
PROTAC Delivery Strategies for Overcoming Physicochemical Properties and Physiological Barriers in Targeted Protein Degradation

 , , and
, , and
Abstract
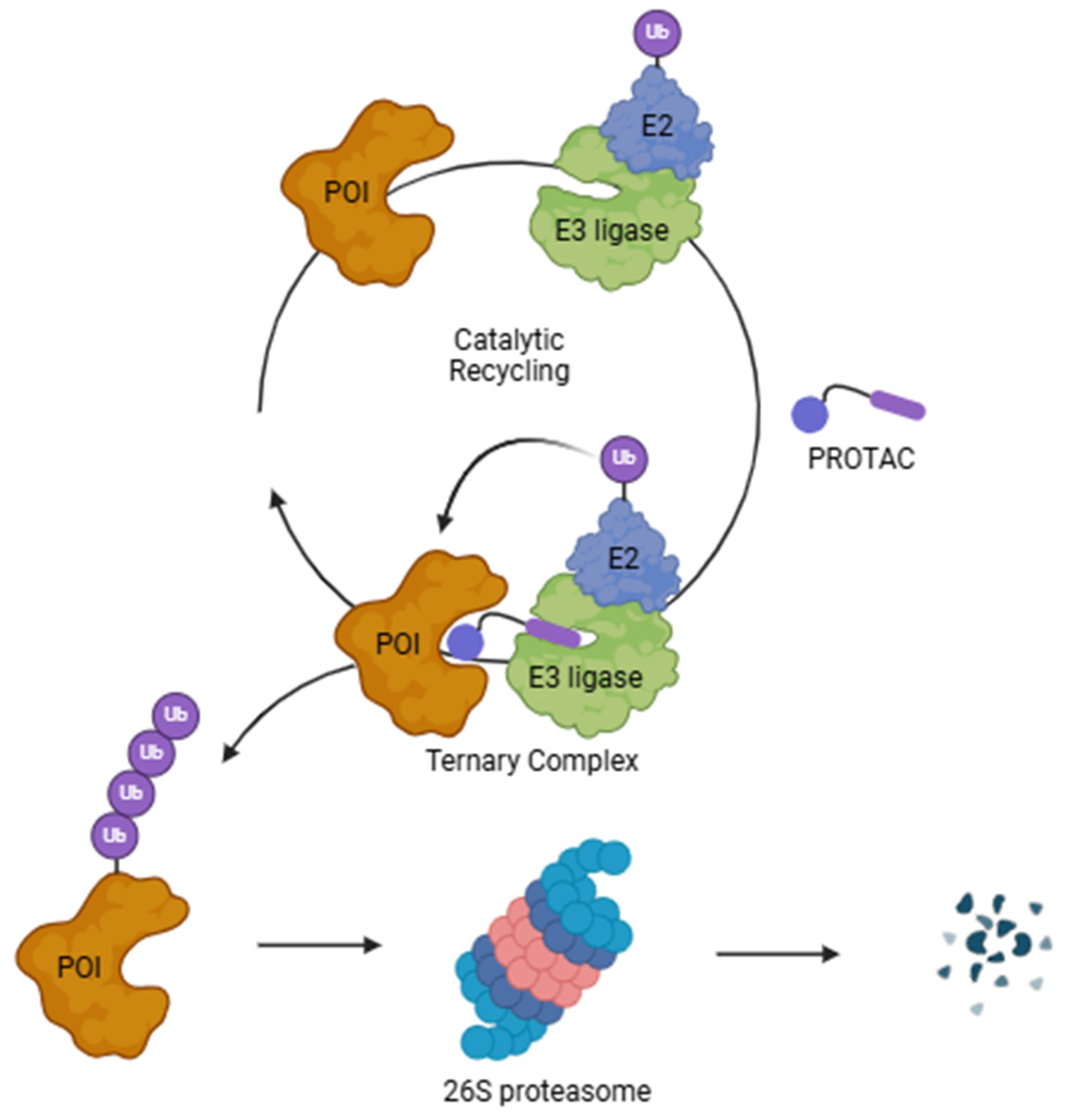
1. Introduction
2. Obstacles and Strategies for Optimal PROTAC Delivery
3. Delivery Systems for PROTACs
3.1. Polymeric Nanoparticle-Based Delivery Systems
3.2. Emulsion-Based Delivery Systems
3.3. Solid Dispersion-Based Delivery Systems
3.4. Lipid Nanoparticle-Bassed Delivery Systems
3.5. Liposome-Based Delivery Systems
3.6. Exosome-Based Delivery Systems
4. Summary
5. Discussion and Future Perspective
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACNPs | Antibody-conjugated nanoparticles |
| AP-NLC | ARV-825-loaded PEGylated NLCs |
| API | Active pharmaceutical ingredient |
| AR | Androgen receptor |
| ARV-SNEP | ARV-825-loaded self-nanoemulsifying preconcentrate |
| ASDs | Amorphous solid dispersion |
| BBB | Blood–brain barrier |
| CME | Camel milk-derived exosome |
| CRBN | Cereblon |
| DS-PLGA | Disulfide-linked poly (lactic-co-glycolic acid) |
| EPR | Enhanced permeability and retention |
| ER | Estrogen receptor |
| GALARV | Galactose-decorated nanoliposomal formulation |
| GSH | Glutathione |
| HCC | Hepatocellular carcinoma |
| LLCM | Lung cancer cell membrane |
| LNP | Lipid nanoparticles |
| MPRO | Folate-PEG-PROTAC micelles |
| MSPM | Mixed-shell polymeric micelle |
| MZ1-NPs | MZ1-loaded polymeric nanoparticles |
| nChap | Nanochaperone-based |
| NLC | Nanostructured lipid carrier |
| NSCLC | Non-small cell lung cancer |
| PCL | Polycaprolactone |
| PDSA | Poly(disulfide amide) |
| PEG | Polyethylene glycol |
| PEI | Polyethyleneimine |
| PEO-PPO-PEO | Poly(ethylene oxide)-poly(propylene oxide)-poly(ethylene oxide) |
| PGDAT | Self-assembling PROTAC nanoparticle |
| PLA | Polylactide |
| PLGA | Poly(lactic-co-glycolic acid) |
| POI | Protein of interest |
| PROTAC | Proteolysis targeting chimeras |
| PVA | Polyvinyl alcohol |
| PVP | Polyvinylpyrrolidone |
| RCNprotac | X-ray radiation responsive PROTAC nanomicelle |
| ROS | Reactive oxygen species |
| SNEDDS | Self-nanoemulsifying drug delivery system |
| SP | Substance P |
| TAMs | Tumor-associated macrophages |
| TNBC | Triple-negative breast cancer |
| UPS | Ubiquitin–proteosome system |
| VHL | Von Hippel–Lindau |
References
- Spradlin, J.N.; Zhang, E.; Nomura, D.K. Reimagining Druggability Using Chemoproteomic Platforms. Acc. Chem. Res. 2021, 54, 1801–1813. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Zhao, J.; Zhong, K.; Tong, A.; Jia, D. Targeted protein degradation: Mechanisms, strategies and application. Signal Transduct. Target. Ther. 2022, 7, 113. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Gao, H.; Yang, Y.; He, M.; Wu, Y.; Song, Y.; Tong, Y.; Rao, Y. PROTACs: Great opportunities for academia and industry. Signal Transduct. Target. Ther. 2019, 4, 64. [Google Scholar] [CrossRef]
- Santos, R.; Ursu, O.; Gaulton, A.; Bento, A.P.; Donadi, R.S.; Bologa, C.G.; Karlsson, A.; Al-Lazikani, B.; Hersey, A.; Oprea, T.I.; et al. A comprehensive map of molecular drug targets. Nat. Rev. Drug Discov. 2017, 16, 19–34. [Google Scholar] [CrossRef]
- Graham, H. The mechanism of action and clinical value of PROTACs: A graphical review. Cell Signal 2022, 99, 110446. [Google Scholar] [CrossRef]
- Xie, X.; Yu, T.; Li, X.; Zhang, N.; Foster, L.J.; Peng, C.; Huang, W.; He, G. Recent advances in targeting the “undruggable” proteins: From drug discovery to clinical trials. Signal Transduct. Target. Ther. 2023, 8, 335. [Google Scholar] [CrossRef]
- Xiao, M.; Zhao, J.; Wang, Q.; Liu, J.; Ma, L. Recent Advances of Degradation Technologies Based on PROTAC Mechanism. Biomolecules 2022, 12, 1257. [Google Scholar] [CrossRef]
- Li, K.; Crews, C.M. PROTACs: Past, present and future. Chem. Soc. Rev. 2022, 51, 5214–5236. [Google Scholar] [CrossRef]
- Ge, J.; Li, S.; Weng, G.; Wang, H.; Fang, M.; Sun, H.; Deng, Y.; Hsieh, C.-Y.; Li, D.; Hou, T. PROTAC-DB 3.0: An updated database of PROTACs with extended pharmacokinetic parameters. Nucleic Acids Res. 2024, 53, D1510–D1515. [Google Scholar] [CrossRef]
- Weng, G.; Cai, X.; Cao, D.; Du, H.; Shen, C.; Deng, Y.; He, Q.; Yang, B.; Li, D.; Hou, T. PROTAC-DB 2.0: An updated database of PROTACs. Nucleic Acids Res. 2023, 51, D1367–D1372. [Google Scholar] [CrossRef]
- Han, X.; Sun, Y. Strategies for the discovery of oral PROTAC degraders aimed at cancer therapy. Cell Rep. Phys. Sci. 2022, 3, 101062. [Google Scholar] [CrossRef]
