
Biofunctional Excipients: Their Emerging Role in Overcoming the Inherent Poor Biopharmaceutical Characteristics of Drugs

 ,
, 
 , ,
, ,  and
and 
Abstract
1. Introduction
2. Biofunctional Excipients
3. Biofunctional Excipients Added to Enhance Drug Solubility and Dissolution
3.1. Functional Polymers

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Uses | BCS Class | Polymer | Formulation 1 | Biofunctional Applications | Ref |
|---|---|---|---|---|---|---|
| Verapamil | Anti-hypertensive | BCS II | HPMC | HME | Enhanced in vivo drug solubilization and drug bioavailability via formation of SD and gel formation upon water contact. | [38] |
| Itraconazole | Anti-fungal | BCS II | SD | [42] | ||
| Nilvadipine | Anti-hypertensive | BCS II | NA | [42] | ||
| Tacrolimus | Immunosuppressant | BCS II | SD | [56] | ||
| Nabilone | Anti-emetic | BCS II | PVP | HME | Enhanced in vivo drug solubilization and stabilization via inhibition of poorly soluble drug crystal growth. | [42] |
| Troglitazone | Anti-diabetic | BCS II | HME | [42] | ||
| Celecoxib | Analgesic | BCS II | NA | [57] | ||
| Griseofulvin | Anti-fungal | BCS II | PEG | HME | Enhanced in vivo drug solubilization via the higher hydrophilicity that allow interactions with hydrophobic drugs. | [58] |
| Fenofibrate | Anti-hyperlipidemia | BCS II | SD | [42] | ||
| Nimodipine | Anti-hypertensive | BCS II | SD | [42] | ||
| Ivacaftor | Cystic fibrosis | BCS II | HPMCAS | SD | Enhanced in vivo drug solubilization and stabilization via the colloidal nature of the polymer when ionized and via the hydrophobic substitutes. | [59] |
| Posaconazole | Anti-fungal | BCS II | HME | [42] | ||
| Ziprasidone | Anti-psychotic | BCS II | Mixing | [32] | ||
| Vemurafenib | Anti-cancer | BCS II | Co-precip. | [60] | ||
| Telaprevir | Anti-viral | BCS II | SD | [42] | ||
| Ritonavir | Anti-viral | BCS IV | PVP-VA | HME | Enhanced in vivo drug solubilization, stabilization, and bioavailability via the formation of micelle-like structures, creating optimal conditions for solubilizing poorly soluble drugs. | [42] |
| Lopinavir | Anti-viral | BCS IV | HME | [42] | ||
| Amenamevir | Anti-viral | BCS IV | PVA | Ball Milling | [25] | |
| Itraconazole | Anti-fungal | BCS II | Eudragit E | SD | Enhanced drug dissolution | [32,61] |
| Ibuprofen | Analgesic | BCS II | PEG-PVP | Evaporation | [62] | |
| Piroxicam | Analgesic | BCS II | Eudragit S | MP | [43] |

3.2. Functional Lipids and Lipid Derivatives
| Drug | Lipid Excipient 1 | BCS Class | Formulation | Biofunctional Application | Route | Ref. |
|---|---|---|---|---|---|---|
| Cyclosporine | Mixed lipids | BCS IV | Soft capsules | Improved drug solubilization | Oral | [78] |
| Ritonavir | Mint oil, PEG–castor | BCS IV | Microemulsion | In vivo drug solubilization | Oral | [82] |
| Cinnarizine | Oleic acid | BCS II | Solution | Enhanced oral bioavailability (4-F) | Oral | [83] |
| Vitamin D3 | LCT, MCT, and SCT | BCS IV | Solution | More BV in LCT>MCT>SCT | Oral | [84] |
| Danazol | Glycerol MO | BCS II | Emulsion | 4-fold increase in bioavailability | Oral | [85] |
| Acyclovir | Labrafac, labrasol | BCS III | Microemulsion | 13-fold increase in BV compared to tablets | Oral | [86] |
| Simvastatin | Capryol 90 | BCS II | SEDDS | 1.5-fold increase in BV | Oral | [87] |
| Carvedilol | Labrafil M1944CS | BCS II | SEDDS | 4-fold increase in BV | Oral | [88] |
| Exenatide | Cre., Labrafil, Cap. | BCS III | SEDDS | 14% enhancement in BV | Oral | [72] |
| Indomethacin | Ethyl oleate | BCS II | SEDDS | 57% increase in AUC | Oral | [71] |
| Ibuprofen | PEG-8 Capryic TGs | BCS I | Microemulsion | Enhanced stability and BV | Oral | [79] |
| Ritonavir | PEG-35 castor oil | BCS II | Microemulsion | Minimal enhancement in BV | Oral | [81] |
| Saquinavir | MCT | BCS II | Microemulsion | Minimal enhancement in BV | Oral | [46] |
| Fenofibrate | Mixed lipids | BCS II | Hard capsules | In vivo drug solubilization | Oral | [70] |
3.3. Functional Amino Acids (AAs)
3.4. Functional Cyclodextrins (CDs)
| Drug | CD Type | BCS Class | Formulation | Route | Biofunctional Applications | Ref. |
|---|---|---|---|---|---|---|
| Repaglinide | HP-β-CD | BCS II | Complex | Oral | Enhanced oral bioavailability | [146] |
| Cilostazol | β-CD | BCS II | Complex | Enhanced oral bioavailability | [147] | |
| Albendazole | BCS IV | Solid matrix | Enhanced dissolution rate | [148] | ||
| Piroxicam | HP-β-CD | BCS II | Gel | Enhanced release and permeation | [149] | |
| Disulfiram | HP-β-CD | BCS IV | Solution | Ocular | Enhanced ocular bioavailability | [150] |
| Tacrolimus | BCS II | Solution | Enhanced in vivo drug solubility | [151] | ||
| Curcumin | BCS IV | Solution | Improved solubility and release | [152] | ||
| Nepafenac | BCS II | Suspension | Increased trans-corneal permeation | [153] | ||
| Alprostadil | α-CD | BCS IV | Powder | Injection | Enhanced drug aqueous solubility and enhanced in vivo bioavailability | [136] |
| Letermovir | HP-β-CD | BCS II | Concentrate | |||
| Amiodarone | SBE-β-CD | BCS II | Solution | |||
| Aripiprazole | BCS II | Solution |
3.5. Functional Surfactants
| Drug | Surfactant 1 | BCS Class | Formulation 1 | Route | Biofunctional Applications | Ref. |
|---|---|---|---|---|---|---|
| Posaconazole | SLS | BCS II | SD | Oral | Enhanced drug bioavailability | [173] |
| Ezetimibe | Tween 80 | BCS II | SD | Oral | Little effect on bioavailability | [174] |
| Ritonavir | Tween 80 | BCS IV | SD | Oral | Enhanced drug bioavailability | [175] |
| Sorafenib | SLS | BCS IV | SD | Oral | Enhanced bioavailability | [176] |
| Ibuprofen | Poloxamer | BCS II | SD | Oral | Enhanced drug bioavailability | [177] |
| Docetaxel | Poloxamer | BCS IV | SD | Oral | Enhanced drug bioavailability | [178] |
| Ibuprofen | Poloxamer | BCS II | SD | In vitro | Enhanced dissolution rate | [179] |
| Desloratadine | Poloxamer | BCS II | SD | In vitro | Enhanced dissolution rate | [180] |
| Celecoxib | SLS | BCS II | Tablets | In vitro | Enhanced dissolution rate | [160] |
| Dutasteride | TPGS | BCS II | SD | Oral | Enhanced bioavailability | [181] |
| Buspirone | SLS | BCS II | Tablets | In vitro | Enhanced dissolution rate | [160] |
4. Biofunctional Excipients Added to Enhance Drug Penetration and Permeability
4.1. Bioadhesive Polymers
| Drug | Polymer 1 | BCS Class | Formulation | Biofunctional Application | Route | Ref. |
|---|---|---|---|---|---|---|
| Doxazocin | Chitosan | BCS III | Hydrogel | Bioadhesive polymeric-based drug delivery systems enable sustained, prolonged, and targeted in vivo drug release through mucoadhesion, diffusion, swelling, and erosion mechanisms. Their mucus-binding properties facilitate controlled drug release across gastric and intestinal environments, enhancing drug permeation, bioactivity, and local persistence while ensuring gradual release from polymeric networks for improved therapeutic efficacy. | Implant | [198] |
| Doxorubicin | BCS IV | Hydrogel | In vitro | [199] | ||
| Gentamicin | BCS III | Hydrogel | Ear | [200] | ||
| L-asparaginase | -- | Nanoparticles | In vitro | [201] | ||
| VEGF1 | Alginate | -- | Hydrogel | Implant | [202] | |
| Curcumin | BCS IV | Nanoparticles | In vitro | [203] | ||
| Doxorubicin | BCS IV | Nanoparticles | In vitro | [204] | ||
| Tilmicosin | BCS III | Nanogel | Buccal | [205] | ||
| Vancomycin | BCS III | Microparticles | In vitro | [205] | ||
| Curcumin | Pectin | BCS IV | Hydrogel beads | Oral | [206] | |
| Vancomycin | BCS III | Hydrogel/scaffold | Oral | [207] | ||
| Ciprofloxacin | Alg-PEG | BCS III | Polymeric system | In vitro | [208] | |
| Cisplatin | Gelatin | BCS III | Microparticles | Injection | [209] |
4.2. Biofunctional Amino Acids, Surfactants, and Cyclodextrins
5. Biofunctional Excipients Added to Enhance Drug Release and Kinetic Profiles
Smart or Intelligent Polymers
| Drug | BCS Class | Polymer 1 | Biofunctional Applications | Formulation | Ref |
|---|---|---|---|---|---|
| Doxorubicin | BCS IV | PNIPAM | Modified and targeted in vivo drug release based on body temperature. PNIPAM polymer collapsed and modified the drug release at temperatures above 32 °C. | Nanoparticles | [243] |
| Doxorubicin | BCS IV | Nanotubes | [249] | ||
| Ibuprofen | BCS II | Hydrogel | [250] | ||
| 5-Fluorouracil | BCS I | Hydrogel | [250] | ||
| Paclitaxel | BCS IV | Liposomes | [251] | ||
| GH | Biologic | PCL-PEG-PCL | Modified in vivo drug release controlled via the balance between the hydrophilic segment (PEG) and hydrophobic segment (PCL) with thermo-sensitive properties. | In situ gel | [252] |
| Silver | Metal | Micelles | [253] | ||
| Diclofenac | BCS II | Hydrogel | [254] | ||
| Doxorubicin | BCS IV | PLGA-PEG-PLGA | Targeted, controlled, and sustained in vivo drug delivery via the benefits from the hydrophobicity of PLGA and the biocompatibility of the PEG segment. | Hydrogel | [255] |
| Dexamethasone | BCS II | Hydrogel | [256] | ||
| Cyclosporine | BCS IV | Hydrogel | [257] | ||
| Naltrexone | BCS I | Hydrogel | [258] | ||
| Simvastatin | BCS II | Hydrogel | [259] | ||
| Ketorolac | BCS II | Hydrogel | [260] | ||
| Metronidazole | BCS I | PVME | Controlled in vivo drug release | Hydrogel | [261] |
| Doxorubicin | BCS IV | PNVCL | Controlled in vivo drug release | In situ gel | [262] |
| 5-Fluorouracil | BCS I | PolyDMAEMA | Modified in vivo drug release | Nanocarriers | [263] |
| Curcumin | BCS IV | PVA | Controlled in vivo drug release | Nanofibrous | [264] |
| Doxorubicin | BCS IV | PNIAM-PCL | Sustained in vivo drug release | Porous film | [265] |
| Drug | BCS Class | Polymer 1 | Biofunctional Applications | Formulation | Ref |
|---|---|---|---|---|---|
| Psoraldin | -- | Eudragit S100 | Enhanced in vivo drug bioavailability, drug release profiles, and drug targeting properties. Eudragit S100 acts as a protective polymer matrix for the drug at acidic pHs, such as in the stomach, and gradually releases the drug at neutral and slightly alkaline pHs, such as in the small intestine and the end of the ileum. These biofunctions allow Eudragit S100 to control drug release, deliver the drug to the colon, protect the drug from the harsh environment of the stomach, and finally enhance drug bioavailability. | Nanocapsules | [271] |
| Zaleplon | BCS II | Microspheres | [272] | ||
| 5-Fluorouracil | BCS I | 3D-Printed Tablets | [273] | ||
| Prednisolone | BCS I | Microsponges | [274] | ||
| Budesonide | BCS II | Nanocapsules | [275] | ||
| Etoricoxib | BCS II | Nanoparticles | [276] | ||
| Insulin | Biologic | Nanoparticles | [277] | ||
| Sulfasalazine | BCS IV | Bilayer tablets | [278] | ||
| Flurbiprofen | BCS II | Coated matrix tablet | [279] | ||
| Mesalamine | BCS IV | Tablets | [280] | ||
| Azithromycin | BCS III | Eudragit L100 | Enhanced delayed, controlled, and ocular drug delivery via drug protection at lower acidic pHs such as in the stomach and release of the drug at higher acidic pHs such as in the upper small intestine. Also, provides precise temporal drug release for ocular drug delivery (ocular pH like pH of Eudragit L100 degradation). | Polymeric Inserts | [281] |
| Dexamethasone | BCS II | Nanoparticles | [282] | ||
| Enoxaparin | Biologic | Nanoparticles | [283] | ||
| Fenofibrate | BCS II | Nanoparticles | [284] | ||
| Rosuvastatin | BCS III | Nanoparticles | [285] | ||
| Duloxetine. HCL | BCS II | Tablets | [286] | ||
| Quercetin | BCS IV | Nanoparticles | [287] | ||
| Senna | -- | Nanoparticles | [288] | ||
| Doxorubicin | BCS IV | PAMAM | Cancer treatment. | Dendrimers | [289] |
| Diclofenac | BCS II | Carbopol | Controlled in vivo drug release. | Hydrogel | [290] |
| Sulfacetamide | BCS III | Ocular drug delivery. | In situ gel | [291] | |
| Naproxen | BCS I | Ocular drug delivery. | In situ gel | [292] | |
| Doxorubicin | BCS IV | DMAEMA | Regulation of in vivo drug release. | Hydrogel | [293] |
6. Favorable and Unfavorable Interactions of Biofunctional Excipients
6.1. Biofunctional Excipients and Drug Metabolism
6.2. Biofunctional Excipients and Intestinal Transporters
6.3. Biofunctional Excipients and pH Modulation
| Drug | BCS Class | pH-Modifier 1 | Formulation 1 | Route | Biofunctional Applications | Ref. |
|---|---|---|---|---|---|---|
| Ketoconazole | BCS II | Citric acid | Granules | Oral | Enhanced oral bioavailability | [322] |
| Ketoconazole | BCS II | Tartaric acid | Granules | Oral | Enhanced oral bioavailability | [322] |
| Paracetamol | BCS II | NaHCO3 | Tablets | Oral | Enhanced absorption rates | [327] |
| Aceclofenac | BCS II | Na2CO3 | Tablets | Oral | Enhanced drug release | [329] |
| Dipyridamole | BCS II | p-toluenesulfonic acid | Granules | Oral | Enhanced dissolution rate | [323] |
| Carvedilol | BCS II | Citric acid | SD | Oral | Consistent pharmacokinetics | [324] |
| Telmisartan | BCS II | Magnesium oxide | SD | In vitro | Enhanced dissolution rate | [326] |
| Paracetamol | BCS II | CaCO3 | Tablets | Oral | Enhanced drug absorption | [327] |
| Aceclofenac | BCS II | Na2CO3 | Tablets | Oral | Enhanced drug release | [328] |
| Diltiazem | BCS I | Citric acid | Tablets | In vitro | Enhanced drug dissolution | [330] |
| Vinpocetine | BCS II | Citric acid | Tablets | In vitro | Enhanced drug dissolution | [331] |
7. Regulatory Aspects of Excipients and Biofunctional Excipients
8. Concluding Remarks and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Elder, D.P.; Kuentz, M.; Holm, R. Pharmaceutical excipients—Quality, regulatory and biopharmaceutical considerations. Eur. J. Pharm. Sci. 2016, 87, 88–99. [Google Scholar] [CrossRef] [PubMed]
- van der Merwe, J.; Steenekamp, J.; Steyn, D.; Hamman, J. The Role of Functional Excipients in Solid Oral Dosage Forms to Overcome Poor Drug Dissolution and Bioavailability. Pharmaceutics 2020, 12, 393. [Google Scholar] [CrossRef] [PubMed]
- Shah, H.; Jain, A.; Laghate, G.; Prabhudesai, D. Chapter 32—Pharmaceutical excipients. In Remington, 23rd ed.; Adejare, A., Ed.; Academic Press: Cambridge, MA, USA, 2021; pp. 633–643. [Google Scholar]
- Ramesh, K.V.; Yadav, H.; Sarheed, O. Safety of pharmaceutical excipients and regulatory issues. Appl. Clin. Res. Clin. Trials Regul. Aff. 2019, 6, 86–98. [Google Scholar] [CrossRef]
- Abrantes, C.G.; Duarte, D.; Reis, C.P. An Overview of Pharmaceutical Excipients: Safe or Not Safe? J. Pharm. Sci. 2016, 105, 2019–2026. [Google Scholar] [CrossRef]
- Ionova, Y.; Wilson, L. Biologic excipients: Importance of clinical awareness of inactive ingredients. PLoS ONE 2020, 15, e0235076. [Google Scholar] [CrossRef]
- Abdelkader, H.; Al-Faease, A.; Wu, Z. Biofunctional Pharmaceutical Additives for Targeted, Improved Bioavailability and Safety of Medicine. Available online: https://www.mdpi.com/journal/pharmaceutics/special_issues/4ZQ48TOTJ1 (accessed on 22 March 2024).
