

Synthesis and Characterization of Poly(Lactic-Co-Glycolic Acid)–Paclitaxel (PLGA-PTX) Nanoparticles Evaluated in Ovarian Cancer Models
 , ,
, ,
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Instruments
- NMR Bruker 400 MHz Ultra Shield instrument (Bruker Corporation, Billerica, MA, USA).
- LC/MS: Ultra High-Performance Liquid Chromatography System Agilent Technologies 1200 series Accurate-Mass TOF LC/MS 6220; Agilent QDB-C18 column, 4.6 × 150 mm, 5 µm (Agilent Technologies, Santa Clara, CA, USA).
- HPLC: High-Performance Liquid Chromatography FLEXAR System Perkin Elmer (Waltham, MA, USA)
- DLS: Dynamic Light Scattering (Brookhaven Instrument Corporation, Holtsville, NY, USA)
- TEM: FEI Titan Themis 200 (Molecular Devices, San Jose, CA, USA)
- Plate reader: SpectraMax M3 (Molecular Devices, San Jose, CA, USA)
- Confocal microscope: LSM 880 Airyscan Fast Live Cell (Carl Zeiss Microscopy GmbH, Jena, Germany)
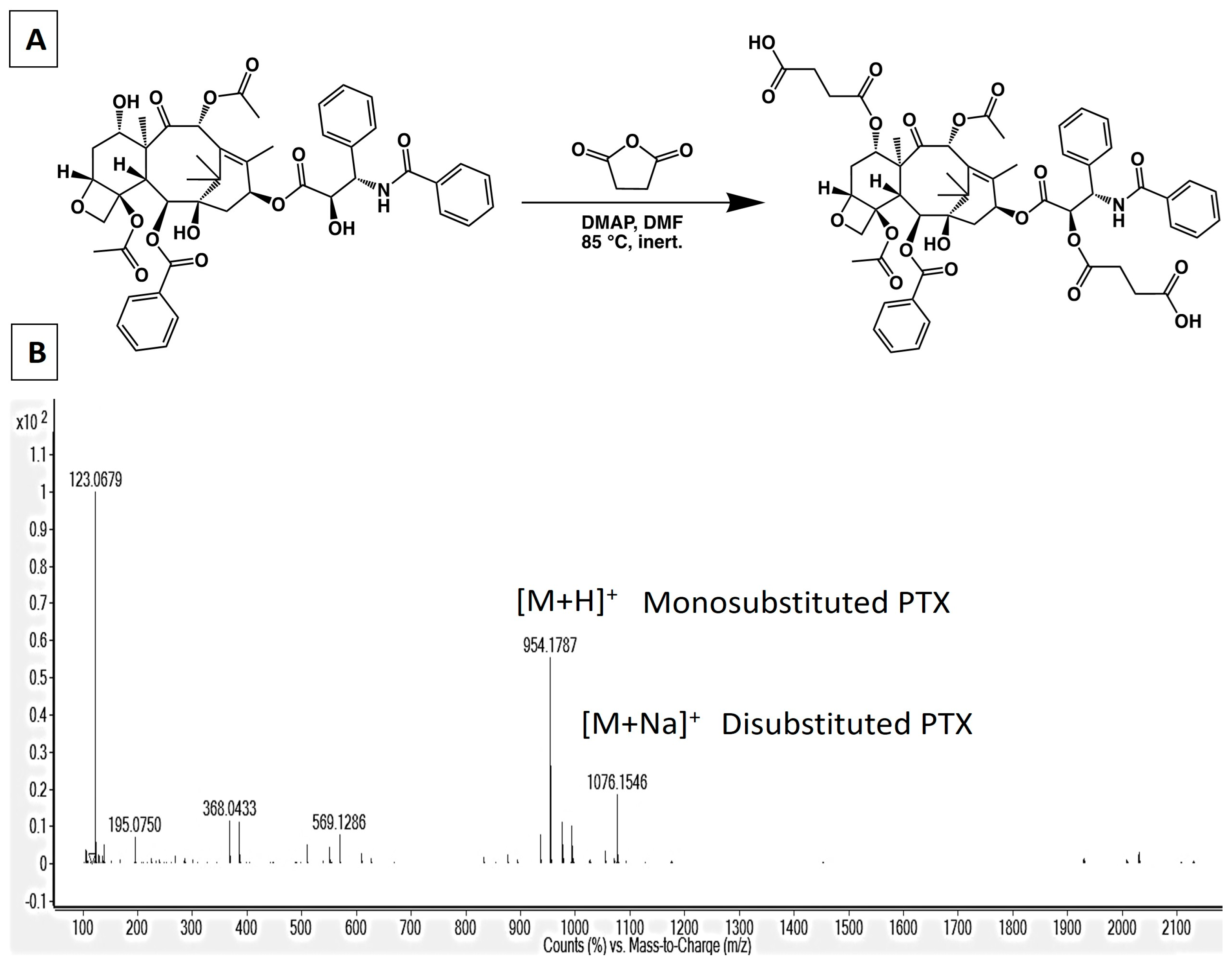
2.3. Synthesis of 2′,7-Disuccinyltaxol
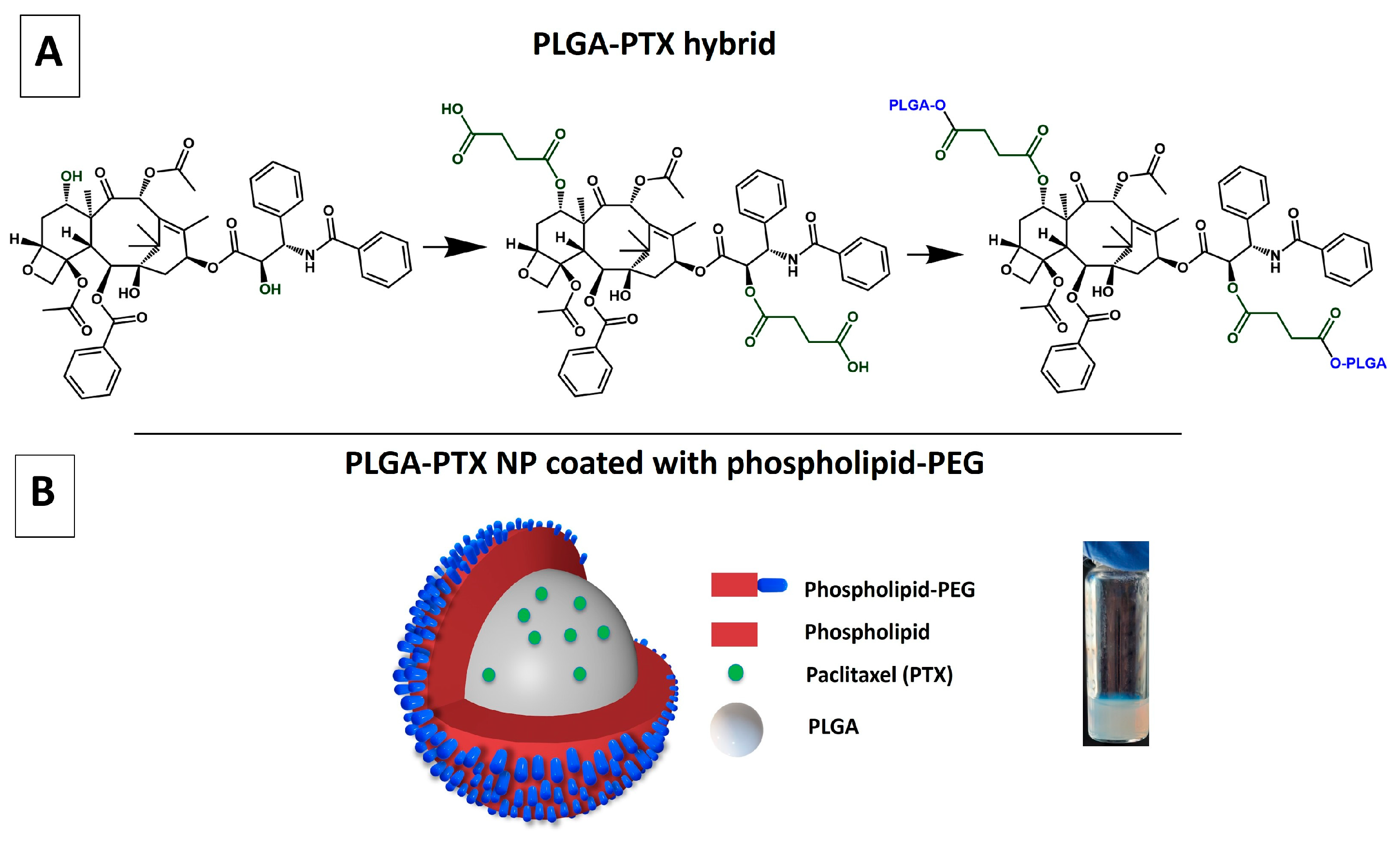
2.4. Synthesis of PTX-PLGA Hybrid with Succinic Acid Linker (Method 1)
2.5. Synthesis of PTX-PLGA Hybrid Without Succinic Acid Linker (Method 2)
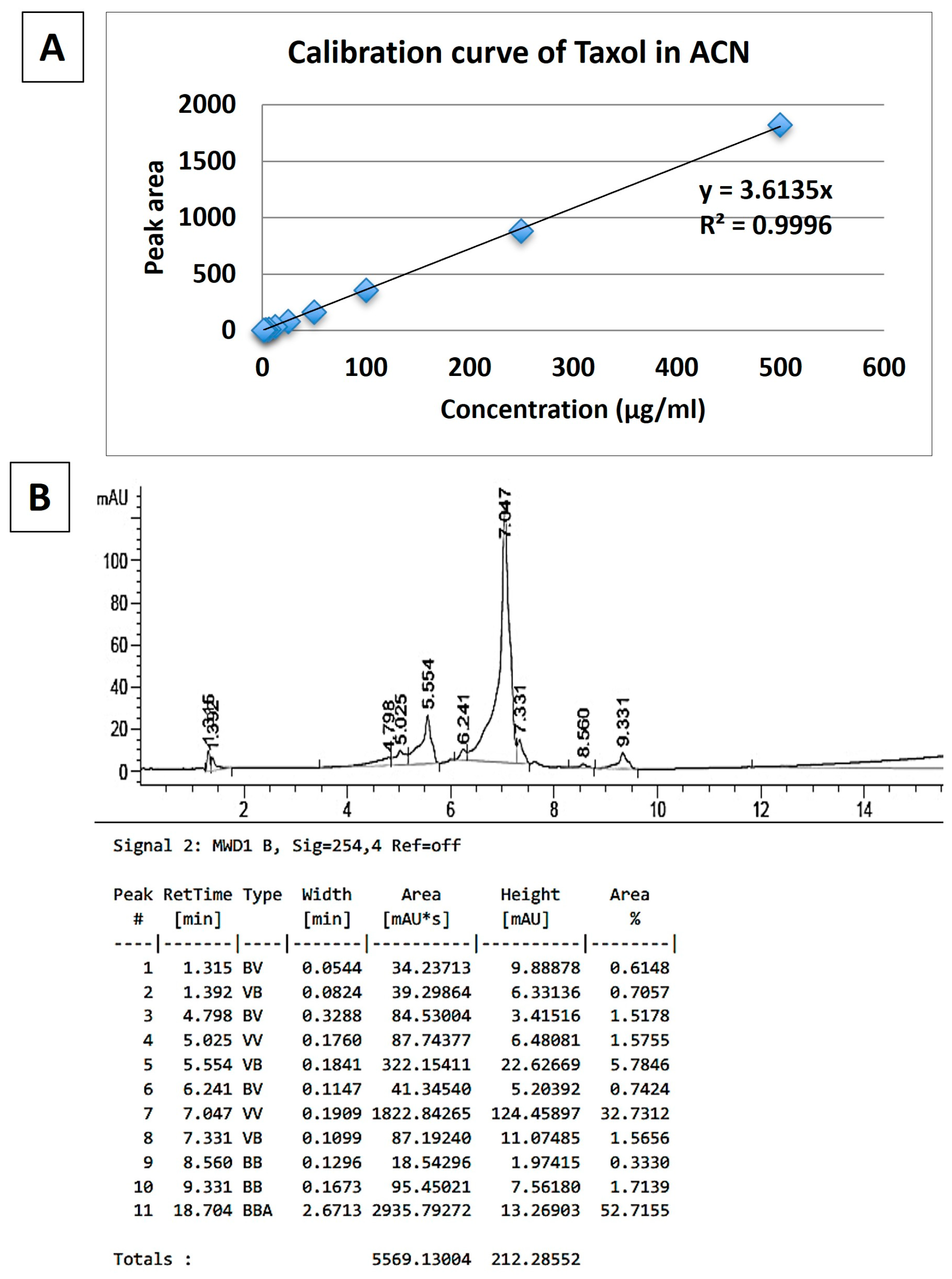
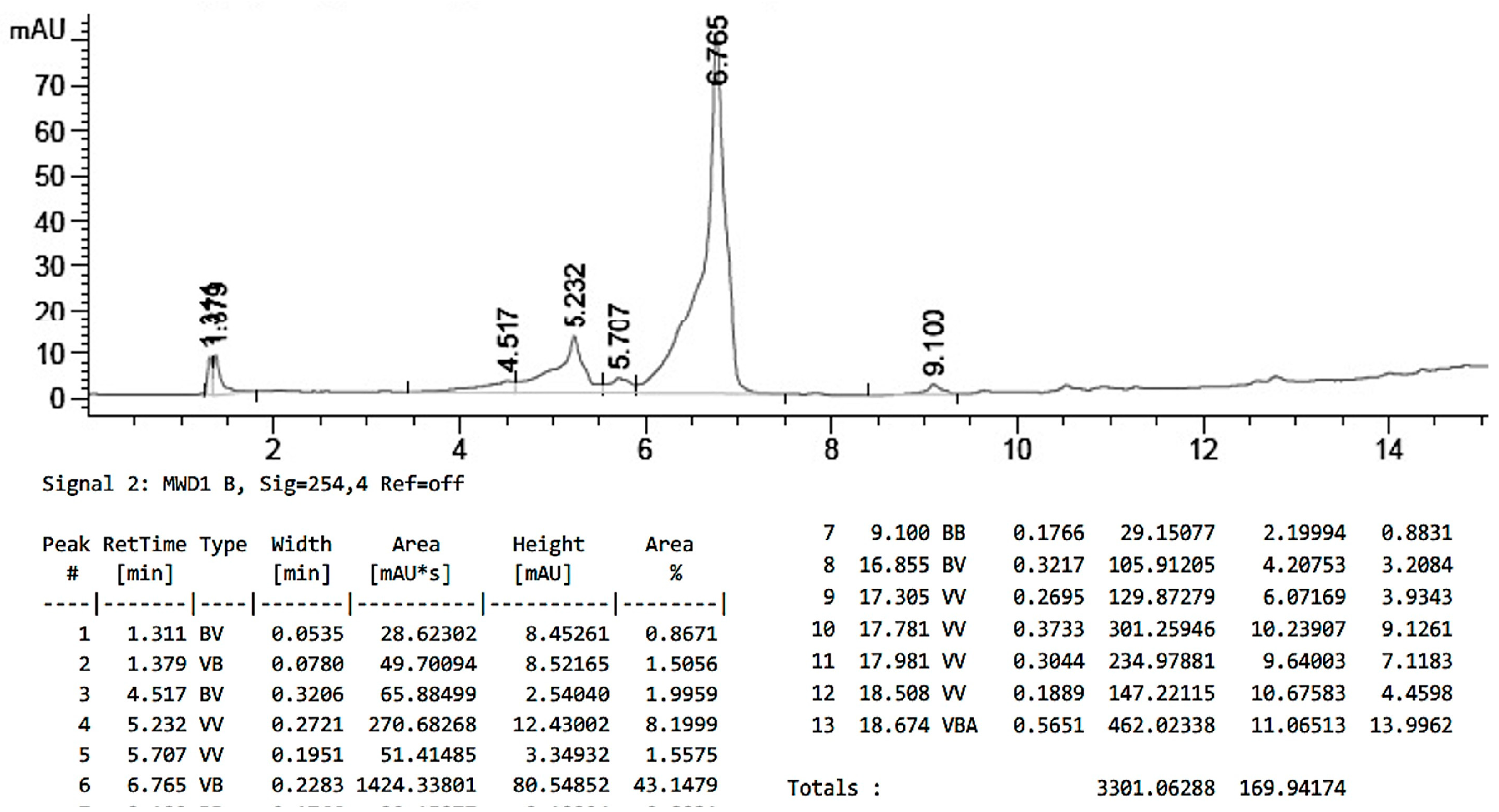
2.6. Method to Calculate PLGA:PTX Ratio
2.7. Synthesis of the PLGA-PTX NPs
2.8. Characterization of Size and Surface Charge of PLGA-PTX NPs
2.8.1. Dynamic Light Scattering (DLS) and Zeta Potential Measurements
2.8.2. Transmission Electron Microscopy (TEM) Imaging
2.9. Characterization of PTX Loading in PLGA-PTX NPs
2.10. Stability of PLGA-PTX NPs
2.11. Drug Release Studies
2.12. In Vitro Cell Culture Models
2.13. Confocal Microscopy Imaging
3. Results and Discussion
3.1. Synthesis of PLGA-PTX Hybrid
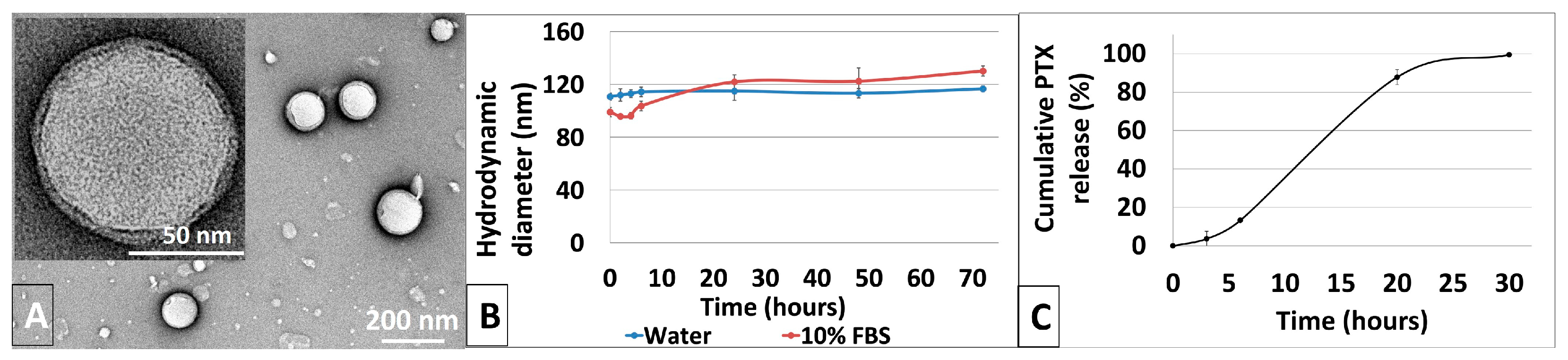
3.2. Synthesis and Characterization of Physicochemical Properties of PLGA-PTX NPs
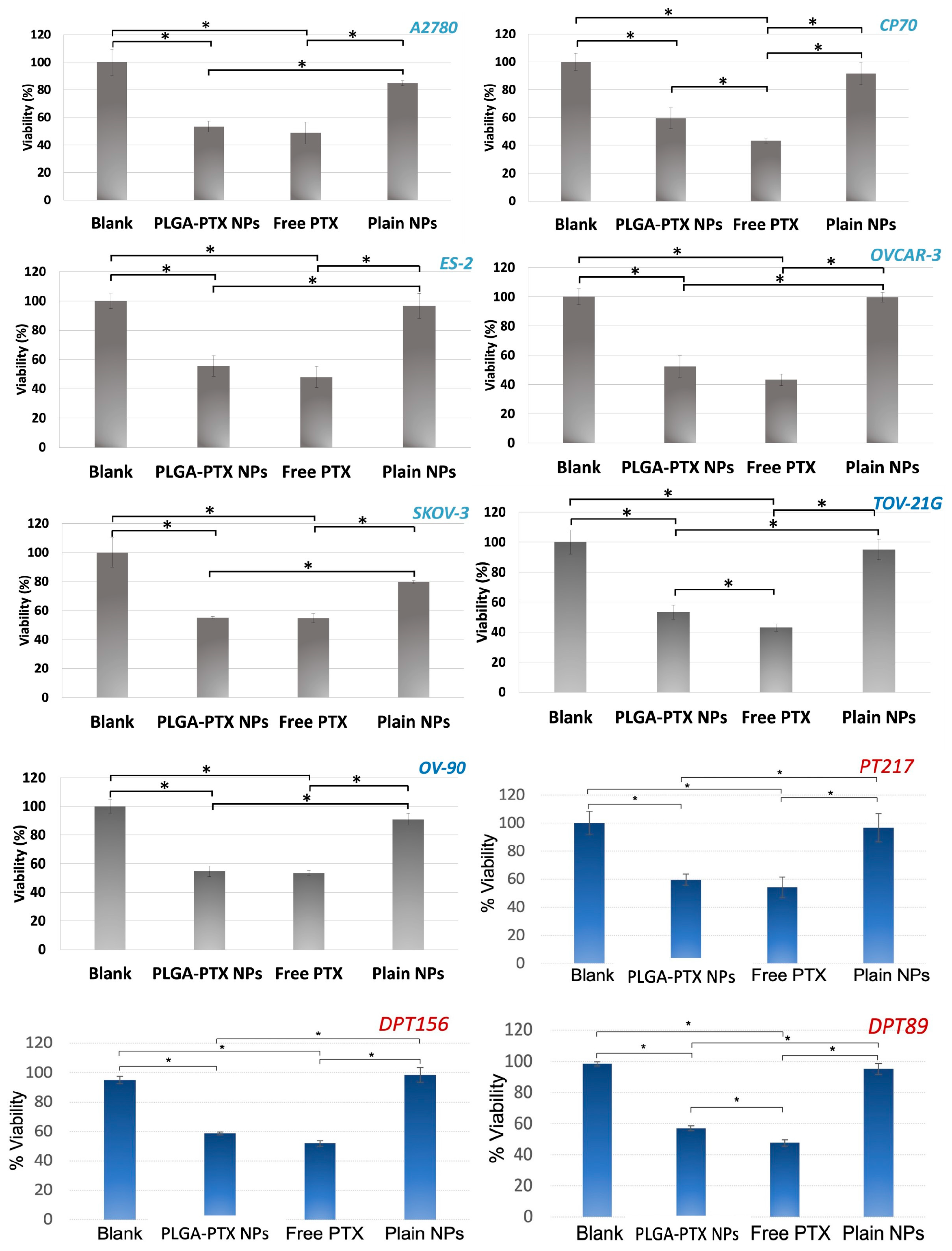
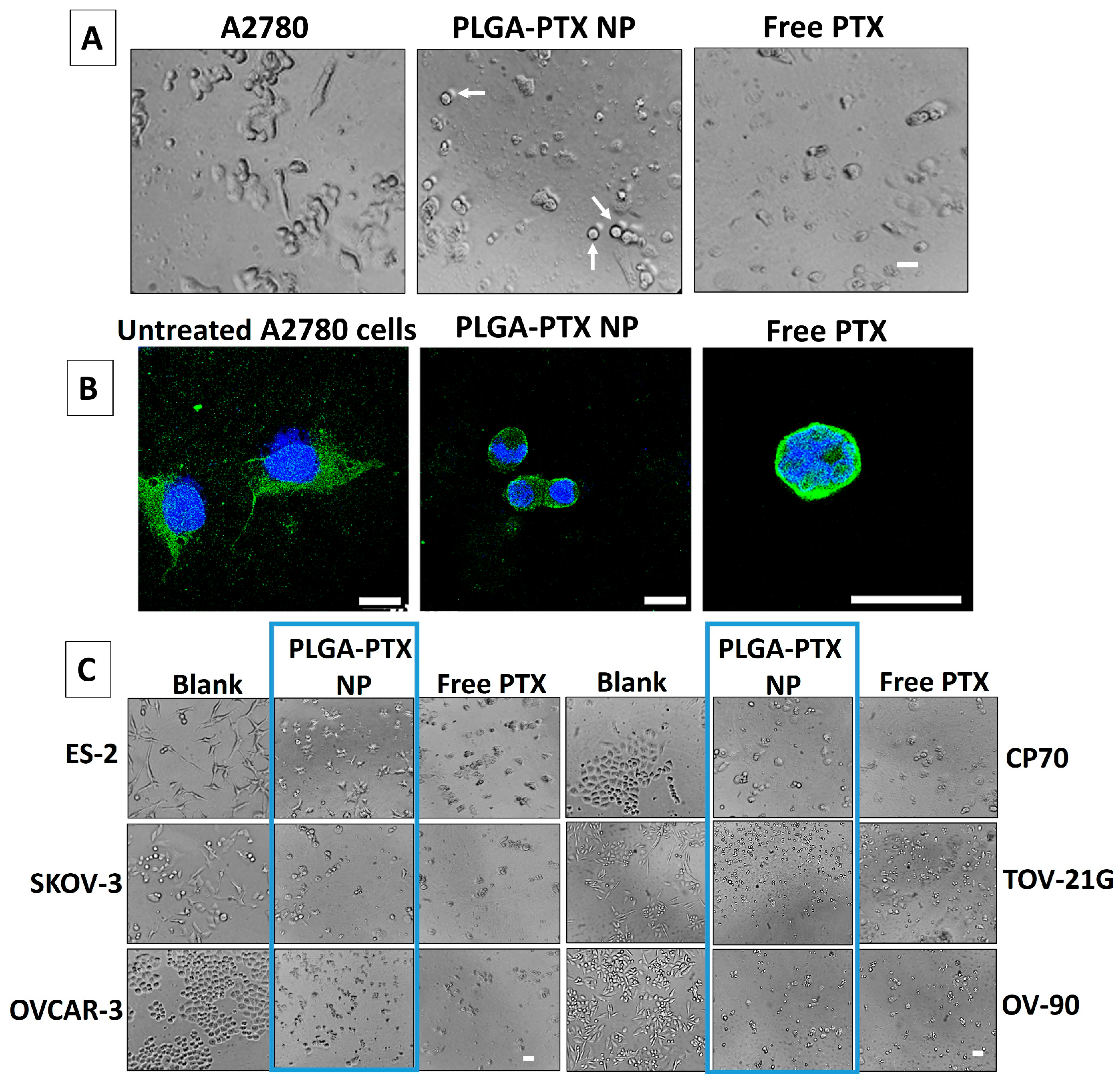
3.3. In Vitro Evaluation of PLGA-PTX NPs
