
Targeted Drug Delivery to the Spleen and Its Implications for the Prevention and Treatment of Cancer


Abstract
1. Introduction
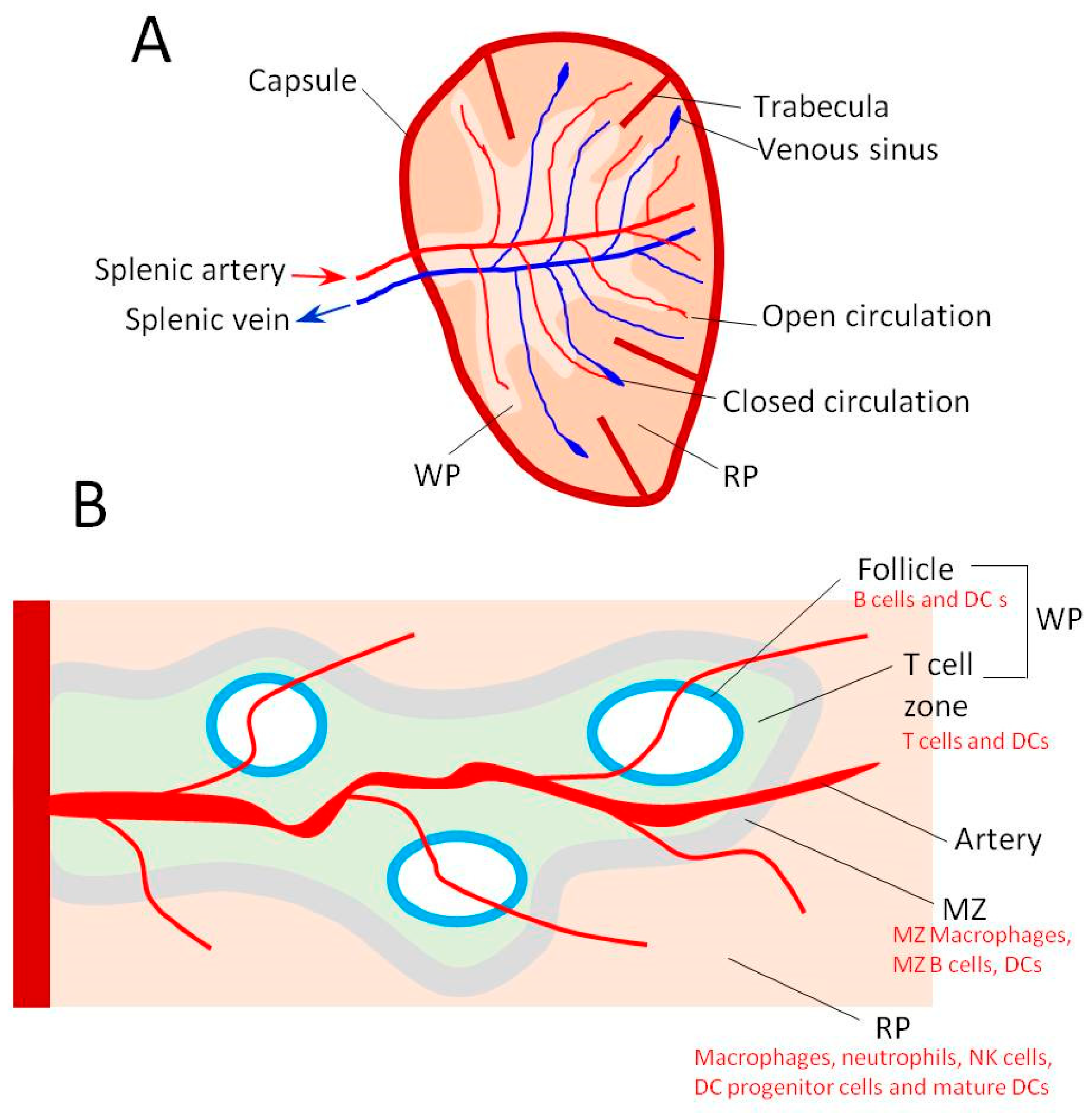
2. Spleen Anatomy
3. Spleen-Targeted Drug Delivery
3.1. Strategies for Spleen Delivery
3.1.1. Manipulation of Physicochemical Properties
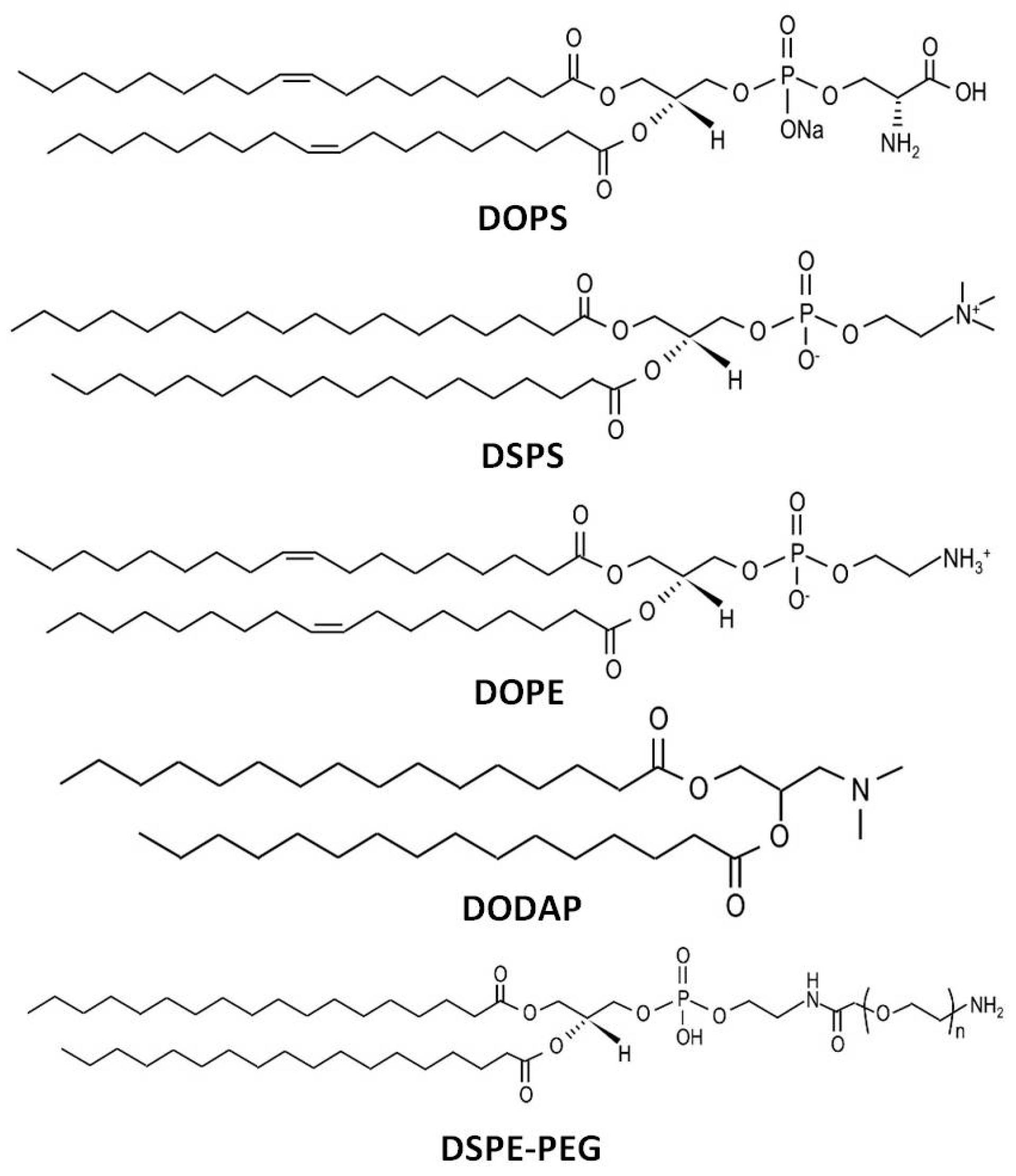
3.1.2. Lipid Composition
3.1.3. Using Specific Ligands
3.1.4. PEG Modification
3.1.5. RBCs as Spleen Delivery Systems
3.2. Cell-Specific Drug Delivery
4. Therapeutic Implications of Spleen Targeting
5. Discussion and Future Directions
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 14PA | 1,2-dimyristoyl-sn-glycero-3-phosphate |
| 18PA | 1,2-dioleoyl-sn-glycero-3-phosphate |
| APCs | Antigen-presenting cells |
| ApoE | Apolipoprotein E |
| Cit-ME | Multiepitope citrullinated peptide |
| CTL | Cytotoxic T lymphocyte |
| DCs | Dendritic cells |
| DODAP | 1,2-dioleoyl-3-dimethylammonium propane |
| DOPE | 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine |
| DOPS | 1,2-dioleoyl-sn-glycero-3-phospho-L-serine |
| DPEG | Dual PEGylation |
| DSPE-PEG | Distearoylphosphatidylethanolamine-polyethylene glycol |
| DSPC | 1,2-distearoyl-sn-glycero-3-phosphocholine |
| EDIT | RBC-Driven Immune Targeting |
| GF | Glomerular filtration |
| iPhos | Multi-tailed ionizable phospholipids |
| LNPs | Lipid nanoparticles |
| LPXs | Lipid polymer complexes |
| mRNA | Messenger RNA |
| miRNA | MicroRNA |
| MZ | Marginal zone |
| NKCs | Nature killer cells |
| NPs | Nanoparticles |
| PCNV | Protein corona-driven nanovaccine |
| pDNA | Plasmid DNA |
| PEG | Polyethylene glycol |
| PEGylation | PEG-modification |
| PEI | Polyethyleneimine |
| PSi | Mesoporous silicon |
| R8 | Octa-arginine peptide |
| RA | Rheumatoid arthritis |
| Rapa | Rapamycin |
| RBCs | Red blood cells |
| RES | Reticuloendothelial system |
| RP | Red pulp |
| SLE | Systemic lupus erythematosus |
| SORT | Selective organ targeting |
| WP | White pulp |
References
- Jindal, A.B. Nanocarriers for spleen targeting: Anatomo-physiological considerations, formulation strategies and therapeutic potential. Drug Deliv. Transl. Res. 2016, 6, 473–485. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Sun, K.; Luo, Z.; Zhang, J.; Zhou, H.; Yin, H.; Liang, Z.; You, J. Spleen-targeted delivery systems and strategies for spleen-related diseases. J. Control. Release 2024, 370, 773–797. [Google Scholar] [CrossRef] [PubMed]
- Khalil, I.A.; Younis, M.A.; Kimura, S.; Harashima, H. Lipid nanoparticles for cell-specific in vivo targeted delivery of nucleic acids. Biol. Pharm. Bull. 2020, 43, 584–595. [Google Scholar] [CrossRef] [PubMed]
- Lewis, S.M.; Williams, A.; Eisenbarth, S.C. Structure and function of the immune system in the spleen. Sci. Immunol. 2019, 4, eaau6085. [Google Scholar] [CrossRef]
- Mebius, R.E.; Kraal, G. Structure and function of the spleen. Nat. Rev. Immunol. 2005, 5, 606–616. [Google Scholar] [CrossRef]
- Pozo, A.L.; Godfrey, E.M.; Bowles, K.M. Splenomegaly: Investigation, diagnosis and management. Blood Rev. 2009, 23, 105–111. [Google Scholar] [CrossRef]
- Borges da Silva, H.; Fonseca, R.; Pereira, R.M.; Cassado, A.D.; Álvarez, J.M.; D’Império Lima, M.R. Splenic macrophage subsets and their function during blood-borne infections. Front. Immunol. 2015, 6, 480. [Google Scholar] [CrossRef]
- Bourquin, J.; Milosevic, A.; Hauser, D.; Lehner, R.; Blank, F.; Petri-Fink, A.; Rothen-Rutishauser, B. Biodistribution, clearance, and long-term fate of clinically relevant nanomaterials. Adv. Mater. 2018, 30, 1704307. [Google Scholar] [CrossRef]
- Li, S.-D.; Huang, L. Pharmacokinetics and biodistribution of nanoparticles. Mol. Pharm. 2008, 5, 496–504. [Google Scholar] [CrossRef]
- Muro, S.; Koval, M.; Muzykantov, V. Endothelial endocytic pathways: Gates for vascular drug delivery. Curr. Vasc. Pharmacol. 2004, 2, 281–299. [Google Scholar] [CrossRef]