- Haigney, S. The Logistics of Developing an Emerging Therapy. BioPharm Int. Emerg. Ther. eBook 2024, 37, 22–26. [Google Scholar]
- Amidon, G.L.; Lennernäs, H.; Shah, V.P.; Crison, J.R. A Theoretical Basis for a Biopharmaceutic Drug Classification: The Correlation of in Vitro Drug Product Dissolution and in Vivo Bioavailability. Pharm. Res. 1995, 12, 413–420. [Google Scholar] [CrossRef]
- Thayer, A.M. Finding solutions. Chem. Eng. News Arch. 2010, 88, 13–18. [Google Scholar] [CrossRef]
- Ting, J.M.; Porter, W.W., III; Mecca, J.M.; Bates, F.S.; Reineke, T.M. Advances in Polymer Design for Enhancing Oral Drug Solubility and Delivery. Bioconjug. Chem. 2018, 29, 939–952. [Google Scholar] [CrossRef]
- Siepmann, J.; Faham, A.; Clas, S.-D.; Boyd, B.J.; Jannin, V.; Bernkop-Schnürch, A.; Zhao, H.; Lecommandoux, S.; Evans, J.C.; Allen, C.; et al. Lipids and polymers in pharmaceutical technology: Lifelong companions. Int. J. Pharm. 2019, 558, 128–142. [Google Scholar] [CrossRef]
- Sarkar, A.; Kellogg, G.E. Hydrophobicity—Shake Flasks, Protein Folding and Drug Discovery. Curr. Top. Med. Chem. 2010, 10, 67–83. [Google Scholar] [CrossRef] [PubMed]
- Sugano, K.; Okazaki, A.; Sugimoto, S.; Tavornvipas, S.; Omura, A.; Mano, T. Solubility and dissolution profile assessment in drug discovery. Drug Metab. Pharmacokinet. 2007, 22, 225–254. [Google Scholar] [CrossRef] [PubMed]
- Löbenberg, R.; Amidon, G.L. Modern bioavailability, bioequivalence and biopharmaceutics classification system. New scientific approaches to international regulatory standards. Eur. J. Pharm. Biopharm. 2000, 50, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Savjani, K.T.; Gajjar, A.K.; Savjani, J.K. Drug Solubility: Importance and Enhancement Techniques. Int. Sch. Res. Not. 2012, 2012, 195727. [Google Scholar] [CrossRef]
- Kumar, A.; Sahoo, S.K.; Padhee, K.; Kochar, P.S.; Sathapathy, A.; Pathak, N. Review on solubility enhancement techniques for hydrophobic drugs. Pharm. Glob. 2011, 3, 1–7. [Google Scholar]
- Yadav, K.; Sachan, A.K.; Kumar, S.; Dubey, A. Techniques for increasing solubility: A review of conventional and new strategies. Asian J. Pharm. Res. Dev. 2022, 10, 144–153. [Google Scholar] [CrossRef]
- D’souza, A.A.; Shegokar, R. Polyethylene glycol (PEG): A versatile polymer for pharmaceutical applications. Expert Opin. Drug Deliv. 2016, 13, 1257–1275. [Google Scholar] [CrossRef]
- Baumann, E.; Stoya, G.; Völkner, A.; Richter, W.; Lemke, C.; Linss, W. Hemolysis of human erythrocytes with saponin affects the membrane structure. Acta Histochem. 2000, 102, 21–35. [Google Scholar] [CrossRef]
- Kalivoda, A.; Fischbach, M.; Kleinebudde, P. Application of mixtures of polymeric carriers for dissolution enhancement of fenofibrate using hot-melt extrusion. Int. J. Pharm. 2012, 429, 58–68. [Google Scholar] [CrossRef]
- Kasselkus, A.; Weiskircher-Hildebrandt, E.; Schornick, E.; Bauer, F.; Zheng, M. Polyvinyl alcohol: Revival of a long lost polymer. Pharma Biopharma Raw Mater. Solut. 2018, 10, 15. [Google Scholar]
- Meijer, G.W.; Kanny, G.; Briois, J.F. Ullmann’s Encyclopedia of Industrial Chemistry; Wiley and Sons: Hoboken, NJ, USA, 2000. [Google Scholar]
- DeMerlis, C.C.; Schoneker, D.R. Review of the oral toxicity of polyvinyl alcohol (PVA). Food Chem. Toxicol. 2003, 41, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Umemoto, Y.; Uchida, S.; Yoshida, T.; Shimada, K.; Kojima, H.; Takagi, A.; Tanaka, S.; Kashiwagura, Y.; Namiki, N. An effective polyvinyl alcohol for the solubilization of poorly water-soluble drugs in solid dispersion formulations. J. Drug Deliv. Sci. Technol. 2020, 55, 101401. [Google Scholar] [CrossRef]
- Brough, C.; Miller, D.A.; Ellenberger, D.; Lubda, D.; Williams, R.O. Use of Polyvinyl Alcohol as a Solubility Enhancing Polymer for Poorly Water-Soluble Drug Delivery (Part 2). AAPS PharmSciTech 2016, 17, 180–190. [Google Scholar] [CrossRef] [PubMed]
- De Jaeghere, W.; De Beer, T.; Van Bocxlaer, J.; Remon, J.P.; Vervaet, C. Hot-melt extrusion of polyvinyl alcohol for oral immediate release applications. Int. J. Pharm. 2015, 492, 1–9. [Google Scholar] [CrossRef]
- Wei, C.; Solanki, N.G.; Vasoya, J.M.; Shah, A.V.; Serajuddin, A.T.M. Development of 3D Printed Tablets by Fused Deposition Modeling Using Polyvinyl Alcohol as Polymeric Matrix for Rapid Drug Release. J. Pharm. Sci. 2020, 109, 1558–1572. [Google Scholar] [CrossRef]
- Saviano, M.; Aquino, R.P.; Del Gaudio, P.; Sansone, F.; Russo, P. Poly(vinyl alcohol) 3D printed tablets: The effect of polymer particle size on drug loading and process efficiency. Int. J. Pharm. 2019, 561, 1–8. [Google Scholar] [CrossRef]
- USP 36-NF 31; U.S. Pharmacopeia and National Formulary. United States Pharmacopeia: Rockville, MD, USA, 2013.
- Ilevbare, G.A.; Liu, H.; Edgar, K.J.; Taylor, L.S. Impact of polymers on crystal growth rate of structurally diverse compounds from aqueous solution. Mol. Pharm. 2013, 10, 2381–2393. [Google Scholar] [CrossRef]
- Liu, H.; Taylor, L.S.; Edgar, K.J. The role of polymers in oral bioavailability enhancement; a review. Polymer 2015, 77, 399–415. [Google Scholar] [CrossRef]
- Rowe, R.C.; Sheskey, P.; Quinn, M. Handbook of Pharmaceutical Excipients; Libros Digitales-Pharmaceutical Press: London, UK, 2009. [Google Scholar]
- Qian, F.; Wang, J.; Hartley, R.; Tao, J.; Haddadin, R.; Mathias, N.; Hussain, M. Solution behavior of PVP-VA and HPMC-AS-Based amorphous solid dispersions and their bioavailability implications. Pharm. Res. 2012, 29, 2766–2776. [Google Scholar] [CrossRef]
- Pasut, G.; Veronese, F.M. State of the art in PEGylation: The great versatility achieved after forty years of research. J. Control. Release 2012, 161, 461–472. [Google Scholar] [CrossRef]
- Nayak, A.K.; Panigrahi, P.P. Solubility Enhancement of Etoricoxib by Cosolvency Approach. Int. Sch. Res. Not. 2012, 2012, 820653. [Google Scholar] [CrossRef]
- Zhu, Q.; Harris, M.T.; Taylor, L.S. Modification of crystallization behavior in drug/polyethylene glycol solid dispersions. Mol. Pharm. 2012, 9, 546–553. [Google Scholar] [CrossRef] [PubMed]
- Williams, H.D.; Trevaskis, N.L.; Charman, S.A.; Shanker, R.M.; Charman, W.N.; Pouton, C.W.; Porter, C.J.H. Strategies to Address Low Drug Solubility in Discovery and Development. Pharmacol. Rev. 2013, 65, 315–499. [Google Scholar] [CrossRef] [PubMed]
- Janssens, S.; Van den Mooter, G. Review: Physical chemistry of solid dispersions. J. Pharm. Pharmacol. 2010, 61, 1571–1586. [Google Scholar] [CrossRef]
- Mishra, D.K.; Dhote, V.; Bhargava, A.; Jain, D.K.; Mishra, P.K. Amorphous solid dispersion technique for improved drug delivery: Basics to clinical applications. Drug Deliv. Transl. Res. 2015, 5, 552–565. [Google Scholar] [CrossRef]
- Li, J.; Patel, D.; Wang, G. Use of Spray-Dried Dispersions in Early Pharmaceutical Development: Theoretical and Practical Challenges. AAPS J. 2017, 19, 321–333. [Google Scholar] [CrossRef]
- Wyttenbach, N.; Kuentz, M. Glass-forming ability of compounds in marketed amorphous drug products. Eur. J. Pharm. Biopharm. 2017, 112, 204–208. [Google Scholar] [CrossRef]
- Maghsoodi, M.; Sadeghpoor, F. Preparation and evaluation of solid dispersions of piroxicam and Eudragit S100 by spherical crystallization technique. Drug Dev. Ind. Pharm. 2010, 36, 917–925. [Google Scholar] [CrossRef]
- Gallardo, D.; Skalsky, B.; Kleinebudde, P. Controlled release solid dosage forms using combinations of (meth) acrylate copolymers. Pharm. Dev. Technol. 2008, 13, 413–423. [Google Scholar] [CrossRef]
- Mehta, K.A.; Kislalioglu, M.S.; Phuapradit, W.; Malick, A.W.; Shah, N.H. Release performance of a poorly soluble drug from a novel, Eudragit®-based multi-unit erosion matrix. Int. J. Pharm. 2001, 213, 7–12. [Google Scholar] [CrossRef]
- Römling, U. Molecular biology of cellulose production in bacteria. Res. Microbiol. 2002, 153, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Klemm, D.; Heublein, B.; Fink, H.-P.; Bohn, A. Cellulose: Fascinating Biopolymer and Sustainable Raw Material. Angew. Chem. Int. Ed. 2005, 44, 3358–3393. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; O’Donnell, K.P.; Keen, J.M.; Rickard, M.A.; McGinity, J.W.; Williams, R.O. A New Extrudable Form of Hypromellose: AFFINISOL™ HPMC HME. AAPS PharmSciTech 2016, 17, 106–119. [Google Scholar] [CrossRef] [PubMed]
- Curatolo, W.; Nightingale, J.A.; Herbig, S.M. Utility of Hydroxypropylmethylcellulose Acetate Succinate (HPMCAS) for Initiation and Maintenance of Drug Supersaturation in the GI Milieu. Pharm. Res. 2009, 26, 1419–1431. [Google Scholar] [CrossRef]
- Ueda, K.; Higashi, K.; Yamamoto, K.; Moribe, K. The effect of HPMCAS functional groups on drug crystallization from the supersaturated state and dissolution improvement. Int. J. Pharm. 2014, 464, 205–213. [Google Scholar] [CrossRef]
- Petruševska, M.; Homar, M.; Petek, B.; Resman, A.; Kocjan, D.; Urleb, U.; Peternel, L. Hydroxypropyl Methylcellulose Mediated Precipitation Inhibition of Sirolimus: From a Screening Campaign to a Proof-of-Concept Human Study. Mol. Pharm. 2013, 10, 2299–2310. [Google Scholar] [CrossRef]
- Friesen, D.T.; Shanker, R.; Crew, M.; Smithey, D.T.; Curatolo, W.J.; Nightingale, J.A.S. Hydroxypropyl Methylcellulose Acetate Succinate-Based Spray-Dried Dispersions: An Overview. Mol. Pharm. 2008, 5, 1003–1019. [Google Scholar] [CrossRef]
- Doelker, E. Cellulose derivatives. In Biopolymers I; Springer: Berlin/Heidelberg, Germany, 1993; pp. 199–265. [Google Scholar]
- Knopp, M.M.; Chourak, N.; Khan, F.; Wendelboe, J.; Langguth, P.; Rades, T.; Holm, R. Effect of polymer type and drug dose on the in vitro and in vivo behavior of amorphous solid dispersions. Eur. J. Pharm. Biopharm. 2016, 105, 106–114. [Google Scholar] [CrossRef]
- O’Donnell, K.P.; Woodward, W.H.H. Dielectric spectroscopy for the determination of the glass transition temperature of pharmaceutical solid dispersions. Drug Dev. Ind. Pharm. 2015, 41, 959–968. [Google Scholar] [CrossRef]
- Yamashita, K.; Nakate, T.; Okimoto, K.; Ohike, A.; Tokunaga, Y.; Ibuki, R.; Higaki, K.; Kimura, T. Establishment of new preparation method for solid dispersion formulation of tacrolimus. Int. J. Pharm. 2003, 267, 79–91. [Google Scholar] [CrossRef]
- Gupta, P.; Kakumanu, V.K.; Bansal, A.K. Stability and Solubility of Celecoxib-PVP Amorphous Dispersions: A Molecular Perspective. Pharm. Res. 2004, 21, 1762–1769. [Google Scholar] [CrossRef] [PubMed]
- Chiou, W.L.; Riegelman, S. Oral Absorption of Griseofulvin in Dogs: Increased Absorption via Solid Dispersion—In Polyethylene Glycol 6000. J. Pharm. Sci. 1970, 59, 937–942. [Google Scholar] [CrossRef] [PubMed]
- Corrie, L.; Ajjarapu, S.; Banda, S.; Parvathaneni, M.; Bolla, P.K.; Kommineni, N. HPMCAS-Based Amorphous Solid Dispersions in Clinic: A Review on Manufacturing Techniques (Hot Melt Extrusion and Spray Drying), Marketed Products and Patents. Materials 2023, 16, 6616. [Google Scholar] [CrossRef]
- Shah, N.; Iyer, R.M.; Mair, H.-J.; Choi, D.; Tian, H.; Diodone, R.; Fahnrich, K.; Pabst-Ravot, A.; Tang, K.; Scheubel, E.; et al. Improved Human Bioavailability of Vemurafenib, a Practically Insoluble Drug, Using an Amorphous Polymer-Stabilized Solid Dispersion Prepared by a Solvent-Controlled Coprecipitation Process. J. Pharm. Sci. 2013, 102, 967–981. [Google Scholar] [CrossRef]
- Jung, J.-Y.; Yoo, S.D.; Lee, S.-H.; Kim, K.-H.; Yoon, D.-S.; Lee, K.-H. Enhanced solubility and dissolution rate of itraconazole by a solid dispersion technique. Int. J. Pharm. 1999, 187, 209–218. [Google Scholar] [CrossRef]
- Hasnain, M.S.; Nayak, A.K. Solubility and dissolution enhancement of ibuprofen by solid dispersion technique using PEG 6000-PVP K 30 combination carrier. J. Chem. 2012, 21, 118–132. [Google Scholar]
- Porter, C.J.H.; Trevaskis, N.L.; Charman, W.N. Lipids and lipid-based formulations: Optimizing the oral delivery of lipophilic drugs. Nat. Rev. Drug Discov. 2007, 6, 231–248. [Google Scholar] [CrossRef]
- Jannin, V.; Musakhanian, J.; Marchaud, D. Approaches for the development of solid and semi-solid lipid-based formulations. Adv. Drug Deliv. Rev. 2008, 60, 734–746. [Google Scholar] [CrossRef]
- Dahan, A.; Hoffman, A. Rationalizing the selection of oral lipid based drug delivery systems by an in vitro dynamic lipolysis model for improved oral bioavailability of poorly water soluble drugs. J. Control. Release 2008, 129, 1–10. [Google Scholar] [CrossRef]
- Yeap, Y.Y.; Trevaskis, N.L.; Porter, C.J.H. Lipid Absorption Triggers Drug Supersaturation at the Intestinal Unstirred Water Layer and Promotes Drug Absorption from Mixed Micelles. Pharm. Res. 2013, 30, 3045–3058. [Google Scholar] [CrossRef]
