

Delivery of PLGA-Loaded Influenza Vaccine Microparticles Using Dissolving Microneedles Induces a Robust Immune Response

 ,
,  , ,
, , 
 and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Formulation and Characterization of Inactivated Influenza A H1N1 and Inactivated Influenza A H3N2 Microparticulate-Based Vaccine
2.2.2. In Vitro Determination of the Immunogenicity of Vaccine Microparticles
2.2.3. Determination of Cytotoxicity of the Vaccine Microparticles Using MTT (3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide Assay
2.2.4. Formulation of Dissolving Microneedles Containing Microparticulate-Based Vaccine
2.2.5. Dosing Schedule and Mice Vaccination Procedure
2.2.6. Evaluation of Humoral Immune Response Using ELISA (Enzyme-Linked Immunosorbent Assay)
2.2.7. Analysis of Specific T Cell Responses in Lymph Node and Spleen
2.2.8. Statistical Analysis and Determination of Mice Sample Size
3. Results
3.1. Characterization of Vaccine Microparticles and Dissolving Microneedles Characterization
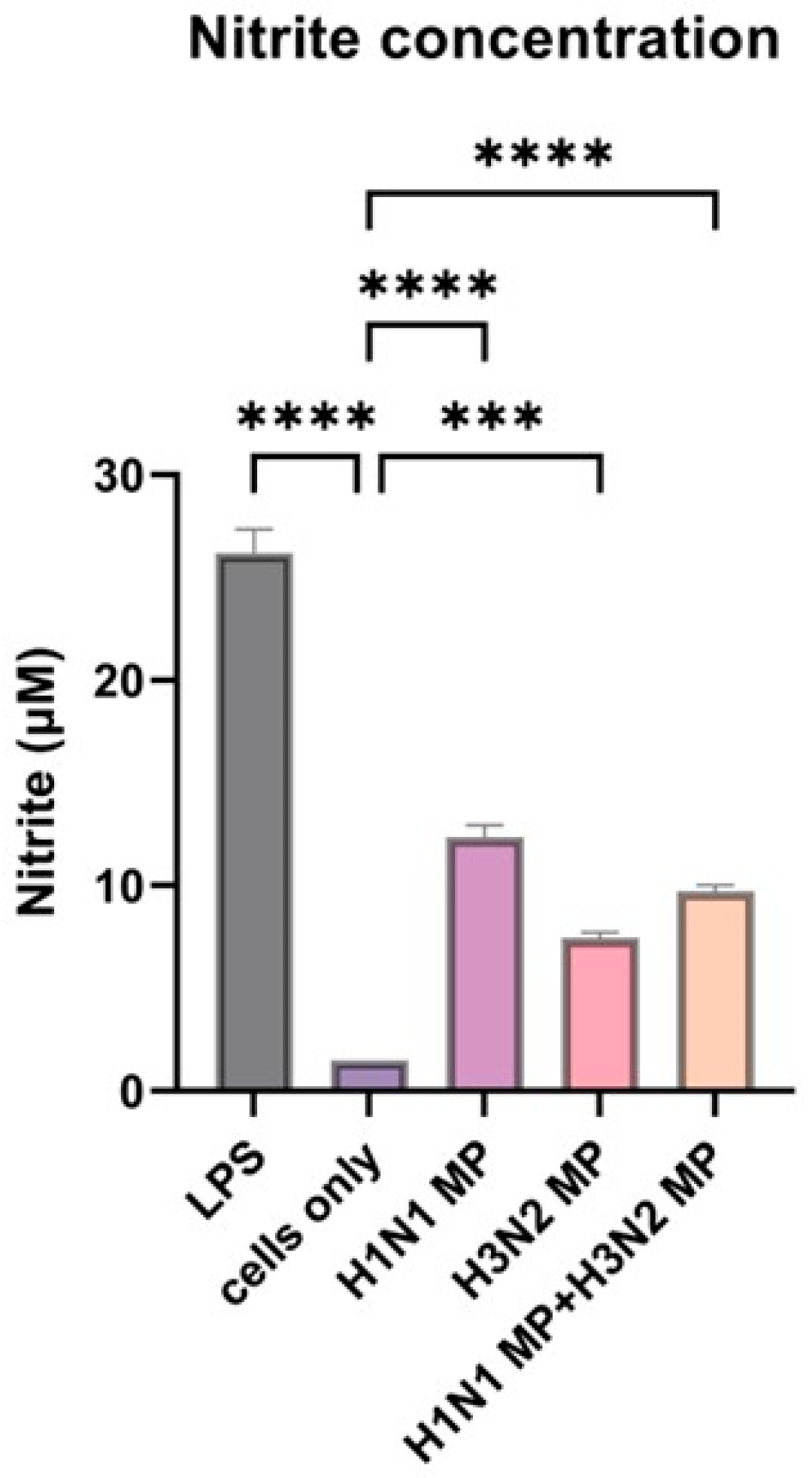
3.2. Vaccine Microparticles Induce Immunogenicity as Evidenced by Nitric Oxide Release in Dendritic Cells
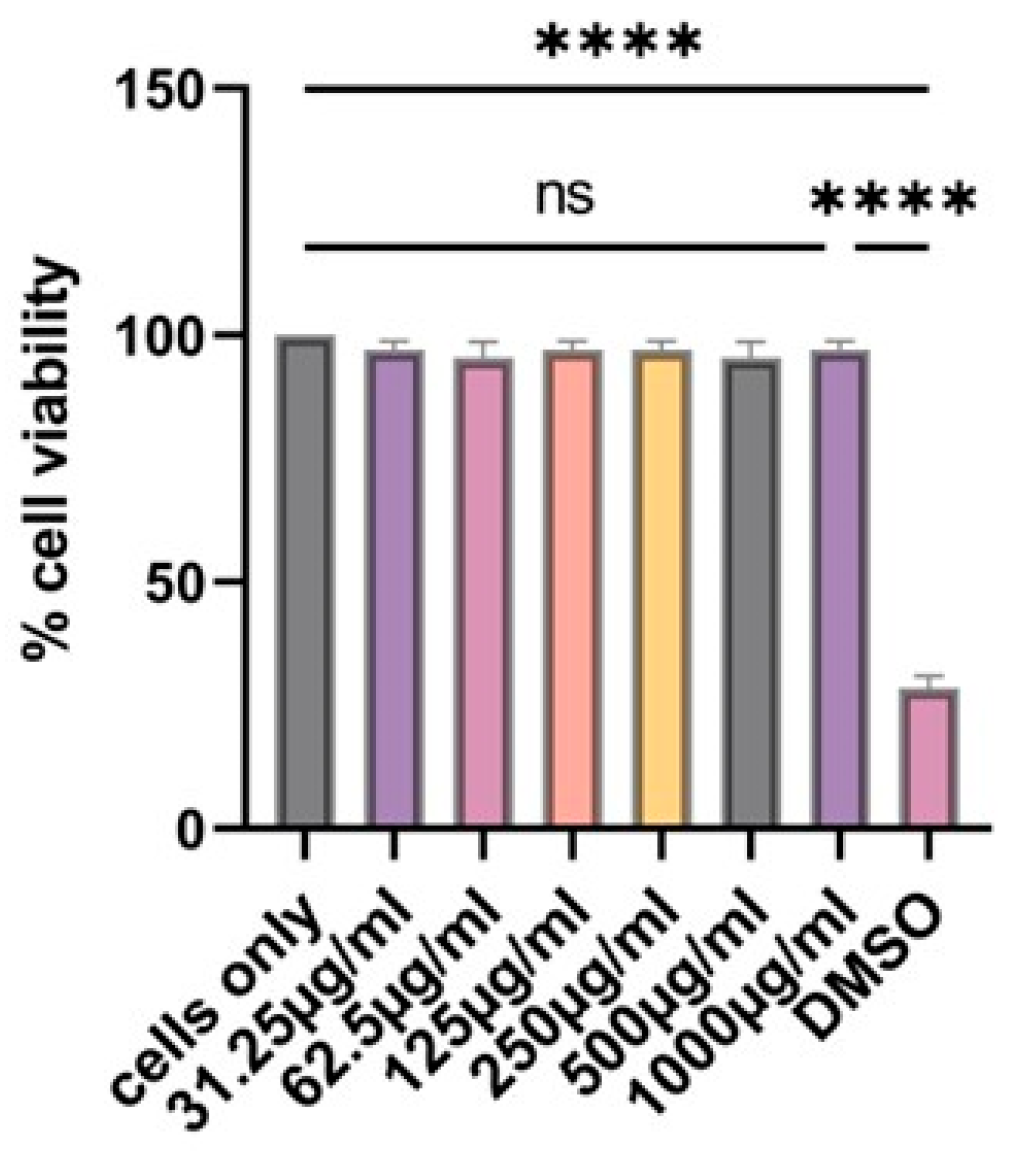
3.3. Determination of Cytotoxicity of the Vaccine Microparticles Using the MTT Assay
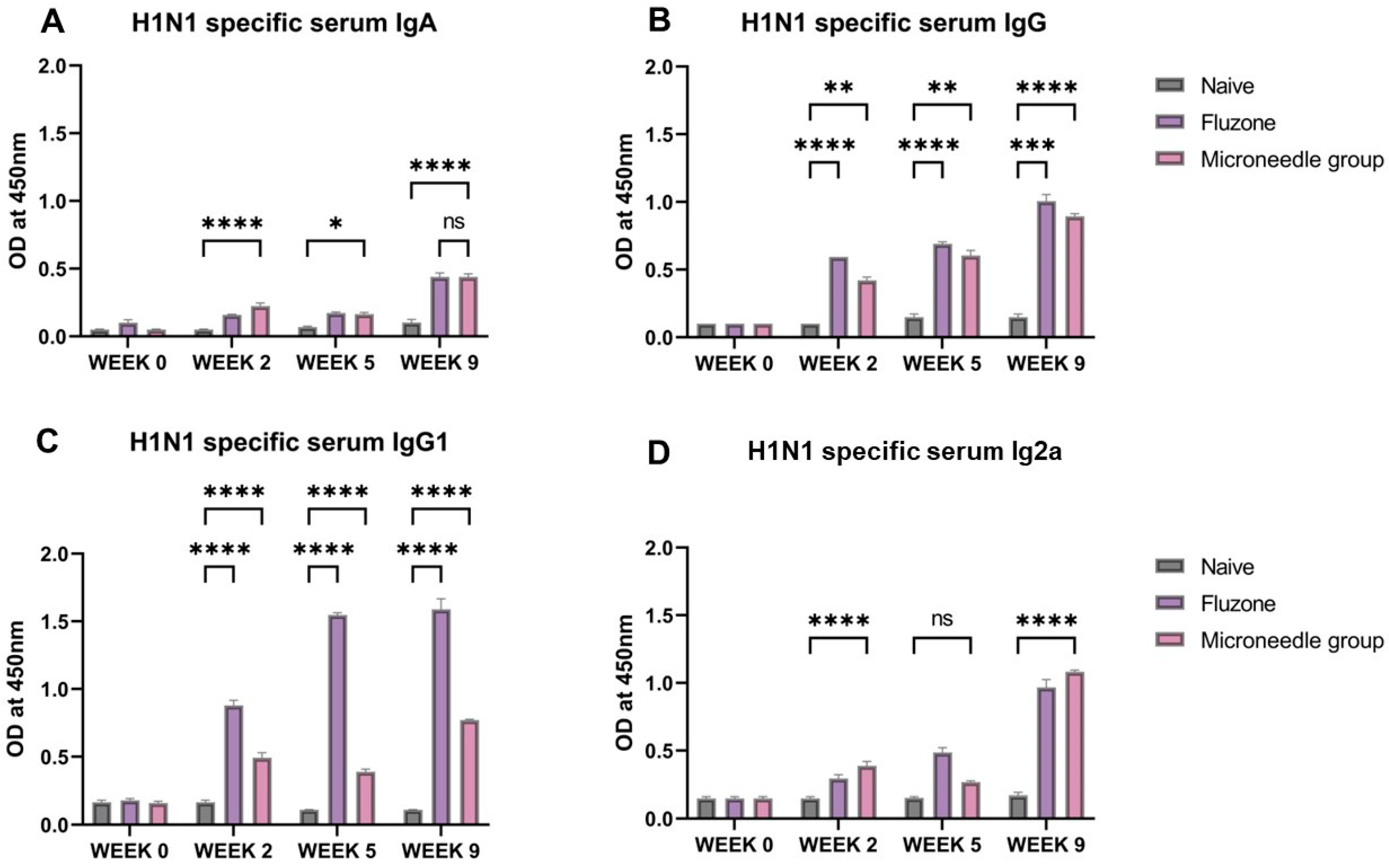
3.4. Vaccine Microparticles Induce High HIN1-Specific Serum Antibodies in Mice
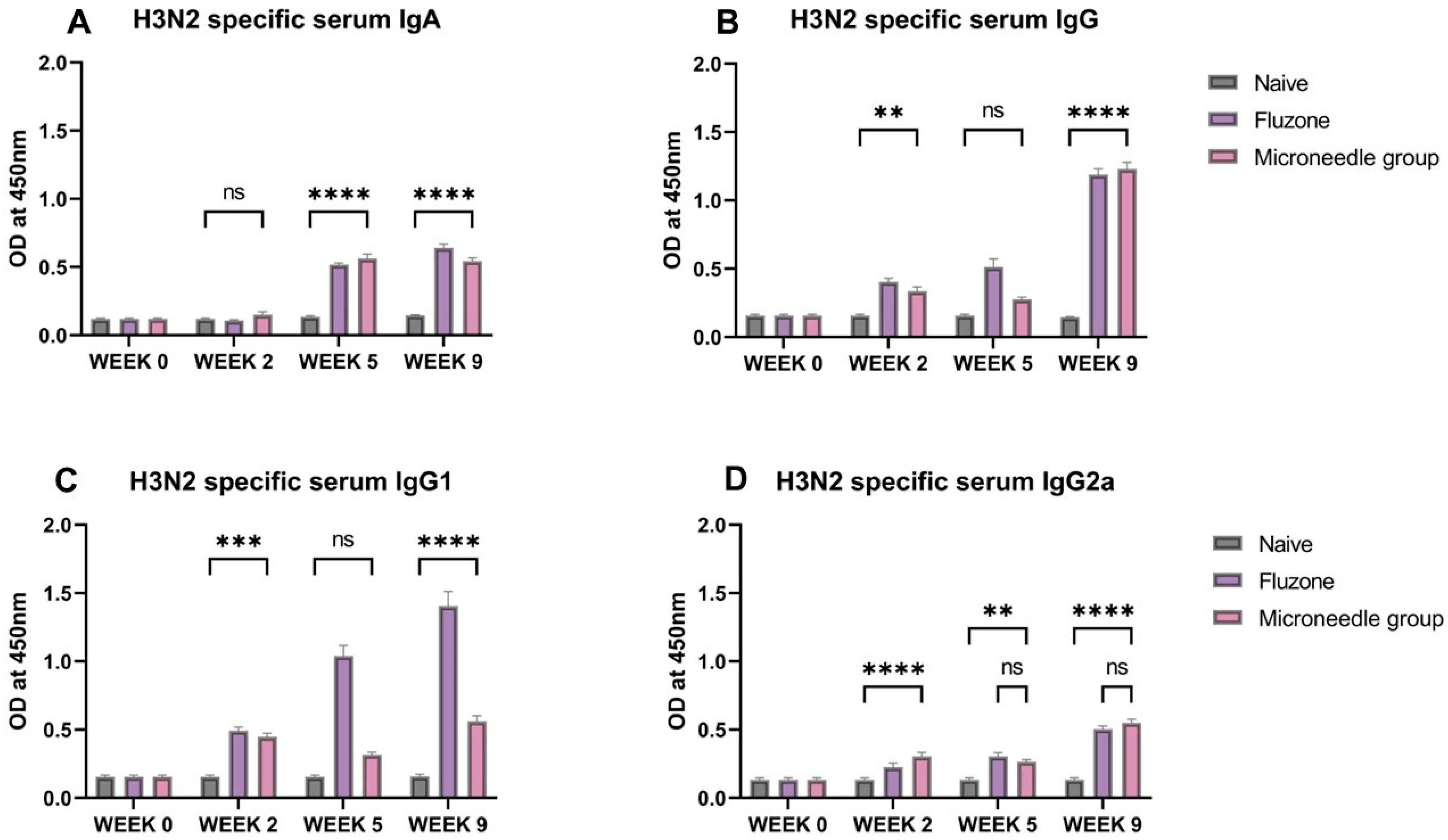
3.5. Vaccine Microparticles Induce High H3N2-Specific Serum Antibodies in Mice