- Sato, Y.; Sakurai, Y.; Kajimoto, K.; Nakamura, T.; Yamada, Y.; Akita, H.; Harashima, H. Innovative technologies in nanomedicines: From passive targeting to active targeting/from controlled pharmacokinetics to controlled intracellular pharmacokinetics. Macromol. Biosci. 2017, 17, 1600179. [Google Scholar] [CrossRef] [PubMed]
- Almeida, J.P.; Chen, A.L.; Foster, A.; Drezek, R. In vivo biodistribution of nanoparticles. Nanomedicine 2011, 6, 815–835. [Google Scholar] [CrossRef] [PubMed]
- Moghimi, S.M.; Hunter, A.C.; Murray, J.C. Long-circulating and target-specific nanoparticles: Theory to practice. Pharmacol. Rev. 2001, 53, 283–318. [Google Scholar] [CrossRef]
- Chen, K.; Wang, J.M.; Yuan, R.; Yi, X.; Li, L.; Gong, W.; Yang, T.; Li, L.; Su, S. Tissue-resident dendritic cells and diseases involving dendritic cell malfunction. Int. Immunopharmacol. 2016, 34, 1–15. [Google Scholar] [CrossRef]
- Shimosakai, R.; Khalil, I.A.; Kimura, S.; Harashima, H. mRNA-loaded lipid nanoparticles targeting immune cells in the spleen for use as cancer vaccines. Pharmaceuticals 2022, 15, 1017. [Google Scholar] [CrossRef]
- Kimura, S.; Khalil, I.A.; Elewa, Y.H.; Harashima, H. Spleen selective enhancement of transfection activities of plasmid DNA driven by octaarginine and an ionizable lipid and its implications for cancer immunization. J. Control. Release 2019, 313, 70–79. [Google Scholar] [CrossRef]
- Kimura, S.; Khalil, I.A.; Elewa, Y.H.; Harashima, H. Novel lipid combination for delivery of plasmid DNA to immune cells in the spleen. J. Control. Release 2021, 330, 753–764. [Google Scholar] [CrossRef]
- Wang, F.; Lou, J.; Gao, X.; Zhang, L.; Sun, F.; Wang, Z.; Ji, T.; Qin, Z. Spleen-targeted nanosystems for immunomodulation. Nano Today 2023, 52, 101943. [Google Scholar] [CrossRef]
- Moghimi, S.; Hunter, A.; Andresen, T. Factors controlling nanoparticle pharmacokinetics: An integrated analysis and perspective. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 481–503. [Google Scholar] [CrossRef]
- Benne, N.; van Duijn, J.; Kuiper, J.; Jiskoot, W.; Slütter, B. Orchestrating immune responses: How size, shape and rigidity affect the immunogenicity of particulate vaccines. J. Control. Release 2016, 234, 124–134. [Google Scholar] [CrossRef]
- Harris, J.M.; Martin, N.E.; Modi, M. Pegylation: A novel process for modifying pharmacokinetics. Clin. Pharmacokinet. 2001, 40, 539–551. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Feng, Z.; Zhang, Y.; Ni, H.; Liu, Z.; Wang, D. pH-responsive Astragalus polysaccharide-loaded PLGA nanoparticles as an adjuvant system to improve immune responses. Int. J. Biol. Macromol. 2022, 222, 1936–1947. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Y.; He, X.; Li, Y.; Han, R.; Ma, Y.; Gao, P.; Qian, Z.; Gu, Y.; Li, S. A splenic-targeted versatile antigen courier: iPSC wrapped in coalescent erythrocyte-liposome as tumor nanovaccine. Sci. Adv. 2021, 7, eabi6326. [Google Scholar] [CrossRef]
- Khalil, I.A.; Kimura, S.; Sato, Y.; Harashima, H. Synergism between a cell penetrating peptide and a pH-sensitive cationic lipid in efficient gene delivery based on double-coated nanoparticles. J. Control. Release 2018, 275, 107–116. [Google Scholar] [CrossRef]
- Fenton, O.S.; Kauffman, K.J.; Kaczmarek, J.C.; McClellan, R.L.; Jhunjhunwala, S.; Tibbitt, M.W.; Zeng, M.D.; Appel, E.A.; Dorkin, J.R.; Mir, F.F.; et al. Synthesis and biological evaluation of ionizable lipid materials for the in vivo delivery of messenger RNA to B lymphocytes. Adv. Mater. 2017, 29, 1606944. [Google Scholar] [CrossRef]
- Bishop, M.B.; Lansing, L.S. The spleen: A correlative overview of normal and pathologic anatomy. Hum. Pathol. 1982, 13, 334–342. [Google Scholar] [CrossRef]
- Steiniger, B.; Barth, P. Microanatomy and Function of the Spleen; Springer Nature: Dordrecht, The Netherlands, 2000. [Google Scholar]
- Liu, D.; Duan, L.; Rodda, L.B.; Lu, E.; Xu, Y.; An, J.; Qiu, L.; Liu, F.; Looney, M.R.; Yang, Z.; et al. CD97 promotes spleen dendritic cell homeostasis through the mechanosensing of red blood cells. Science 2022, 375, eabi5965. [Google Scholar] [CrossRef]