- Li, L.; Yi, T.; Lam, C.W.-K. Interactions between human multidrug resistance related protein (MRP2; ABCC2) and excipients commonly used in self-emulsifying drug delivery systems (SEDDS). Int. J. Pharm. 2013, 447, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Werle, M. Natural and Synthetic Polymers as Inhibitors of Drug Efflux Pumps. Pharm. Res. 2008, 25, 500–511. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Zhang, H.; Wang, X.; He, S.; Zhai, G. Development and evaluation of ibuprofen loaded mixed micelles preparations for topical delivery. J. Drug Deliv. Sci. Technol. 2018, 48, 363–371. [Google Scholar] [CrossRef]
- Nanjwade, B.K.; Patel, D.J.; Udhani, R.A.; Manvi, F.V. Functions of Lipids for Enhancement of Oral Bioavailability of Poorly Water-Soluble Drugs. Sci. Pharm. 2011, 79, 705–728. [Google Scholar] [CrossRef]
- Kim, J.Y.; Ku, Y.S. Enhanced absorption of indomethacin after oral or rectal administration of a self-emulsifying system containing indomethacin to rats. Int. J. Pharm. 2000, 194, 81–89. [Google Scholar] [CrossRef]
- Menzel, C.; Holzeisen, T.; Laffleur, F.; Zaichik, S.; Abdulkarim, M.; Gumbleton, M.; Bernkop-Schnürch, A. In vivo evaluation of an oral self-emulsifying drug delivery system (SEDDS) for exenatide. J. Control. Release 2018, 277, 165–172. [Google Scholar] [CrossRef]
- Prince, L. Microemulsions Theory and Practice; Elsevier: Amsterdam, The Netherlands, 2012. [Google Scholar]
- Damasceno, B.P.G.L.; Dominici, V.A.; Urbano, I.A.; Silva, J.A.; Araújo, I.B.; Santos-Magalhães, N.S.; Silva, A.K.A.; Medeiros, A.C.; Oliveira, A.G.; Egito, E.S.T. Amphotericin B Microemulsion Reduces Toxicity and Maintains the Efficacy as an Antifungal Product. J. Biomed. Nanotechnol. 2012, 8, 290–300. [Google Scholar] [CrossRef]
- Date, A.A.; Nagarsenker, M.S. Parenteral microemulsions: An overview. Int. J. Pharm. 2008, 355, 19–30. [Google Scholar] [CrossRef]
- Gatto, H.; Rème, C.; Kennedy, J.F. Designing a New Range of Topical Products: The Allermyl® Story; VIRBAC Labo: Carros, France, 2005. [Google Scholar]
- van Doren, H.A.; Smits, E.; Pestman, J.M.; Engberts, J.B.; Kellogg, R.M. Mesogenic sugars. From aldoses to liquid crystals and surfactants. Chem. Soc. Rev. 2000, 29, 183–199. [Google Scholar] [CrossRef]
- Yalavarthi, P.; Prasanna, Y.; Basaveswara, M.; Sundaresan, C. Insights of microemulsions—A thermodynamic comprehension. Jordan J. Pharm. Sci. 2017, 10, 23–40. [Google Scholar] [CrossRef]
- Djekic, L.; Primorac, M. The influence of cosurfactants and oils on the formation of pharmaceutical microemulsions based on PEG-8 caprylic/capric glycerides. Int. J. Pharm. 2008, 352, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, J.; Kortegaard, K.; Neil Pinder, D.; Rades, T.; Singh, H. Solubilisation of soybean oil in microemulsions using various surfactants. Food Hydrocoll. 2006, 20, 253–260. [Google Scholar] [CrossRef]
- Gibaud, S.; Attivi, D. Microemulsions for oral administration and their therapeutic applications. Expert Opin. Drug Deliv. 2012, 9, 937–951. [Google Scholar] [CrossRef] [PubMed]
- Egito, E.S.T.; Amaral-Machado, L.; Alencar, E.N.; Oliveira, A.G. Microemulsion systems: From the design and architecture to the building of a new delivery system for multiple-route drug delivery. Drug Deliv. Transl. Res. 2021, 11, 2108–2133. [Google Scholar] [CrossRef]
- Tokumura, T.; Tsushima, Y.; Tatsuishi, K.; Kayano, M.; Machida, Y.; Nagai, T. Enhancement of the Oral Bioavailability of Cinnarizine in Oleic Acid in Beagle Dogs. J. Pharm. Sci. 1987, 76, 286–288. [Google Scholar] [CrossRef]
- Dahan, A.; Hoffman, A. Use of a Dynamic in Vitro Lipolysis Model to Rationalize Oral Formulation Development for Poor Water Soluble Drugs: Correlation with in Vivo Data and the Relationship to Intra-Enterocyte Processes in Rats. Pharm. Res. 2006, 23, 2165–2174. [Google Scholar] [CrossRef]
- Charman, W.N.; Rogge, M.C.; Boddy, A.W.; Berger, B.M. Effect of Food and a Monoglyceride Emulsion Formulation on Danazol Bioavailability. J. Clin. Pharmacol. 1993, 33, 381–386. [Google Scholar] [CrossRef]
- Ghosh, P.K.; Majithiya, R.J.; Umrethia, M.L.; Murthy, R.S.R. Design and development of microemulsion drug delivery system of acyclovir for improvement of oral bioavailability. AAPS PharmSciTech 2017, 7, 77. [Google Scholar] [CrossRef]
- Kang, B.K.; Lee, J.S.; Chon, S.K.; Jeong, S.Y.; Yuk, S.H.; Khang, G.; Lee, H.B.; Cho, S.H. Development of self-microemulsifying drug delivery systems (SMEDDS) for oral bioavailability enhancement of simvastatin in beagle dogs. Int. J. Pharm. 2004, 274, 65–73. [Google Scholar] [CrossRef]
- Wei, L.; Sun, P.; Nie, S.; Pan, W. Preparation and Evaluation of SEDDS and SMEDDS Containing Carvedilol. Drug Dev. Ind. Pharm. 2005, 31, 785–794. [Google Scholar] [CrossRef]
- Bongioanni, A.; Bueno, M.S.; Mezzano, B.A.; Longhi, M.R.; Garnero, C. Amino acids and its pharmaceutical applications: A mini review. Int. J. Pharm. 2022, 613, 121375. [Google Scholar] [CrossRef] [PubMed]
- Tilborg, A.; Norberg, B.; Wouters, J. Pharmaceutical salts and cocrystals involving amino acids: A brief structural overview of the state-of-art. Eur. J. Med. Chem. 2014, 74, 411–426. [Google Scholar] [CrossRef] [PubMed]
- Kamei, N.; Khafagy, E.-S.; Hirose, J.; Takeda-Morishita, M. Potential of single cationic amino acid molecule “Arginine” for stimulating oral absorption of insulin. Int. J. Pharm. 2017, 521, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Jensen, K.T.; Löbmann, K.; Rades, T.; Grohganz, H. Improving Co-Amorphous Drug Formulations by the Addition of the Highly Water Soluble Amino Acid, Proline. Pharmaceutics 2014, 6, 416–435. [Google Scholar] [CrossRef]
- Kasten, G.; Löbmann, K.; Grohganz, H.; Rades, T. Co-former selection for co-amorphous drug-amino acid formulations. Int. J. Pharm. 2019, 557, 366–373. [Google Scholar] [CrossRef]
- Vong, L.B.; Trinh, N.-T.; Nagasaki, Y. Design of amino acid-based self-assembled nano-drugs for therapeutic applications. J. Control. Release 2020, 326, 140–149. [Google Scholar] [CrossRef]
- Alsalhi, M.S.; Royall, P.G.; Chan, K.L.A. Mechanistic study of the solubilization effect of basic amino acids on a poorly water-soluble drug. RSC Adv. 2022, 12, 19040–19053. [Google Scholar] [CrossRef]
- Valizadeh, H.; Mahdinloo, S.; Zakeri, N.; Sarfraz, M.; Nezafat, S.; Zakeri-Milani, P. Investigating the Effect of Basic Amino Acids and Glucosamine on the Solubility of Ibuprofen and Piroxicam. Adv. Pharm. Bull. 2023, 13, 532–538. [Google Scholar] [CrossRef]
- Shoaib, A.; Mangla, B.; Javed, S.; Sultan, M.H.; Alqahtani, S.S.; Shakeel, F. Vicissitudes of liquid crystals for solubility enhancement of poorly soluble drugs. J. Mol. Liq. 2021, 321, 114924. [Google Scholar] [CrossRef]
- Miranda, J.A.; Garnero, C.; Zoppi, A.; Sterren, V.; Ayala, A.P.; Longhi, M.R. Characterization of systems with amino-acids and oligosaccharides as modifiers of biopharmaceutical properties of furosemide. J. Pharm. Biomed. Anal. 2018, 149, 143–150. [Google Scholar] [CrossRef]
- Grohganz, H.; Priemel, P.A.; Löbmann, K.; Nielsen, L.H.; Laitinen, R.; Mullertz, A.; Van den Mooter, G.; Rades, T. Refining stability and dissolution rate of amorphous drug formulations. Expert Opin. Drug Deliv. 2014, 11, 977–989. [Google Scholar] [CrossRef] [PubMed]
- ElShaer, A.; Khan, S.; Perumal, D.; Hanson, P.; Mohammed, A.R. Use of Amino Acids as Counterions Improves the Solubility of the BCS II Model Drug, Indomethacin. Curr. Drug Deliv. 2011, 8, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.; Wang, Y.W. Initial Salt Screening Procedures for Manufacturing Ibuprofen. Drug Dev. Ind. Pharm. 2009, 35, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Abdelkader, H.; Fathalla, Z. Investigation into the Emerging Role of the Basic Amino Acid L-Lysine in Enhancing Solubility and Permeability of BCS Class II and BCS Class IV Drugs. Pharm. Res. 2018, 35, 160. [Google Scholar] [CrossRef]
- Abou-Taleb, H.A.; Fathalla, Z.; Abdelkader, H. Comparative studies of the effects of novel excipients amino acids with cyclodextrins on enhancement of dissolution and oral bioavailability of the non-ionizable drug carbamazepine. Eur. J. Pharm. Sci. 2020, 155, 105562. [Google Scholar] [CrossRef]
- Mura, P.; Maestrelli, F.; Cirri, M. Ternary systems of naproxen with hydroxypropyl-β-cyclodextrin and aminoacids. Int. J. Pharm. 2003, 260, 293–302. [Google Scholar] [CrossRef]
- Samiei, N.; Mangas-Sanjuan, V.; González-Álvarez, I.; Foroutan, M.; Shafaati, A.; Zarghi, A.; Bermejo, M. Ion-pair strategy for enabling amifostine oral absorption: Rat in situ and in vivo experiments. Eur. J. Pharm. Sci. 2013, 49, 499–504. [Google Scholar] [CrossRef]
- Ivaturi, V.D.; Kim, S.K. Enhanced permeation of methotrexate in vitro by ion pair formation with L-arginine. J. Pharm. Sci. 2009, 98, 3633–3639. [Google Scholar] [CrossRef]
- ElShaer, A.; Hanson, P.; Mohammed, A.R. A novel concentration dependent amino acid ion pair strategy to mediate drug permeation using indomethacin as a model insoluble drug. Eur. J. Pharm. Sci. 2014, 62, 124–131. [Google Scholar] [CrossRef]
- Iyire, A.; Alaayedi, M.; Mohammed, A.R. Pre-formulation and systematic evaluation of amino acid assisted permeability of insulin across in vitro buccal cell layers. Sci. Rep. 2016, 6, 32498. [Google Scholar] [CrossRef]
- Gunnam, A.; Nangia, A.K. Solubility improvement of curcumin with amino acids. CrystEngComm 2021, 23, 3398–3410. [Google Scholar] [CrossRef]
- Löbmann, K.; Laitinen, R.; Strachan, C.; Rades, T.; Grohganz, H. Amino acids as co-amorphous stabilizers for poorly water-soluble drugs—Part 2: Molecular interactions. Eur. J. Pharm. Biopharm. 2013, 85, 882–888. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Wu, J.; Yin, T. Solubility and permeation enhancement of poor soluble drug by cholinium-amino acid based ionic liquids. J. Drug Deliv. Sci. Technol. 2020, 60, 102037. [Google Scholar] [CrossRef]
- Huang, Y.; Zhang, Q.; Wang, J.-R.; Lin, K.-L.; Mei, X. Amino acids as co-amorphous excipients for tackling the poor aqueous solubility of valsartan. Pharm. Dev. Technol. 2017, 22, 69–76. [Google Scholar] [CrossRef]
- Garbiec, E.; Rosiak, N.; Zalewski, P.; Tajber, L.; Cielecka-Piontek, J. Genistein Co-Amorphous Systems with Amino Acids: An Investigation into Enhanced Solubility and Biological Activity. Pharmaceutics 2023, 15, 2653. [Google Scholar] [CrossRef]
- Khanfar, M.; Al-Remawi, M.; Al-Akayleh, F.; Hmouze, S. Preparation and Evaluation of Co-amorphous Formulations of Telmisartan—Amino Acids as a Potential Method for Solubility and Dissolution Enhancement. AAPS PharmSciTech 2021, 22, 112. [Google Scholar] [CrossRef]
- Mishra, J.; Löbmann, K.; Grohganz, H.; Rades, T. Influence of preparation technique on co-amorphization of carvedilol with acidic amino acids. Int. J. Pharm. 2018, 552, 407–413. [Google Scholar] [CrossRef]
- Sormunen, H.; Ruponen, M.; Laitinen, R. The effect of co-amorphization of glibenclamide on its dissolution properties and permeability through an MDCKII-MDR1 cell layer. Int. J. Pharm. 2019, 570, 118653. [Google Scholar] [CrossRef]
- Wu, W.; Grohganz, H.; Rades, T.; Löbmann, K. Comparison of co-former performance in co-amorphous formulations: Single amino acids, amino acid physical mixtures, amino acid salts and dipeptides as co-formers. Eur. J. Pharm. Sci. 2021, 156, 105582. [Google Scholar] [CrossRef]
- Bongioanni, A.; Araújo, B.S.; de Oliveira, Y.S.; Longhi, M.R.; Ayala, A.; Garnero, C. Improving Properties of Albendazole Desmotropes by Supramolecular Systems with Maltodextrin and Glutamic Acid. AAPS PharmSciTech 2018, 19, 1468–1476. [Google Scholar] [CrossRef]
- França, M.T.; Marcos, T.M.; Pereira, R.N.; Stulzer, H.K. Could the small molecules such as amino acids improve aqueous solubility and stabilize amorphous systems containing Griseofulvin? Eur. J. Pharm. Sci. 2020, 143, 105178. [Google Scholar] [CrossRef] [PubMed]
- Abdelkader, H.; Fatease, A.A.; Fathalla, Z.; Shoman, M.E.; Abou-Taleb, H.A. Meloxicam-amino acids salts/ion pair complexes with advanced solubility, dissolution, and gastric safety. Pharm. Dev. Technol. 2024, 29, 1075–1083. [Google Scholar] [CrossRef] [PubMed]
- Fatease, A.A.; Shoman, M.E.; Abourehab, M.A.S.; Abou-Taleb, H.A.; Abdelkader, H. A Novel Curcumin Arginine Salt: A Solution for Poor Solubility and Potential Anticancer Activities. Molecules 2023, 28, 262. [Google Scholar] [CrossRef] [PubMed]
- Abou-Taleb, H.A.; Shoman, M.E.; Makram, T.S.; Abdel-Aleem, J.A.; Abdelkader, H. Exploration of the Safety and Solubilization, Dissolution, Analgesic Effects of Common Basic Excipients on the NSAID Drug Ketoprofen. Pharmaceutics 2023, 15, 713. [Google Scholar] [CrossRef]