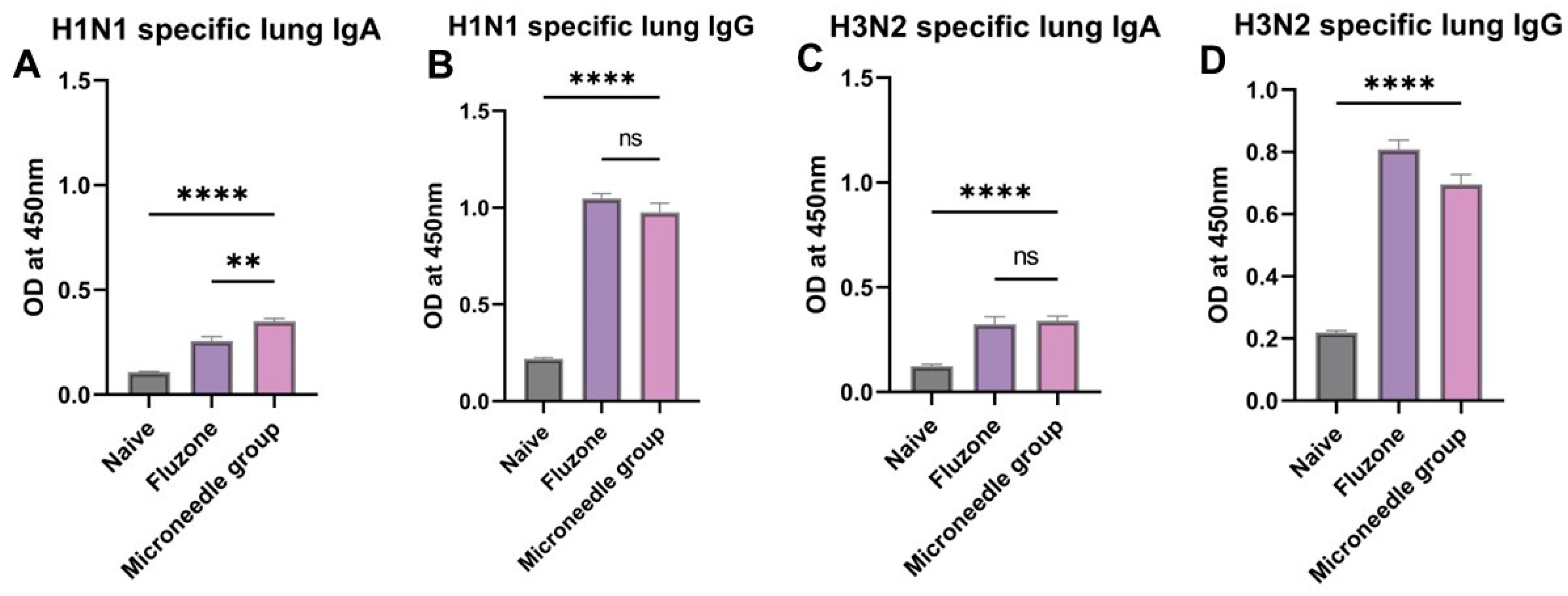
3.6. Elevated Antigen-Specific Antibody Levels in Lung Supernatant Representing Excellent Mucosal Responses
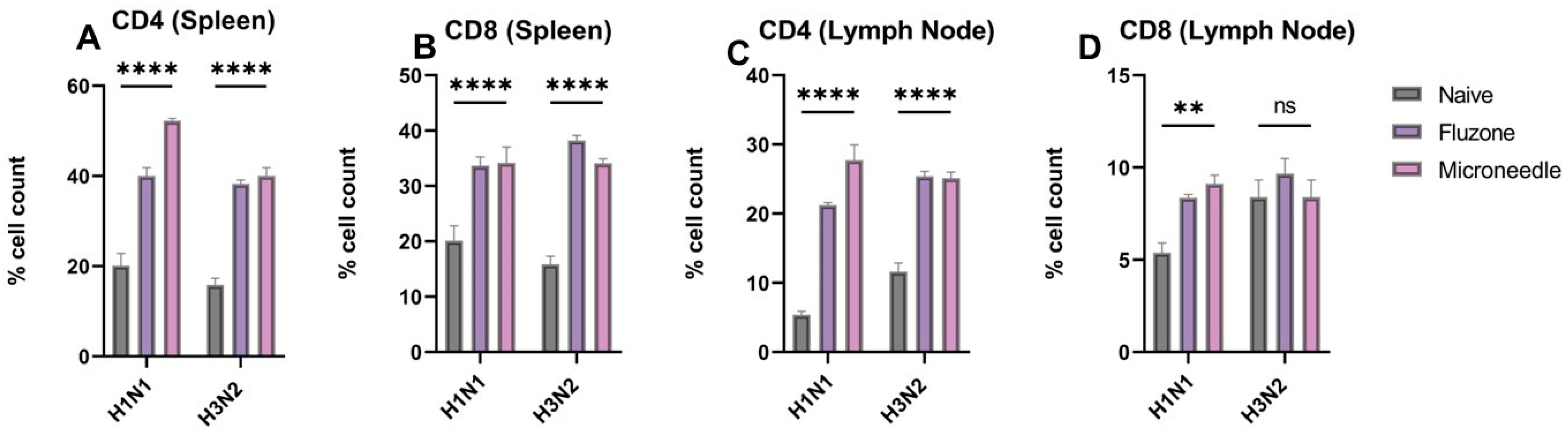
3.7. Vaccine Microparticles Induce Cellular Responses in Mice
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Deng, L.; Wang, B.Z. A Perspective on Nanoparticle Universal Influenza Vaccines. ACS Infect. Dis. 2018, 4, 1656–1665. [Google Scholar] [CrossRef]
- Dou, D.; Revol, R.; Östbye, H.; Wang, H.; Daniels, R. Influenza A Virus Cell Entry, Replication, Virion Assembly and Movement. Front. Immunol. 2018, 9, 1581. [Google Scholar] [CrossRef] [PubMed]
- Krammer, F. Emerging influenza viruses and the prospect of a universal influenza virus vaccine. Biotechnol. J. 2015, 10, 690–701. [Google Scholar] [CrossRef] [PubMed]
- Brankston, G.; Gitterman, L.; Hirji, Z.; Lemieux, C.; Gardam, M. Transmission of influenza A in human beings. Lancet Infect. Dis. 2007, 7, 257–265. [Google Scholar] [CrossRef]
- Bischoff, W.E.; Swett, K.; Leng, I.; Peters, T.R. Exposure to Influenza Virus Aerosols During Routine Patient Care. J. Infect. Dis. 2013, 207, 1037–1046. [Google Scholar] [CrossRef] [PubMed]
- Yeolekar, L.R.; Guilfoyle, K.; Ganguly, M.; Tyagi, P.; Stittelaar, K.J.; van Amerongen, G.; Dhere, R.M.; BerlandaScorza, F.; Mahmood, K. Immunogenicity and efficacy comparison of MDCK cell-based and egg-based live attenuated influenza vaccines of H5 and H7 subtypes in ferrets. Vaccine 2020, 38, 6280–6290. [Google Scholar] [CrossRef]