- Goralsky, L.P.; Dunaievska, O.F.; Kolesnik, N.L.; Stechenko, L.A.; Grynevych, N.E.; Kaminsky, R.F.; Sokurenko, L. Ultramicroscopic features of cells and vessels of the spleen (experimental study). Wiad. Lek. 2018, 71, 1019–1025. [Google Scholar]
- Drenckhahn, D.; Wagner, J. Stress fibers in the splenic sinus endothelium in situ: Molecular structure, relationship to the extracellular matrix, and contractility. J. Cell Biol. 1986, 102, 1738–1747. [Google Scholar] [CrossRef]
- He, H.; Yuan, Q.; Bie, J.; Wallace, R.L.; Yannie, P.J.; Wang, J.; Lancina, M.G.; Zolotarskaya, O.Y.; Korzun, W.; Yang, H.; et al. Development of mannose functionalized dendrimeric nanoparticles for targeted delivery to macrophages: Use of this platform to modulate atherosclerosis. Transl. Res. 2018, 193, 13–30. [Google Scholar] [CrossRef]
- Bertrand, N.; Leroux, J.-C. The journey of a drug-carrier in the body: An anatomo-physiological perspective. J. Control. Release 2012, 161, 152–163. [Google Scholar] [CrossRef] [PubMed]
- Blue, J.; Weiss, L. Vascular pathways in nonsinusal red pulp—An electron microscope study of the cat spleen. Am. J. Anat. 1981, 161, 135–168. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Zhao, L. Passive lung-targeted drug delivery systems via intravenous administration. Pharm. Dev. Technol. 2014, 19, 129–136. [Google Scholar] [CrossRef]
- Kusumoto, K.; Akita, H.; Ishitsuka, T.; Matsumoto, Y.; Nomoto, T.; Furukawa, R.; El-Sayed, A.; Hatakeyama, H.; Kajimoto, K.; Yamada, Y.; et al. Lipid envelope-type nanoparticle incorporating a multifunctional peptide for systemic siRNA delivery to the pulmonary endothelium. ACS Nano 2013, 7, 7534–7541. [Google Scholar] [CrossRef]
- Khalil, I.A.; Harashima, H. The gala peptide: Multiple roles in drug and gene delivery. Bull. Pharm. Sci. Assiut Univ. 2020, 43, 39–51. [Google Scholar] [CrossRef]
- Braet, F.; Wisse, E.; Bomans, P.; Frederik, P.; Geerts, W.; Koster, A.; Soon, L.; Ringer, S. Contribution of high-resolution correlative imaging techniques in the study of the liver sieve in three-dimensions. Microsc. Res. Tech. 2007, 70, 230–242. [Google Scholar] [CrossRef]
- Younis, M.A.; Khalil, I.A.; Elewa, Y.H.; Kon, Y.; Harashima, H. Ultra-small lipid nanoparticles encapsulating sorafenib and midkine-siRNA selectively-eradicate sorafenib-resistant hepatocellular carcinoma in vivo. J. Control. Release 2021, 331, 335–349. [Google Scholar] [CrossRef]
- Younis, M.A.; Khalil, I.A.; Harashima, H. Gene Therapy for hepatocellular carcinoma: Highlighting the journey from theory to clinical applications. Adv. Ther. 2020, 3, 2000087. [Google Scholar] [CrossRef]
- Hayashi, Y.; Mizuno, R.; Ikramy, K.A.; Akita, H.; Harashima, H. Pretreatment of hepatocyte growth factor gene transfer mediated by octaarginine peptide-modified nanoparticles ameliorates LPS/D-galactosamine-induced hepatitis. Nucleic Acid Ther. 2012, 22, 360–363. [Google Scholar] [CrossRef]
- Hirn, S.; Semmler-Behnke, M.; Schleh, C.; Wenk, A.; Lipka, J.; Schäffler, M.; Takenaka, S.; Möller, W.; Schmid, G.; Simon, U.; et al. Particle size-dependent and surface charge-dependent biodistribution of gold nanoparticles after intravenous administration. Eur. J. Pharm. Biopharm. 2011, 77, 407–416. [Google Scholar] [CrossRef]
- Choi, H.S.; Liu, W.; Misra, P.; Tanaka, E.; Zimmer, J.P.; Ipe, B.I.; Bawendi, M.G.; Frangioni, J.V. Renal clearance of quantum dots. Nat. Biotechnol. 2007, 25, 1165–1170. [Google Scholar] [CrossRef] [PubMed]
- Khalil, I.A.; Sato, Y.; Harashima, H. Recent advances in the targeting of systemically administered non-viral gene delivery systems. Expert Opin. Drug Deliv. 2019, 16, 1037–1050. [Google Scholar] [CrossRef] [PubMed]
- Decuzzi, P.; Ferrari, M. Design maps for nanoparticles targeting the diseased microvasculature. Biomaterials 2008, 29, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Moghimi, S.; Porter, C.; Muir, I.; Illum, L.; Davis, S. Non-phagocytic uptake of intravenously injected microspheres in rat spleen: Influence of particle size and hydrophilic coating. Biochem. Biophys. Res. Commun. 1991, 177, 861–866. [Google Scholar] [CrossRef]