- Sincere, N.I.; Anand, K.; Ashique, S.; Yang, J.; You, C. PROTACs: Emerging Targeted Protein Degradation Approaches for Advanced Druggable Strategies. Molecules 2023, 28, 4014. [Google Scholar] [CrossRef] [PubMed]
- Békés, M.; Langley, D.R.; Crews, C.M. PROTAC targeted protein degraders: The past is prologue. Nat. Rev. Drug Discov. 2022, 21, 181–200. [Google Scholar] [CrossRef] [PubMed]
- Mullard, A. Targeted protein degraders crowd into the clinic. Nat. Rev. Drug Discov. 2021, 20, 247–250. [Google Scholar] [CrossRef]
- Moon, Y.; Jeon, S.I.; Shim, M.K.; Kim, K. Cancer-Specific Delivery of Proteolysis-Targeting Chimeras (PROTACs) and Their Application to Cancer Immunotherapy. Pharmaceutics 2023, 15, 411. [Google Scholar] [CrossRef]
- Ezike, T.C.; Okpala, U.S.; Onoja, U.L.; Nwike, C.P.; Ezeako, E.C.; Okpara, O.J.; Okoroafor, C.C.; Eze, S.C.; Kalu, O.L.; Odoh, E.C.; et al. Advances in drug delivery systems, challenges and future directions. Heliyon 2023, 9, e17488. [Google Scholar] [CrossRef]
- Chen, Y.; Tandon, I.; Heelan, W.; Wang, Y.; Tang, W.; Hu, Q. Proteolysis-targeting chimera (PROTAC) delivery system: Advancing protein degraders towards clinical translation. Chem. Soc. Rev. 2022, 51, 5330–5350. [Google Scholar] [CrossRef]
- Fan, L.; Tong, W.; Wei, A.; Mu, X. Progress of proteolysis-targeting chimeras (PROTACs) delivery system in tumor treatment. Int. J. Biol. Macromol. 2024, 275, 133680. [Google Scholar] [CrossRef]
- An, S.; Fu, L. Small-molecule PROTACs: An emerging and promising approach for the development of targeted therapy drugs. eBioMedicine 2018, 36, 553–562. [Google Scholar] [CrossRef]
- Kumbhar, P.; Kolekar, K.; Kamble, V.; Umeyor, C.E.; Disouza, J.; Patravale, V.B. Future of Trends in the Design and Development of PROTAC. In PROTAC-Mediated Protein Degradation: A Paradigm Shift in Cancer Therapeutics; Nandave, M., Jain, P., Eds.; Springer Nature: Singapore, 2024; pp. 117–134. [Google Scholar]
- Pei, H.; Peng, Y.; Zhao, Q.; Chen, Y. Small molecule PROTACs: An emerging technology for targeted therapy in drug discovery. RSC Adv. 2019, 9, 16967–16976. [Google Scholar] [CrossRef]
- Cecchini, C.; Pannilunghi, S.; Tardy, S.; Scapozza, L. From Conception to Development: Investigating PROTACs Features for Improved Cell Permeability and Successful Protein Degradation. Front. Chem. 2021, 9, 672267. [Google Scholar] [CrossRef] [PubMed]
- Pu, C.; Wang, S.; Liu, L.; Feng, Z.; Zhang, H.; Gong, Q.; Sun, Y.; Guo, Y.; Li, R. Current strategies for improving limitations of proteolysis targeting chimeras. Chin. Chem. Lett. 2023, 34, 107927. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, Y.; Chen, W.; Wu, Y.; Xing, D. New-generation advanced PROTACs as potential therapeutic agents in cancer therapy. Mol. Cancer 2024, 23, 110. [Google Scholar] [CrossRef]
- Abeje, Y.E.; Wieske, L.H.E.; Poongavanam, V.; Maassen, S.; Atilaw, Y.; Cromm, P.; Lehmann, L.; Erdelyi, M.; Meibom, D.; Kihlberg, J. Impact of Linker Composition on VHL PROTAC Cell Permeability. J. Med. Chem. 2025, 68, 638–657. [Google Scholar] [CrossRef]
- Ciulli, A.; Trainor, N. A beginner’s guide to PROTACs and targeted protein degradation. Biochemist 2021, 43, 74–79. [Google Scholar] [CrossRef]
- Nalawansha, D.A.; Crews, C.M. PROTACs: An Emerging Therapeutic Modality in Precision Medicine. Cell Chem. Biol. 2020, 27, 998–1014. [Google Scholar] [CrossRef]
- Benowitz, A.B.; Scott-Stevens, P.T.; Harling, J.D. Challenges and opportunities for in vivo PROTAC delivery. Future Med. Chem. 2022, 14, 119–121. [Google Scholar] [CrossRef]
- Yang, W.; Saboo, S.; Zhou, L.; Askin, S.; Bak, A. Early evaluation of opportunities in oral delivery of PROTACs to overcome their molecular challenges. Drug Discov. Today 2024, 29, 103865. [Google Scholar] [CrossRef]
- O’ Donovan, D.H.; De Fusco, C.; Kuhnke, L.; Reichel, A. Trends in Molecular Properties, Bioavailability, and Permeability across the Bayer Compound Collection. J. Med. Chem. 2023, 66, 2347–2360. [Google Scholar] [CrossRef]
- Kim, C.H.; Lee, S.G.; Kang, M.J.; Lee, S.; Choi, Y.W. Surface modification of lipid-based nanocarriers for cancer cell-specific drug targeting. J. Pharm. Investig. 2017, 47, 203–227. [Google Scholar] [CrossRef]
- Hessa, T.; Sharma, A.; Mariappan, M.; Eshleman, H.D.; Gutierrez, E.; Hegde, R.S. Protein targeting and degradation are coupled for elimination of mislocalized proteins. Nature 2011, 475, 394–397. [Google Scholar] [CrossRef] [PubMed]