- Vemula, S.V.; Sayedahmed, E.E.; Sambhara, S.; Mittal, S.K. Vaccine approaches conferring cross-protection against influenza viruses. Expert Rev. Vaccines 2017, 16, 1141–1154. [Google Scholar] [CrossRef]
- Durando, P.; Iudici, R.; Alicino, C.; Alberti, M.; De Florentis, D.; Ansaldi, F.; Icardi, G. Adjuvants and alternative routes of administration towards the development of the ideal influenza vaccine. Hum. Vaccines 2011, 7 (Suppl. 1), 29–40. [Google Scholar] [CrossRef]
- Tian, Y.; Bhide, Y.C.; Woerdenbag, H.J.; Huckriede, A.L.W.; Frijlink, H.W.; Hinrichs, W.L.J.; Visser, J.C. Development of an Orodispersible Film Containing Stabilized Influenza Vaccine. Pharmaceutics 2020, 12, 245. [Google Scholar] [CrossRef]
- Burgess, T.H.; Murray, C.K.; Bavaro, M.F.; Landrum, M.L.; O’bryan, T.A.; Rosas, J.G.; Cammarata, S.M.; Martin, N.J.; Ewing, D.; Raviprakash, K.; et al. Self-administration of intranasal influenza vaccine: Immunogenicity and volunteer acceptance. Vaccine 2015, 33, 3894–3899. [Google Scholar] [CrossRef]
- Shae, D.; Postma, A.; Wilson, J.T. Vaccine delivery: Where polymer chemistry meets immunology. Ther. Deliv. 2016, 7, 193–196. [Google Scholar] [CrossRef] [PubMed]
- O’hagan, D.T.; Singh, M.; Ulmer, J.B. Microparticle-based technologies for vaccines. Methods 2006, 40, 10–19. [Google Scholar] [CrossRef]
- Kanke, M.; Sniecinski, I.; DeLuca, P.P. Interaction of Microspheres with Blood Constituents: I. Uptake of Polystyrene Spheres by Monocytes and Granulocytes and Effect on Immune Responsiveness of Lymphocytes. PDA J. Pharm. Sci. Technol. 1983, 37, 210–217. [Google Scholar]