- Klibanov, A.L.; Maruyama, K.; Beckerleg, A.M.; Torchilin, V.P.; Huang, L. Activity of amphipathic poly(ethylene glycol) 5000 to prolong the circulation time of liposomes depends on the liposome size and is unfavorable for immunoliposome binding to target. Biochim. Biophys. Acta (BBA) Biomembr. 1991, 1062, 142–148. [Google Scholar] [CrossRef]
- Nakamura, M.; Mochizuki, C.; Kuroda, C.; Shiohama, Y.; Nakamura, J. Size effect of fluorescent thiol-organosilica particles on their distribution in the mouse spleen. Colloids Surf. B Biointerfaces 2023, 228, 113397. [Google Scholar] [CrossRef]
- Albanese, A.; Tang, P.S.; Chan, W.C. The effect of nanoparticle size, shape, and surface chemistry on biological systems. Annu. Rev. Biomed. Eng. 2012, 14, 1–16. [Google Scholar] [CrossRef]
- Roser, M.; Fischer, D.; Kissel, T. Surface-modified biodegradable albumin nano- and microspheres. II: Effect of surface charges on in vitro phagocytosis and biodistribution in rats. Eur. J. Pharm. Biopharm. 1998, 46, 255–263. [Google Scholar] [CrossRef]
- Kranz, L.M.; Diken, M.; Haas, H.; Kreiter, S.; Loquai, C.; Reuter, K.C.; Meng, M.; Fritz, D.; Vascotto, F.; Hefesha, H.; et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature 2016, 534, 396–401. [Google Scholar] [CrossRef]
- Kurosaki, T.; Kodama, Y.; Muro, T.; Higuchi, N.; Nakamura, T.; Kitahara, T.; Miyakoda, M.; Yui, K.; Sasaki, H. Secure Splenic Delivery of Plasmid DNA and Its Application to DNA Vaccine. Biol. Pharm. Bull. 2013, 36, 1800–1806. [Google Scholar] [CrossRef]
- Dilliard, S.A.; Cheng, Q.; Siegwart, D.J. On the mechanism of tissue-specific mRNA delivery by selective organ targeting nanoparticles. Proc. Natl. Acad. Sci. USA 2021, 118, e2109256118. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.; Wei, T.; Farbiak, L.; Johnson, L.T.; Dilliard, S.A.; Siegwart, D.J. Selective organ targeting (SORT) nanoparticles for tissue-specific mRNA delivery and CRISPR–Cas gene editing. Nat. Nanotechnol. 2020, 15, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Kedmi, R.; Ben-Arie, N.; Peer, D. The systemic toxicity of positively charged lipid nanoparticles and the role of Toll-like receptor 4 in immune activation. Biomaterials 2010, 31, 6867–6875. [Google Scholar] [CrossRef] [PubMed]
- Owens, D.E.; Peppas, N.A. Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int. J. Pharm. 2006, 307, 93–102. [Google Scholar] [CrossRef]
- Sodipo, B.K.; Mohammed, Z.K. Advances in biodistribution of gold nanoparticles: The influence of size, surface charge, and route of administration. Biomed. Mater. 2024, 19, 042010. [Google Scholar] [CrossRef]
- Xu, M.; Qi, Y.; Liu, G.; Song, Y.; Jiang, X.; Du, B. Size-Dependent In Vivo Transport of Nanoparticles: Implications for Delivery, Targeting, and Clearance. ACS Nano 2023, 17, 20825–20849. [Google Scholar] [CrossRef]
- Moghimi, S.; Patel, H.M. Tissue specific opsonins for phagocytic cells and their different affinity for cholesterol-rich liposomes. FEBS Lett. 1988, 233, 143–147. [Google Scholar] [CrossRef]
- Kong, S.M.; Costa, D.F.; Jagielska, A.; Van Vliet, K.J.; Hammond, P.T. Stiffness of targeted layer-by-layer nanoparticles impacts elimination half-life, tumor accumulation, and tumor penetration. Proc. Natl. Acad. Sci. USA 2021, 118, e2104826118. [Google Scholar] [CrossRef]
- Gupta, A.S. Role of particle size, shape, and stiffness in design of intravascular drug delivery systems: Insights from computations, experiments, and nature. WIREs Nanomed. Nanobiotechnol. 2015, 8, 255–270. [Google Scholar] [CrossRef]
- Walkey, C.D.; Olsen, J.B.; Song, F.; Liu, R.; Guo, H.; Olsen, D.W.H.; Cohen, Y.; Emili, A.; Chan, W.C.W. Protein Corona Fingerprinting Predicts the Cellular Interaction of Gold and Silver Nanoparticles. ACS Nano 2014, 8, 2439–2455. [Google Scholar] [CrossRef]