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| NP | Nanoparticle |
| PTX | Paclitaxel, Taxol |
| PLGA | Poly(lactic)co-(glycolic)acid |
| PEG | Polyethylene glycol |
| DLS | Dynamic Light Scattering |
| TEM | Transmission Electron Microscopy |
| FBS | Fetal Bovine Serum |
| HPLC | High Performance Liquid Chromatography |
| LC-MS | Ultra High-Performance Liquid Chromatography |
| 1H NMR | Proton Nuclear Magnetic Resonance |
References
- Colombo, N.; Van Gorp, T.; Parma, G.; Amant, F.; Gatta, G.; Sessa, C.; Vergote, I. Ovarian cancer. Crit. Rev. Oncol. Hematol. 2006, 60, 159–179. [Google Scholar] [CrossRef]
- Micek, H.M.; Visetsouk, M.R.; Fleszar, A.J.; Kreeger, P.K. The Many Microenvironments of Ovarian Cancer. Adv. Exp. Med. Biol. 2020, 1296, 199–213. [Google Scholar]
- Konstantinopoulos, P.A.; Matulonis, U.A. Clinical and translational advances in ovarian cancer therapy. Nat. Cancer 2023, 4, 1239–1257. [Google Scholar] [CrossRef]
- Penny, S.M. Ovarian Cancer: An Overview. Radiol. Technol. 2020, 91, 561–575. [Google Scholar] [PubMed]
- Li, S.S.; Ma, J.; Wong, A.S.T. Chemoresistance in ovarian cancer: Exploiting cancer stem cell metabolism. J. Gynecol. Oncol. 2018, 29, e32. [Google Scholar] [CrossRef] [PubMed]
- Galic, V.L.; Wright, J.D.; Lewin, S.N.; Herzog, T.J. Paclitaxel poliglumex for ovarian cancer. Expert. Opin. Investig. Drugs 2011, 20, 813–821. [Google Scholar] [CrossRef]
- Gong, W.; Yu, R.; Cao, C.; Fang, Y.; Zhao, X.; Gao, Q. Dose-dense regimen versus conventional three-weekly paclitaxel combination with carboplatin chemotherapy in first-line ovarian cancer treatment: A systematic review and meta-analysis. J. Ovarian Res. 2023, 16, 136. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Li, P.; Jiang, R.; Meng, E.; Wu, H. ADC: A deadly killer of platinum resistant ovarian cancer. J. Ovarian Res. 2024, 17, 196. [Google Scholar] [CrossRef]
- Richardson, D.L.; Eskander, R.N.; O’Malley, D.M. Advances in Ovarian Cancer Care and Unmet Treatment Needs for Patients with Platinum Resistance: A Narrative Review. JAMA Oncol. 2023, 9, 851–859. [Google Scholar] [CrossRef]
