

Amphiphilic Celecoxib-Polysaccharide Delivery System for Enhanced Colon-Targeted Colitis Therapy

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis and Characterization of CSA-CXB Polymers
2.3. Preparation and Characterization of CSA-CXB Nanoprodrug
2.4. In Vitro Drug Release Test
2.5. In Vitro Antioxidant Capability Study
2.6. Hemolysis Test
2.7. In Vitro Cytotoxicity
2.8. Measurement of Intracellular ROS
2.9. Determination of Anti-Inflammatory Effect In Vitro
2.10. Western Blot Analysis
2.11. Evaluation of Anti-Colitis Effect In Vivo
2.12. Bioimaging of Colon-Targeting Effect
2.13. In Vivo Inflammatory Cytokine Test
2.14. Statistical Analysis
3. Results and Discussion
3.1. Characterization of CSA-CXB Polymers
3.2. Characterization of CSA-CXB Nanoparticles
3.3. In Vitro Drug Release Behavior
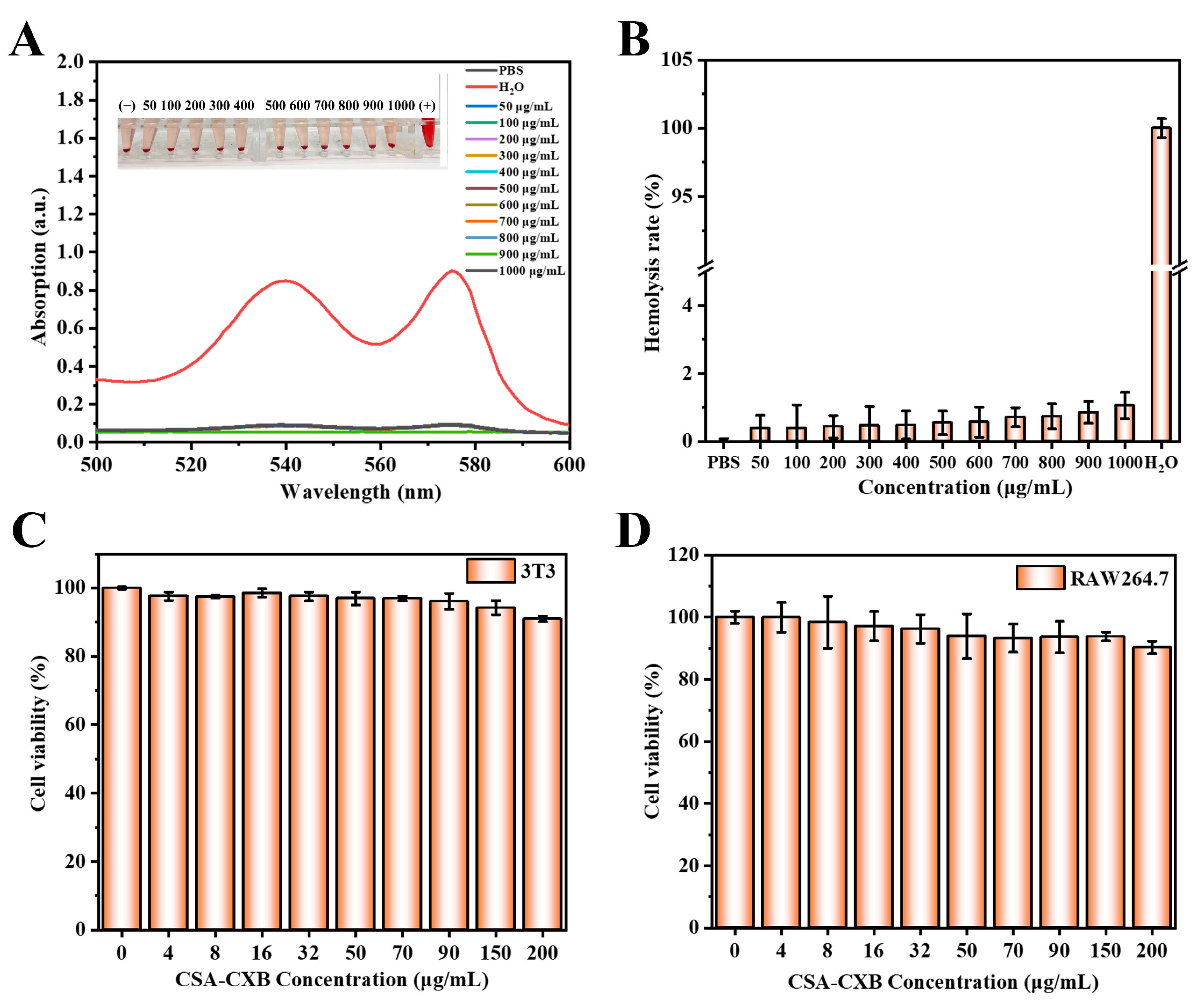
3.4. Biocompatibility Assessment of CSA-CXB
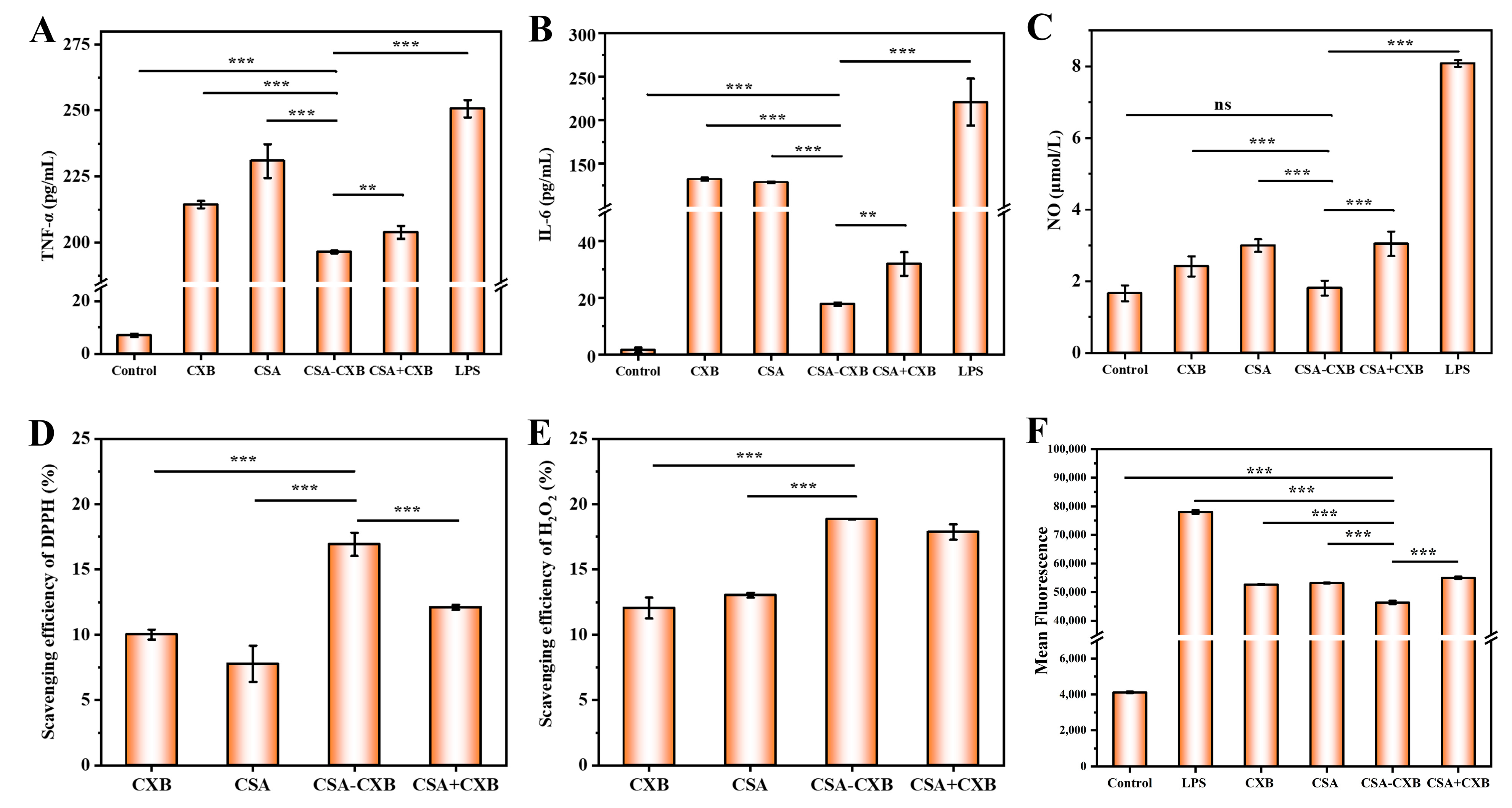
3.5. Anti-Inflammatory and Antioxidant Activities
3.6. Inhibitory Effect of COX-2 Protein Expression
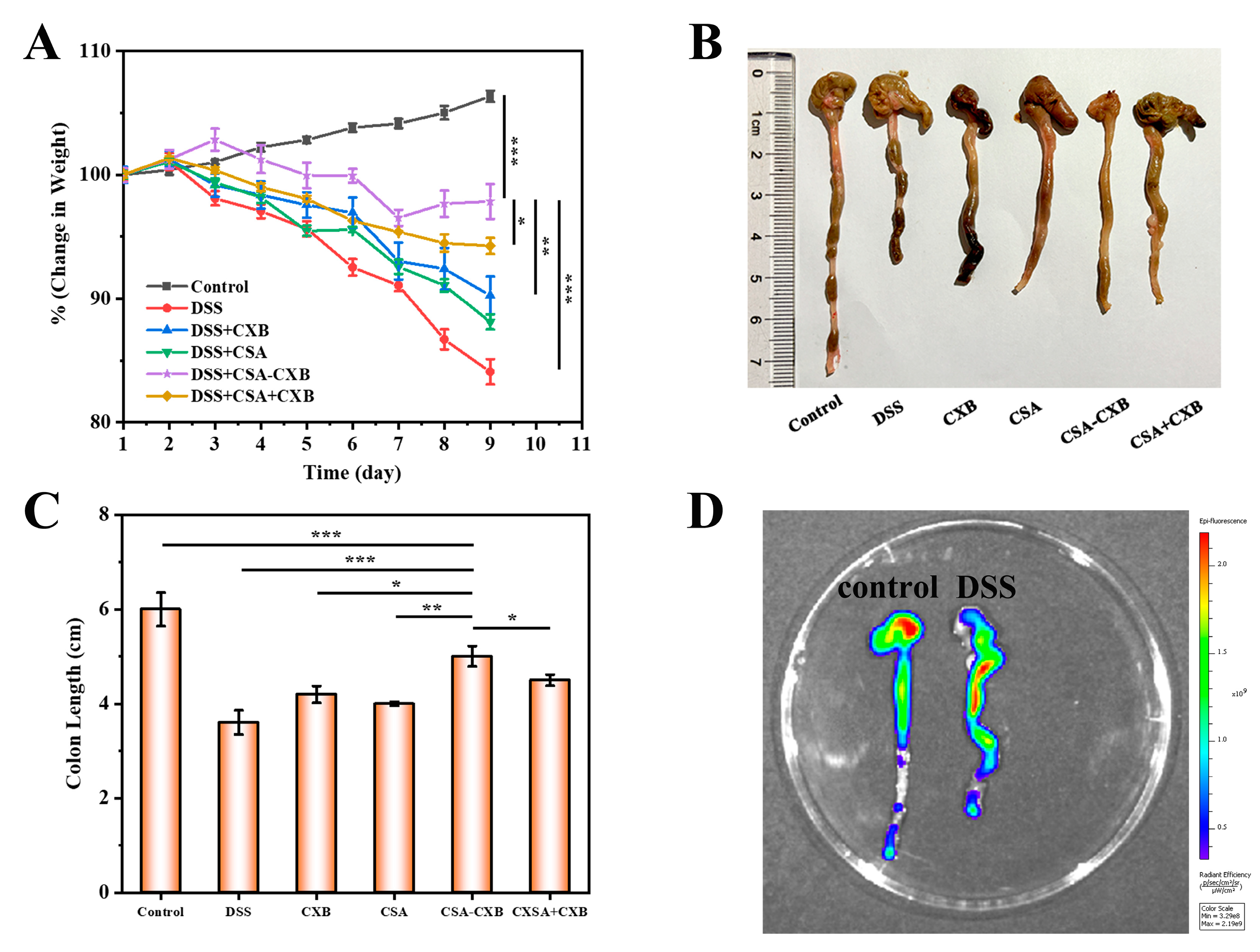
3.7. In Vivo Therapeutic Effect of CSA-CXB
3.8. Investigation of Anti-Inflammatory Mechanism In Vivo
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Le Berre, C.; Honap, S.; Peyrin-Biroulet, L. Ulcerative Colitis. Lancet 2023, 402, 571–584. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Mao, T.; Zhou, H.; Jiang, X.; Zhao, Z.; Zhang, X. Global Trends and Hotspots of Ulcerative Colitis Based on Bibliometric and Visual Analysis from 1993 to 2022. Medicine 2023, 103, e37095. [Google Scholar] [CrossRef]