- Nissinen, T.; Näkki, S.; Laakso, H.; Kučiauskas, D.; Kaupinis, A.; Kettunen, M.I.; Liimatainen, T.; Hyvönen, M.T.; Valius, M.; Gröhn, O.; et al. Tailored Dual PEGylation of Inorganic Porous Nanocarriers for Extremely Long Blood Circulation in Vivo. ACS Appl. Mater. Interfaces 2016, 8, 32723–32731. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Zhou, H.; Su, G.; Ma, M.; Liu, Y. Protein corona-driven nanovaccines improve antigen intracellular release and immunotherapy efficacy. J. Control. Release 2022, 345, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Monteiro-Riviere, N.A. Mechanisms of Cell Uptake, Inflammatory Potential and Protein Corona Effects with Gold Nanoparticles. Nanomedicine 2016, 11, 3185–3203. [Google Scholar] [CrossRef] [PubMed]
- Ritz, S.; Schöttler, S.; Kotman, N.; Baier, G.; Musyanovych, A.; Kuharev, J.; Landfester, K.; Schild, H.; Jahn, O.; Tenzer, S.; et al. Protein Corona of Nanoparticles: Distinct Proteins Regulate the Cellular Uptake. Biomacromolecules 2015, 16, 1311–1321. [Google Scholar] [CrossRef]
- Kurosaki, T.; Nakasone, C.; Kodama, Y.; Egashira, K.; Harasawa, H.; Muro, T.; Nakagawa, H.; Kitahara, T.; Higuchi, N.; Nakamura, T.; et al. Splenic Gene Delivery System Using Self-assembling Nano-complex with Phosphatidylserine Analog. Biol. Pharm. Bull. 2015, 38, 23–29. [Google Scholar] [CrossRef]
- Kauffman, K.J.; Dorkin, J.R.; Yang, J.H.; Heartlein, M.W.; DeRosa, F.; Mir, F.F.; Fenton, O.S.; Anderson, D.G. Optimization of Lipid Nanoparticle Formulations for mRNA Delivery in Vivo with Fractional Factorial and Definitive Screening Designs. Nano Lett. 2015, 15, 7300–7306. [Google Scholar] [CrossRef]
- Sabnis, S.; Kumarasinghe, E.S.; Salerno, T.; Mihai, C.; Ketova, T.; Senn, J.J.; Lynn, A.; Bulychev, A.; McFadyen, I.; Chan, J.; et al. A Novel Amino Lipid Series for mRNA Delivery: Improved Endosomal Escape and Sustained Pharmacology and Safety in Non-human Primates. Mol. Ther. 2018, 26, 1509–1519. [Google Scholar] [CrossRef]
- Chen, D.; Parayath, N.; Ganesh, S.; Wang, W.; Amiji, M. The role of apolipoprotein- and vitronectin-enriched protein corona on lipid nanoparticles for in vivo targeted delivery and transfection of oligonucleotides in murine tumor models. Nanoscale 2019, 11, 18806–18824. [Google Scholar] [CrossRef]
- Liu, S.; Cheng, Q.; Wei, T.; Yu, X.; Johnson, L.T.; Farbiak, L.; Siegwart, D.J. Membrane-destabilizing ionizable phospholipids for organ-selective mRNA delivery and CRISPR–Cas gene editing. Nat. Mater. 2021, 20, 701–710. [Google Scholar] [CrossRef]
- Wang, F.; Zhang, M.; Tian, M.; Lou, J.; Pan, L.; Gao, X.; Zhang, L.; Lou, X.; Zhu, L.; Sheng, Y.; et al. Natural long-chain saturated fatty acids doped LNPs enabling spleen selective mRNA translation and potent cancer immunotherapy. Nano Res. 2024, 17, 1804–1817. [Google Scholar] [CrossRef]
- Younis, M.A.; Sato, Y.; Elewa, Y.H.A.; Harashima, H. Harnessing the composition of lipid nanoparticles to selectively deliver mRNA to splenic immune cells for anticancer vaccination. Drug Deliv. Transl. Res. 2025, 15, 1–16. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Wang, J.; Tang, Y.; Chiang, S.T.; Han, T.; Chen, Q.; Qian, C.; Shen, X.; Li, R.; Ai, X. Recent Advances of Emerging Spleen-Targeting Nanovaccines for Immunotherapy. Adv. Health Mater. 2023, 12, e2300351. [Google Scholar] [CrossRef] [PubMed]
- Macri, C.; Paxman, M.; Jenika, D.; Lin, X.P.; Elahi, Z.; Gleeson, P.A.; Caminschi, I.; Lahoud, M.H.; Villadangos, J.A.; Mintern, J.D. FcRn regulates antigen presentation in dendritic cells downstream of DEC205-targeted vaccines. npj Vaccines 2024, 9, 76. [Google Scholar] [CrossRef] [PubMed]
- Yuba, E.; Fukaya, Y.; Yanagihara, S.; Kasho, N.; Harada, A. Development of Mannose-Modified Carboxylated Curdlan-Coated Liposomes for Antigen Presenting Cell Targeted Antigen Delivery. Pharmaceutics 2020, 12, 754. [Google Scholar] [CrossRef]
