
Recent Advances in Vitamin E TPGS-Based Organic Nanocarriers for Enhancing the Oral Bioavailability of Active Compounds: A Systematic Review

 , , , , and
, , , , and
Abstract
:
1. Introduction
2. Method
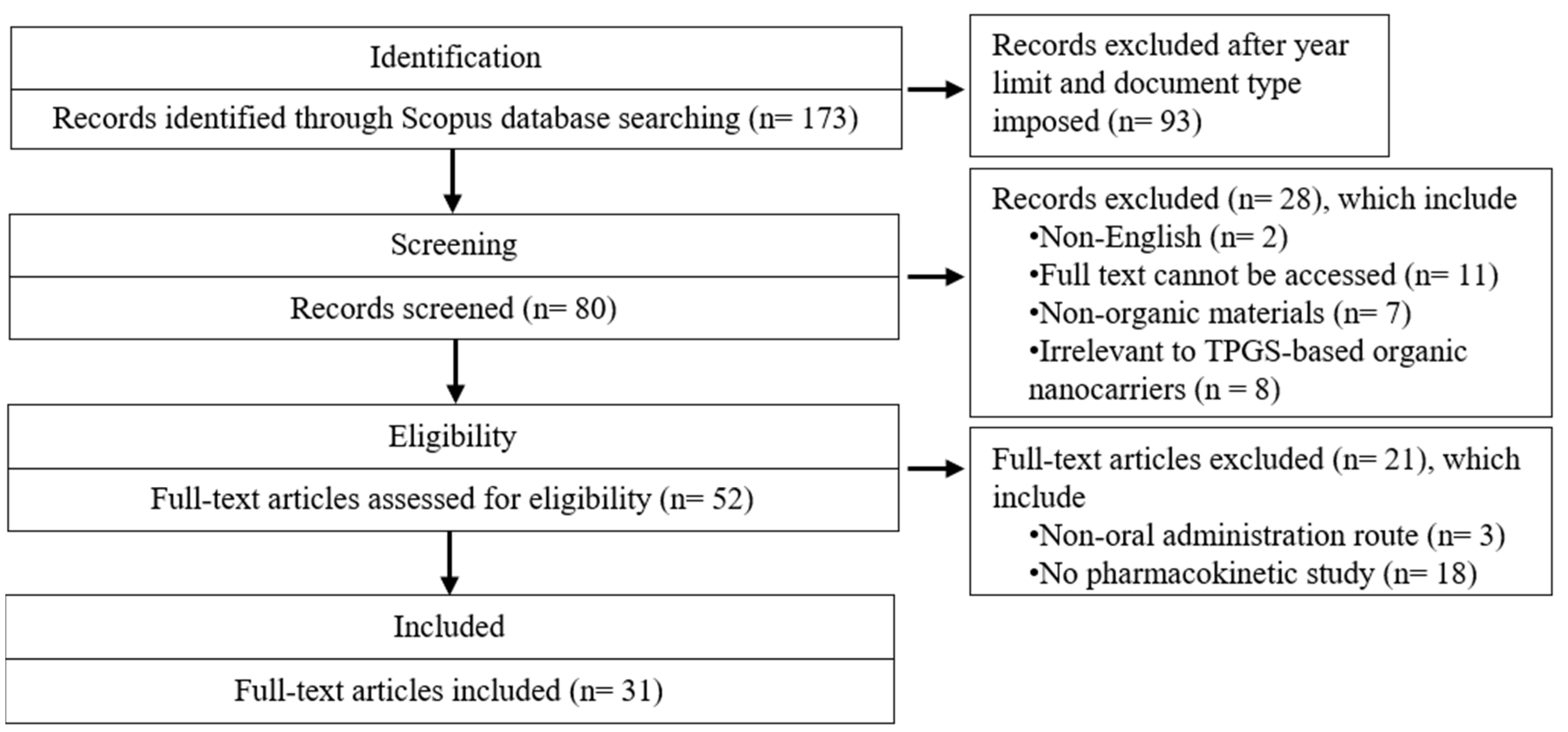
2.1. Search Strategy
2.2. Eligibility Criteria
2.3. Data Extraction and Variables of Interest
3. Main Content
3.1. Polymer-Based Nanocarrier and TPGS
3.1.1. Micelle and TPGS
3.1.2. Polymersome and TPGS
3.1.3. Amorphous Solid Dispersion and TPGS
3.2. Lipid-Based Nanocarrier and TPGS
3.2.1. Liposome and TPGS
3.2.2. Polymer–Lipid Hybrid Nanoparticle and TPGS
3.2.3. Niosome and TPGS
3.2.4. Solid Lipid Nanoparticle and TPGS
3.2.5. Lipid Nanocapsule and TPGS
3.2.6. Self-Emulsifying Drug Delivery System and TPGS
3.3. Nanocrystals and TPGS
3.4. Nanosuspensions and TPGS
4. Discussion
5. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Azman, M.; Sabri, A.H.; Anjani, Q.K.; Mustaffa, M.F.; Hamid, K.A. Intestinal Absorption Study: Challenges and Absorption Enhancement Strategies in Improving Oral Drug Delivery. Pharmaceuticals 2022, 15, 975. [Google Scholar] [CrossRef] [PubMed]
- Mir, M.; Ali, A.; Ahamad, R.; Khan, Z. Chemical Stability of Drugs: Unlocking the Future of Pharmacy and Nursing. In Futuristic Trends in Pharmacy & Nursing; IIP Series: Chikkamagaluru, India, 2024; pp. 59–65. [Google Scholar]
- Khadka, P.; Ro, J.; Kim, H.; Kim, I.; Kim, J.T.; Kim, H.; Cho, J.M.; Yun, G.; Lee, J. Pharmaceutical particle technologies: An approach to improve drug solubility, dissolution and bioavailability. Asian J. Pharm. Sci. 2014, 9, 304–316. [Google Scholar] [CrossRef]
- Lu, H.; Zhang, S.; Wang, J.; Chen, Q. A Review on Polymer and Lipid-Based Nanocarriers and Its Application to Nano-Pharmaceutical and Food-Based Systems. Front. Nutr. 2021, 8, 783831. [Google Scholar] [CrossRef] [PubMed]
- Uttreja, P.; Karnik, I.; Adel Ali Youssef, A.; Narala, N.; Elkanayati, R.M.; Baisa, S.; Alshammari, N.D.; Banda, S.; Vemula, S.K.; Repka, M.A. Self-Emulsifying Drug Delivery Systems (SEDDS): Transition from Liquid to Solid—A Comprehensive Review of Formulation, Characterization, Applications, and Future Trends. Pharmaceutics 2025, 17, 63. [Google Scholar] [CrossRef]
- Liu, Y.; Liang, Y.; Yuhong, J.; Xin, P.; Han, J.L.; Du, Y.; Yu, X.; Zhu, R.; Zhang, M.; Chen, W.; et al. Advances in Nanotechnology for Enhancing the Solubility and Bioavailability of Poorly Soluble Drugs. Drug Des. Devel Ther. 2024, 18, 1469–1495. [Google Scholar] [CrossRef]
- Mod Razif, M.R.F.; Chan, S.Y.; Chew, Y.-L.; Hassan, M.; Ahmad Hisham, S.; Abdul Rahman, S.; Mai, C.-W.; Teo, M.Y.M.; Kee, P.E.; Khoo, K.S.; et al. Recent Developments in Luteolin-Loaded Nanoformulations for Enhanced Anti-Carcinogenic Activities: Insights from In Vitro and In Vivo Studies. Science 2024, 6, 68. [Google Scholar] [CrossRef]