- Tabata, Y.; Ikada, Y. Effect of the size and surface charge of polymer microspheres on their phagocytosis by macrophage. Biomaterials 1988, 9, 356–362. [Google Scholar] [CrossRef]
- O’Hagan, D. Poly(lactide-co-glycolide) microparticles for the development of single-dose controlled-release vaccines. Adv. Drug Deliv. Rev. 1998, 32, 225–246. [Google Scholar] [CrossRef]
- Shah, S.; Patel, P.; Ferguson, A.; Bagwe, P.; Kale, A.; Adediran, E.; Singh, R.; Arte, T.; Pasupuleti, D.; Uddin, M.N.; et al. Buccal Administration of a Zika Virus Vaccine Utilizing 3D-Printed Oral Dissolving Films in a Mouse Model. Vaccines 2024, 12, 720. [Google Scholar] [CrossRef]
- Menon, I.; Kang, S.M.; D’Souza, M. Nanoparticle formulation of the fusion protein virus like particles of respiratory syncytial virus stimulates enhanced in vitro antigen presentation and autophagy. Int. J. Pharm. 2022, 623, 121919. [Google Scholar] [CrossRef]
- Gomes, K.B.; Vijayanand, S.; Bagwe, P.; Menon, I.; Kale, A.; Patil, S.; Kang, S.-M.; Uddin, M.N.; D’souza, M.J. Vaccine-Induced Immunity Elicited by Microneedle Delivery of Influenza Ectodomain Matrix Protein 2 Virus-like Particle (M2e VLP)-Loaded PLGA Nanoparticles. IJMS 2023, 24, 10612. [Google Scholar] [CrossRef]
- Vijayanand, S.; Patil, S.; Menon, I.; Gomes, K.B.; Kale, A.; Bagwe, P.; Uddin, M.N.; Zughaier, S.M.; D’souza, M.J. An Adjuvanted Inactivated SARS-CoV-2 Microparticulate Vaccine Delivered Using Microneedles Induces a Robust Immune Response in Vaccinated Mice. Pharmaceutics 2023, 15, 895. [Google Scholar] [CrossRef]