- Van der Jeught, K.; De Koker, S.; Bialkowski, L.; Heirman, C.; Tjok Joe, P.; Perche, F.; Maenhout, S.; Bevers, S.; Broos, K.; Deswarte, K.; et al. Dendritic Cell Targeting mRNA Lipopolyplexes Combine Strong Antitumor T-Cell Immunity with Improved Inflammatory Safety. ACS Nano 2018, 12, 9815–9829. [Google Scholar] [CrossRef]
- Bondioli, L.; Ruozi, B.; Belletti, D.; Forni, F.; Vandelli, M.A.; Tosi, G. Sialic acid as a potential approach for the protection and targeting of nanocarriers. Expert Opin. Drug Deliv. 2011, 8, 921–937. [Google Scholar] [CrossRef]
- Ding, C.; Wang, L.; Marroquin, J.; Yan, J. Targeting of antigens to B cells augments antigen-specific T-cell responses and breaks immune tolerance to tumor-associated antigen MUC1. Blood 2008, 112, 2817–2825. [Google Scholar] [CrossRef]
- Kang, D.; Zhang, Y.; Yu, D.-G.; Kim, I.; Song, W. Integrating synthetic polypeptides with innovative material forming techniques for advanced biomedical applications. J. Nanobiotechnol. 2025, 23, 101. [Google Scholar] [CrossRef]
- Alexis, F.; Pridgen, E.; Molnar, L.K.; Farokhzad, O.C. Factors Affecting the Clearance and Biodistribution of Polymeric Nanoparticles. Mol. Pharm. 2008, 5, 505–515. [Google Scholar] [CrossRef]
- Laverman, P.; Dams, E.T.; Storm, G.; Hafmans, T.G.; Croes, H.J.; Oyen, W.J.; Corstens, F.H.; Boerman, O.C. Microscopic localization of PEG-liposomes in a rat model of focal infection. J. Control. Release 2001, 75, 347–355. [Google Scholar] [CrossRef]
- Oh, C.; Lee, W.; Park, J.; Choi, J.; Lee, S.; Li, S.; Na Jung, H.; Lee, J.-S.; Hwang, J.-E.; Park, J.; et al. Development of Spleen Targeting H2S Donor Loaded Liposome for the Effective Systemic Immunomodulation and Treatment of Inflammatory Bowel Disease. ACS Nano 2023, 17, 4327–4345. [Google Scholar] [CrossRef] [PubMed]
- Abu-Dief, A.M.; Alsehli, M.; Awaad, A. A higher dose of PEGylated gold nanoparticles reduces the accelerated blood clearance phenomenon effect and induces spleen B lymphocytes in albino mice. Histochem. Cell Biol. 2022, 157, 641–656. [Google Scholar] [CrossRef] [PubMed]
- Hamidi, M.; Tajerzadeh, H. Carrier Erythrocytes: An Overview. Drug Deliv. 2003, 10, 9–20. [Google Scholar] [CrossRef]
- Zarrin, A.; Foroozesh, M.; Hamidi, M. Carrier erythrocytes: Recent advances, present status, current trends and future horizons. Expert Opin. Drug Deliv. 2014, 11, 433–447. [Google Scholar] [CrossRef]
- Brenner, J.S.; Pan, D.C.; Myerson, J.W.; Marcos-Contreras, O.A.; Villa, C.H.; Patel, P.; Hekierski, H.; Chatterjee, S.; Tao, J.-Q.; Parhiz, H.; et al. Red blood cell-hitchhiking boosts delivery of nanocarriers to chosen organs by orders of magnitude. Nat. Commun. 2018, 9, 2684. [Google Scholar] [CrossRef]
- Wang, L.; Wang, X.; Yang, F.; Liu, Y.; Meng, L.; Pang, Y.; Zhang, M.; Chen, F.; Pan, C.; Lin, S.; et al. Systemic antiviral immunization by virus-mimicking nanoparticles-decorated erythrocytes. Nano Today 2021, 40, 101280. [Google Scholar] [CrossRef]
- Han, X.; Shen, S.; Fan, Q.; Chen, G.; Archibong, E.; Dotti, G.; Liu, Z.; Gu, Z.; Wang, C. Red blood cell–derived nanoerythrosome for antigen delivery with enhanced cancer immunotherapy. Sci. Adv. 2019, 5, eaaw6870. [Google Scholar] [CrossRef]
- Ukidve, A.; Zhao, Z.; Fehnel, A.; Krishnan, V.; Pan, D.C.; Gao, Y.; Mandal, A.; Muzykantov, V.; Mitragotri, S. Erythrocyte-driven immunization via biomimicry of their natural antigen-presenting function. Proc. Natl. Acad. Sci. USA 2020, 117, 17727–17736. [Google Scholar] [CrossRef]
- Shimizu, T.; Kawaguchi, Y.; Ando, H.; Ishima, Y.; Ishida, T. Development of an Antigen Delivery System for a B Cell-Targeted Vaccine as an Alternative to Dendritic Cell-Targeted Vaccines. Chem. Pharm. Bull. 2022, 70, 341–350. [Google Scholar] [CrossRef]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA vaccines—A new era in vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef]