- Thapa, R.K.; Kim, J.O. Nanomedicine-based commercial formulations: Current developments and future prospects. J. Pharm. Investig. 2023, 53, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Sim, T.; Lim, C.; Hoang, N.H.; Joo, H.; Lee, J.W.; Kim, D.-w.; Lee, E.S.; Youn, Y.S.; Kim, J.O.; Oh, K.T. Nanomedicines for oral administration based on diverse nanoplatform. J. Pharm. Investig. 2016, 46, 351–362. [Google Scholar] [CrossRef]
- Haider, M.; Abdin, S.M.; Kamal, L.; Orive, G. Nanostructured Lipid Carriers for Delivery of Chemotherapeutics: A Review. Pharmaceutics 2020, 12, 288. [Google Scholar] [CrossRef]
- Yang, K.; Yu, G.; Yang, Z.; Yue, L.; Zhang, X.; Sun, C.; Wei, J.; Rao, L.; Chen, X.; Wang, R. Supramolecular Polymerization-Induced Nanoassemblies for Self-Augmented Cascade Chemotherapy and Chemodynamic Therapy of Tumor. Angew. Chem. Int. Ed. 2021, 60, 17570–17578. [Google Scholar] [CrossRef]
- Sánchez-Iglesias, A.; Grzelczak, M.; Altantzis, T.; Goris, B.; Pérez-Juste, J.; Bals, S.; Van Tendeloo, G.; Donaldson, S.H., Jr.; Chmelka, B.F.; Israelachvili, J.N.; et al. Hydrophobic Interactions Modulate Self-Assembly of Nanoparticles. ACS Nano 2012, 6, 11059–11065. [Google Scholar] [CrossRef]
- Tao, J.; Yi, C.; Dong, W.; Zhang, Y.; He, H.; Yang, Y.; Ye, S.; Wu, Q.; Shen, X.; Yang, F.; et al. Competitive hydrogen bonding and electrostatic interactions-mediated alternating nanoparticles copolymerization. Nano Res. 2025, 18, 94907086. [Google Scholar] [CrossRef]
- Droumaguet, B.L.; Grande, D. Diblock and Triblock Copolymers as Nanostructured Precursors to Functional Nanoporous Materials: From Design to Application. ACS Appl. Mater. Interfaces 2023, 15, 58023–58040. [Google Scholar] [CrossRef]
- Ma, X.; Williams, R.O. Polymeric nanomedicines for poorly soluble drugs in oral delivery systems: An update. J. Pharm. Investig. 2018, 48, 61–75. [Google Scholar] [CrossRef]
- Hoang, N.H.; Lim, C.; Sim, T.; Oh, K.T. Triblock copolymers for nano-sized drug delivery systems. J. Pharm. Investig. 2017, 47, 27–35. [Google Scholar] [CrossRef]
- Zhang, K.; Tang, X.; Zhang, J.; Lu, W.; Lin, X.; Zhang, Y.; Tian, B.; Yang, H.; He, H. PEG–PLGA copolymers: Their structure and structure-influenced drug delivery applications. J. Control. Release 2014, 183, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Behl, A.; Parmar, V.S.; Malhotra, S.; Chhillar, A.K. Biodegradable diblock copolymeric PEG-PCL nanoparticles: Synthesis, characterization and applications as anticancer drug delivery agents. Polymer 2020, 207, 122901. [Google Scholar] [CrossRef]
- Locatelli, E.; Comes Franchini, M. Biodegradable PLGA-b-PEG polymeric nanoparticles: Synthesis, properties, and nanomedical applications as drug delivery system. J. Nanoparticle Res. 2012, 14, 1316. [Google Scholar] [CrossRef]
- Saraswat, A.; Patki, M.; Fu, Y.; Barot, S.; Dukhande, V.V.; Patel, K. Nanoformulation of PROteolysis TArgeting Chimera targeting ’undruggable’ c-Myc for the treatment of pancreatic cancer. Nanomedicine 2020, 15, 1761–1777. [Google Scholar] [CrossRef]
- Yang, T.; Hu, Y.; Miao, J.; Chen, J.; Liu, J.; Cheng, Y.; Gao, X. A BRD4 PROTAC nanodrug for glioma therapy via the intervention of tumor cells proliferation, apoptosis and M2 macrophages polarization. Acta Pharm. Sin. B 2022, 12, 2658–2671. [Google Scholar] [CrossRef]
- Ruan, C.; Liu, L.; Lu, Y.; Zhang, Y.; He, X.; Chen, X.; Zhang, Y.; Chen, Q.; Guo, Q.; Sun, T.; et al. Substance P-modified human serum albumin nanoparticles loaded with paclitaxel for targeted therapy of glioma. Acta Pharm. Sin. B 2018, 8, 85–96. [Google Scholar] [CrossRef]
- Mohamed, S.; Parayath, N.N.; Taurin, S.; Greish, K. Polymeric nano-micelles: Versatile platform for targeted delivery in cancer. Ther. Deliv. 2014, 5, 1101–1121. [Google Scholar] [CrossRef]
- Aw, M.S.; Kurian, M.; Losic, D. Polymeric Micelles for Multidrug Delivery and Combination Therapy. Chem. Eur. J. 2013, 19, 12586–12601. [Google Scholar] [CrossRef]
- Shrestha, B.; Tang, L.; Romero, G. Nanoparticles-Mediated Combination Therapies for Cancer Treatment. Adv. Ther. 2019, 2, 1900076. [Google Scholar] [CrossRef]
- Choi, J.Y.; Thapa, R.K.; Yong, C.S.; Kim, J.O. Nanoparticle-based combination drug delivery systems for synergistic cancer treatment. J. Pharm. Investig. 2016, 46, 325–339. [Google Scholar] [CrossRef]