- Liang, Z.; Zhu, H.; Wang, X.; Jing, B.; Li, Z.; Xia, X.; Sun, H.; Yang, Y.; Zhang, W.; Shi, L.; et al. Adjuvants for Coronavirus Vaccines. Front Immunol. 2020, 11, 589833. [Google Scholar] [CrossRef]
- Nian, X.; Zhang, J.; Deng, T.; Liu, J.; Gong, Z.; Lv, C.; Yao, L.; Li, J.; Huang, S.; Yang, X. AddaVax Formulated with PolyI:C as a Potential Adjuvant of MDCK-based Influenza Vaccine Enhances Local, Cellular, and Antibody Protective Immune Response in Mice. Aaps Pharmscitech 2021, 22, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, A.L.; Kazmin, D.; Napolitani, G.; Clutterbuck, E.A.; Pulendran, B.; Siegrist, C.-A.; Pollard, A.J. AS03- and MF59-Adjuvanted Influenza Vaccines in Children. Front. Immunol. 2017, 8, 1760. [Google Scholar] [CrossRef]
- Kim, M.-C.; Lee, J.W.; Choi, H.-J.; Lee, Y.-N.; Hwang, H.S.; Lee, J.; Kim, C.; Lee, J.S.; Montemagno, C.; Prausnitz, M.R.; et al. Microneedle patch delivery to the skin of virus-like particles containing heterologous M2e extracellular domains of influenza virus induces broad heterosubtypic cross-protection. J. Control. Release 2015, 210, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Marshall, S.; Sahm, L.J.; Moore, A.C. The success of microneedle-mediated vaccine delivery into skin. Hum. Vaccines Immunother. 2016, 12, 2975–2983. [Google Scholar] [CrossRef]