- Mennini, N.; Maestrelli, F.; Cirri, M.; Mura, P. Analysis of physicochemical properties of ternary systems of oxaprozin with randomly methylated-ß-cyclodextrin and l-arginine aimed to improve the drug solubility. J. Pharm. Biomed. Anal. 2016, 129, 350–358. [Google Scholar] [CrossRef]
- Bilensoy, E. Cyclodextrins in Pharmaceutics, Cosmetics, and Biomedicine: Current and Future Industrial Applications; John Wiley & Sons: Hoboken, NJ, USA, 2011. [Google Scholar]
- Loftsson, T.; Brewster, M.E. Cyclodextrins as Functional Excipients: Methods to Enhance Complexation Efficiency. J. Pharm. Sci. 2012, 101, 3019–3032. [Google Scholar] [CrossRef]
- Schlüter, F.; Bela, M.M.; Glikman, D.; Braunschweig, B.; Ravoo, B.J. A cyclodextrin surfactant for stable emulsions with an accessible cavity for host–guest complexation. Chem. Commun. 2020, 56, 15434–15437. [Google Scholar] [CrossRef]
- Hoang Thi, T.T.; Lee, Y.; Ryu, S.B.; Sung, H.J.; Park, K.D. Oxidized cyclodextrin-functionalized injectable gelatin hydrogels as a new platform for tissue-adhesive hydrophobic drug delivery. RSC Adv. 2017, 7, 34053–34062. [Google Scholar] [CrossRef]
- Kasal, P.; Jindřich, J. Mono-6-Substituted Cyclodextrins—Synthesis and Applications. Molecules 2021, 26, 5065. [Google Scholar] [CrossRef]
- Bálint, M.; Darcsi, A.; Benkovics, G.; Varga, E.; Malanga, M.; Béni, S. Synthesis of the chiral selector heptakis(6-O-methyl)-β-cyclodextrin by phase-transfer catalysis and hydrazine-mediated transfer-hydrogenation. Electrophoresis 2019, 40, 1941–1950. [Google Scholar] [CrossRef]
- Kurkov, S.V.; Loftsson, T. Cyclodextrins. Int. J. Pharm. 2013, 453, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Adeoye, O.; Cabral-Marques, H. Cyclodextrin nanosystems in oral drug delivery: A mini review. Int. J. Pharm. 2017, 531, 521–531. [Google Scholar] [CrossRef] [PubMed]
- Lantz, A.W.; Rodriguez, M.A.; Wetterer, S.M.; Armstrong, D.W. Estimation of association constants between oral malodor components and various native and derivatized cyclodextrins. Anal. Chim. Acta 2006, 557, 184–190. [Google Scholar] [CrossRef]
- Vyas, A.; Saraf, S.; Saraf, S. Cyclodextrin based novel drug delivery systems. J. Incl. Phenom. Macrocycl. Chem. 2008, 62, 23–42. [Google Scholar] [CrossRef]
- Stella, V.J.; He, Q. Cyclodextrins. Toxicol. Pathol. 2008, 36, 30–42. [Google Scholar] [CrossRef]
- Dhiman, P.; Bhatia, M. Pharmaceutical applications of cyclodextrins and their derivatives. J. Incl. Phenom. Macrocycl. Chem. 2020, 98, 171–186. [Google Scholar] [CrossRef]
- Ferreira, L.; Campos, J.; Veiga, F.; Cardoso, C.; Paiva-Santos, A.C. Cyclodextrin-based delivery systems in parenteral formulations: A critical update review. Eur. J. Pharm. Biopharm. 2022, 178, 35–52. [Google Scholar] [CrossRef]
- Davis, M.E.; Brewster, M.E. Cyclodextrin-based pharmaceutics: Past, present and future. Nat. Rev. Drug Discov. 2004, 3, 1023–1035. [Google Scholar] [CrossRef]
- Serno, T.; Geidobler, R.; Winter, G. Protein stabilization by cyclodextrins in the liquid and dried state. Adv. Drug Deliv. Rev. 2011, 63, 1086–1106. [Google Scholar] [CrossRef]
- Strickley, R.G. Solubilizing Excipients in Oral and Injectable Formulations. Pharm. Res. 2004, 21, 201–230. [Google Scholar] [CrossRef]
- Shelley, H.; Babu, R.J. Role of Cyclodextrins in Nanoparticle-Based Drug Delivery Systems. J. Pharm. Sci. 2018, 107, 1741–1753. [Google Scholar] [CrossRef] [PubMed]
- Fülöp, Z.; Kurkov, S.V.; Nielsen, T.T.; Larsen, K.L.; Loftsson, T. Self-assembly of cyclodextrins: Formation of cyclodextrin polymer based nanoparticles. J. Drug Deliv. Sci. Technol. 2012, 22, 215–221. [Google Scholar] [CrossRef]
- Roy, M.N.; Roy, A.; Saha, S. Probing inclusion complexes of cyclodextrins with amino acids by physicochemical approach. Carbohydr. Polym. 2016, 151, 458–466. [Google Scholar] [CrossRef] [PubMed]
- Rey-Rico, A.; Cucchiarini, M. Supramolecular Cyclodextrin-Based Hydrogels for Controlled Gene Delivery. Polymers 2019, 11, 514. [Google Scholar] [CrossRef] [PubMed]
- Bai, H.; Wang, J.; Li, Z.; Tang, G. Macrocyclic Compounds for Drug and Gene Delivery in Immune-Modulating Therapy. Int. J. Mol. Sci. 2019, 20, 2097. [Google Scholar] [CrossRef]
- Zhang, J.; Ma, P.X. Host–guest interactions mediated nano-assemblies using cyclodextrin-containing hydrophilic polymers and their biomedical applications. Nano Today 2010, 5, 337–350. [Google Scholar] [CrossRef]
- Liu, M.; Cao, W.; Sun, Y.; He, Z. Preparation, characterization and in vivo evaluation of formulation of repaglinide with hydroxypropyl-β-cyclodextrin. Int. J. Pharm. 2014, 477, 159–166. [Google Scholar] [CrossRef]
- Patel, S.G.; Rajput, S.J. Enhancement of oral bioavailability of cilostazol by forming its inclusion complexes. AAPS PharmSciTech 2009, 10, 660–669. [Google Scholar] [CrossRef]
- Chattah, A.K.; Pfund, L.Y.; Zoppi, A.; Longhi, M.R.; Garnero, C. Toward novel antiparasitic formulations: Complexes of Albendazole desmotropes and β-cyclodextrin. Carbohydr. Polym. 2017, 164, 379–385. [Google Scholar] [CrossRef]
- Doliwa, A.; Santoyo, S.; Ygartua, P. Transdermal lontophoresis and skin retention of piroxicam from gels containing piroxicam: Hydroxypropyl-beta-cyclodextrin complexes. Drug Dev. Ind. Pharm. 2001, 27, 751–758. [Google Scholar] [CrossRef]
- Wang, S.; Li, D.; Ito, Y.; Nabekura, T.; Wang, S.; Zhang, J.; Wu, C. Bioavailability and anticataract effects of a topical ocular drug delivery system containing disulfiram and hydroxypropyl-beta-cyclodextrin on selenite-treated rats. Curr. Eye Res. 2004, 29, 51–58. [Google Scholar] [CrossRef] [PubMed]
- García-Otero, X.; Díaz-Tomé, V.; Varela-Fernández, R.; Martín-Pastor, M.; González-Barcia, M.; Blanco-Méndez, J.; Mondelo-García, C.; Bermudez, M.A.; Gonzalez, F.; Aguiar, P.; et al. Development and Characterization of a Tacrolimus/Hydroxypropyl-β-Cyclodextrin Eye Drop. Pharmaceutics 2021, 13, 149. [Google Scholar] [CrossRef] [PubMed]
- Maria, D.N.; Mishra, S.R.; Wang, L.; Abd-Elgawad, A.-E.H.; Soliman, O.A.-E.; El-Dahan, M.S.; Jablonski, M.M. Water-soluble Complex of Curcumin with Cyclodextrins: Enhanced Physical Properties For Ocular Drug Delivery. Curr. Drug Deliv. 2017, 14, 875–886. [Google Scholar] [CrossRef] [PubMed]
- Shelley, H.; Grant, M.; Smith, F.T.; Abarca, E.M.; Jayachandra Babu, R. Improved Ocular Delivery of Nepafenac by Cyclodextrin Complexation. AAPS PharmSciTech 2018, 19, 2554–2563. [Google Scholar] [CrossRef]
- Zhang, D.; Sha, M.; Pan, R.; Lin, X.; Xing, P.; Jiang, B. Synthesis and properties study of novel fluorinated surfactants with perfluorinated branched ether chain. J. Fluor. Chem. 2019, 219, 62–69. [Google Scholar] [CrossRef]
- Hussain, S.M.S.; Kamal, M.S.; Fogang, L.T. Synthesis and physicochemical investigation of betaine type polyoxyethylene zwitterionic surfactants containing different ionic headgroups. J. Mol. Struct. 2019, 1178, 83–88. [Google Scholar] [CrossRef]
- Shaban, S.M.; Kang, J.; Kim, D.-H. Surfactants: Recent advances and their applications. Compos. Commun. 2020, 22, 100537. [Google Scholar] [CrossRef]
- Ainurofiq, A.; Putro, D.S.; Ramadhani, D.A.; Putra, G.M.; Do Espirito Santo, L.D.C. A Review on Solubility Enhancement Methods for Poorly Water-Soluble Drugs. J. Rep. Pharm. Sci. 2021, 10, 137–147. [Google Scholar] [CrossRef]
- Lu, Y.; Park, K. Polymeric micelles and alternative nanonized delivery vehicles for poorly soluble drugs. Int. J. Pharm. 2013, 453, 198–214. [Google Scholar] [CrossRef]
- Gaucher, G.; Dufresne, M.H.; Sant, V.P.; Kang, N.; Maysinger, D.; Leroux, J.C. Block copolymer micelles: Preparation, characterization and application in drug delivery. J. Control. Release 2005, 109, 169–188. [Google Scholar] [CrossRef]
- Dun, J.; Osei-Yeboah, F.; Boulas, P.; Lin, Y.; Sun, C.C. A systematic evaluation of dual functionality of sodium lauryl sulfate as a tablet lubricant and wetting enhancer. Int. J. Pharm. 2018, 552, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh, M.N.; Shayanfar, A.; Jouyban, A. Solubilization of drugs using sodium lauryl sulfate: Experimental data and modeling. J. Mol. Liq. 2018, 268, 410–414. [Google Scholar] [CrossRef]
- Dave, R.H.; Patel, H.H.; Donahue, E.; Patel, A.D. To evaluate the change in release from solid dispersion using sodium lauryl sulfate and model drug sulfathiazole. Drug Dev. Ind. Pharm. 2013, 39, 1562–1572. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, S.P.; Dugar, R.P. Application of surfactants in solid dispersion technology for improving solubility of poorly water soluble drugs. J. Drug Deliv. Sci. Technol. 2017, 41, 68–77. [Google Scholar] [CrossRef]
- Lang, B.; McGinity, J.W.; Williams, R.O., III. Dissolution Enhancement of Itraconazole by Hot-Melt Extrusion Alone and the Combination of Hot-Melt Extrusion and Rapid Freezing—Effect of Formulation and Processing Variables. Mol. Pharm. 2014, 11, 186–196. [Google Scholar] [CrossRef]
- Pouton, C.W. Formulation of poorly water-soluble drugs for oral administration: Physicochemical and physiological issues and the lipid formulation classification system. Eur. J. Pharm. Sci. 2006, 29, 278–287. [Google Scholar] [CrossRef]
- Liu, C.; Chen, Z.; Chen, Y.; Lu, J.; Li, Y.; Wang, S.; Wu, G.; Qian, F. Improving Oral Bioavailability of Sorafenib by Optimizing the “Spring” and “Parachute” Based on Molecular Interaction Mechanisms. Mol. Pharm. 2016, 13, 599–608. [Google Scholar] [CrossRef]
- Medarević, D.P.; Kachrimanis, K.; Mitrić, M.; Djuriš, J.; Djurić, Z.; Ibrić, S. Dissolution rate enhancement and physicochemical characterization of carbamazepine-poloxamer solid dispersions. Pharm. Dev. Technol. 2016, 21, 268–276. [Google Scholar] [CrossRef]
- Morris, K.R.; Knipp, G.T.; Serajuddin, A.T.M. Structural Properties of Polyethylene Glycol-Polysorbate 80 Mixture, a Solid Dispersion Vehicle. J. Pharm. Sci. 1992, 81, 1185–1188. [Google Scholar] [CrossRef]
- Serajuddin, A.T.; Sheen, P.-C.; Augustine, M.A. Improved Dissolution of a Poorly Water-Soluble Drug from Solid Dispersions in Polyethylene Glyco1: Polysorbate 80 Mixtures. J. Pharm. Sci. 1990, 79, 463–464. [Google Scholar] [CrossRef]
- Law, S.L.; Lo, W.Y.; Lin, F.M.; Chaing, C.H. Dissolution and absorption of nifedipine in polyethylene glycol solid dispersion containing phosphatidylcholine. Int. J. Pharm. 1992, 84, 161–166. [Google Scholar] [CrossRef]
- Rege, B.D.; Yu, L.X.; Hussain, A.S.; Polli, J.E. Effect of common excipients on Caco-2 transport of low-permeability drugs. J. Pharm. Sci. 2001, 90, 1776–1786. [Google Scholar] [CrossRef] [PubMed]
- Varma, M.V.; Panchagnula, R. Enhanced oral paclitaxel absorption with vitamin E-TPGS: Effect on solubility and permeability in vitro, in situ and in vivo. Eur. J. Pharm. 2005, 25, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, S.; Wang, S.; Liu, C.; Su, C.; Hageman, M.; Hussain, M.; Haskell, R.; Stefanski, K.; Qian, F. Sodium Lauryl Sulfate Competitively Interacts with HPMC-AS and Consequently Reduces Oral Bioavailability of Posaconazole/HPMC-AS Amorphous Solid Dispersion. Mol. Pharm. 2016, 13, 2787–2795. [Google Scholar] [CrossRef]
- Rashid, R.; Kim, D.W.; Din, F.u.; Mustapha, O.; Yousaf, A.M.; Park, J.H.; Kim, J.O.; Yong, C.S.; Choi, H.-G. Effect of hydroxypropylcellulose and Tween 80 on physicochemical properties and bioavailability of ezetimibe-loaded solid dispersion. Carbohydr. Polym. 2015, 130, 26–31. [Google Scholar] [CrossRef]
- Sinha, S.; Ali, M.; Baboota, S.; Ahuja, A.; Kumar, A.; Ali, J. Solid Dispersion as an Approach for Bioavailability Enhancement of Poorly Water-Soluble Drug Ritonavir. AAPS PharmSciTech 2010, 11, 518–527. [Google Scholar] [CrossRef]
- Truong, D.H.; Tran, T.H.; Ramasamy, T.; Choi, J.Y.; Choi, H.-G.; Yong, C.S.; Kim, J.O. Preparation and characterization of solid dispersion using a novel amphiphilic copolymer to enhance dissolution and oral bioavailability of sorafenib. Powder Technol. 2015, 283, 260–265. [Google Scholar] [CrossRef]
- Newa, M.; Bhandari, K.H.; Oh, D.H.; Kim, Y.R.; Sung, J.H.; Kim, J.O.; Woo, J.S.; Choi, H.G.; Yong, C.S. Enhanced dissolution of ibuprofen using solid dispersion with poloxamer 407. Arch. Pharmacal Res. 2008, 31, 1497–1507. [Google Scholar] [CrossRef]
- Song, C.K.; Yoon, I.-S.; Kim, D.-D. Poloxamer-based solid dispersions for oral delivery of docetaxel: Differential effects of F68 and P85 on oral docetaxel bioavailability. Int. J. Pharm. 2016, 507, 102–108. [Google Scholar] [CrossRef]