- Yang, L.; Xie, H.J.; Li, Y.Y.; Wang, X.; Liu, X.X.; Mai, J. Molecular mechanisms of platinum-based chemotherapy resistance in ovarian cancer (Review). Oncol. Rep. 2022, 47, 82. [Google Scholar] [CrossRef]
- Mekhail, T.M.; Markman, M. Paclitaxel in cancer therapy. Expert. Opin. Pharmacother. 2002, 3, 755–766. [Google Scholar] [PubMed]
- Ma, W.W.; Hidalgo, M. The winning formulation: The development of paclitaxel in pancreatic cancer. Clin. Cancer Res. 2013, 19, 5572–5579. [Google Scholar] [CrossRef] [PubMed]
- Abu Samaan, T.M.; Samec, M.; Liskova, A.; Kubatka, P.; Büsselberg, D. Paclitaxel’s Mechanistic and Clinical Effects on Breast Cancer. Biomolecules 2019, 9, 789. [Google Scholar] [CrossRef]
- Shi, X.; Sun, X. Regulation of paclitaxel activity by microtubule-associated proteins in cancer chemotherapy. Cancer Chemother. Pharmacol. 2017, 80, 909–917. [Google Scholar] [CrossRef]
- Zhao, S.; Tang, Y.; Wang, R.; Najafi, M. Mechanisms of cancer cell death induction by paclitaxel: An updated review. Apoptosis 2022, 27, 647–667. [Google Scholar] [CrossRef]
- Gelderblom, H.; Verweij, J.; Nooter, K.; Sparreboom, A. Cremophor EL: The drawbacks and advantages of vehicle selection for drug formulation. Eur. J. Cancer 2001, 37, 1590–1598. [Google Scholar] [CrossRef] [PubMed]
- Singla, A.K.; Garg, A.; Aggarwal, D. Paclitaxel and its formulations. Int. J. Pharm. 2002, 235, 179–192. [Google Scholar] [CrossRef]
- Wang, F.; Zhang, D.; Zhang, Q.; Chen, Y.; Zheng, D.; Hao, L.; Duan, C.; Jia, L.; Liu, G.; Liu, Y. Synergistic effect of folate-mediated targeting and verapamil-mediated P-gp inhibition with paclitaxel -polymer micelles to overcome multi-drug resistance. Biomaterials 2011, 32, 9444–9456. [Google Scholar] [CrossRef]
- Peer, D.; Karp, J.M.; Hong, S.; Farokhzad, O.C.; Margalit, R.; Langer, R. Nanocarriers as an emerging platform for cancer therapy. Nat. Nanotechnol. 2007, 2, 751–760. [Google Scholar] [CrossRef]