- Cimas, F.J.; Niza, E.; Juan, A.; Noblejas-López, M.D.; Bravo, I.; Lara-Sanchez, A.; Alonso-Moreno, C.; Ocaña, A. Controlled Delivery of BET-PROTACs: In Vitro Evaluation of MZ1-Loaded Polymeric Antibody Conjugated Nanoparticles in Breast Cancer. Pharmaceutics 2020, 12, 986. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Ma, F.; Zhang, Y.; Xu, M.; Xu, L.; Liu, Y.; Ma, R.; Shi, L. Synergizing CXCL9 with BRD4-PROTAC Using Nanochaperone Boosts Robust T Cell-Dependent Antitumor Immune Responses for Cancer Immunotherapy. Adv. Funct. Mater. 2024, 34, 2314203. [Google Scholar] [CrossRef]
- He, Y.; Ju, Y.; Hu, Y.; Wang, B.; Che, S.; Jian, Y.; Zhuo, W.; Fu, X.; Cheng, Y.; Zheng, S.; et al. Brd4 proteolysis-targeting chimera nanoparticles sensitized colorectal cancer chemotherapy. J. Control. Release 2023, 354, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Yu, J.; Hao, T.; Wang, W.; Wei, M.; Li, G. Advances in Polymeric Micelles: Responsive and Targeting Approaches for Cancer Immunotherapy in the Tumor Microenvironment. Pharmaceutics 2023, 15, 2622. [Google Scholar] [CrossRef]
- Dai, Y.; Chen, X.; Zhang, X. Recent advances in stimuli-responsive polymeric micelles via click chemistry. Polym. Chem. 2019, 10, 34–44. [Google Scholar] [CrossRef]
- Gao, J.; Jiang, X.; Lei, S.; Cheng, W.; Lai, Y.; Li, M.; Yang, L.; Liu, P.; Chen, X.-h.; Huang, M.; et al. A region-confined PROTAC nanoplatform for spatiotemporally tunable protein degradation and enhanced cancer therapy. Nat. Commun. 2024, 15, 6608. [Google Scholar] [CrossRef]
- Liu, H.-J.; Chen, W.; Wu, G.; Zhou, J.; Liu, C.; Tang, Z.; Huang, X.; Gao, J.; Xiao, Y.; Kong, N.; et al. Glutathione-Scavenging Nanoparticle-Mediated PROTACs Delivery for Targeted Protein Degradation and Amplified Antitumor Effects. Adv. Sci. 2023, 10, 2207439. [Google Scholar] [CrossRef]
- Zhang, H.-T.; Peng, R.; Chen, S.; Shen, A.; Zhao, L.; Tang, W.; Wang, X.-H.; Li, Z.-Y.; Zha, Z.-G.; Yi, M.; et al. Versatile Nano-PROTAC-Induced Epigenetic Reader Degradation for Efficient Lung Cancer Therapy. Adv. Sci. 2022, 9, 2202039. [Google Scholar] [CrossRef]
- Guan, X.; Xu, X.; Tao, Y.; Deng, X.; He, L.; Lin, Z.; Chang, J.; Huang, J.; Zhou, D.; Yu, X.; et al. Dual targeting and bioresponsive nano-PROTAC induced precise and effective lung cancer therapy. J. Nanobiotechnol. 2024, 22, 692. [Google Scholar] [CrossRef]
- Ma, J.; Fang, L.; Sun, Z.; Li, M.; Fan, T.; Xiang, G.; Ma, X. Folate-PEG-PROTAC Micelles for Enhancing Tumor-Specific Targeting Proteolysis In Vivo. Adv. Healthc. Mater. 2024, 13, 2400109. [Google Scholar] [CrossRef]
- Schaue, D.; McBride, W.H. Opportunities and challenges of radiotherapy for treating cancer. Nat. Rev. Clin. Oncol. 2015, 12, 527–540. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Yun, Y.; Li, C.; Ruan, Y.; Muraoka, O.; Xie, W.; Sun, X. Radiation responsive PROTAC nanoparticles for tumor-specific proteolysis enhanced radiotherapy. J. Mater. Chem. B 2024, 12, 3240–3248. [Google Scholar] [CrossRef] [PubMed]
- Kalepu, S.; Manthina, M.; Padavala, V. Oral lipid-based drug delivery systems—An overview. Acta Pharm. Sin. B 2013, 3, 361–372. [Google Scholar] [CrossRef]
- Kalepu, S.; Nekkanti, V. Insoluble drug delivery strategies: Review of recent advances and business prospects. Acta Pharm. Sin. B 2015, 5, 442–453. [Google Scholar] [CrossRef]
- Sripriya, R.; Raja, K.M.; Santhosh, G.; Chandrasekaran, M.; Noel, M. The effect of structure of oil phase, surfactant and co-surfactant on the physicochemical and electrochemical properties of bicontinuous microemulsion. J. Colloid Interface Sci. 2007, 314, 712–717. [Google Scholar] [CrossRef]
- Kumar, G.P.; Rajeshwarrao, P. Nonionic surfactant vesicular systems for effective drug delivery—An overview. Acta Pharm. Sin. B 2011, 1, 208–219. [Google Scholar] [CrossRef]
- Taher, S.; Al-Kinani, K.; Hammoudi, Z.; Ghareeb, M. Co-surfactant effect of polyethylene glycol 400 on microemulsion using BCS class II model drug. J. Adv. Pharm. Educ. Res. 2022, 12, 63–69. [Google Scholar] [CrossRef]
- Lim, C.; Lee, D.; Kim, M.; Lee, S.; Shin, Y.; Ramsey, J.D.; Choi, H.-G.; Lee, E.S.; Youn, Y.S.; Oh, K.T. Development of a sorafenib-loaded solid self-nanoemulsifying drug delivery system: Formulation optimization and characterization of enhanced properties. J. Drug Deliv. Sci. Technol. 2023, 82, 104374. [Google Scholar] [CrossRef]
- Mudassir, J.; Raza, A.; Khan, M.A.; Hameed, H.; Shazly, G.A.; Irfan, A.; Rana, S.J.; Abbas, K.; Arshad, M.S.; Muhammad, S.; et al. Design and Evaluation of Hydrophobic Ion Paired Insulin Loaded Self Micro-Emulsifying Drug Delivery System for Oral Delivery. Pharmaceutics 2023, 15, 1973. [Google Scholar] [CrossRef]