- Rouphael, N.G.; Paine, M.; Mosley, R.; Henry, S.; McAllister, D.V.; Kalluri, H.; Pewin, W.; Frew, P.M.; Yu, T.; Thornburg, N.J.; et al. The safety, immunogenicity, and acceptability of inactivated influenza vaccine delivered by microneedle patch (TIV-MNP 2015): A randomised, partly blinded, placebo-controlled, phase 1 trial. Lancet 2017, 390, 649–658. [Google Scholar] [CrossRef]
- Sekiya, T.; Ohno, M.; Nomura, N.; Handabile, C.; Shingai, M.; Jackson, D.C.; Brown, L.E.; Kida, H. Selecting and Using the Appropriate Influenza Vaccine for Each Individual. Viruses 2021, 13, 971. [Google Scholar] [CrossRef]
- Calzas, C.; Chevalier, C. Innovative Mucosal Vaccine Formulations Against Influenza A Virus Infections. Front. Immunol. 2019, 10, 1605. [Google Scholar] [CrossRef] [PubMed]
- Gomes, K.B.; D’sa, S.; Allotey-Babington, G.L.; Kang, S.-M.; D’souza, M.J. Transdermal Vaccination with the Matrix-2 Protein Virus-like Particle (M2e VLP) Induces Immunity in Mice against Influenza A Virus. Vaccines 2021, 9, 1324. [Google Scholar] [CrossRef]
- Vijayanand, S.; Patil, S.; Joshi, D.; Menon, I.; Gomes, K.B.; Kale, A.; Bagwe, P.; Yacoub, S.; Uddin, M.N.; D’souza, M.J. Microneedle Delivery of an Adjuvanted Microparticulate Vaccine Induces High Antibody Levels in Mice Vaccinated against Coronavirus. Vaccines 2022, 10, 1491. [Google Scholar] [CrossRef]
- Piacentini, E. Encapsulation Efficiency. In Encyclopedia of Membranes [Internet]; Drioli, E., Giorno, L., Eds.; Springer: Berlin/Heidelberg, Germany, 2016; pp. 706–707. Available online: http://link.springer.com/10.1007/978-3-662-44324-8_1945 (accessed on 25 February 2025).