- Ali, W.; Williams, A.C.; Rawlinson, C.F. Stochiometrically governed molecular interactions in drug: Poloxamer solid dispersions. Int. J. Pharm. 2010, 391, 162–168. [Google Scholar] [CrossRef]
- Kolašinac, N.; Kachrimanis, K.; Homšek, I.; Grujić, B.; Đurić, Z.; Ibrić, S. Solubility enhancement of desloratadine by solid dispersion in poloxamers. Int. J. Pharm. 2012, 436, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.-S.; Lee, S.-E.; Jang, W.S.; Byeon, J.C.; Park, J.-S. Solid dispersion of dutasteride using the solvent evaporation method: Approaches to improve dissolution rate and oral bioavailability in rats. Mater. Sci. Eng. C 2018, 90, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Luo, Z.; Chen, T.; Ouyang, Y.; Xiao, L.; Liang, S.; Peng, Z.; Liu, Y.; Deng, Y. Bioadhesive Nanoparticles for Local Drug Delivery. Int. J. Mol. Sci. 2022, 23, 2370. [Google Scholar] [CrossRef] [PubMed]
- Jawadi, Z.; Yang, C.; Haidar, Z.S.; Santa Maria, P.L.; Massa, S. Bio-Inspired Muco-Adhesive Polymers for Drug Delivery Applications. Polymers 2022, 14, 5459. [Google Scholar] [CrossRef]
- Ugoeze, K.C. Bioadhesive polymers for drug delivery applications. In Bioadhesives in Drug Delivery; John Wiley & Sons: Hoboken, NJ, USA, 2020; pp. 29–56. [Google Scholar]
- Asati, S.; Jain, S.; Choubey, A. Bioadhesive or mucoadhesive drug delivery system: A potential alternative to conventional therapy. J. Drug Deliv. Ther. 2019, 9, 858–867. [Google Scholar]
- Forooshani, P.K.; Lee, B.P. Recent approaches in designing bioadhesive materials inspired by mussel adhesive protein. J. Polym. Sci. Part A Polym. Chem. 2017, 55, 9–33. [Google Scholar] [CrossRef]
- Sogias, I.A.; Williams, A.C.; Khutoryanskiy, V.V. Why is Chitosan Mucoadhesive? Biomacromolecules 2008, 9, 1837–1842. [Google Scholar] [CrossRef]
- Lehr, C.-M.; Bouwstra, J.A.; Schacht, E.H.; Junginger, H.E. In vitro evaluation of mucoadhesive properties of chitosan and some other natural polymers. Int. J. Pharm. 1992, 78, 43–48. [Google Scholar] [CrossRef]
- He, P.; Davis, S.S.; Illum, L. In vitro evaluation of the mucoadhesive properties of chitosan microspheres. Int. J. Pharm. 1998, 166, 75–88. [Google Scholar] [CrossRef]
- Xu, J.; Soliman, G.M.; Barralet, J.; Cerruti, M. Mollusk Glue Inspired Mucoadhesives for Biomedical Applications. Langmuir 2012, 28, 14010–14017. [Google Scholar] [CrossRef]
- Kim, K.; Kim, K.; Ryu, J.H.; Lee, H. Chitosan-catechol: A polymer with long-lasting mucoadhesive properties. Biomaterials 2015, 52, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.P.; Dalsin, J.L.; Messersmith, P.B. Biomimetic adhesive polymers based on mussel adhesive proteins. In Biological Adhesives; Springer: Berlin/Heidelberg, Germany, 2006; pp. 257–278. [Google Scholar]
- Kesavan, K.; Nath, G.; Pandit, J.K. Sodium alginate based mucoadhesive system for gatifloxacin and its in vitro antibacterial activity. Sci. Pharm. 2010, 78, 941–957. [Google Scholar] [CrossRef] [PubMed]
- Davidovich-Pinhas, M.; Bianco-Peled, H. Alginate–PEGAc: A new mucoadhesive polymer. Acta Biomater. 2011, 7, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Zia, K.M.; Zia, F.; Zuber, M.; Rehman, S.; Ahmad, M.N. Alginate based polyurethanes: A review of recent advances and perspective. Int. J. Biol. Macromol. 2015, 79, 377–387. [Google Scholar] [CrossRef]
- Gupta, V.; Hwang, B.H.; Lee, J.; Anselmo, A.C.; Doshi, N.; Mitragotri, S. Mucoadhesive intestinal devices for oral delivery of salmon calcitonin. J. Control. Release 2013, 172, 753–762. [Google Scholar] [CrossRef]
- Takeuchi, H.; Matsui, Y.; Yamamoto, H.; Kawashima, Y. Mucoadhesive properties of carbopol or chitosan-coated liposomes and their effectiveness in the oral administration of calcitonin to rats. J. Control. Release 2003, 86, 235–242. [Google Scholar] [CrossRef]
- Jamal, A.; Shahzadi, L.; Ahtzaz, S.; Zahid, S.; Chaudhry, A.A.; Rehman, I.u.; Yar, M. Identification of anti-cancer potential of doxazocin: Loading into chitosan based biodegradable hydrogels for on-site delivery to treat cervical cancer. Mater. Sci. Eng. C 2018, 82, 102–109. [Google Scholar] [CrossRef]
- Wu, J.; Su, Z.-G.; Ma, G.-H. A thermo- and pH-sensitive hydrogel composed of quaternized chitosan/glycerophosphate. Int. J. Pharm. 2006, 315, 1–11. [Google Scholar] [CrossRef]
- Lajud, S.A.; Han, Z.; Chi, F.-L.; Gu, R.; Nagda, D.A.; Bezpalko, O.; Sanyal, S.; Bur, A.; Han, Z.; O’Malley, B.W.; et al. A regulated delivery system for inner ear drug application. J. Control. Release 2013, 166, 268–276. [Google Scholar] [CrossRef]
- Bahreini, E.; Aghaiypour, K.; Abbasalipourkabir, R.; Mokarram, A.R.; Goodarzi, M.T.; Saidijam, M. Preparation and nanoencapsulation of l-asparaginase II in chitosan-tripolyphosphate nanoparticles and in vitro release study. Nanoscale Res. Lett. 2014, 9, 340. [Google Scholar] [CrossRef]
- DeVolder, R.; Antoniadou, E.; Kong, H. Enzymatically cross-linked injectable alginate-g-pyrrole hydrogels for neovascularization. J. Control. Release 2013, 172, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Saralkar, P.; Dash, A.K. Alginate Nanoparticles Containing Curcumin and Resveratrol: Preparation, Characterization, and In Vitro Evaluation Against DU145 Prostate Cancer Cell Line. AAPS PharmSciTech 2017, 18, 2814–2823. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Liu, C.-J.; Zhuo, R.-X.; Cheng, S.-X. Alginate/CaCO3 Hybrid Nanoparticles for Efficient Codelivery of Antitumor Gene and Drug. Mol. Pharm. 2012, 9, 2887–2893. [Google Scholar] [CrossRef] [PubMed]
- Zhou, K.; Wang, X.; Chen, D.; Yuan, Y.; Wang, S.; Li, C.; Yan, Y.; Liu, Q.; Shao, L.; Huang, L.; et al. Enhanced Treatment Effects of Tilmicosin Against Staphylococcus aureus Cow Mastitis by Self-Assembly Sodium Alginate-Chitosan Nanogel. Pharmaceutics 2019, 11, 524. [Google Scholar] [CrossRef]
- Zhou, M.; Hu, Q.; Wang, T.; Xue, J.; Luo, Y. Alginate hydrogel beads as a carrier of low density lipoprotein/pectin nanogels for potential oral delivery applications. Int. J. Biol. Macromol. 2018, 120, 859–864. [Google Scholar] [CrossRef]
- Ahadi, F.; Khorshidi, S.; Karkhaneh, A. A hydrogel/fiber scaffold based on silk fibroin/oxidized pectin with sustainable release of vancomycin hydrochloride. Eur. Polym. J. 2019, 118, 265–274. [Google Scholar] [CrossRef]
- Sarwar, M.S.; Ghaffar, A.; Huang, Q.; Zafar, M.S.; Usman, M.; Latif, M. Controlled-release behavior of ciprofloxacin from a biocompatible polymeric system based on sodium alginate/poly(ethylene glycol) mono methyl ether. Int. J. Biol. Macromol. 2020, 165, 1047–1054. [Google Scholar] [CrossRef]
- Ohta, S.; Nitta, N.; Sonoda, A.; Seko, A.; Tanaka, T.; Takahashi, M.; Takemura, S.; Tabata, Y.; Murata, K. Prolonged local persistence of cisplatin-loaded gelatin microspheres and their chemoembolic anti-cancer effect in rabbits. Eur. J. Radiol. 2009, 72, 534–540. [Google Scholar] [CrossRef]
- Choonara, B.F.; Choonara, Y.E.; Kumar, P.; Bijukumar, D.; du Toit, L.C.; Pillay, V. A review of advanced oral drug delivery technologies facilitating the protection and absorption of protein and peptide molecules. Biotechnol. Adv. 2014, 32, 1269–1282. [Google Scholar] [CrossRef]
- Maestrelli, F.; Zerrouk, N.; Chemtob, C.; Mura, P. Influence of chitosan and its glutamate and hydrochloride salts on naproxen dissolution rate and permeation across Caco-2 cells. Int. J. Pharm. 2004, 271, 257–267. [Google Scholar] [CrossRef]
- Cirri, M.; Maestrelli, F.; Mennini, N.; Mura, P. Combined use of bile acids and aminoacids to improve permeation properties of acyclovir. Int. J. Pharm. 2015, 490, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.K.; Park, K.-S.; Yeo, W.-S.; Choo, H.; Chong, Y. In vitro solubility, stability and permeability of novel quercetin–amino acid conjugates. Bioorg. Med. Chem. 2009, 17, 1164–1171. [Google Scholar] [CrossRef] [PubMed]
- Landowski, C.P.; Song, X.; Lorenzi, P.L.; Hilfinger, J.M.; Amidon, G.L. Floxuridine Amino Acid Ester Prodrugs: Enhancing Caco-2 Permeability and Resistance to Glycosidic Bond Metabolism. Pharm. Res. 2005, 22, 1510–1518. [Google Scholar] [CrossRef] [PubMed]
- ElShaer, A.; Hanson, P.; Worthington, T.; Lambert, P.; Mohammed, A.R. Preparation and Characterization of Amino Acids-Based Trimethoprim Salts. Pharmaceutics 2012, 4, 179–196. [Google Scholar] [CrossRef]
- Latif, S.; Ijaz, Q.A.; Hameed, M.; Shoaib, Q.-u.-a.; Fatima, K.; Hussain, A.; Arshad, M.S.; Abbas, N. Improvement of Physico-mechanical and pharmacokinetic attributes of naproxen by cocrystallization with l-alanine. J. Drug Deliv. Sci. Technol. 2021, 61, 102236. [Google Scholar] [CrossRef]
- Shete, A.; Murthy, S.; Korpale, S.; Yadav, A.; Sajane, S.; Sakhare, S.; Doijad, R. Cocrystals of itraconazole with amino acids: Screening, synthesis, solid state characterization, in vitro drug release and antifungal activity. J. Drug Deliv. Sci. Technol. 2015, 28, 46–55. [Google Scholar] [CrossRef]
- Wu, W.; Dong, Y.; Gao, J.; Gong, M.; Zhang, X.; Kong, W.; Li, Y.; Zeng, Y.; Si, D.; Wei, Z.; et al. Aspartate-modified doxorubicin on its N-terminal increases drug accumulation in 1-overexpressing tumors. Cancer Sci. 2015, 106, 747–756. [Google Scholar] [CrossRef]
- Türker, S.; Onur, E.; Ozer, Y. Nasal route and drug delivery systems. Pharm. World Sci. 2004, 26, 137–142. [Google Scholar] [CrossRef]
- Marttin, E.; Verhoef, J.C.; Merkus, F.W. Efficacy, safety and mechanism of cyclodextrins as absorption enhancers in nasal delivery of peptide and protein drugs. J. Drug Target 1998, 6, 17–36. [Google Scholar] [CrossRef]
- Merkus, F.W.; Verhoef, J.C.; Marttin, E.; Romeijn, S.G.; van der Kuy, P.H.; Hermens, W.A.; Schipper, N.G. Cyclodextrins in nasal drug delivery. Adv. Drug Deliv. Rev. 1999, 36, 41–57. [Google Scholar] [CrossRef]
- Schwarz, D.H.; Engelke, A.; Wenz, G. Solubilizing steroidal drugs by β-cyclodextrin derivatives. Int. J. Pharm. 2017, 531, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Kristinsson, J.K.; Fridriksdóttir, H.; Thórisdóttir, S.; Sigurdardóttir, A.M.; Stefánsson, E.; Loftsson, T. Dexamethasone-cyclodextrin-polymer co-complexes in aqueous eye drops. Aqueous humor pharmacokinetics in humans. Investig. Ophthalmol. Vis. Sci. 1996, 37, 1199–1203. [Google Scholar]
- Fridriksdóttir, H.; Loftsson, T.; Stefánsson, E. Formulation and testing of methazolamide cyclodextrin eye drop solutions. J. Control. Release 1997, 44, 95–99. [Google Scholar] [CrossRef]
- Tiwari, G.; Tiwari, R.; Rai, A.K. Cyclodextrins in delivery systems: Applications. J. Pharm. Bioallied Sci. 2010, 2, 72–79. [Google Scholar] [CrossRef]
- Uekama, K.; Masaki, K.; Arimori, K.; Irie, T.; Hirayama, F. Effects of beta- and dimethyl beta-cyclodextrins on release and percutaneous absorption behaviors of prednisolone from some ointment bases. Yakugaku Zasshi J. Pharm. Soc. Jpn. 1987, 107, 449–456. [Google Scholar] [CrossRef]
- Bentley, M.V.; Vianna, R.F.; Wilson, S.; Collett, J.H. Characterization of the influence of some cyclodextrins on the stratum corneum from the hairless mouse. J. Pharm. Pharmacol. 1997, 49, 397–402. [Google Scholar] [CrossRef]
- Maestrelli, F.; González-Rodríguez, M.L.; Rabasco, A.M.; Mura, P. Effect of preparation technique on the properties of liposomes encapsulating ketoprofen-cyclodextrin complexes aimed for transdermal delivery. Int. J. Pharm. 2006, 312, 53–60. [Google Scholar] [CrossRef]
- Ananthapadmanabhan, K.; Yu, K.; Meyers, C.; Aronson, M. Binding of surfactants to stratum corneum. J. Soc. Cosmet. Chem. 1996, 47, 185–200. [Google Scholar]
- Som, I.; Bhatia, K.; Yasir, M. Status of surfactants as penetration enhancers in transdermal drug delivery. J. Pharm. Bioallied Sci. 2012, 4, 2–9. [Google Scholar] [CrossRef]
- Jantharaprapap, R.; Stagni, G. Effects of penetration enhancers on in vitro permeability of meloxicam gels. Int. J. Pharm. 2007, 343, 26–33. [Google Scholar] [CrossRef]
- Patil, S.; Singh, P.; Maibach, H. Radial spread of sodium lauryl sulfate after topical application. Pharm. Res. 1995, 12, 2018–2023. [Google Scholar] [CrossRef] [PubMed]
- Hamman, J.; Steenekamp, J. Excipients with specialized functions for effective drug delivery. Expert Opin. Drug Deliv. 2012, 9, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Lakkadwala, S.; Nguyen, S.; Nesamony, J.; Narang, A.S.; Boddu, S.H. Smart Polymers in Drug Delivery. In Excipient Applications in Formulation Design and Drug Delivery; Narang, A.S., Boddu, S.H.S., Eds.; Springer International Publishing: Cham, Switzerland, 2015; pp. 169–199. [Google Scholar]
- Karolewicz, B. A review of polymers as multifunctional excipients in drug dosage form technology. Saudi Pharm. J. 2016, 24, 525–536. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Srivastava, A.; Galaev, I.Y.; Mattiasson, B. Smart polymers: Physical forms and bioengineering applications. Prog. Polym. Sci. 2007, 32, 1205–1237. [Google Scholar] [CrossRef]