- Deng, J.; Xian, D.; Cai, X.; Liao, S.; Lei, S.; Han, F.; An, Y.; He, Q.; Quan, G.; Wu, C.; et al. Surface-Engineered Vanadium Carbide MXenzyme for Anti-Inflammation and Photoenhanced Antitumor Therapy of Colon Diseases. Adv. Funct. Mater. 2023, 33, 2211846. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, Inflammation, and Cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef]
- Zhang, M.; Li, X.; Zhang, Q.; Yang, J.; Liu, G. Roles of Macrophages on Ulcerative Colitis and Colitis-Associated Colorectal Cancer. Front. Immunol. 2023, 14, 1103617. [Google Scholar] [CrossRef]
- Porter, R.J.; Kalla, R.; Ho, G.-T. Ulcerative Colitis: Recent Advances in the Understanding of Disease Pathogenesis. F1000Research 2020, 9, 294. [Google Scholar] [CrossRef]
- Kimura, H.; Miura, S.; Shigematsu, T.; Ohkubo, N.; Tsuzuki, Y.; Kurose, I.; Higuchi, H.; Akiba, Y.; Hokari, R.; Hirokawa, M.; et al. Increased Nitric Oxide Production and Inducible Nitric Oxide Synthase Activity in Colonic Mucosa of Patients with Active Ulcerative Colitis and Crohn’s Disease. Dig. Dis. Sci. 1997, 42, 1047–1054. [Google Scholar]
- Gajendran, M.; Loganathan, P.; Jimenez, G.; Catinella, A.P.; Ng, N.; Umapathy, C.; Ziade, N.; Hashash, J.G. A Comprehensive Review and Update on Ulcerative Colitis. Dis.-A-Mon. 2019, 65, 100851. [Google Scholar] [CrossRef]
- Aslam, N.; Lo, S.W.; Sikafi, R.; Barnes, T.; Segal, J.; Smith, P.J.; Limdi, J.K. A Review of the Therapeutic Management of Ulcerative Colitis. Ther. Adv. Gastroenterol. 2022, 15, 175628482211381. [Google Scholar] [CrossRef]
- McCormack, P.L. Celecoxib: A Review of Its Use for Symptomatic Relief in the Treatment of Osteoarthritis, Rheumatoid Arthritis and Ankylosing Spondylitis. Drugs 2011, 71, 2457–2489. [Google Scholar] [CrossRef]