- Patra, J.K.; Das, G.; Fraceto, L.F.; Campos, E.V.R.; Rodriguez-Torres, M.d.P.; Acosta-Torres, L.S.; Diaz-Torres, L.A.; Grillo, R.; Swamy, M.K.; Sharma, S.; et al. Nano based drug delivery systems: Recent developments and future prospects. J. Nanobiotechnology 2018, 16, 71. [Google Scholar] [CrossRef]
- Mitchell, M.J.; Billingsley, M.M.; Haley, R.M.; Wechsler, M.E.; Peppas, N.A.; Langer, R. Engineering precision nanoparticles for drug delivery. Nat. Rev. Drug Discov. 2021, 20, 101–124. [Google Scholar] [CrossRef]
- Bartenstein, J.E.; Robertson, J.; Battaglia, G.; Briscoe, W.H. Stability of polymersomes prepared by size exclusion chromatography and extrusion. Colloids Surf. A Physicochem. Eng. Asp. 2016, 506, 739–746. [Google Scholar] [CrossRef]
- Li, J.; Wang, Z.; Zhang, H.; Gao, J.; Zheng, A. Progress in the development of stabilization strategies for nanocrystal preparations. Drug Deliv. 2021, 28, 19–36. [Google Scholar] [CrossRef]
- Guan, W.; Ma, Y.; Ding, S.; Liu, Y.; Song, Z.; Liu, X.; Tang, L.; Wang, Y. The technology for improving stability of nanosuspensions in drug delivery. J. Nanoparticle Res. 2022, 24, 14. [Google Scholar] [CrossRef]
- Kumbhar, P.S.; Nadaf, S.; Manjappa, A.S.; Jha, N.K.; Shinde, S.S.; Chopade, S.S.; Shete, A.S.; Disouza, J.I.; Sambamoorthy, U.; Kumar, S.A. D-ɑ-tocopheryl polyethylene glycol succinate: A review of multifarious applications in nanomedicines. OpenNano 2022, 6, 100036. [Google Scholar] [CrossRef]
- Yang, C.; Wu, T.; Qi, Y.; Zhang, Z. Recent Advances in the Application of Vitamin E TPGS for Drug Delivery. Theranostics 2018, 8, 464–485. [Google Scholar] [CrossRef]
- Pooja, D.; Kulhari, H.; Singh, M.K.; Mukherjee, S.; Rachamalla, S.S.; Sistla, R. Dendrimer–TPGS mixed micelles for enhanced solubility and cellular toxicity of taxanes. Colloids Surf. B Biointerfaces 2014, 121, 461–468. [Google Scholar] [CrossRef]
- Mod Razif, M.R.F.; Chan, S.Y.; Widodo, R.T.; Chew, Y.L.; Hassan, M.; Hisham, S.A.; Rahman, S.A.; Ming, L.C.; Tan, C.S.; Lee, S.K.; et al. Optimization of a Luteolin-Loaded TPGS/Poloxamer 407 Nanomicelle: The Effects of Copolymers, Hydration Temperature and Duration, and Freezing Temperature on Encapsulation Efficiency, Particle Size, and Solubility. Cancers 2023, 15, 3741. [Google Scholar] [CrossRef]
- Kesarwani, K.; Gupta, R.; Mukerjee, A. Bioavailability enhancers of herbal origin: An overview. Asian Pac. J. Trop. Biomed. 2013, 3, 253–266. [Google Scholar] [CrossRef]
- Ghadiri, M.; Young, P.M.; Traini, D. Strategies to Enhance Drug Absorption via Nasal and Pulmonary Routes. Pharmaceutics 2019, 11, 113. [Google Scholar] [CrossRef]