- Lindsay, K.E.; Bhosle, S.M.; Zurla, C.; Beyersdorf, J.; Rogers, K.A.; Vanover, D.; Xiao, P.; Araínga, M.; Shirreff, L.M.; Pitard, B.; et al. Visualization of early events in mRNA vaccine delivery in non-human primates via PET–CT and near-infrared imaging. Nat. Biomed. Eng. 2019, 3, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Ma, J.; Shen, R.; Lin, J.; Li, S.; Lu, X.; Stelzel, J.L.; Kong, J.; Cheng, L.; Vuong, I.; et al. Screening for lipid nanoparticles that modulate the immune activity of helper T cells towards enhanced antitumour activity. Nat. Biomed. Eng. 2023, 8, 544–560. [Google Scholar] [CrossRef] [PubMed]
- Bonifaz, L.; Bonnyay, D.; Mahnke, K.; Rivera, M.; Nussenzweig, M.C.; Steinman, R.M. Efficient Targeting of Protein Antigen to the Dendritic Cell Receptor DEC-205 in the Steady State Leads to Antigen Presentation on Major Histocompatibility Complex Class I Products and Peripheral CD8+ T Cell Tolerance. J. Exp. Med. 2002, 196, 1627–1638. [Google Scholar] [CrossRef]
- Wu, M.; Luo, Z.; Cai, Z.; Mao, Q.; Li, Z.; Li, H.; Zhang, C.; Zhang, Y.; Zhong, A.; Wu, L.; et al. Spleen-targeted neoantigen DNA vaccine for personalized immunotherapy of hepatocellular carcinoma. EMBO Mol. Med. 2023, 15, e16836. [Google Scholar] [CrossRef]
- Oberli, M.A.; Reichmuth, A.M.; Dorkin, J.R.; Mitchell, M.J.; Fenton, O.S.; Jaklenec, A.; Anderson, D.G.; Langer, R.; Blankschtein, D. Lipid Nanoparticle Assisted mRNA Delivery for Potent Cancer Immunotherapy. Nano Lett. 2017, 17, 1326–1335. [Google Scholar] [CrossRef]
- Jain, S.; Tran, T.-H.; Amiji, M. Macrophage repolarization with targeted alginate nanoparticles containing IL-10 plasmid DNA for the treatment of experimental arthritis. Biomaterials 2015, 61, 162–177. [Google Scholar] [CrossRef]
- Nie, S.; Yang, B.; Ma, R.; Zha, L.; Qin, Y.; Ou, L.; Chen, X.; Li, L. Synthetic nanomaterials for spleen-specific mRNA delivery. Biomaterials 2024, 314, 122859. [Google Scholar] [CrossRef]
- Boraschi, D.; Italiani, P. From antigen delivery system to adjuvanticy: The board application of nanoparticles in vaccinology. Vaccines 2015, 3, 930–939. [Google Scholar] [CrossRef]
- Furlan, R. A tolerizing mRNA vaccine against autoimmunity? Mol. Ther. 2021, 29, 896–897. [Google Scholar] [CrossRef]
- Pishesha, N.; Bilate, A.M.; Wibowo, M.C.; Huang, N.-J.; Li, Z.; Deshycka, R.; Bousbaine, D.; Li, H.; Patterson, H.C.; Dougan, S.K.; et al. Engineered erythrocytes covalently linked to antigenic peptides can protect against autoimmune disease. Proc. Natl. Acad. Sci. USA 2017, 114, 3157–3162. [Google Scholar] [CrossRef]
- Krienke, C.; Kolb, L.; Diken, E.; Streuber, M.; Kirchhoff, S.; Bukur, T.; Akilli-Öztürk, Ö.; Kranz, L.M.; Berger, H.; Petschenka, J.; et al. A noninflammatory mRNA vaccine for treatment of experimental autoimmune encephalomyelitis. Science 2021, 371, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Chen, C.; Fu, H.; Yu, J.; Sun, Y.; Huang, H.; Tang, Y.; Shen, N.; Duan, Y. MicroRNA-125a-Loaded Polymeric Nanoparticles Alleviate Systemic Lupus Erythematosus by Restoring Effector/Regulatory T Cells Balance. ACS Nano 2020, 14, 4414–4429. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Du, G.; Bai, S.; Dijia, L.; Li, C.; Hou, Y.; Zhang, Y.; Zhang, Z.; Gong, T.; Fu, Y.; et al. Restoring immunological tolerance in established experimental arthritis by combinatorial citrullinated peptides and immunomodulatory signals. Nano Today 2021, 41, 101307. [Google Scholar] [CrossRef]
- Rauchhaus, U.; Kinne, R.W.; Pohlers, D.; Wiegand, S.; Wölfert, A.; Gajda, M.; Bräuer, R.; Panzner, S. Targeted delivery of liposomal dexamethasone phosphate to the spleen provides a persistent therapeutic effect in rat antigen-induced arthritis. Ann. Rheum. Dis. 2009, 68, 1933–1934. [Google Scholar] [CrossRef]
- Sahin, U.; Oehm, P.; Derhovanessian, E.; Jabulowsky, R.A.; Vormehr, M.; Gold, M.; Maurus, D.; Schwarck-Kokarakis, D.; Kuhn, A.N.; Omokoko, T.; et al. An RNA vaccine drives immunity in checkpoint-inhibitor-treated melanoma. Nature 2020, 585, 107–112. [Google Scholar] [CrossRef]
- Khalil, I.A.; Kogure, K.; Akita, H.; Harashima, H. Uptake pathways and subsequent intracellular trafficking in nonviral gene delivery. Pharmacol. Rev. 2006, 58, 32–45. [Google Scholar] [CrossRef]