- Lin, X.; Sun, Q.; Zhou, L.; He, M.; Dong, X.; Lai, M.; Liu, M.; Su, Y.; Jia, C.; Han, Z.; et al. Colonic Epithelial mTORC1 Promotes Ulcerative Colitis through COX-2-Mediated Th17 Responses. Mucosal Immunol. 2018, 11, 1663–1673. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ma, M.; Wang, X.; Li, J.; Fang, Z.; Li, J.; Yang, B.; Lu, Y.; Xu, X.; Li, Y. Celecoxib Alleviates the DSS-Induced Ulcerative Colitis in Mice by Enhancing Intestinal Barrier Function, Inhibiting Ferroptosis and Suppressing Apoptosis. Immunopharmacol. Immunotoxicol. 2024, 46, 240–254. [Google Scholar] [CrossRef] [PubMed]
- Song, W.H.; Yeom, D.W.; Lee, D.H.; Lee, K.M.; Yoo, H.J.; Chae, B.R.; Song, S.H.; Choi, Y.W. In Situ Intestinal Permeability and in Vivo Oral Bioavailability of Celecoxib in Supersaturating Self-Emulsifying Drug Delivery System. Arch. Pharm. Res. 2014, 37, 626–635. [Google Scholar] [CrossRef] [PubMed]
- Abdul Khader, A.H.S.; Singh, M. Celecoxib-Induced Acute Generalized Exanthematous Pustulosis: Uncommon and under-Recognized Side Effect. EXCLI J. 2024, 23, 108–113. [Google Scholar] [CrossRef]
- Cruz, J.V.; Rosa, J.M.C.; Kimani, N.M.; Giuliatti, S.; Dos Santos, C.B.R. The Role of Celecoxib as a Potential Inhibitor in the Treatment ofInflammatory Diseases—A Review. CMC 2022, 29, 3028–3049. [Google Scholar] [CrossRef]
- Barclay, T.G.; Day, C.M.; Petrovsky, N.; Garg, S. Review of Polysaccharide Particle-Based Functional Drug Delivery. Carbohydr. Polym. 2019, 221, 94–112. [Google Scholar] [CrossRef]
- Wu, Q.; Hu, Y.; Yu, B.; Hu, H.; Xu, F.-J. Polysaccharide-Based Tumor Microenvironment-Responsive Drug Delivery Systems for Cancer Therapy. J. Control. Release 2023, 362, 19–43. [Google Scholar] [CrossRef]