- Farokhzad, O.C.; Langer, R. Impact of nanotechnology on drug delivery. ACS Nano 2009, 3, 16–20. [Google Scholar] [CrossRef]
- Shi, J.; Xiao, Z.; Kamaly, N.; Farokhzad, O.C. Self-assembled targeted nanoparticles: Evolution of technologies and bench to bedside translation. Acc. Chem. Res. 2011, 44, 1123–1134. [Google Scholar] [CrossRef] [PubMed]
- Moghimi, S.M.; Hunter, A.C.; Murray, J.C. Long-circulating and target-specific nanoparticles: Theory to practice. Pharmacol. Rev. 2001, 53, 283–318. [Google Scholar] [CrossRef]
- Sanhai, W.R.; Sakamoto, J.H.; Canady, R.; Ferrari, M. Seven challenges for nanomedicine. Nat. Nanotechnol. 2008, 3, 242–244. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Song, L.; Li, N.; Qiu, Z.; Zhou, S.; Li, C.; Zhao, J.; Song, H.; Chen, X. Pharmacokinetics and biodistribution study of paclitaxel liposome in Sprague-Dawley rats and Beagle dogs by liquid chromatography-tandem mass spectrometry. Drug Res. 2013, 63, 603–606. [Google Scholar] [CrossRef]
- Zhang, Q.; Huang, X.E.; Gao, L.L. A clinical study on the premedication of paclitaxel liposome in the treatment of solid tumors. Biomed. Pharmacother. 2009, 63, 603–607. [Google Scholar] [CrossRef]
- Zhang, J.A.; Anyarambhatla, G.; Ma, L.; Ugwu, S.; Xuan, T.; Sardone, T.; Ahmad, I. Development and characterization of a novel Cremophor EL free liposome-based paclitaxel (LEP-ETU) formulation. Eur. J. Pharm. Biopharm. 2005, 59, 177–187. [Google Scholar] [CrossRef]
- Borgå, O.; Lilienberg, E.; Bjermo, H.; Hansson, F.; Heldring, N.; Dediu, R. Pharmacokinetics of Total and Unbound Paclitaxel After Administration of Paclitaxel Micellar or Nab-Paclitaxel: An Open, Randomized, Cross-Over, Explorative Study in Breast Cancer Patients. Adv. Ther. 2019, 36, 2825–2837. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Tan, T.; Zhu, D.; Yu, H.; Liu, Y.; Zhou, H.; Jin, Y.; Xia, Q. Paclitaxel-Loaded Macrophage Membrane Camouflaged Albumin Nanoparticles for Targeted Cancer Therapy. Int. J. Nanomed. 2020, 15, 1915–1928. [Google Scholar] [CrossRef]