- Mitchell, D.A.; Nair, S.K. RNA-transfected dendritic cells in cancer immunotherapy. J. Clin. Investig. 2000, 106, 1065–1069. [Google Scholar] [CrossRef]
- Weissig, V.; D’Souza, G.G. Organelle-Specific Pharmaceutical Nanotechnology; John Wiley & Sons: Hoboken, NJ, USA, 2011. [Google Scholar]

{kind=link}
{kind=link}
| Strategies | Examples | Description |
|---|---|---|
| Particle size | Particles > 200 nm are preferred for spleen delivery [46]. |
| Surface charge | Slightly negatively charged NPs are delivered preferentially to the spleen [51]. | |
| Particle rigidity | Rigid NPs show greater retention in the spleen [58]. | |
| Binding to protein corona | Binding to protein corona in the circulation can either enhance or suppress spleen delivery, depending on its composition [64]. | |
| DOPS | An anionic lipid that facilitates recognition by spleen macrophages and DCs [66]. |
| DSPC | A helper lipid that exhibits higher spleen delivery by inhibiting the interaction of NPs with serum apolipoprotein [67]. | |
| DODAP | An ionizable lipid that showed spleen-targeting properties only when combined with the helper lipid DOPE [17]. | |
| OF-Deg-Lin | An ionizable lipid that showed predominant gene expression in B cells of the spleen [25]. | |
| Mannose-modified nanocarriers are frequently used for targeting mannose receptors expressed in DCs and macrophages [75]. | |
| Increasing the PEG molecular weight improved spleen accumulation, particularly through binding to WP macrophages [81]. | |
| RBCs can serve as carriers for spleen targeting due to their natural spleen-mediated clearance. RBCs are frequently modified with other NPs, ligands, or fused with lipid systems for enhancing spleen targeting [23]. | |
| Challenge | Potential Solutions |
|---|---|
| Optimize the particle size, surface charge, and rigidity of NPs. Use spleen-specific lipids and ligands to enhance spleen uptake. Develop lipids that are stable in the spleen but degrade in the liver. Design spleen-specific delivery systems. Improve cellular uptake in spleen immune cells. Apply PEG-coating to reduce liver uptake. |
| Optimize surface charge. Apply PEG coating. Enhance cellular uptake in spleen immune cells. |
| Optimize NP particle size and surface charge. |
| Develop novel lipids and ligands that target specific spleen cells. Design smart systems that are activated in specific spleen cells. |
| Use novel pH-sensitive lipids with high endosomal escape ability. Incorporate fusogenic helper lipids. Apply pH-responsive polymers or peptides. |
| Apply anti-inflammatory coatings. Use cytokine inhibitors and agents to modulate immune reactions. Develop controlled release formulations. |
| Develop stable formulations with proper lipids and polymers. Optimize PEG coating. Use chemically modified nucleic acids for increased stability. |
| Apply microfluidic technology for the precise and reproducible fabrication of NPs. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khalil, I.A.; Faheem, A.; El-Tanani, M. Targeted Drug Delivery to the Spleen and Its Implications for the Prevention and Treatment of Cancer. Pharmaceutics 2025, 17, 651. https://doi.org/10.3390/pharmaceutics17050651
Khalil IA, Faheem A, El-Tanani M. Targeted Drug Delivery to the Spleen and Its Implications for the Prevention and Treatment of Cancer. Pharmaceutics. 2025; 17(5):651. https://doi.org/10.3390/pharmaceutics17050651
Chicago/Turabian StyleKhalil, Ikramy A., Ahmed Faheem, and Mohamed El-Tanani. 2025. "Targeted Drug Delivery to the Spleen and Its Implications for the Prevention and Treatment of Cancer" Pharmaceutics 17, no. 5: 651. https://doi.org/10.3390/pharmaceutics17050651
APA StyleKhalil, I. A., Faheem, A., & El-Tanani, M. (2025). Targeted Drug Delivery to the Spleen and Its Implications for the Prevention and Treatment of Cancer. Pharmaceutics, 17(5), 651. https://doi.org/10.3390/pharmaceutics17050651