R.; El Guerdaoui, A.; Haounati, R.; Amaterz, E.; Addi, A.A.; Akbal, F.; Taha, M.L. Experimental and Molecular Dynamics Simulation Study on the Adsorption of Rhodamine B Dye on Magnetic Montmorillonite Composite γ-Fe2O3@Mt. J. Mol. Liq. 2020, 309, 113142. [Google Scholar] [CrossRef]
- Baig, M.M.; Yousuf, M.A.; Alsafari, I.A.; Ali, M.; Agboola, P.O.; Shakir, I.; Haider, S.; Warsi, M.F. New Mesostructured Origami Silica Matrix: A Nano-Platform for Highly Retentive and pH-Controlled Delivery System. J. Taibah Univ. Sci. 2021, 15, 133–144. [Google Scholar] [CrossRef]
- D’Souza, S.S.; DeLuca, P.P. Methods to Assess in Vitro Drug Release from Injectable Polymeric Particulate Systems. Pharm. Res. 2006, 23, 460–474. [Google Scholar] [CrossRef]
- Nothnagel, L.; Wacker, M.G. How to Measure Release from Nanosized Carriers? Eur. J. Pharm. Sci. 2018, 120, 199–211. [Google Scholar] [CrossRef]
- Brown, C.K.; Friedel, H.D.; Barker, A.R.; Buhse, L.F.; Keitel, S.; Cecil, T.L.; Kraemer, J.; Morris, J.M.; Reppas, C.; Stickelmeyer, M.P.; et al. FIP/AAPS Joint Workshop Report: Dissolution/In Vitro Release Testing of Novel/Special Dosage Forms. AAPS PharmSciTech 2011, 12, 782–794. [Google Scholar] [CrossRef]
- Shah, V.P.; Gurbarg, M.; Noory, A.; Dighe, S.; Skelly, J.P. Influence of Higher Rates of Agitation on Release Patterns of Immediate-Release Drug Products. J. Pharm. Sci. 1992, 81, 500–503. [Google Scholar] [CrossRef]
- Kamba, M.; Seta, Y.; Takeda, N.; Hamaura, T.; Kusai, A.; Nakane, H.; Nishimura, K. Measurement of Agitation Force in Dissolution Test and Mechanical Destructive Force in Disintegration Test. Int. J. Pharm. 2003, 250, 99–109. [Google Scholar] [CrossRef]
- Seeger, N.; Lange, S.; Klein, S. Impact of Vibration and Agitation Speed on Dissolution of USP Prednisone Tablets RS and Various IR Tablet Formulations. AAPS PharmSciTech 2015, 16, 759–766. [Google Scholar] [CrossRef]
- Costa, P.; Lobo, J.M.S. Influence of Dissolution Medium Agitation on Release Profiles of Sustained-Release Tablets. Drug Dev. Ind. Pharm. 2001, 27, 811–817. [Google Scholar] [CrossRef]
- Kincl, M.; Turk, S.; Vrečer, F. Application of Experimental Design Methodology in Development and Optimization of Drug Release Method. Int. J. Pharm. 2005, 291, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Ferrero, C.; Jiménez-Castellanos, M.R. In Vitro Release Testing of Matrices Based on Starch–Methyl Methacrylate Copolymers: Effect of Tablet Crushing Force, Dissolution Medium pH and Stirring Rate. Int. J. Pharm. 2014, 461, 270–279. [Google Scholar] [CrossRef]
- Baxter, J.L.; Kukura, J.; Muzzio, F.J. Hydrodynamics-Induced Variability in the USP Apparatus II Dissolution Test. Int. J. Pharm. 2005, 292, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Bai, G.; Armenante, P.M.; Plank, R.V.; Gentzler, M.; Ford, K.; Harmon, P. Hydrodynamic Investigation of USP Dissolution Test Apparatus II. J. Pharm. Sci. 2007, 96, 2327–2349. [Google Scholar] [CrossRef] [PubMed]