- Mohammed, A.S.A.; Naveed, M.; Jost, N. Polysaccharides; Classification, Chemical Properties, and Future Perspective Applications in Fields of Pharmacology and Biological Medicine (A Review of Current Applications and Upcoming Potentialities). J. Polym. Environ. 2021, 29, 2359–2371. [Google Scholar] [CrossRef]
- Sood, A.; Gupta, A.; Agrawal, G. Recent Advances in Polysaccharides Based Biomaterials for Drug Delivery and Tissue Engineering Applications. Carbohydr. Polym. Technol. Appl. 2021, 2, 100067. [Google Scholar] [CrossRef]
- Mohan, T.; Kleinschek, K.S.; Kargl, R. Polysaccharide Peptide Conjugates: Chemistry, Properties and Applications. Carbohydr. Polym. 2022, 280, 118875. [Google Scholar] [CrossRef]
- Wang, J.; Zhao, W.; Chen, H.; Qin, A.; Zhu, P. Anti-Tumor Study of Chondroitin Sulfate-Methotrexate Nanogels. Nanoscale Res. Lett. 2017, 12, 572. [Google Scholar] [CrossRef] [PubMed]
- Iovu, M.; Dumais, G.; du Souich, P. Anti-Inflammatory Activity of Chondroitin Sulfate. Osteoarthr. Cartil. 2008, 16, S14–S18. [Google Scholar] [CrossRef]
- du Souich, P.; García, A.G.; Vergés, J.; Montell, E. Immunomodulatory and Anti-Inflammatory Effects of Chondroitin Sulphate. J. Cell. Mol. Med. 2009, 13, 1451–1463. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Xu, W.; Jin, Z.; Zhao, K. Chondroitin Sulfate Functionalized Palmitic Acid and Cysteine Cografted-Quaternized Chitosan for CD44 and Gut Microbiota Dual-Targeted Delivery of Curcumin. Mater. Today Bio 2023, 20, 100617. [Google Scholar] [CrossRef]
- Rosenberg, W.M.C.; Jackson, D.G.; Trowell, J.M.; Bell, J.I.; Rosenberg, W.M.C.; Prince, C.; Chapman, R.W.; Trowell, J.M.; Jewel, D.P.; Kaklamanis, L.; et al. Increased Expression of CD44v6 and CD44v3 in Ulcerative Colitis but Not Colonic Crohn’s Disease. Lancet 1995, 345, 1205–1209. [Google Scholar] [CrossRef]