- Tran, P.; Park, J.-S. Recent trends of self-emulsifying drug delivery system for enhancing the oral bioavailability of poorly water-soluble drugs. J. Pharm. Investig. 2021, 51, 439–463. [Google Scholar] [CrossRef]
- Salawi, A. Self-emulsifying drug delivery systems: A novel approach to deliver drugs. Drug Deliv. 2022, 29, 1811–1823. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Kesarla, R.; Omri, A. Formulation strategies to improve the bioavailability of poorly absorbed drugs with special emphasis on self-emulsifying systems. Int. Sch. Res. Not. 2013, 2013, 848043. [Google Scholar] [CrossRef] [PubMed]
- Buya, A.B.; Beloqui, A.; Memvanga, P.B.; Préat, V. Self-Nano-Emulsifying Drug-Delivery Systems: From the Development to the Current Applications and Challenges in Oral Drug Delivery. Pharmaceutics 2020, 12, 1194. [Google Scholar] [CrossRef] [PubMed]
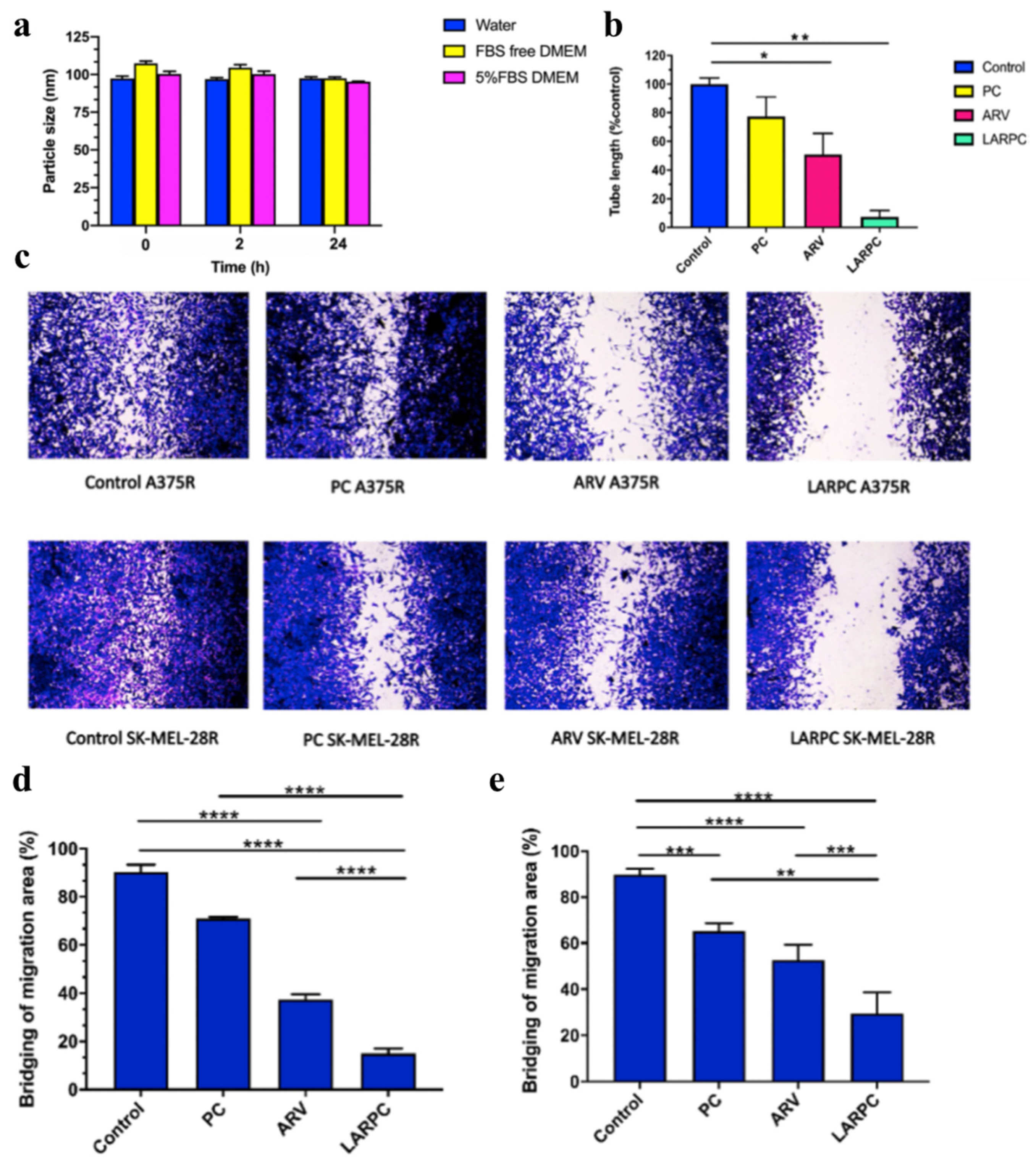
- Rathod, D.; Fu, Y.; Patel, K. BRD4 PROTAC as a novel therapeutic approach for the treatment of vemurafenib resistant melanoma: Preformulation studies, formulation development and in vitro evaluation. Eur. J. Pharm. Sci. 2019, 138, 105039. [Google Scholar] [CrossRef]
- Saraswat, A.; Vartak, R.; Hegazy, R.; Fu, Y.; Rao, T.J.R.; Billack, B.; Patel, K. Oral lipid nanocomplex of BRD4 PROteolysis TArgeting Chimera and vemurafenib for drug-resistant malignant melanoma. Biomed. Pharmacother. 2023, 168, 115754. [Google Scholar] [CrossRef]
- Zhang, J.; Guo, M.; Luo, M.; Cai, T. Advances in the development of amorphous solid dispersions: The role of polymeric carriers. Asian J. Pharm. Sci. 2023, 18, 100834. [Google Scholar] [CrossRef]
- He, Y.; Ho, C. Amorphous Solid Dispersions: Utilization and Challenges in Drug Discovery and Development. J. Pharm. Sci. 2015, 104, 3237–3258. [Google Scholar] [CrossRef]
- Van Duong, T.; Van den Mooter, G. The role of the carrier in the formulation of pharmaceutical solid dispersions. Part II: Amorphous carriers. Expert Opin. Drug Deliv. 2016, 13, 1681–1694. [Google Scholar] [CrossRef]
- Arpagaus, C. PLA/PLGA nanoparticles prepared by nano spray drying. J. Pharm. Investig. 2019, 49, 405–426. [Google Scholar] [CrossRef]
- Bapat, P.; Paul, S.; Tseng, Y.-C.; Taylor, L.S. Interplay of Drug–Polymer Interactions and Release Performance for HPMCAS-Based Amorphous Solid Dispersions. Mol. Pharm. 2024, 21, 1466–1478. [Google Scholar] [CrossRef]
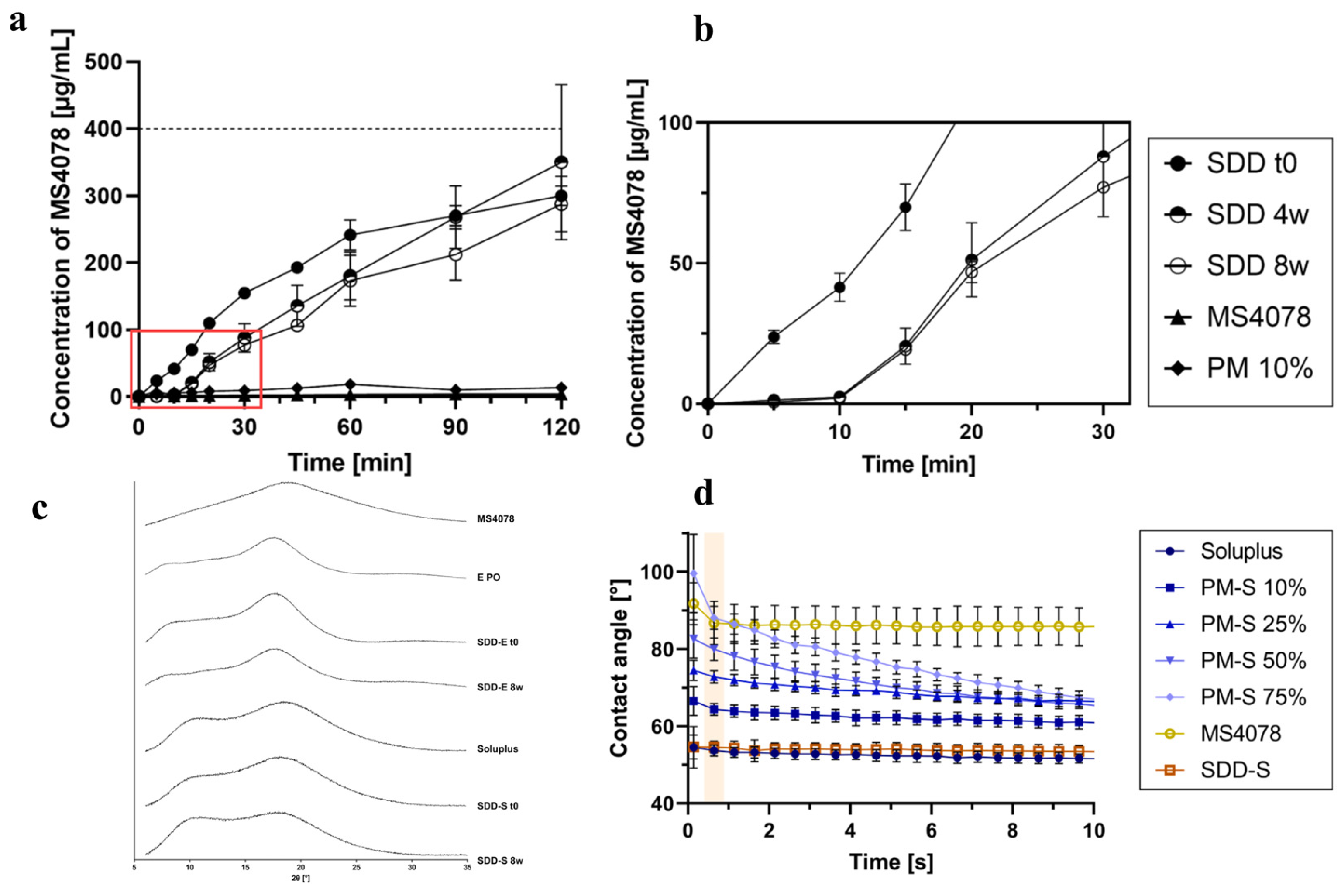
- Pöstges, F.; Kayser, K.; Appelhaus, J.; Monschke, M.; Gütschow, M.; Steinebach, C.; Wagner, K.G. Solubility Enhanced Formulation Approaches to Overcome Oral Delivery Obstacles of PROTACs. Pharmaceutics 2023, 15, 156. [Google Scholar] [CrossRef] [PubMed]
- Mareczek, L.; Mueller, L.K.; Halstenberg, L.; Geiger, T.M.; Walz, M.; Zheng, M.; Hausch, F. Use of Poly(vinyl alcohol) in Spray-Dried Dispersions: Enhancing Solubility and Stability of Proteolysis Targeting Chimeras. Pharmaceutics 2024, 16, 924. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, N.; Harms, M.; Mäder, K. ASDs of PROTACs: Spray-dried solid dispersions as enabling formulations. Int. J. Pharm. 2024, 650, 123725. [Google Scholar] [CrossRef] [PubMed]
- Mehta, M.; Bui, T.A.; Yang, X.; Aksoy, Y.; Goldys, E.M.; Deng, W. Lipid-Based Nanoparticles for Drug/Gene Delivery: An Overview of the Production Techniques and Difficulties Encountered in Their Industrial Development. ACS Mater. Au 2023, 3, 600–619. [Google Scholar] [CrossRef]
- Gupta, B.; Kim, J.O. Recent progress in cancer immunotherapy approaches based on nanoparticle delivery devices. J. Pharm. Investig. 2021, 51, 399–412. [Google Scholar] [CrossRef]
- Allen, T.M.; Cullis, P.R. Liposomal drug delivery systems: From concept to clinical applications. Adv. Drug Deliv. Rev. 2013, 65, 36–48. [Google Scholar] [CrossRef]
- Alfutaimani, A.S.; Alharbi, N.K.; Alahmari, A.S.; Alqabbani, A.A.; Aldayel, A.M. Exploring the landscape of Lipid Nanoparticles (LNPs): A comprehensive review of LNPs types and biological sources of lipids. Int. J. Pharm. X 2024, 8, 100305. [Google Scholar] [CrossRef]
- Lee, M.-K. Clinical usefulness of liposomal formulations in cancer therapy: Lessons from the experiences of doxorubicin. J. Pharm. Investig. 2019, 49, 203–214. [Google Scholar] [CrossRef]
- Liu, Y.; Bravo, K.M.C.; Liu, J. Targeted liposomal drug delivery: A nanoscience and biophysical perspective. Nanoscale Horiz. 2021, 6, 78–94. [Google Scholar] [CrossRef]