- Salehi, N.; Al-Gousous, J.; Hens, B.; Amidon, G.L.; Ziff, R.M.; Amidon, G.E. Comparative Evaluation of Dissolution Performance in a USP 2 Setup and Alternative Stirrers and Vessel Designs: A Systematic Computational Investigation. Mol. Pharm. 2024, 21, 2406–2414. [Google Scholar] [CrossRef]
- Salehi, N.; Al-Gousous, J.; Mudie, D.M.; Amidon, G.L.; Ziff, R.M.; Amidon, G.E. Hierarchical Mass Transfer Analysis of Drug Particle Dissolution, Highlighting the Hydrodynamics, pH, Particle Size, and Buffer Effects for the Dissolution of Ionizable and Nonionizable Drugs in a Compendial Dissolution Vessel. Mol. Pharm. 2020, 17, 3870–3884. [Google Scholar] [CrossRef]
- Gibaldi, M.; Feldman, S. Establishment of Sink Conditions in Dissolution Rate Determinations. Theoretical Considerations and Application to Nondisintegrating Dosage Forms. J. Pharm. Sci. 1967, 56, 1238–1242. [Google Scholar] [CrossRef]
- Guy, R.H.; Hadgraft, J. Theoretical Comparison of Release Rates of Drugs into Sink and Nonsink Conditions. J. Pharm. Sci. 1981, 70, 1243–1245. [Google Scholar] [CrossRef]
- Shen, J.; Choi, S.; Qu, W.; Wang, Y.; Burgess, D.J. In Vitro-in Vivo Correlation of Parenteral Risperidone Polymeric Microspheres. J. Control. Release 2015, 218, 2–12. [Google Scholar] [CrossRef]
- Siepmann, J.; Siepmann, F. Sink Conditions Do Not Guarantee the Absence of Saturation Effects. Int. J. Pharm. 2020, 577, 119009. [Google Scholar] [CrossRef]
- Hate, S.S.; Thompson, S.A.; Singaraju, A.B. Impact of Sink Conditions on Drug Release Behavior of Controlled-Release Formulations. J. Pharm. Sci. 2025, 114, 520–529. [Google Scholar] [CrossRef] [PubMed]
- Ng, J.B.S.; Kamali-Zare, P.; Brismar, H.; Bergström, L. Release and Molecular Transport of Cationic and Anionic Fluorescent Molecules in Mesoporous Silica Spheres. Langmuir 2008, 24, 11096–11102. [Google Scholar] [CrossRef] [PubMed]
- DeMuth, P.; Hurley, M.; Wu, C.; Galanie, S.; Zachariah, M.R.; DeShong, P. Mesoscale Porous Silica as Drug Delivery Vehicles: Synthesis, Characterization, and pH-Sensitive Release Profiles. Microporous Mesoporous Mater. 2011, 141, 128–134. [Google Scholar] [CrossRef]
- Pepin, X.; Goetschy, M.; Abrahmsén-Alami, S. Mechanistic Models for USP2 Dissolution Apparatus, Including Fluid Hydrodynamics and Sedimentation. J. Pharm. Sci. 2022, 111, 185–196. [Google Scholar] [CrossRef]
- Zambito, Y.; Pedreschi, E.; Di Colo, G. Is Dialysis a Reliable Method for Studying Drug Release from Nanoparticulate Systems?—A Case Study. Int. J. Pharm. 2012, 434, 28–34. [Google Scholar] [CrossRef]
- Billings, C.; Anderson, D.E. Comparison of Three Methods for Assessment of Drug Elution: In Vitro Elution of Gentamicin from a Collagen-Based Scaffold. Front. Drug Deliv. 2022, 2, 958731. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | f2 | f1 | |
|---|---|---|---|
| Independent batch | Shaking vs. Stirring | - | - |