- Harris, A.J.; Dean, D.; Burge, S.; Wojnarowska, F. Changes in CD44 Isoform Expression during Inflammatory Skin Disease. Clin. Exp. Dermatol. 1997, 22, 128–133. [Google Scholar] [CrossRef]
- Azimijou, N.; Karimi-Soflou, R.; Karkhaneh, A. CD44 Targeted-Chondroitin Sulfate Nanoparticles: Fine-Tuning Hydrophobic Groups to Enhance in Vitro pH-Responsiveness and in Vivo Efficacy for Advanced Breast Cancer Treatment. Biomater. Adv. 2024, 158, 213776. [Google Scholar] [CrossRef] [PubMed]
- Kesharwani, P.; Chadar, R.; Sheikh, A.; Rizg, W.Y.; Safhi, A.Y. CD44-Targeted Nanocarrier for Cancer Therapy. Front. Pharmacol. 2022, 12, 800481. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Sun, J.; Zhang, W.; Zhao, Y.; Zhang, S.; Zhang, S. Drug Delivery Systems Based on CD44-Targeted Glycosaminoglycans for Cancer Therapy. Carbohydr. Polym. 2021, 251, 117103. [Google Scholar] [CrossRef]
- Li, Y.; Hou, H.; Liu, Z.; Tang, W.; Wang, J.; Lu, L.; Fu, J.; Gao, D.; Zhao, F.; Gao, X.; et al. CD44 Targeting Nanodrug Based on Chondroitin Sulfate for Melanoma Therapy by Inducing Mitochondrial Apoptosis Pathways. Carbohydr. Polym. 2023, 320, 121255. [Google Scholar] [CrossRef]
- Liu, P.; Chen, N.; Yan, L.; Gao, F.; Ji, D.; Zhang, S.; Zhang, L.; Li, Y.; Xiao, Y. Preparation, Characterisation and in Vitro and in Vivo Evaluation of CD44-Targeted Chondroitin Sulphate-Conjugated Doxorubicin PLGA Nanoparticles. Carbohydr. Polym. 2019, 213, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Brusini, R.; Varna, M.; Couvreur, P. Advanced Nanomedicines for the Treatment of Inflammatory Diseases. Adv. Drug Deliv. Rev. 2020, 157, 161–178. [Google Scholar] [CrossRef]