- Bawa, P.; Pillay, V.; Choonara, Y.E.; du Toit, L.C. Stimuli-responsive polymers and their applications in drug delivery. Biomed. Mater. 2009, 4, 022001. [Google Scholar] [CrossRef]
- James, H.P.; John, R.; Alex, A.; Anoop, K. Smart polymers for the controlled delivery of drugs—A concise overview. Acta Pharm. Sin. B 2014, 4, 120–127. [Google Scholar] [CrossRef]
- Jeong, B.; Gutowska, A. Lessons from nature: Stimuli-responsive polymers and their biomedical applications. Trends Biotechnol. 2002, 20, 305–311. [Google Scholar] [CrossRef]
- Zarrintaj, P.; Jouyandeh, M.; Ganjali, M.R.; Hadavand, B.S.; Mozafari, M.; Sheiko, S.S.; Vatankhah-Varnoosfaderani, M.; Gutiérrez, T.J.; Saeb, M.R. Thermo-sensitive polymers in medicine: A review. Eur. Polym. J. 2019, 117, 402–423. [Google Scholar] [CrossRef]
- Singh, S.; Webster, D.C.; Singh, J. Thermosensitive polymers: Synthesis, characterization, and delivery of proteins. Int. J. Pharm. 2007, 341, 68–77. [Google Scholar] [CrossRef]
- Rahimi, M.; Kilaru, S.; Hajj Sleiman, G.E.L.; Saleh, A.; Rudkevich, D.; Nguyen, K. Synthesis and Characterization of Thermo-Sensitive Nanoparticles for Drug Delivery Applications. J. Biomed. Nanotechnol. 2008, 4, 482–490. [Google Scholar] [CrossRef]
- He, W.; Ma, Y.; Gao, X.; Wang, X.; Dai, X.; Song, J. Application of Poly(N-isopropylacrylamide) As Thermosensitive Smart Materials. J. Phys. Conf. Ser. 2020, 1676, 012063. [Google Scholar] [CrossRef]
- Dethe, M.R.; Prabakaran, A.; Ahmed, H.; Agrawal, M.; Roy, U.; Alexander, A. PCL-PEG copolymer based injectable thermosensitive hydrogels. J. Control. Release 2022, 343, 217–236. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Zhang, C.; Shen, W.; Cheng, Z.; Yu, L.; Ping, Q. Poly(N-isopropylacrylamide)–chitosan as thermosensitive in situ gel-forming system for ocular drug delivery. J. Control. Release 2007, 120, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-Z.; Zhuo, R.-X.; Cui, J.-Z.; Zhang, J.-T. A novel thermo-responsive drug delivery system with positive controlled release. Int. J. Pharm. 2002, 235, 43–50. [Google Scholar] [CrossRef]
- Huang, Z.; Xiao, H.; Lu, X.; Yan, W.; Ji, Z. Enhanced photo/chemo combination efficiency against bladder tumor by encapsulation of DOX and ZnPC into in situ-formed thermosensitive polymer hydrogel. Int. J. Nanomed. 2018, 13, 7623–7631. [Google Scholar] [CrossRef]
- Thévenot, J.; Oliveira, H.; Sandre, O.; Lecommandoux, S. Magnetic responsive polymer composite materials. Chem. Soc. Rev. 2013, 42, 7099–7116. [Google Scholar] [CrossRef]
- Pasban, S.; Raissi, H. PNIPAM/Hexakis as a thermosensitive drug delivery system for biomedical and pharmaceutical applications. Sci. Rep. 2022, 12, 14363. [Google Scholar] [CrossRef]
- Mohan, A.; Santhamoorthy, M.; Phan, T.T.; Kim, S.-C. pNIPAm-Based pH and Thermoresponsive Copolymer Hydrogel for Hydrophobic and Hydrophilic Drug Delivery. Gels 2024, 10, 184. [Google Scholar] [CrossRef]
- Xi, L.; Li, C.; Wang, Y.; Gong, Y.; Su, F.; Li, S. Novel Thermosensitive Polymer-Modified Liposomes as Nano-Carrier of Hydrophobic Antitumor Drugs. J. Pharm. Sci. 2020, 109, 2544–2552. [Google Scholar] [CrossRef]
- Khodaverdi, E.; Delroba, K.; Mohammadpour, F.; Khameneh, B.; Sajadi Tabassi, S.A.; Tafaghodi, M.; Kamali, H.; Hadizadeh, F. In-vitro Release Evaluation of Growth Hormone from an Injectable In-Situ Forming Gel Using PCL-PEG-PCL Thermosensitive Triblock. Curr. Drug Deliv. 2020, 17, 174–183. [Google Scholar] [CrossRef]
- Hosseinzadeh, F.; Tabesh, H.; Farzaneh, F. Nano drug delivery platform based on thermosensitive PEG-PCL hydrogel encapsulated in silver-bearing micelles and its antifungal activity investigation against vaginal candidiasis. Front. Mater. 2023, 10, 1210542. [Google Scholar] [CrossRef]
- Patel, P.; Mandal, A.; Gote, V.; Pal, D.; Mitra, A.K. Thermosensitive hydrogel-based drug delivery system for sustained drug release. J. Polym. Res. 2019, 26, 131. [Google Scholar] [CrossRef]
- Yang, Z.; Yu, S.; Li, D.; Gong, Y.; Zang, J.; Liu, J.; Chen, X. The effect of PLGA-based hydrogel scaffold for improving the drug maximum-tolerated dose for in situ osteosarcoma treatment. Colloids Surf. B Biointerfaces 2018, 172, 387–394. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Shen, W.; Luan, J.; Yang, D.; Wei, G.; Yu, L.; Lu, W.; Ding, J. Sustained intravitreal delivery of dexamethasone using an injectable and biodegradable thermogel. Acta Biomater. 2015, 23, 271–281. [Google Scholar] [CrossRef]
- Sun, J.; Liu, X.; Lei, Y.; Tang, M.; Dai, Z.; Yang, X.; Yu, X.; Yu, L.; Sun, X.; Ding, J. Sustained subconjunctival delivery of cyclosporine A using thermogelling polymers for glaucoma filtration surgery. J. Mater. Chem. B 2017, 5, 6400–6411. [Google Scholar] [CrossRef]
- Mohajeri, S.A.; Yaghoubi, S.; Abdollahi, E.; Tekie, F.S.M.; Kamali, H.; Khodaverdi, E.; Hadizadeh, F. In-vivo study of naltrexone hydrochloride release from an in-situ forming PLGA-PEG-PLGA system in the rabbit. J. Drug Deliv. Sci. Technol. 2016, 36, 156–160. [Google Scholar] [CrossRef]
- Yan, Q.; Xiao, L.-Q.; Tan, L.; Sun, W.; Wu, T.; Chen, L.-W.; Mei, Y.; Shi, B. Controlled release of simvastatin-loaded thermo-sensitive PLGA-PEG-PLGA hydrogel for bone tissue regeneration: In vitro and in vivo characteristics. J. Biomed. Mater. Res. Part A 2015, 103, 3580–3589. [Google Scholar] [CrossRef]
- López-Cano, J.J.; Sigen, A.; Andrés-Guerrero, V.; Tai, H.; Bravo-Osuna, I.; Molina-Martínez, I.T.; Wang, W.; Herrero-Vanrell, R. Thermo-Responsive PLGA-PEG-PLGA Hydrogels as Novel Injectable Platforms for Neuroprotective Combined Therapies in the Treatment of Retinal Degenerative Diseases. Pharmaceutics 2021, 13, 234. [Google Scholar] [CrossRef]
- Torres-Figueroa, A.V.; Pérez-Martínez, C.J.; Encinas, J.C.; Burruel-Ibarra, S.; Silvas-García, M.I.; García Alegría, A.M.; del Castillo-Castro, T. Thermosensitive Bioadhesive Hydrogels Based on Poly(N-isopropylacrilamide) and Poly(methyl vinyl ether-alt-maleic anhydride) for the Controlled Release of Metronidazole in the Vaginal Environment. Pharmaceutics 2021, 13, 1284. [Google Scholar] [CrossRef]
- Ribeiro, L.S.; Sala, R.L.; Robeldo, T.A.; Borra, R.C.; Camargo, E.R. Injectable Thermosensitive Nanocomposites Based on Poly(N-vinylcaprolactam) and Silica Particles for Localized Release of Hydrophilic and Hydrophobic Drugs. Langmuir 2023, 39, 2380–2388. [Google Scholar] [CrossRef]
- Işıklan, N.; Polat, S. Synthesis and characterization of thermo/pH-sensitive pectin-graft-poly(dimethylaminoethyl methacrylate) coated magnetic nanoparticles. Int. J. Biol. Macromol. 2020, 164, 4499–4515. [Google Scholar] [CrossRef] [PubMed]
- Ju, G.; Liu, X.; Li, R.; Li, M.; Qin, Z.; Yin, X. Temperature-controlled release of curcumin from thermosensitive PVA/CurM nanofibrous membranes with antibacterial activity. Colloid Polym. Sci. 2021, 299, 1955–1966. [Google Scholar] [CrossRef]
- Amgoth, C.; Patra, S.; Wasnik, K.; Maity, P.; Paik, P. Controlled synthesis of thermosensitive tunable porous film of (pNIPAM)-b-(PCL) copolymer for sustain drug delivery. J. Appl. Polym. Sci. 2023, 140, e53854. [Google Scholar] [CrossRef]
- Purohit, A.; Jain, S.; Nema, P.; Vishwakarma, H.; Jain, P.K. Intelligent or smart polymers: Advance in novel drug delivery. J. Drug Deliv. Ther. 2022, 12, 208–216. [Google Scholar] [CrossRef]
- Kocak, G.; Tuncer, C.; Bütün, V. pH-Responsive polymers. Polym. Chem. 2017, 8, 144–176. [Google Scholar] [CrossRef]
- Ofridam, F.; Tarhini, M.; Lebaz, N.; Gagnière, É.; Mangin, D.; Elaissari, A. pH-sensitive polymers: Classification and some fine potential applications. Polym. Adv. Technol. 2021, 32, 1455–1484. [Google Scholar] [CrossRef]
- Ji, W.; Wu, Q.; Han, X.; Zhang, W.; Wei, W.; Chen, L.; Li, L.; Huang, W. Photosensitive hydrogels: From structure, mechanisms, design to bioapplications. Sci. China Life Sci. 2020, 63, 1813–1828. [Google Scholar] [CrossRef]
- Ali, Z.H.; Alkotaji, M. Smart hydrogel polymers for drug delivery. Mil. Med. Sci. Lett. 2022, 91, 105–118. [Google Scholar] [CrossRef]
- Yin, J.; Xiang, C.; Song, X. Nanoencapsulation of psoralidin via chitosan and Eudragit S100 for enhancement of oral bioavailability. Int. J. Pharm. 2016, 510, 203–209. [Google Scholar] [CrossRef]
- Jablan, J.; Jug, M. Development of Eudragit® S100 based pH-responsive microspheres of zaleplon by spray-drying: Tailoring the drug release properties. Powder Technol. 2015, 283, 334–343. [Google Scholar] [CrossRef]
- Gioumouxouzis, C.I.; Chatzitaki, A.T.; Karavasili, C.; Katsamenis, O.L.; Tzetzis, D.; Mystiridou, E.; Bouropoulos, N.; Fatouros, D.G. Controlled Release of 5-Fluorouracil from Alginate Beads Encapsulated in 3D Printed pH-Responsive Solid Dosage Forms. AAPS PharmSciTech 2018, 19, 3362–3375. [Google Scholar] [CrossRef] [PubMed]
- Kumari, A.; Jain, A.; Hurkat, P.; Tiwari, A.; Jain, S.K. Eudragit S100 coated microsponges for Colon targeting of prednisolone. Drug Dev. Ind. Pharm. 2018, 44, 902–913. [Google Scholar] [CrossRef] [PubMed]
- Qelliny, M.R.; Aly, U.F.; Elgarhy, O.H.; Khaled, K.A. Budesonide-Loaded Eudragit S 100 Nanocapsules for the Treatment of Acetic Acid-Induced Colitis in Animal Model. AAPS PharmSciTech 2019, 20, 237. [Google Scholar] [CrossRef] [PubMed]
- El-Maghawry, E.; Tadros, M.I.; Elkheshen, S.A.; Abd-Elbary, A. Eudragit®-S100 Coated PLGA Nanoparticles for Colon Targeting of Etoricoxib: Optimization and Pharmacokinetic Assessments in Healthy Human Volunteers. Int. J. Nanomed. 2020, 15, 3965–3980. [Google Scholar] [CrossRef]
- Chen, S.; Guo, F.; Deng, T.; Zhu, S.; Liu, W.; Zhong, H.; Yu, H.; Luo, R.; Deng, Z. Eudragit S100-Coated Chitosan Nanoparticles Co-loading Tat for Enhanced Oral Colon Absorption of Insulin. AAPS PharmSciTech 2017, 18, 1277–1287. [Google Scholar] [CrossRef]
- Jadiya, S.; Upmanyu, N.; Sathiyanarayanan, A.; Jain, V.; Dubey, R.; Buwade, P. Formulation and Development of Novel Sulfasalazine Bilayer Tablets for The Treatment of Arthritis Associated With IBD: In-vitro and In-vivo Investigations. J. Pharm. Sci. 2024, 113, 1919–1926. [Google Scholar] [CrossRef]
- Vemula, S.K.; Daravath, B.; Gummadi, S.B.; Repka, M. Formulation and Development of Flurbiprofen Colon-Specific Eudragit Coated Matrix Tablets: Use of a Novel Crude Banana Peel Powder as a Time-Dependent Polymer. AAPS PharmSciTech 2023, 24, 189. [Google Scholar] [CrossRef]
- Sen, S.; Das, M.; Ghosh, B.; Ghosh, L.K. Design, development and in vitro evaluation of sequentially optimized mesalamine tablets for optimum colonic delivery. Future J. Pharm. Sci. 2018, 4, 8–13. [Google Scholar] [CrossRef]
- Taghe, S.; Mirzaeei, S.; Alany, R.G.; Nokhodchi, A. Polymeric Inserts Containing Eudragit® L100 Nanoparticle for Improved Ocular Delivery of Azithromycin. Biomedicines 2020, 8, 466. [Google Scholar] [CrossRef]
- Dong, P.; Sahle, F.F.; Lohan, S.B.; Saeidpour, S.; Albrecht, S.; Teutloff, C.; Bodmeier, R.; Unbehauen, M.; Wolff, C.; Haag, R.; et al. pH-sensitive Eudragit® L 100 nanoparticles promote cutaneous penetration and drug release on the skin. J. Control. Release 2019, 295, 214–222. [Google Scholar] [CrossRef]
- Patriota, Y.B.G.; Arruda, I.E.S.; de Jesus Oliveira, A.C.; de Oliveira, T.C.; de Lemos Vasconcelos Silva, E.; Chaves, L.L.; de Oliveira Silva Ribeiro, F.; da Silva, D.A.; de La Roca Soares, M.F.; Soares-Sobrinho, J.L. Synthesis of Eudragit® L100-coated chitosan-based nanoparticles for oral enoxaparin delivery. Int. J. Biol. Macromol. 2021, 193, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Torres-Flores, G.; Türeli Nazende, G.; Akif Emre, T. Preparation of Fenofibrate loaded Eudragit L100 nanoparticles by nanoprecipitation method. Mater. Today Proc. 2019, 13, 428–435. [Google Scholar] [CrossRef]
- Saadallah, M. Eudragit® L100 nanoparticles for transdermal delivery of rosuvastatin calcium. J. Excip. Food Chem. 2022, 13, 2022. [Google Scholar]
- Yasmin Begum, M.; Alqahtani, A.; Ghazwani, M.; Alhamood, N.A.; Hani, U.; Jajala, A.; Rahamathulla, M. Development of Duloxetine Hydrochloride Tablets for Delayed and Complete Release Using Eudragit L 100. Int. J. Polym. Sci. 2021, 2021, 8890503. [Google Scholar] [CrossRef]
- Ahmad, M.Z.; Pathak, K.; Das, R.J.; Saikia, R.; Sarma, H.; Gogoi, N.; Gogoi, U.; Das, A.; Alasiri, A.S.; Abdel-Wahab, B.A.; et al. Design and Optimization of Quercetin-Loaded Polymeric Eudragit L-100 Nanoparticles for Anti-diabetic Activity with Improved Oral Delivery: In-Vitro and In-Vivo Evaluation. J. Inorg. Organomet. Polym. Mater. 2023, 33, 2411–2428. [Google Scholar] [CrossRef]