- Guo, S.; Vieweger, M.; Zhang, K.; Yin, H.; Wang, H.; Li, X.; Li, S.; Hu, S.; Sparreboom, A.; Evers, B.M.; et al. Ultra-thermostable RNA nanoparticles for solubilizing and high-yield loading of paclitaxel for breast cancer therapy. Nat. Commun. 2020, 11, 972. [Google Scholar] [CrossRef]
- Shu, Y.; Yin, H.; Rajabi, M.; Li, H.; Vieweger, M.; Guo, S.; Shu, D.; Guo, P. RNA-based micelles: A novel platform for paclitaxel loading and delivery. J. Control. Release 2018, 276, 17–29. [Google Scholar] [CrossRef]
- Liu, Y.; Zhao, F.; Wang, Q.; Zhao, Q.; Hou, G.; Meng, Q. Current Perspectives on Paclitaxel: Focus on Its Production, Delivery and Combination Therapy. Mini Rev. Med. Chem. 2023, 23, 1780–1796. [Google Scholar] [CrossRef] [PubMed]
- Jagur-Grodzinski, J. Biomedical application of functional polymers. React. Funct. Polym. 1999, 39, 99–138. [Google Scholar] [CrossRef]
- Cryan, S.A. Carrier-based strategies for targeting protein and peptide drugs to the lungs. AAPS J. 2005, 7, E20–E41. [Google Scholar] [CrossRef]
- Danhier, F.; Ansorena, E.; Silva, J.M.; Coco, R.; Le Breton, A.; Préat, V. PLGA-based nanoparticles: An overview of biomedical applications. J. Control. Release 2012, 161, 505–522. [Google Scholar] [CrossRef]
- Rigon, L.; Salvalaio, M.; Pederzoli, F.; Legnini, E.; Duskey, J.T.; D’Avanzo, F.; De Filippis, C.; Ruozi, B.; Marin, O.; Vandelli, M.A.; et al. Targeting Brain Disease in MPSII: Preclinical Evaluation of IDS-Loaded PLGA Nanoparticles. Int. J. Mol. Sci. 2019, 20, 2014. [Google Scholar] [CrossRef] [PubMed]
- Yang, X. Design and optimization of crocetin loaded PLGA nanoparticles against diabetic nephropathy via suppression of inflammatory biomarkers: A formulation approach to preclinical study. Drug Deliv. 2019, 26, 849–859. [Google Scholar] [CrossRef]
- Godara, S.; Lather, V.; Kirthanashri, S.V.; Awasthi, R.; Pandita, D. Lipid-PLGA hybrid nanoparticles of paclitaxel: Preparation, characterization, in vitro and in vivo evaluation. Mater. Sci. Eng. C Mater. Biol. Appl. 2020, 109, 110576. [Google Scholar] [CrossRef]