- Hegde, M.M.; Prabhu, S.; Mutalik, S.; Chatterjee, A.; Goda, J.S.; Satish Rao, B.S. Multifunctional lipidic nanocarriers for effective therapy of glioblastoma: Recent advances in stimuli-responsive, receptor and subcellular targeted approaches. J. Pharm. Investig. 2022, 52, 49–74. [Google Scholar] [CrossRef]
- Kiio, T.M.; Park, S. Physical properties of nanoparticles do matter. J. Pharm. Investig. 2021, 51, 35–51. [Google Scholar] [CrossRef]
- Vartak, R.; Saraswat, A.; Yang, Y.; Chen, Z.S.; Patel, K. Susceptibility of Lung Carcinoma Cells to Nanostructured Lipid Carrier of ARV-825, a BRD4 Degrading Proteolysis Targeting Chimera. Pharm. Res. 2022, 39, 2745–2759. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Qiu, M.; Ma, F.; Yang, L.; Glass, Z.; Xu, Q. Enhanced protein degradation by intracellular delivery of pre-fused PROTACs using lipid-like nanoparticles. J. Control. Release 2021, 330, 1244–1249. [Google Scholar] [CrossRef] [PubMed]
- Saraswat, A.; Vemana, H.P.; Dukhande, V.; Patel, K. Novel gene therapy for drug-resistant melanoma: Synergistic combination of PTEN plasmid and BRD4 PROTAC-loaded lipid nanocarriers. Mol. Ther. Nucleic Acids 2024, 35, 102292. [Google Scholar] [CrossRef]
- Liu, P.; Chen, G.; Zhang, J. A Review of Liposomes as a Drug Delivery System: Current Status of Approved Products, Regulatory Environments, and Future Perspectives. Molecules 2022, 27, 1372. [Google Scholar] [CrossRef]
- Singh, D.; Bedi, N.; Tiwary, A.K. Enhancing solubility of poorly aqueous soluble drugs: Critical appraisal of techniques. J. Pharm. Investig. 2018, 48, 509–526. [Google Scholar] [CrossRef]
- Akbarzadeh, A.; Rezaei-Sadabady, R.; Davaran, S.; Joo, S.W.; Zarghami, N.; Hanifehpour, Y.; Samiei, M.; Kouhi, M.; Nejati-Koshki, K. Liposome: Classification, preparation, and applications. Nanoscale Res. Lett. 2013, 8, 102. [Google Scholar] [CrossRef]
- Noh, G.; Keum, T.; Bashyal, S.; Seo, J.-E.; Shrawani, L.; Kim, J.H.; Lee, S. Recent progress in hydrophobic ion-pairing and lipid-based drug delivery systems for enhanced oral delivery of biopharmaceuticals. J. Pharm. Investig. 2022, 52, 75–93. [Google Scholar] [CrossRef]
- Nsairat, H.; Khater, D.; Sayed, U.; Odeh, F.; Al Bawab, A.; Alshaer, W. Liposomes: Structure, composition, types, and clinical applications. Heliyon 2022, 8, e09394. [Google Scholar] [CrossRef]
- Almeida, B.; Nag, O.; Rogers, K.; Delehanty, J. Recent Progress in Bioconjugation Strategies for Liposome-Mediated Drug Delivery. Molecules 2020, 25, 5672. [Google Scholar] [CrossRef]
- Lee, M.-K. Liposomes for Enhanced Bioavailability of Water-Insoluble Drugs: In Vivo Evidence and Recent Approaches. Pharmaceutics 2020, 12, 264. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, B.; Pu, C.; Cui, J.; Huang, K.; Wang, H.; Zhao, Y. Nanoliposomal Bcl-xL proteolysis-targeting chimera enhances anti-cancer effects on cervical and breast cancer without on-target toxicities. Adv. Compos. Hybrid Mater. 2023, 6, 78. [Google Scholar] [CrossRef]
- Saraswat, A.; Vemana, H.P.; Dukhande, V.V.; Patel, K. Galactose-decorated liver tumor-specific nanoliposomes incorporating selective BRD4-targeted PROTAC for hepatocellular carcinoma therapy. Heliyon 2022, 8, e08702. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Jin, Y.; Wang, J.; Gu, M.; Wang, Y.; Zhang, X.; Zhang, Y.; Yu, W.; Liu, Y.; Yuan, W.-E.; et al. Co-delivery of PROTAC and siRNA via novel liposomes for the treatment of malignant tumors. J. Colloid Interface Sci. 2025, 678, 896–907. [Google Scholar] [CrossRef]
- Fu, Y.; Rathod, D.; Patel, K. Protein kinase C inhibitor anchored BRD4 PROTAC PEGylated nanoliposomes for the treatment of vemurafenib-resistant melanoma. Exp. Cell Res. 2020, 396, 112275. [Google Scholar] [CrossRef]
- Fu, Y.; Saraswat, A.; Wei, Z.; Agrawal, M.Y.; Dukhande, V.V.; Reznik, S.E.; Patel, K. Development of Dual ARV-825 and Nintedanib-Loaded PEGylated Nano-Liposomes for Synergistic Efficacy in Vemurafnib-Resistant Melanoma. Pharmaceutics 2021, 13, 1005. [Google Scholar] [CrossRef]
- Chen, X.; Li, F.; Cui, B.; Yan, Q.; Qiu, C.; Zhu, Z.; Wen, L.; Chen, W. Liposomes-mediated enhanced antitumor effect of docetaxel with BRD4-PROTAC as synergist for breast cancer chemotherapy/immunotherapy. Int. J. Pharm. 2025, 668, 124973. [Google Scholar] [CrossRef]
- Mukerjee, N.; Maitra, S.; Ghosh, A.; Alexiou, A.; Thorat, N.D. Exosome-mediated PROTAC delivery for treatment of RNA viral infections and zoonosis. Drug Discov. Today 2024, 29, 104044. [Google Scholar] [CrossRef]
- Mukerjee, N.; Maitra, S.; Ghosh, A.; Subramaniyan, V.; Sharma, R. Exosome-mediated PROTACs delivery to target viral infections. Drug Dev. Res. 2023, 84, 1031–1036. [Google Scholar] [CrossRef]