- Pawar, A.Y.; Tapkir, A.D.; Rao, J.B.; Dayama, R.P. Formulation and Evaluation of Eudragit L-100 based Nanoparticles of Senna for Treatment of Constipation. Int. J. Pharm. Investig. 2022, 12, 317–322. [Google Scholar] [CrossRef]
- Zhang, M.; Zhu, J.; Zheng, Y.; Guo, R.; Wang, S.; Mignani, S.; Caminade, A.-M.; Majoral, J.-P.; Shi, X. Doxorubicin-Conjugated PAMAM Dendrimers for pH-Responsive Drug Release and Folic Acid-Targeted Cancer Therapy. Pharmaceutics 2018, 10, 162. [Google Scholar] [CrossRef]
- Suhail, M.; Chiu, I.H.; Liu, J.-Y.; Ullah, H.; Lin, I.L.; Minhas, M.U.; Tsai, M.-J.; Wu, P.-C. Fabrication and In vitro Evaluation of Carbopol/Polyvinyl Alcohol-based pH-sensitive Hydrogels for Controlled Drug Delivery. Curr. Pharm. Des. 2023, 29, 2489–2500. [Google Scholar] [CrossRef]
- Sheshala, R.; Ming, N.J.; Kok, Y.Y.; Singh, T.R.R.; Dua, K. Formulation and characterization of pH induced in situ gels containing sulfacetamide sodium for ocular drug delivery: A combination of Carbopol®/HPMC polymer. Indian J. Pharm. Educ. Res. 2019, 53, 654–662. [Google Scholar] [CrossRef]
- Dawood, B.Y.; Kassab, H. Preparation and in vitro evaluation of naproxen as a pH sensitive ocular in-situ gel. Int. J. Appl. Pharm. 2019, 11, 37–44. [Google Scholar] [CrossRef]
- Lee, S.-H.; You, J.-O. Poly (Dimethylaminoethyl Methacrylate)-Based pH-Responsive Hydrogels Regulate Doxorubicin Release at Acidic Condition. J. Korean Appl. Sci. Technol. 2015, 32, 202–214. [Google Scholar] [CrossRef]
- Ganetsky, M.; Böhlke, M.; Pereira, L.; Williams, D.; LeDuc, B.; Guatam, S.; Salhanick, S.D. Effect of excipients on acetaminophen metabolism and its implications for prevention of liver injury. J. Clin. Pharmacol. 2013, 53, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Sekhon, B.S. Surfactants: Pharmaceutical and medicinal aspects. J. Pharm. Technol. Res. Manag. 2013, 1, 43–68. [Google Scholar] [CrossRef]
- Mudra, D.R.; Borchardt, R.T. Absorption Barriers in the Rat Intestinal Mucosa. 3: Effects of Polyethoxylated Solubilizing Agents on Drug Permeation and Metabolism. J. Pharm. Sci. 2010, 99, 1016–1027. [Google Scholar] [CrossRef]
- Christiansen, A.; Backensfeld, T.; Denner, K.; Weitschies, W. Effects of non-ionic surfactants on cytochrome P450-mediated metabolism in vitro. Eur. J. Pharm. Biopharm. 2011, 78, 166–172. [Google Scholar] [CrossRef]
- Randall, K.; Cheng, S.W.; Kotchevar, A.T. Evaluation of surfactants as solubilizing agents in microsomal metabolism reactions with lipophilic substrates. Vitr. Cell. Dev. Biol. Anim. 2011, 47, 631–639. [Google Scholar] [CrossRef]
- Bravo González, R.C.; Huwyler, J.; Boess, F.; Walter, I.; Bittner, B. In vitro investigation on the impact of the surface-active excipients Cremophor EL, Tween 80 and Solutol HS 15 on the metabolism of midazolam. Biopharm. Drug Dispos. 2004, 25, 37–49. [Google Scholar] [CrossRef]
- Rao, Z.; Si, L.; Guan, Y.; Pan, H.; Qiu, J.; Li, G. Inhibitive effect of cremophor RH40 or tween 80-based self-microemulsiflying drug delivery system on cytochrome P450 3A enzymes in murine hepatocytes. J. Huazhong Univ. Sci. Technol. Med. Sci. 2010, 30, 562–568. [Google Scholar] [CrossRef]
- Ren, S.; Park, M.-J.; Kim, A.; Lee, B.-J. In vitro metabolic stability of moisture-sensitive rabeprazole in human liver microsomes and its modulation by pharmaceutical excipients. Arch. Pharmacal Res. 2008, 31, 406–413. [Google Scholar] [CrossRef]
- Da Silva, M.E.F.; Meirelles, N.C. Interaction of non-ionic surfactants with hepatic CYP in Prochilodus scrofa. Toxicol. Vitr. 2004, 18, 859–867. [Google Scholar] [CrossRef]
- Qiu, L.; Li, Q.; Huang, J.; Wu, Q.; Tu, K.; Wu, Y.; Zhang, X.; Qian, J.; Zhang, R.; Li, G.; et al. In vitro effect of mPEG2k-PCLx micelles on rat liver cytochrome P450 enzymes. Int. J. Pharm. 2018, 552, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Martin, P.; Giardiello, M.; McDonald, T.O.; Rannard, S.P.; Owen, A. Mediation of in Vitro Cytochrome P450 Activity by Common Pharmaceutical Excipients. Mol. Pharm. 2013, 10, 2739–2748. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.; Barker, J.; ElShaer, A. Pharmaceutical Excipients and Drug Metabolism: A Mini-Review. Int. J. Mol. Sci. 2020, 21, 8224. [Google Scholar] [CrossRef] [PubMed]
- Goole, J.; Lindley, D.J.; Roth, W.; Carl, S.M.; Amighi, K.; Kauffmann, J.-M.; Knipp, G.T. The effects of excipients on transporter mediated absorption. Int. J. Pharm. 2010, 393, 17–31. [Google Scholar] [CrossRef]
- Zhang, H.; Yao, M.; Morrison, R.A.; Chong, S. Commonly used surfactant, Tween 80, improves absorption of P-glycoprotein substrate, digoxin, in rats. Arch. Pharmacal Res. 2003, 26, 768–772. [Google Scholar] [CrossRef]
- Lo, Y.-L. Relationships between the hydrophilic–lipophilic balance values of pharmaceutical excipients and their multidrug resistance modulating effect in Caco-2 cells and rat intestines. J. Control. Release 2003, 90, 37–48. [Google Scholar] [CrossRef]
- Shen, Q.; Lin, Y.; Handa, T.; Doi, M.; Sugie, M.; Wakayama, K.; Okada, N.; Fujita, T.; Yamamoto, A. Modulation of intestinal P-glycoprotein function by polyethylene glycols and their derivatives by in vitro transport and in situ absorption studies. Int. J. Pharm. 2006, 313, 49–56. [Google Scholar] [CrossRef]
- Batrakova, E.V.; Li, S.; Li, Y.; Alakhov, V.Y.; Kabanov, A.V. Effect of Pluronic P85 on ATPase Activity of Drug Efflux Transporters. Pharm. Res. 2004, 21, 2226–2233. [Google Scholar] [CrossRef]
- Sawangrat, K.; Yamashita, S.; Tanaka, A.; Morishita, M.; Kusamori, K.; Katsumi, H.; Sakane, T.; Yamamoto, A. Modulation of Intestinal Transport and Absorption of Topotecan, a BCRP Substrate, by Various Pharmaceutical Excipients and Their Inhibitory Mechanisms of BCRP Transporter. J. Pharm. Sci. 2019, 108, 1315–1325. [Google Scholar] [CrossRef]
- Arima, H.; Yunomae, K.; Morikawa, T.; Hirayama, F.; Uekama, K. Contribution of Cholesterol and Phospholipids to Inhibitory Effect of Dimethyl-β-Cyclodextrin on Efflux Function of P-Glycoprotein and Multidrug Resistance-Associated Protein 2 in Vinblastine-Resistant Caco-2 Cell Monolayers. Pharm. Res. 2004, 21, 625–634. [Google Scholar] [CrossRef]
- Ferté, J. Analysis of the tangled relationships between P-glycoprotein-mediated multidrug resistance and the lipid phase of the cell membrane. Eur. J. Biochem. 2000, 267, 277–294. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Komura, H.; Yoshikawa, T.; Enya, S.; Nagao, A.; Takubo, H.; Kogayu, M. Characterization of gastrointestinal absorption of digoxin involving influx and efflux transporter in rats: Application of mdr1a knockout (−/−) rats into absorption study of multiple transporter substrate. Xenobiotica 2014, 44, 1039–1045. [Google Scholar] [CrossRef] [PubMed]
- McCartney, F.; Gleeson, J.P.; Brayden, D.J. Safety concerns over the use of intestinal permeation enhancers: A mini-review. Tissue Barriers 2016, 4, e1176822. [Google Scholar] [CrossRef]
- Maher, S.; Wang, X.; Bzik, V.; McClean, S.; Brayden, D.J. Evaluation of intestinal absorption and mucosal toxicity using two promoters. II. Rat instillation and perfusion studies. Eur. J. Pharm. Sci. 2009, 38, 301–311. [Google Scholar] [CrossRef]
- Sharma, P.; Varma, M.V.S.; Chawla, H.P.S.; Panchagnula, R. Absorption enhancement, mechanistic and toxicity studies of medium chain fatty acids, cyclodextrins and bile salts as peroral absorption enhancers. Il Farm. 2005, 60, 884–893. [Google Scholar] [CrossRef]
- Lucarini, S.; Fagioli, L.; Cavanagh, R.; Liang, W.; Perinelli, D.R.; Campana, M.; Stolnik, S.; Lam, J.K.W.; Casettari, L.; Duranti, A. Synthesis, Structure–Activity Relationships and In Vitro Toxicity Profile of Lactose-Based Fatty Acid Monoesters as Possible Drug Permeability Enhancers. Pharmaceutics 2018, 10, 81. [Google Scholar] [CrossRef]
- Jambhekar, S.S.; Breen, P.J. Drug dissolution: Significance of physicochemical properties and physiological conditions. Drug Discov. Today 2013, 18, 1173–1184. [Google Scholar] [CrossRef]
- Martins, G.B.; Tarouco, F.; Rosa, C.E.; Robaldo, R.B. The utilization of sodium bicarbonate, calcium carbonate or hydroxide in biofloc system: Water quality, growth performance and oxidative stress of Nile tilapia (Oreochromis niloticus). Aquaculture 2017, 468, 10–17. [Google Scholar] [CrossRef]
- Kataoka, M.; Fukahori, M.; Ikemura, A.; Kubota, A.; Higashino, H.; Sakuma, S.; Yamashita, S. Effects of gastric pH on oral drug absorption: In vitro assessment using a dissolution/permeation system reflecting the gastric dissolution process. Eur. J. Pharm. Biopharm. 2016, 101, 103–111. [Google Scholar] [CrossRef]
- Adachi, M.; Hinatsu, Y.; Kusamori, K.; Katsumi, H.; Sakane, T.; Nakatani, M.; Wada, K.; Yamamoto, A. Improved dissolution and absorption of ketoconazole in the presence of organic acids as pH-modifiers. Eur. J. Pharm. Sci. 2015, 76, 225–230. [Google Scholar] [CrossRef]
- Taniguchi, C.; Inoue, R.; Kawabata, Y.; Yamashita, K.; Wada, K.; Yamauchi, Y.; Yamada, S.; Onoue, S. Novel formulations of dipyridamole with microenvironmental pH-modifiers for improved dissolution and bioavailability under hypochlorhydria. Int. J. Pharm. 2012, 434, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Halder, S.; Tabata, A.; Seto, Y.; Sato, H.; Onoue, S. Amorphous solid dispersions of carvedilol along with pH-modifiers improved pharmacokinetic properties under hypochlorhydoria. Biopharm. Drug Dispos. 2018, 39, 232–242. [Google Scholar] [CrossRef] [PubMed]
- Tran, P.H.-L.; Tran, T.T.-D.; Lee, K.-H.; Kim, D.-J.; Lee, B.-J. Dissolution-modulating mechanism of pH modifiers in solid dispersion containing weakly acidic or basic drugs with poor water solubility. Expert Opin. Drug Deliv. 2010, 7, 647–661. [Google Scholar] [CrossRef] [PubMed]
- Tran, P.H.L.; Tran, H.T.T.; Lee, B.-J. Modulation of microenvironmental pH and crystallinity of ionizable telmisartan using alkalizers in solid dispersions for controlled release. J. Control. Release 2008, 129, 59–65. [Google Scholar] [CrossRef]
- Grattan, T.; Hickman, R.; Darby-Dowman, A.; Hayward, M.; Boyce, M.; Warrington, S. A five way crossover human volunteer study to compare the pharmacokinetics of paracetamol following oral administration of two commercially available paracetamol tablets and three development tablets containing paracetamol in combination with sodium bicarbonate or calcium carbonate. Eur. J. Pharm. Biopharm. 2000, 49, 225–229. [Google Scholar] [CrossRef]
- Tran, T.T.; Tran, P.H.; Lee, B.J. Dissolution-modulating mechanism of alkalizers and polymers in a nanoemulsifying solid dispersion containing ionizable and poorly water-soluble drug. Eur. J. Pharm. Biopharm. 2009, 72, 83–90. [Google Scholar] [CrossRef]
- Kang, W.H.; Nguyen, H.V.; Park, C.; Choi, Y.W.; Lee, B.J. Modulation of microenvironmental pH for dual release and reduced in vivo gastrointestinal bleeding of aceclofenac using hydroxypropyl methylcellulose-based bilayered matrix tablet. Eur. J. Pharm. Sci. 2017, 102, 85–93. [Google Scholar] [CrossRef]
- Schilling, S.U.; Bruce, C.D.; Shah, N.H.; Malick, A.W.; McGinity, J.W. Citric acid monohydrate as a release-modifying agent in melt extruded matrix tablets. Int. J. Pharm. 2008, 361, 158–168. [Google Scholar] [CrossRef]
- Nie, S.; Pan, W.; Li, X.; Wu, X. The Effect of Citric Acid Added to Hydroxypropyl Methylcellulose (HPMC) Matrix Tablets on the Release Profile of Vinpocetine. Drug Dev. Ind. Pharm. 2004, 30, 627–635. [Google Scholar] [CrossRef]
- Yu, Y.B.; Taraban, M.B.; Briggs, K.T.; Brinson, R.G.; Marino, J.P. Excipient Innovation Through Precompetitive Research. Pharm. Res. 2021, 38, 2179–2184. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. Guidance for Industry: Nonclinical Studies for the Safety Evaluation of Pharmaceutical Excipients; Food and Drug Administration: Rockville, MD, USA, 2005.
- Ismail, R.; Baaity, Z.; Csóka, I. Regulatory status quo and prospects for biosurfactants in pharmaceutical applications. Drug Discov. Today 2021, 26, 1929–1935. [Google Scholar] [CrossRef]
- El-Emam, G.A.; Girgis, G.N.S.; Hamed, M.F.; El-Azeem Soliman, O.A.; Abd El Gawad, A. Formulation and Pathohistological Study of Mizolastine-Solid Lipid Nanoparticles-Loaded Ocular Hydrogels. Int. J. Nanomed. 2021, 16, 7775–7799. [Google Scholar] [CrossRef]
- CHMP. Guideline on Excipients in the Dossier for Application for Marketing Authorisation of a Medicinal Product. 2007. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-excipients-dossier-application-marketing-authorisation-medicinal-product-revision-2_en.pdf (accessed on 1 May 2024).