- Roberts, J.M.; Milo, S.; Metcalf, D.G. Harnessing the Power of Our Immune System: The Antimicrobial and Antibiofilm Properties of Nitric Oxide. Microorganisms 2024, 12, 2543. [Google Scholar] [CrossRef]
- Çelïk, I.; Saatçï, E.; Eyüboğlu, F.Ö. Emerging and reemerging respiratory viral infections up to COVID-19. Turk. J. Med Sci. 2020, 50, 557–562. [Google Scholar] [CrossRef]
- Olson, S.M.; Newhams, M.M.; Halasa, N.B.; Feldstein, L.R.; Novak, T.; Weiss, S.L.; Coates, B.M.; E Schuster, J.; Schwarz, A.J.; Maddux, A.B.; et al. Vaccine Effectiveness Against Life-Threatening Influenza Illness in US Children. Clin. Infect. Dis. 2022, 75, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Oyewumi, M.O.; Kumar, A.; Cui, Z. Nano-microparticles as immune adjuvants: Correlating particle sizes and the resultant immune responses. Expert Rev. Vaccines 2010, 9, 1095–1107. [Google Scholar] [CrossRef] [PubMed]
- Nakaoka, R.; Inoue, Y.; Tabata, Y.; Ikada, Y. Size effect on the antibody production induced by biodegradable microspheres containing antigen. Vaccine 1996, 14, 1251–1256. [Google Scholar] [CrossRef]
- Xiang, S.D.; Scholzen, A.; Minigo, G.; David, C.; Apostolopoulos, V.; Mottram, P.L.; Plebanski, M. Pathogen recognition and development of particulate vaccines: Does size matter? Methods 2006, 40, 1–9. [Google Scholar] [CrossRef]
- Liu, Z.; Roche, P.A. Macropinocytosis in phagocytes: Regulation of MHC class-II-restricted antigen presentation in dendritic cells. Front. Physiol. 2015, 6, 1. [Google Scholar] [CrossRef]
- El-Hammadi, M.M.; Arias, J.L. Recent Advances in the Surface Functionalization of PLGA-Based Nanomedicines. Nanomaterials 2022, 12, 354. [Google Scholar] [CrossRef]
- Haque, S.; Boyd, B.J.; McIntosh, M.P.; Pouton, C.W.; Kaminskas, L.M.; Whittaker, M.R. Suggested Procedures for the Reproducible Synthesis of Poly(d,l-lactideco-glycolide) Nanoparticles Using the Emulsification Solvent Diffusion Platform. Curr. Nanosci. 2018, 14, 448–453. [Google Scholar] [CrossRef]
- Kilbourne, E. Purified influenza A virus N2 neuraminidase vaccine is immunogenic and non-toxic in humans. Vaccine 1995, 13, 1799–1803. [Google Scholar] [CrossRef]
- Saha, I.; Rai, V.K. Hyaluronic acid based microneedle array: Recent applications in drug delivery and cosmetology. Carbohydr. Polym. 2021, 267, 118168. [Google Scholar] [CrossRef]
- Vassilieva, E.V.; Kalluri, H.; McAllister, D.; Taherbhai, M.T.; Esser, E.S.; Pewin, W.P.; Pulit-Penaloza, J.A.; Prausnitz, M.R.; Compans, R.W.; Skountzou, I. Improved immunogenicity of individual influenza vaccine components delivered with a novel dissolving microneedle patch stable at room temperature. Drug Deliv. Transl. Res. 2015, 5, 360–371. [Google Scholar] [CrossRef] [PubMed]
- Aleebrahim-Dehkordi, E.; Molavi, B.; Mokhtari, M.; Deravi, N.; Fathi, M.; Fazel, T.; Mohebalizadeh, M.; Koochaki, P.; Shobeiri, P.; Hasanpour-Dehkordi, A.; et al. T helper type (Th1/Th2) responses to SARS-CoV-2 and influenza A (H1N1) virus: From cytokines produced to immune responses. Transpl. Immunol. 2022, 70, 101495. [Google Scholar] [CrossRef]
- Trincado, V.; Gala, R.P.; Morales, J.O. Buccal and Sublingual Vaccines: A Review on Oral Mucosal Immunization and Delivery Systems. Vaccines 2021, 9, 1177. [Google Scholar] [CrossRef] [PubMed]
- Carty, S.A.; Riese, M.J.; Koretzky, G.A. T-Cell Immunity. In Hematology [Internet]; Elsevier: Amsterdam, The Netherlands, 2018; pp. 221–239. Available online: https://linkinghub.elsevier.com/retrieve/pii/B9780323357623000214 (accessed on 27 February 2025).