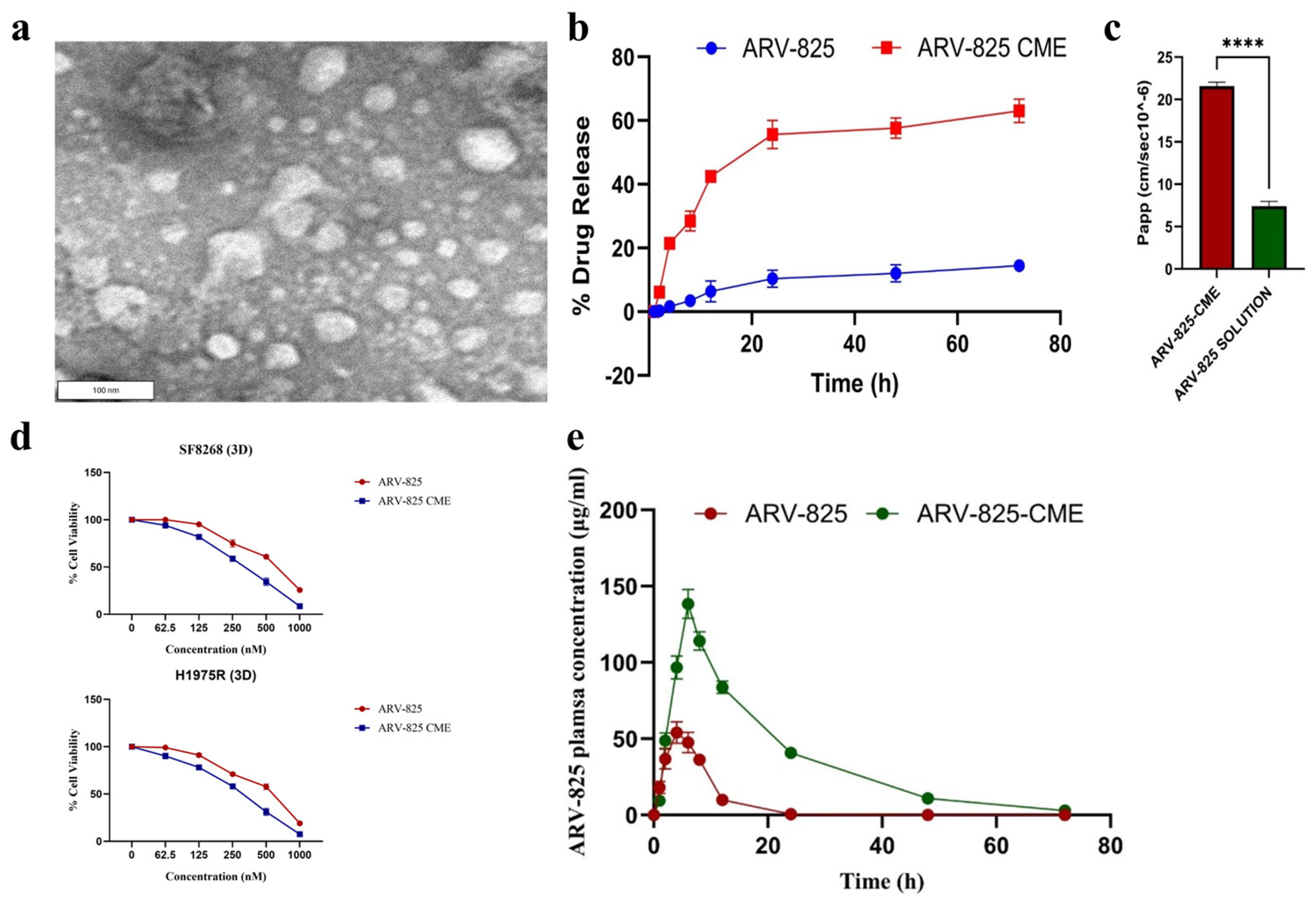
- Nathani, A.; Aare, M.; Sun, L.; Bagde, A.; Li, Y.; Rishi, A.; Singh, M. Unlocking the Potential of Camel Milk-Derived Exosomes as Novel Delivery Systems: Enhanced Bioavailability of ARV-825 PROTAC for Cancer Therapy. Pharmaceutics 2024, 16, 1070. [Google Scholar] [CrossRef]
- Aparna, T.N.; Kumar, R.; Ali, S.R.; Patel, D.J.; Julekha, K.; Begum, T.; Bala, J.; Kumar, P. Silica Nanoparticles: A Promising Vehicle for Anti-Cancer Drugs Delivery. AAPS PharmSciTech 2025, 26, 33. [Google Scholar] [CrossRef] [PubMed]
- Shabnum, S.S.; Siranjeevi, R.; Raj, C.K.; Nivetha, P.; Benazir, K. A Comprehensive Review on Recent Progress in Carbon Nanotubes for Biomedical Application. Environ. Qual. Manag. 2025, 34, e70040. [Google Scholar] [CrossRef]
- Henning, N.J.; Boike, L.; Spradlin, J.N.; Ward, C.C.; Liu, G.; Zhang, E.; Belcher, B.P.; Brittain, S.M.; Hesse, M.J.; Dovala, D.; et al. Deubiquitinase-targeting chimeras for targeted protein stabilization. Nat. Chem. Biol. 2022, 18, 412–421. [Google Scholar] [CrossRef]
- Kabir, M.; Sun, N.; Hu, X.; Martin, T.C.; Yi, J.; Zhong, Y.; Xiong, Y.; Kaniskan, H.Ü.; Gu, W.; Parsons, R.; et al. Acetylation Targeting Chimera Enables Acetylation of the Tumor Suppressor p53. J. Am. Chem. Soc. 2023, 145, 14932–14944. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.-H.; Hu, Z.; An, E.; Okeke, I.; Zheng, S.; Luo, X.; Gong, A.; Jaime-Figueroa, S.; Crews, C.M. Modulation of Phosphoprotein Activity by Phosphorylation Targeting Chimeras (PhosTACs). ACS Chem. Biol. 2021, 16, 2808–2815. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PROTAC | POI | E3 Ligase | Clinical Phase | ROA | Diseases | Target Type | Clinical Trial Number |
|---|---|---|---|---|---|---|---|
| ARV-110 | AR | CRBN | Phase II/III | Oral | Prostate cancer | Nuclear receptor | NCT03888612 |
| ARV-471 | ER | CRBN | Phase III | Oral | Breast cancer | Nuclear receptor | NCT04072952 |
| ARV-766 | AR | - | Phase I/II | Oral | Prostate cancer | Nuclear receptor | NCT05067140 |
| AC682 | ER | CRBN | Phase I | Oral | Breast cancer | Nuclear receptor | NCT05080842 |
| CC-94676 | AR | CRBN | Phase I | Oral | Prostate cancer | Nuclear receptor | NCT04428788 |
| DT2216 | Bcl-xL | VHL | Phase I | I.V | Liquid and solid tumors | Anti-apoptotic protein | NCT04886622 |
| FHD-609 | BRD9 | CRBN | Phase I | I.V | Synovial sarcoma | Nuclear protein | NCT04965753 |
| KT-333 | STAT3 | - | Phase I | I.V | Liquid and solid tumors | Nuclear protein | NCT05225584 |
| KT-413 | IRAK4 | CRBN | Phase I | I.V | DLBCL (MYD88-mutant) | Serine/threonine kinase | NCT05233033 |
| KT-474 | IRAK4 | CRBN | Phase I | Oral | Autoimmune diseases | Serine/threonine kinase | NCT04772885 |
| NX-2127 | BTK | CRBN | Phase I | Oral | B cell malignancies | Non-receptor tyrosine kinase | NCT04830137 |
| NX-5948 | BTK | CRBN | Phase I | Oral | B cell malignancies and autoimmune diseases | Non-receptor tyrosine kinase | NCT05131022 |
| CFT8634 | BRD9 | CRBN | Phase I/II | Oral | Synovial sarcoma | Nuclear protein | NCT05355573 |
| CFT1946 | BRAF-V600X | CRBN | Phase I/II | Oral | Solid tumors | Serine/threonine kinase | NCT05668585 |
| CFT8919 | EGFR-L858R | CRBN | Phase I | Oral | Non-small cell lung cancer (NSCLC) | Receptor tyrosine kinase | NCT06641609 |
| CG001419 | TRK | CRBN | Phase I | Oral | Cancer and other indications | Receptor tyrosine kinase | NCT06636500 |
| PROTAC | POI | E3 Ligase | Diseases/Cell Lines | Particle Size (nm) | Zeta Potential (mV) | Delivery System | Improvements | Limitations | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| ARV-825 | BRD4 | - | Pancreatic cancer | 89.63 ± 16.39 | - | Polymeric nanoparticle | Prolonged half-life, enhanced cell permeability | - | [45] |
| ARV-825 | BRD4 | - | Glioma | 26.3 ± 0.7 | −13.3 ± 8.0 | Polymeric nanoparticle | Penetrates BBB, increased stability, and reduced toxicity | Slow drug release (26.68% at 96 h) | [46] |
| MZ1 | BRD4 | - | HER2-positive breast cancer | 114 ± 2.3 | 31.8 ± 0.5 | Polymeric nanoparticle | Targeted delivery | - | [52] |
| dBET6 | BRD4 | CRBN | CD8+ T cells | - | - | Polymeric nanoparticle | Minimized toxicity and enhanced stability | No formulation stability data | [53] |
| ARV-825 | BRD4 | CRBN | Colorectal cancer | 59.31 | −0.64 | Polymeric nanoparticle | Enhanced cell permeability and EPR effects | Drug release depends on redox-responsive release | [54] |