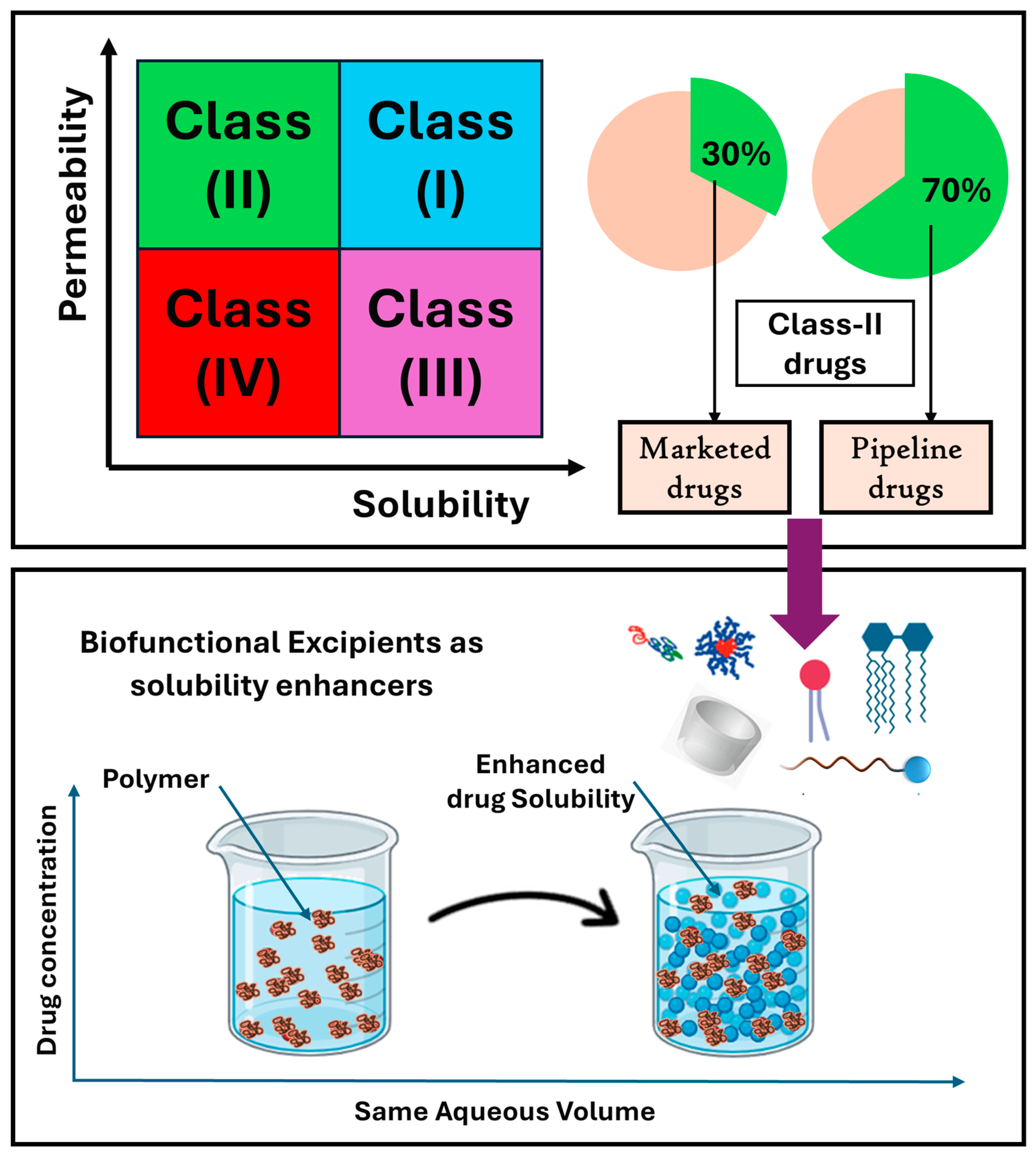
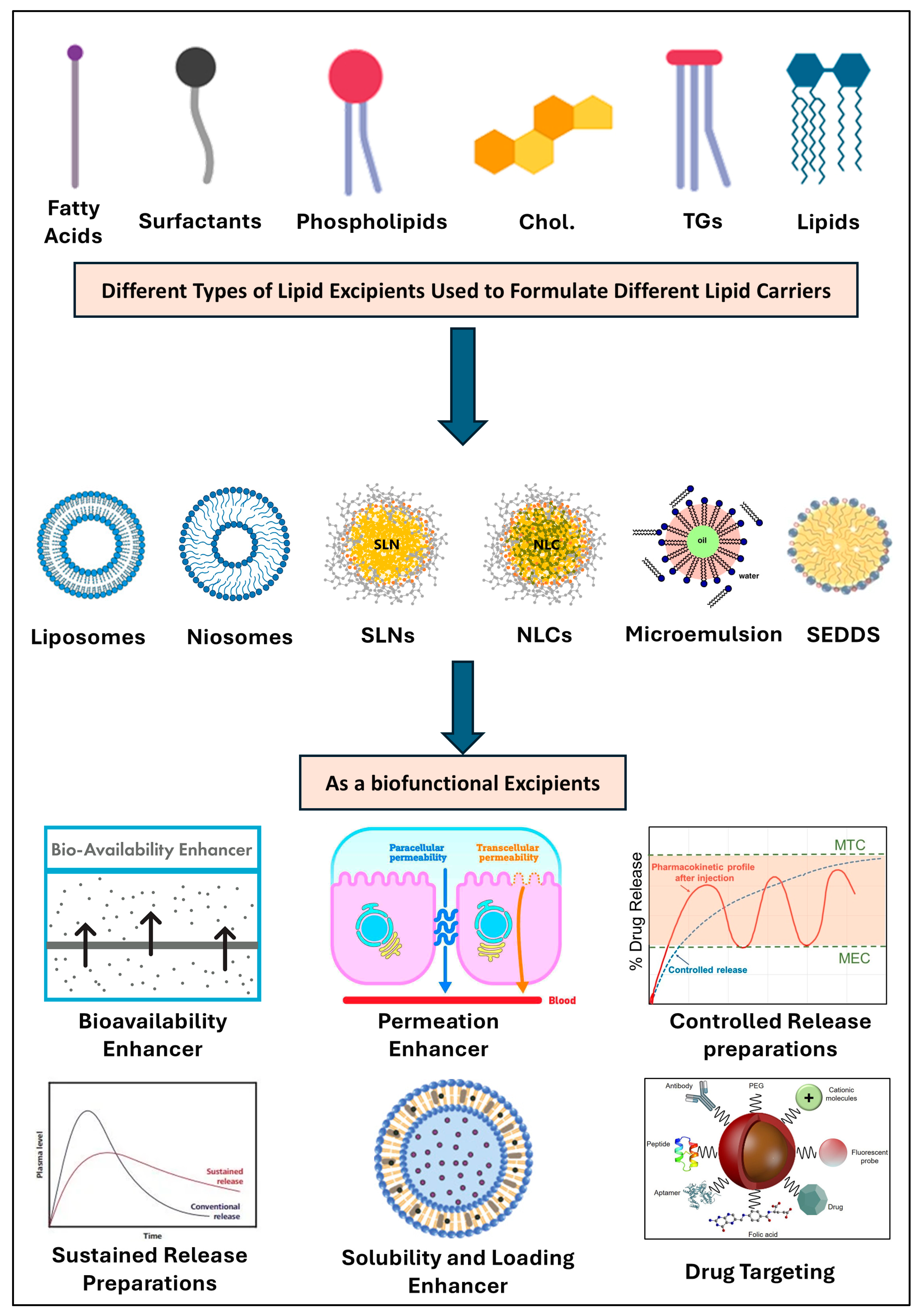
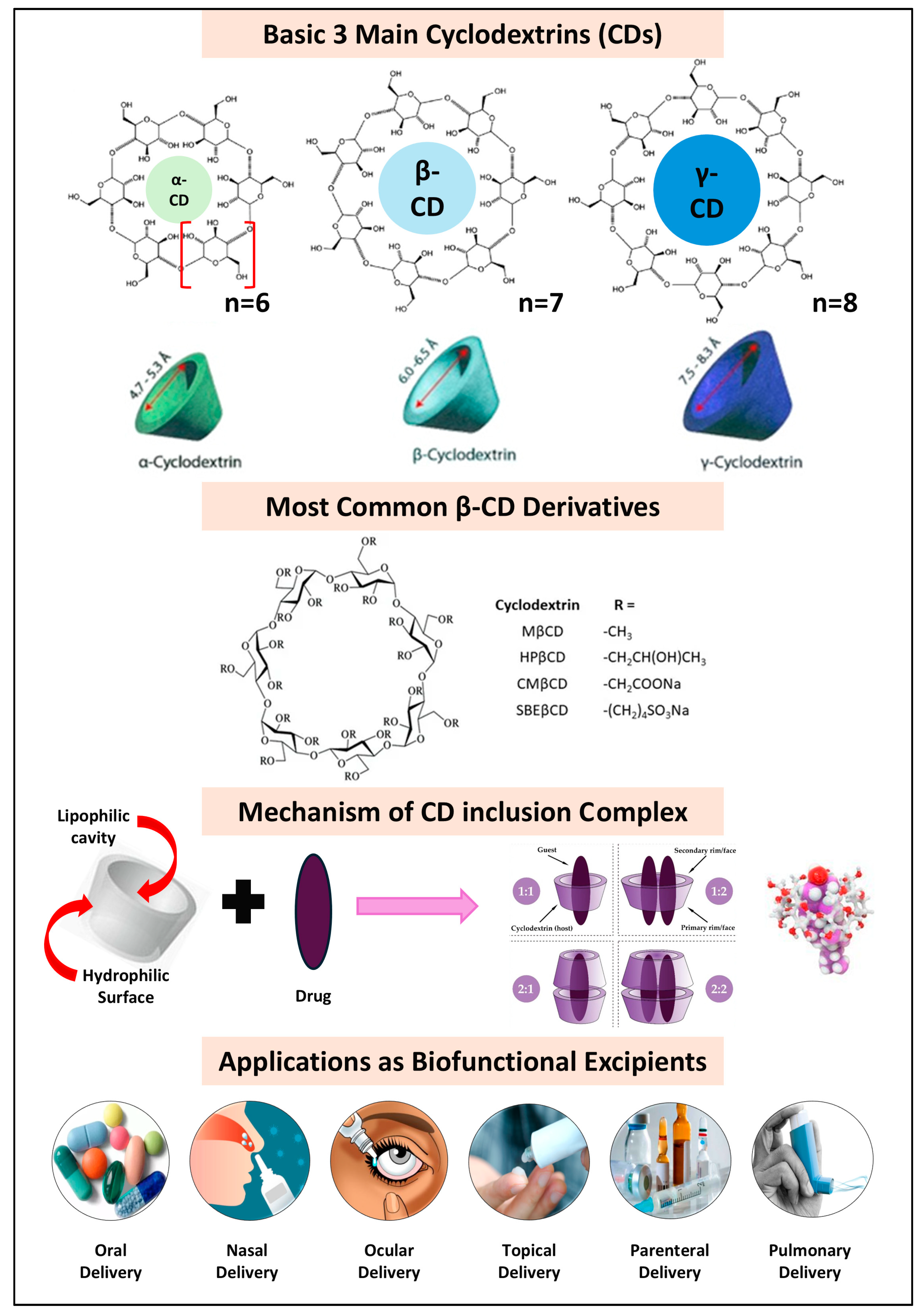
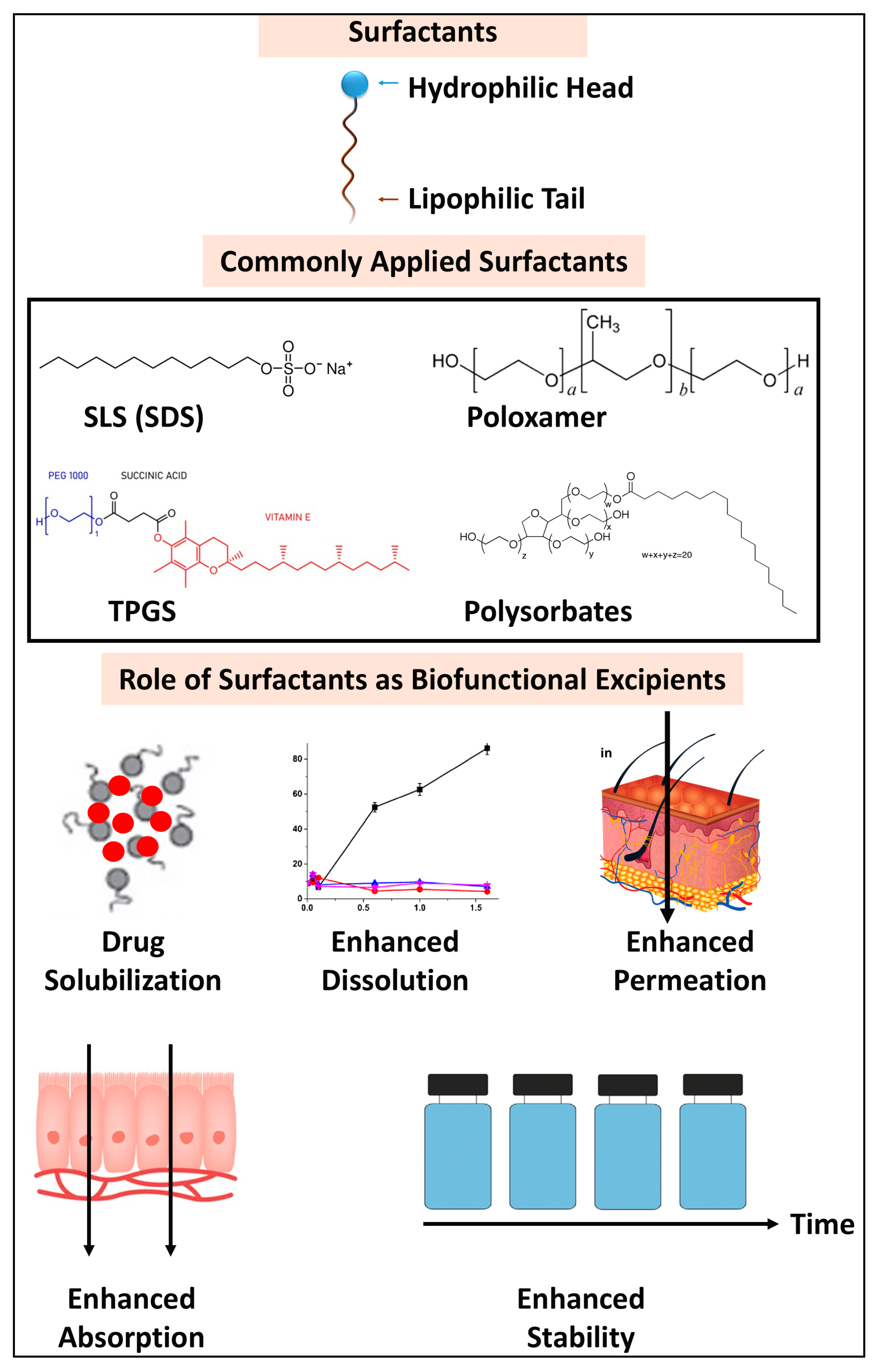

| Drug | AA Excipients 1,2 | BCS Class | Formulation | Biofunctional Applications | Ref. |
|---|---|---|---|---|---|
| Curcumin | TYR, TRP, ARG | BCS IV | Binary Mixture | AAs show enhanced in vivo drug solubilization, permeation, stabilization, and bioavailability. AAs play a vital role in improving drug solubility, permeation, and stability, addressing key challenges in drug formulation, such as poor absorption and low bioavailability. AAs act as hydrophilic carriers and form ion pairs, enhancing the solubility and permeability of small molecules through AA carriers. They also improve drug stability by forming stable cocrystals and co-amorphous systems that prevent the recrystallization of poorly soluble drugs. Moreover, AAs facilitate drug transport across biological membranes, enhancing permeability and bioavailability. These properties make AAs effective solubility, permeation, and stability enhancers, crucial for optimizing drug formulations and therapeutic outcomes. | [109] |
| Carbamazepine | PHE, TRP | BCS II | Co-Amorphous | [110] | |
| Naproxen | ARG | BCS II | Co-Amorphous | [104] | |
| Ibuprofen | Cholinium | BCS II | Ionic liquids | [111] | |
| Valsartan | HIS, ARG, LYS | BCS IV | Co-Amorphous | [112] | |
| Genistein | ARG, GLU, LYS | BCS IV | Co-Amorphous | [113] | |
| Telmisartan | ARG | BCS II | Co-Amorphous | [114] | |
| Carvedilol | ASP, GLU | BCS II | Co-Amorphous | [115] | |
| Glipizide | ARG, SER | BCS II | Co-Amorphous | [116] | |
| Mebendazole | ARG, GLU | BCS IV | Co-Amorphous | [117] | |
| Albendazole | GLU | BCS IV | Supramolecular | [118] | |
| Griseofulvin | ASP, TRP, VAL | BCS II | Co-Amorphous | [119] | |
| Insulin | ARG | BCS IV | Mixture | [91] | |
| Meloxicam | SER, ARG | BCS II | Solid Dispersion | [120] | |
| Curcumin | L-ARG | BCS IV | Salt Formation | [121] | |
| Ketoprofen | L-LYS, L-ARG | BCS II | Salt Formation | [122] | |
| Oxaprozin | ARG | BCS II | Ternary System | [123] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qelliny, M.R.; Mustafa, W.W.; Fatease, A.A.; Alamri, A.H.; Alany, R.; Abdelkader, H. Biofunctional Excipients: Their Emerging Role in Overcoming the Inherent Poor Biopharmaceutical Characteristics of Drugs. Pharmaceutics 2025, 17, 598. https://doi.org/10.3390/pharmaceutics17050598
Qelliny MR, Mustafa WW, Fatease AA, Alamri AH, Alany R, Abdelkader H. Biofunctional Excipients: Their Emerging Role in Overcoming the Inherent Poor Biopharmaceutical Characteristics of Drugs. Pharmaceutics. 2025; 17(5):598. https://doi.org/10.3390/pharmaceutics17050598
Chicago/Turabian StyleQelliny, Milad Reda, Wesam W. Mustafa, Adel Al Fatease, Ali H. Alamri, Raid Alany, and Hamdy Abdelkader. 2025. "Biofunctional Excipients: Their Emerging Role in Overcoming the Inherent Poor Biopharmaceutical Characteristics of Drugs" Pharmaceutics 17, no. 5: 598. https://doi.org/10.3390/pharmaceutics17050598
APA StyleQelliny, M. R., Mustafa, W. W., Fatease, A. A., Alamri, A. H., Alany, R., & Abdelkader, H. (2025). Biofunctional Excipients: Their Emerging Role in Overcoming the Inherent Poor Biopharmaceutical Characteristics of Drugs. Pharmaceutics, 17(5), 598. https://doi.org/10.3390/pharmaceutics17050598