- Duan, T.; Xu, Z.; Sun, F.; Wang, Y.; Zhang, J.; Luo, C.; Wang, M. HPA aptamer functionalized paclitaxel-loaded PLGA nanoparticles for enhanced anticancer therapy through targeted effects and microenvironment modulation. Biomed. Pharmacother. 2019, 117, 109121. [Google Scholar] [CrossRef]
- Rivas, C.J.M.; Tarhini, M.; Badri, W.; Miladi, K.; Greige-Gerges, H.; Nazari, Q.A.; Rodríguez, S.A.G.; Román, R.Á.; Fessi, H.; Elaissari, A. Nanoprecipitation process: From encapsulation to drug delivery. Int. J. Pharm. 2017, 532, 66–81. [Google Scholar] [CrossRef]
- Liu, X.; Krawczyk, E.; Suprynowicz, F.A.; Palechor-Ceron, N.; Yuan, H.; Dakic, A.; Simic, V.; Zheng, Y.L.; Sripadhan, P.; Chen, C.; et al. Conditional reprogramming and long-term expansion of normal and tumor cells from human biospecimens. Nat. Protoc. 2017, 12, 439–451. [Google Scholar] [CrossRef]
- Zhang, D.; Liu, L.; Wang, J.; Zhang, H.; Zhang, Z.; Xing, G.; Wang, X.; Liu, M. Drug-loaded PEG-PLGA nanoparticles for cancer treatment. Front. Pharmacol. 2022, 13, 990505. [Google Scholar] [CrossRef] [PubMed]
- Rezvantalab, S.; Drude, N.I.; Moraveji, M.K.; Güvener, N.; Koons, E.K.; Shi, Y.; Lammers, T.; Kiessling, F. PLGA-Based Nanoparticles in Cancer Treatment. Front. Pharmacol. 2018, 9, 1260. [Google Scholar] [CrossRef] [PubMed]
- Makadia, H.K.; Siegel, S.J. Poly Lactic-co-Glycolic Acid (PLGA) as Biodegradable Controlled Drug Delivery Carrier. Polymers 2011, 3, 1377–1397. [Google Scholar] [CrossRef]
- Houchin, M.L.; Topp, E.M. Physical properties of PLGA films during polymer degradation. J. Appl. Polym. Sci. 2009, 114, 2848–2854. [Google Scholar] [CrossRef]
- Elmowafy, E.M.; Tiboni, M.; Soliman, M.E. Biocompatibility, biodegradation and biomedical applications of poly(lactic acid)/poly(lactic-co-glycolic acid) micro and nanoparticles. J. Pharm. Investig. 2019, 49, 347–380. [Google Scholar] [CrossRef]