| Shaking vs. Stirring (small stir bar) | 87.8 | 4.6 | |
| Stirring vs. Stirring (small stir bar) | - | - | |
| Sample and separate with renewal | Shaking vs. Stirring | 55.9 | 20.6 |
| Shaking vs. Stirring (small stir bar) | 91.0 | 2.1 | |
| Stirring vs. Stirring (small stir bar) | 55.3 | 21.9 | |
| Dialysis bag | Shaking vs. Stirring | - | - |
| Shaking vs. Stirring (small stir bar) | 76.8 | 12.6 | |
| Stirring vs. Stirring (small stir bar) | - | - | |
| Method | Zero Order | First Order | Power Law | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| k (% h−1) ± SD | R2 | AIC | k (h−1) ± SD | R2 | AIC | n ± SD | R2 | AIC | ||
| Independent batch a | Shaking | 17.07 ± 0.23 | 0.5870 | 69.8 | 0.59 ± 0.05 | 0.9740 | 45.7 | 0.83 ± 0.19 | 0.9904 | 10.7 |
| Stirring | 14.79 ± 1.81 | 0.4969 | 67.5 | 0.44 ± 0.09 | 0.9453 | 46.4 | 0.52 ± 0.10 | 0.9777 | 16.6 | |
| Stirring (small stir bar) | 16.11 ± 0.82 | 0.6457 | 64.2 | 0.47 ± 0.06 | 0.9658 | 48.2 | 0.77 ± 0.10 | 0.9666 | 21.2 | |
| Sample and separate with renewal b | Shaking | 2.68 ± 0.02 | 0.3049 | 90.3 | 0.14 ± 0.01 | 0.9814 | 53.9 | 0.64 ± 0.05 | 0.9931 | 21.3 |
| Stirring | 2.74 ± 0.05 | 0.1247 | 92.9 | 0.16 ± 0.02 | 0.9816 | 52.9 | 0.65 ± 0.03 | 0.9929 | 24.3 | |
| Stirring (small stir bar) | 2.71 ± 0.02 | 0.2459 | 91.6 | 0.15 ± 0.01 | 0.9874 | 50.5 | 0.63 ± 0.03 | 0.9898 | 23.1 | |
| Dialysis bag b | Shaking | 2.56 ± 0.02 | 0.7109 | 82.5 | 0.083 ± 0.003 | 0.9977 | 32.7 | 0.94 ± 0.14 | 0.9923 | 26.4 |
| Stirring | 2.75 ± 0.01 | 0.3004 | 92.6 | 0.16 ± 0.02 | 0.9805 | 55.6 | 0.83 ± 0.21 | 0.9633 | 37.4 | |
| Stirring (small stir bar) | 2.61 ± 0.06 | 0.6174 | 85.7 | 0.10 ± 0.01 | 0.9851 | 51.3 | 1.08 ± 0.07 | 0.9839 | 35.2 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rozas, R.; Ortiz, A.C.; Peñaloza, S.; Lizama, S.; Flores, M.E.; Morales, J.; Arriagada, F. Kinetic and Methodological Insights into Hydrophilic Drug Release from Mesoporous Silica Nanocarriers. Pharmaceutics 2025, 17, 694. https://doi.org/10.3390/pharmaceutics17060694
Rozas R, Ortiz AC, Peñaloza S, Lizama S, Flores ME, Morales J, Arriagada F. Kinetic and Methodological Insights into Hydrophilic Drug Release from Mesoporous Silica Nanocarriers. Pharmaceutics. 2025; 17(6):694. https://doi.org/10.3390/pharmaceutics17060694
Chicago/Turabian StyleRozas, Rodrigo, Andrea C. Ortiz, Sofía Peñaloza, Sebastián Lizama, Mario E. Flores, Javier Morales, and Francisco Arriagada. 2025. "Kinetic and Methodological Insights into Hydrophilic Drug Release from Mesoporous Silica Nanocarriers" Pharmaceutics 17, no. 6: 694. https://doi.org/10.3390/pharmaceutics17060694
APA StyleRozas, R., Ortiz, A. C., Peñaloza, S., Lizama, S., Flores, M. E., Morales, J., & Arriagada, F. (2025). Kinetic and Methodological Insights into Hydrophilic Drug Release from Mesoporous Silica Nanocarriers. Pharmaceutics, 17(6), 694. https://doi.org/10.3390/pharmaceutics17060694