- Crielaard, B.J.; Lammers, T.; Schiffelers, R.M.; Storm, G. Drug Targeting Systems for Inflammatory Disease: One for All, All for One. J. Control. Release 2012, 161, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Li, J.; Luo, Z.; Wang, H.; Ma, Z. Nanoparticle-Based Drug Delivery Systems for Inflammatory Bowel Disease Treatment. DDDT 2024, 18, 2921–2949. [Google Scholar] [CrossRef]
- Lee, Y.; Kim, J.; Kim, H.; Kang, S.; Yoon, J.-H.; Kim, D.-D.; Kim, Y.M.; Jung, Y. N-Succinylaspart-1-Yl Celecoxib Is a Potential Colon-Specific Prodrug of Celecoxib with Improved Therapeutic Properties. J. Pharm. Sci. 2012, 101, 1831–1842. [Google Scholar] [CrossRef] [PubMed]
- Seetharaman, G.; Kallar, A.R.; Vijayan, V.M.; Muthu, J.; Selvam, S. Design, Preparation and Characterization of pH-Responsive Prodrug Micelles with Hydrolyzable Anhydride Linkages for Controlled Drug Delivery. J. Colloid Interface Sci. 2017, 492, 61–72. [Google Scholar] [CrossRef]
- Dr. Reddy’s Laboratories Ltd. Process for Preparation of Celecoxib Crystalline Form. EP2363395, 7 September 2011. [Google Scholar]
- Ferro, L.J.; Miyake, P.S. Polymorphic Crystalline Forms of Celecoxib. US7476744, 13 January 2009. [Google Scholar]
- Liu, H.; Wu, S.; Yu, J.; Fan, D.; Ren, J.; Zhang, L.; Zhao, J. Reduction-Sensitive Micelles Self-Assembled from Amphiphilic Chondroitin Sulfate A-Deoxycholic Acid Conjugate for Triggered Release of Doxorubicin. Mater. Sci. Eng. C 2017, 75, 55–63. [Google Scholar] [CrossRef]
- Grisham, M.B. Oxidants and Free Radicals in Inflammatory Bowel Disease. Lancet 1994, 344, 859–861. [Google Scholar] [CrossRef]