- Ustariz-Peyret, C.; Coudane, J.; Vert, M.; Kaltsatos, V.; Boisrame, B. Labile conjugation of a hydrophilic drug to PLA oligomers to modify a drug delivery system: Cephradin in a PLAGA matrix. J. Microencapsul. 2000, 17, 615–624. [Google Scholar] [CrossRef]
- Yoo, J.Y.; Kim, J.M.; Seo, K.S.; Jeong, Y.K.; Lee, H.B.; Khang, G. Characterization of degradation behavior for PLGA in various pH condition by simple liquid chromatography method. Biomed. Mater. Eng. 2005, 15, 279–288. [Google Scholar]
- Kato, Y.; Ozawa, S.; Miyamoto, C.; Maehata, Y.; Suzuki, A.; Maeda, T.; Baba, Y. Acidic extracellular microenvironment and cancer. Cancer Cell Int. 2013, 13, 89. [Google Scholar] [CrossRef]
- Surapaneni, M.S.; Das, S.K.; Das, N.G. Designing Paclitaxel drug delivery systems aimed at improved patient outcomes: Current status and challenges. ISRN Pharmacol. 2012, 2012, 623139. [Google Scholar] [CrossRef]
- Zhang, T.; Tang, M.; Yao, Y.; Ma, Y.; Pu, Y. MWCNT interactions with protein: Surface-induced changes in protein adsorption and the impact of protein corona on cellular uptake and cytotoxicity. Int. J. Nanomed. 2019, 14, 993–1009. [Google Scholar] [CrossRef]
- Fam, S.Y.; Chee, C.F.; Yong, C.Y.; Ho, K.L.; Mariatulqabtiah, A.R.; Tan, W.S. Stealth Coating of Nanoparticles in Drug-Delivery Systems. Nanomaterials 2020, 10, 787. [Google Scholar] [CrossRef] [PubMed]
- Shao, X.R.; Wei, X.Q.; Song, X.; Hao, L.Y.; Cai, X.X.; Zhang, Z.R.; Peng, Q.; Lin, Y.F. Independent effect of polymeric nanoparticle zeta potential/surface charge, on their cytotoxicity and affinity to cells. Cell Prolif. 2015, 48, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Kim, C.S.; Saylor, D.M.; Koo, D. Polymer degradation and drug delivery in PLGA-based drug-polymer applications: A review of experiments and theories. J. Biomed. Mater. Res. B Appl. Biomater. 2017, 105, 1692–1716. [Google Scholar] [CrossRef] [PubMed]
- Goodson, H.V.; Jonasson, E.M. Microtubules and Microtubule-Associated Proteins. Cold Spring Harb. Perspect. Biol. 2018, 10, a022608. [Google Scholar] [CrossRef]