- Vijayanand, S.; Patil, S.; Bagwe, P.; Singh, R.; Adediran, E.; D’Souza, M.J. Evaluating the Immunogenicity of an Intranasal Microparticle Combination Vaccine for COVID-19 and Influenza. Vaccines 2025, 13, 282. [Google Scholar] [CrossRef]
- Domnich, A.; Manini, I.; Panatto, D.; Calabrò, G.E.; Montomoli, E. Immunogenicity Measures of Influenza Vaccines: A Study of 1164 Registered Clinical Trials. Vaccines 2020, 8, 325. [Google Scholar] [CrossRef]
- Kommareddy, S.; Baudner, B.C.; Oh, S.; Kwon, S.Y.; Singh, M.; O’Hagan, D.T. Dissolvable microneedle patches for the delivery of cell-culture-derived influenza vaccine antigens. J. Pharm. Sci. 2012, 101, 1021–1027. [Google Scholar] [CrossRef]
- Howard, F.H.N.; Kwan, A.; Winder, N.; Mughal, A.; Collado-Rojas, C.; Muthana, M. Understanding Immune Responses to Viruses—Do Underlying Th1/Th2 Cell Biases Predict Outcome? Viruses 2022, 14, 1493. [Google Scholar] [CrossRef]
- Kim, B.M.; Kim, Y.-H.; Ngo, H.V.; Nguyen, H.D.; Park, C.; Lee, B.-J. Enhanced and Prolonged Immunogenicity in Mice of Thermally Stabilized Fatty Acid-Conjugated Vaccine Antigen. Vaccines 2025, 13, 168. [Google Scholar] [CrossRef]
- Hinkula, J.; Nyström, S.; Devito, C.; Bråve, A.; Applequist, S.E. Long-Lasting Mucosal and Systemic Immunity against Influenza A Virus Is Significantly Prolonged and Protective by Nasal Whole Influenza Immunization with Mucosal Adjuvant N3 and DNA-Plasmid Expressing Flagellin in Aging In- and Outbred Mice. Vaccines 2019, 7, 64. [Google Scholar] [CrossRef]
- Kasten-Jolly, J.; Lawrence, D.A. Cellular and Molecular Immunity to Influenza Viruses and Vaccines. Vaccines 2024, 12, 389. [Google Scholar] [CrossRef]
- Tsang, T.K.; Lam, K.T.; Liu, Y.; Fang, V.J.; Mu, X.; Leung, N.H.L.; Peiris, J.S.M.; Leung, G.M.; Cowling, B.J.; Tu, W. Investigation of CD4 and CD8 T cell-mediated protection against influenza A virus in a cohort study. BMC Med. 2022, 20, 230. [Google Scholar] [CrossRef] [PubMed]
- Patil, S.; Vijayanand, S.; Menon, I.; Gomes, K.B.; Kale, A.; Bagwe, P.; Yacoub, S.; Uddin, M.N.; D’Souza, M.J. Adjuvanted-SARS-CoV-2 Spike Protein-Based Microparticulate Vaccine Delivered by Dissolving Microneedles Induces Humoral, Mucosal, and Cellular Immune Responses in Mice. Pharmaceuticals 2023, 16, 1131. [Google Scholar] [CrossRef]
- DeMuth, P.C.; Garcia-Beltran, W.F.; Ai-Ling, M.L.; Hammond, P.T.; Irvine, D.J. Composite Dissolving Microneedles for Coordinated Control of Antigen and Adjuvant Delivery Kinetics in Transcutaneous Vaccination. Adv. Funct. Mater. 2023, 23, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Neustrup, M.A.; Slütter, B.; O’Mahony, C.; Bouwstra, J.A.; van der Maaden, K. Intradermal Vaccination with PLGA Nanoparticles via Dissolving Microneedles and Classical Injection Needles. Pharm. Res. 2024, 41, 305–319. [Google Scholar] [CrossRef] [PubMed]
- Mazzara, J.M.; Ochyl, L.J.; Hong, J.K.Y.; Moon, J.J.; Prausnitz, M.R.; Schwendeman, S.P. Self-healing encapsulation and controlled release of vaccine antigens from PLGA microparticles delivered by microneedle patches. Bioeng. Transl. Med. 2019, 4, 116–128. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Properties | H1N1 MP | H3N2 MP |
|---|---|---|
| Size | 1470 nm ± 108.77 | 1413 nm ± 117.1 |
| Polydispersity Index | 0.4 ± 0.1 | 0.4 ± 0.1 |
| Zeta Potential | −26.4 mv ± 4.609 | −23.8 mv ± 3.5 |
| Encapsulation Efficiency | 92% ± 5% | 91% ± 5% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adediran, E.; Arte, T.; Pasupuleti, D.; Vijayanand, S.; Singh, R.; Patel, P.; Gulani, M.; Ferguson, A.; Uddin, M.; Zughaier, S.M.; et al. Delivery of PLGA-Loaded Influenza Vaccine Microparticles Using Dissolving Microneedles Induces a Robust Immune Response. Pharmaceutics 2025, 17, 510. https://doi.org/10.3390/pharmaceutics17040510
Adediran E, Arte T, Pasupuleti D, Vijayanand S, Singh R, Patel P, Gulani M, Ferguson A, Uddin M, Zughaier SM, et al. Delivery of PLGA-Loaded Influenza Vaccine Microparticles Using Dissolving Microneedles Induces a Robust Immune Response. Pharmaceutics. 2025; 17(4):510. https://doi.org/10.3390/pharmaceutics17040510
Chicago/Turabian StyleAdediran, Emmanuel, Tanisha Arte, Dedeepya Pasupuleti, Sharon Vijayanand, Revanth Singh, Parth Patel, Mahek Gulani, Amarae Ferguson, Mohammad Uddin, Susu M. Zughaier, and et al. 2025. "Delivery of PLGA-Loaded Influenza Vaccine Microparticles Using Dissolving Microneedles Induces a Robust Immune Response" Pharmaceutics 17, no. 4: 510. https://doi.org/10.3390/pharmaceutics17040510
APA StyleAdediran, E., Arte, T., Pasupuleti, D., Vijayanand, S., Singh, R., Patel, P., Gulani, M., Ferguson, A., Uddin, M., Zughaier, S. M., & D’Souza, M. J. (2025). Delivery of PLGA-Loaded Influenza Vaccine Microparticles Using Dissolving Microneedles Induces a Robust Immune Response. Pharmaceutics, 17(4), 510. https://doi.org/10.3390/pharmaceutics17040510