- Middleton, S.J.; Shorthouse, M.; Hunter, J.O. Increased Nitric Oxide Synthesis in Ulcerative Colitis. Lancet 1993, 341, 465–466. [Google Scholar] [CrossRef]
- Singer, I.I.; Kawka, D.W.; Schloemann, S.; Tessner, T.; Riehl, T.; Stenson, W.F. Cyclooxygenase 2 Is Induced in Colonic Epithelial Cells in Inflammatory Bowel Disease. Gastroenterology 1998, 115, 297–306. [Google Scholar] [CrossRef]
- Cañas, N.; Gorina, R.; Planas, A.M.; Vergés, J.; Montell, E.; García, A.G.; López, M.G. Chondroitin Sulfate Inhibits Lipopolysaccharide-Induced Inflammation in Rat Astrocytes by Preventing Nuclear Factor Kappa B Activation. Neuroscience 2010, 167, 872–879. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qiao, Q.; Wan, X.; Li, J.; Chen, W.; Li, E.; Qiu, L.; Tu, H. Amphiphilic Celecoxib-Polysaccharide Delivery System for Enhanced Colon-Targeted Colitis Therapy. Pharmaceutics 2025, 17, 511. https://doi.org/10.3390/pharmaceutics17040511
Qiao Q, Wan X, Li J, Chen W, Li E, Qiu L, Tu H. Amphiphilic Celecoxib-Polysaccharide Delivery System for Enhanced Colon-Targeted Colitis Therapy. Pharmaceutics. 2025; 17(4):511. https://doi.org/10.3390/pharmaceutics17040511
Chicago/Turabian StyleQiao, Qiao, Xian Wan, Jie Li, Weijun Chen, Enxuan Li, Lipeng Qiu, and Huiming Tu. 2025. "Amphiphilic Celecoxib-Polysaccharide Delivery System for Enhanced Colon-Targeted Colitis Therapy" Pharmaceutics 17, no. 4: 511. https://doi.org/10.3390/pharmaceutics17040511
APA StyleQiao, Q., Wan, X., Li, J., Chen, W., Li, E., Qiu, L., & Tu, H. (2025). Amphiphilic Celecoxib-Polysaccharide Delivery System for Enhanced Colon-Targeted Colitis Therapy. Pharmaceutics, 17(4), 511. https://doi.org/10.3390/pharmaceutics17040511