| ARV-771 | BRD4 | - | TNBC cells | Polymeric nanoparticle | Cell permeability enhancement | - | [57] | ||
| ARV-771 | BRD4 | VHL | HeLa and B16F10 cells | 118 | −32.1 | Polymeric nanoparticle | Improved solubility and intracellular delivery | Unstable particle size, low reproducibility | [58] |
| dBET6 | BRD4 | CRBN | Lung cancer | 229.71 ± 72.1 | −29.0 | Stimuli-responsive and NPs | Targeted delivery | Particle size is over 200 nm, high dependency on pH and GSH level | [59] |
| dBET6 | BRD4 | - | Lung cancer | - | - | Stimuli-responsive and NPs | High drug-loading capacity, improved stability | - | [60] |
| MS39 | EGFR | VHL | HCC-827 and PC-9 cells | 202 ± 1.7 | −7.1 ± 0.12 | Self-assembled NPs | Increased stability and cell permeability | Reproducibility issue | [61] |
| MZ1 | BRD4 | VHL | - | 141.80 ± 5.66 | - | Stimuli-responsive delivery | Enhanced permeability and EPR effect | Long and pricey development process | [62] |
| ARV-825 | BRD4 | CRBN | Vemurafenib-resistant melanoma cells | 45.02 | -3.78 | SNEDDS | Enhanced solubility | Rapid precipitation, high concentration of DMA, stability relies on the selection and balance of excipients | [75] |
| ARV-825 | BRD4 | - | Vemurafenib-resistant melanoma cells | - | - | Emulsion | Enhanced solubility | - | [76] |
| ARCC-4 | AR | VHL | - | - | - | ASDs | Enhanced solubility and stability | Low drug loading, high dependency on polymer and its concentration | [82] |
| ARV-110 and SelDeg51 | - | - | - | - | - | ASDs | Enhanced solubility and stability | Long-term stability issues, low drug loading, pH-dependent dissolution profiles | [83] |
| MS4078 | - | - | - | - | - | ASDs | Enhanced solubility and stability | Long-term stability issues | [84] |
| ARV-825 | BRD4 | CRBN | NSCLC | 56.33 ± 0.42 | −21 ± 1.24 | LNPs | Improved solubility, stability, and intracellular delivery | Long-term stability issues | [93] |
| ARV-771 | BRD4 | VHL | HeLa cells | - | - | LNPs | Cell permeability enhancement | Low encapsulation efficiency | [94] |
| ARV-825 | BRD4 | - | BRAFi-resistant melanoma cells | 100 | - | LNPs | Lowered dosing and improved safety | - | [95] |
| DT2216 | Bcl-xL | - | Cervical and breast cancer | ~100 | - | Liposome | Good bioavailability in cells, reduced off-target and side effect | systemic toxicity concerns due to long-term released (up to 120 h) | [103] |
| ARV-825 | BRD4 | CRBN | Vemurafenib-resistant melanoma cells | 93.83 ± 10.05 | −27.30 | Liposome | Improved solubility and stability | Low apoptotic effect (<50%) | [104] |
| DT2216 | Bcl-xL | - | - | 200–300 | - | Liposome | Improved solubility | Low encapsulation efficiency, formulation stability | [105] |
| ARV-825 | BRD4 | - | Vemurafenib-resistant melanoma cells | 105.25 ± 2.76 | 26.6 | Liposome | Enhanced stability and minimized side effects | - | [106] |
| ARV-825 | BRD4 | - | Vemurafenib-resistant melanoma cells | 111.1 ± 6.55 | 13.9 ± 6.62 | Liposome | Improved cell permeability and stability | Long-term stability issues | [107] |
| ARV-825 | BRD4 | - | Breast cancer | ~115.7 | −17.1 | Liposome | Increased solubility, reduced systemic toxicity | - | [108] |
| ARV-825 | - | - | - | 136.8 ± 1.94 | - | Exosome | Improved cellular uptake and cell permeability | Entrapment efficiency below 50% | [111] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Syahputra, E.W.; Lee, H.; Cho, H.; Park, H.J.; Park, K.-S.; Hwang, D. PROTAC Delivery Strategies for Overcoming Physicochemical Properties and Physiological Barriers in Targeted Protein Degradation. Pharmaceutics 2025, 17, 501. https://doi.org/10.3390/pharmaceutics17040501
Syahputra EW, Lee H, Cho H, Park HJ, Park K-S, Hwang D. PROTAC Delivery Strategies for Overcoming Physicochemical Properties and Physiological Barriers in Targeted Protein Degradation. Pharmaceutics. 2025; 17(4):501. https://doi.org/10.3390/pharmaceutics17040501
Chicago/Turabian StyleSyahputra, Endry Wahyu, Hyunji Lee, Hyukjun Cho, Hyun Jin Park, Kwang-Su Park, and Duhyeong Hwang. 2025. "PROTAC Delivery Strategies for Overcoming Physicochemical Properties and Physiological Barriers in Targeted Protein Degradation" Pharmaceutics 17, no. 4: 501. https://doi.org/10.3390/pharmaceutics17040501
APA StyleSyahputra, E. W., Lee, H., Cho, H., Park, H. J., Park, K.-S., & Hwang, D. (2025). PROTAC Delivery Strategies for Overcoming Physicochemical Properties and Physiological Barriers in Targeted Protein Degradation. Pharmaceutics, 17(4), 501. https://doi.org/10.3390/pharmaceutics17040501