- Horwitz, S.B. Taxol (paclitaxel): Mechanisms of action. Ann. Oncol. 1994, 5 (Suppl. 6), S3–S6. [Google Scholar]
- Kiwanuka, M.; Higgins, G.; Ngcobo, S.; Nagawa, J.; Lang, D.M.; Zaman, M.H.; Davies, N.H.; Franz, T. Effect of paclitaxel treatment on cellular mechanics and morphology of human oesophageal squamous cell carcinoma in 2D and 3D environments. Integr. Biol. 2022, 14, 137–149. [Google Scholar]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dragulska, S.A.; Acosta Santiago, M.; Swierczek, S.; Chuang, L.; Camacho-Vanegas, O.; Camacho, S.C.; Padron-Rhenals, M.M.; Martignetti, J.A.; Mieszawska, A.J. Synthesis and Characterization of Poly(Lactic-Co-Glycolic Acid)–Paclitaxel (PLGA-PTX) Nanoparticles Evaluated in Ovarian Cancer Models. Pharmaceutics 2025, 17, 689. https://doi.org/10.3390/pharmaceutics17060689
Dragulska SA, Acosta Santiago M, Swierczek S, Chuang L, Camacho-Vanegas O, Camacho SC, Padron-Rhenals MM, Martignetti JA, Mieszawska AJ. Synthesis and Characterization of Poly(Lactic-Co-Glycolic Acid)–Paclitaxel (PLGA-PTX) Nanoparticles Evaluated in Ovarian Cancer Models. Pharmaceutics. 2025; 17(6):689. https://doi.org/10.3390/pharmaceutics17060689
Chicago/Turabian StyleDragulska, Sylwia A., Maxier Acosta Santiago, Sabina Swierczek, Linus Chuang, Olga Camacho-Vanegas, Sandra Catalina Camacho, Maria M. Padron-Rhenals, John A. Martignetti, and Aneta J. Mieszawska. 2025. "Synthesis and Characterization of Poly(Lactic-Co-Glycolic Acid)–Paclitaxel (PLGA-PTX) Nanoparticles Evaluated in Ovarian Cancer Models" Pharmaceutics 17, no. 6: 689. https://doi.org/10.3390/pharmaceutics17060689
APA StyleDragulska, S. A., Acosta Santiago, M., Swierczek, S., Chuang, L., Camacho-Vanegas, O., Camacho, S. C., Padron-Rhenals, M. M., Martignetti, J. A., & Mieszawska, A. J. (2025). Synthesis and Characterization of Poly(Lactic-Co-Glycolic Acid)–Paclitaxel (PLGA-PTX) Nanoparticles Evaluated in Ovarian Cancer Models. Pharmaceutics, 17(6), 689. https://doi.org/10.3390/pharmaceutics17060689