- Eckford, P.D.; Sharom, F.J. Interaction of the P-glycoprotein multidrug efflux pump with cholesterol: Effects on ATPase activity, drug binding and transport. Biochemistry 2008, 47, 13686–13698. [Google Scholar] [CrossRef]
- Liu, T.; Liu, X.; Xiong, H.; Xu, C.; Yao, J.; Zhu, X.; Zhou, J.; Yao, J. Mechanisms of TPGS and its derivatives inhibiting P-glycoprotein efflux pump and application for reversing multidrug resistance in hepatocellular carcinoma. Polym. Chem. 2018, 9, 1827–1839. [Google Scholar] [CrossRef]
- Ziyad, B.; Afsaneh, L. P-glycoprotein Inhibition as a Therapeutic Approach for Overcoming Multidrug Resistance in Cancer: Current Status and Future Perspectives. Curr. Cancer Drug Targets 2013, 13, 326–346. [Google Scholar] [CrossRef]
- Li, D.; Liu, S.; Zhu, J.; Shen, L.; Zhang, Q.y.; Zhu, H. Folic acid modified TPGS as a novel nano-micelle for delivery of nitidine chloride to improve apoptosis induction in Huh7 human hepatocellular carcinoma. BMC Pharmacol. Toxicol. 2021, 22, 1. [Google Scholar] [CrossRef]
- Rathod, S.; Bahadur, P.; Tiwari, S. Nanocarriers based on vitamin E-TPGS: Design principle and molecular insights into improving the efficacy of anticancer drugs. Int. J. Pharm. 2021, 592, 120045. [Google Scholar] [CrossRef] [PubMed]
- Shahab, M.S.; Rizwanullah, M.; Sarim Imam, S. Formulation, optimization and evaluation of vitamin E TPGS emulsified dorzolamide solid lipid nanoparticles. J. Drug Deliv. Sci. Technol. 2022, 68, 103062. [Google Scholar] [CrossRef]
- Farooq, M.A.; Trevaskis, N.L. TPGS Decorated Liposomes as Multifunctional Nano-Delivery Systems. Pharm. Res. 2023, 40, 245–263. [Google Scholar] [CrossRef]
- Antares Health Products, Inc. TPGS Regulatory Compliance. Available online: https://www.tpgs.com/tpgs-technical-info/tpgs-regulatory-status#:~:text=EFSA%20%2D%20The%20Scientific%20Panel%20on,special%20medical%20purposes%20(FSMP) (accessed on 25 February 2025).
- U.S. Food & Drug Administration. Inactive Ingredient Search for Approved Drug Products. Available online: https://www.accessdata.fda.gov/scripts/cder/iig/index.cfm?event=BasicSearch.page (accessed on 30 January 2025).
- European Food Safety Authority. Opinion of the Scientific Panel on food additives, flavourings, processing aids and materials in contact with food (AFC) related to D-alpha-tocopheryl polyethylene glycol 1000 succinate (TPGS) in use for food for particular nutritional purposes. EFSA J. 2007, 5, 490. [Google Scholar] [CrossRef]
- Government of Canada. The Safety of Vitamin E Supplements. Available online: https://www.canada.ca/en/health-canada/services/healthy-living/your-health/food-nutrition/safety-vitamin-supplements.html?utm_source=chatgpt.com (accessed on 30 January 2025).
- Mehata, A.K.; Setia, A.; Vikas; Malik, A.K.; Hassani, R.; Dailah, H.G.; Alhazmi, H.A.; Albarraq, A.A.; Mohan, S.; Muthu, M.S. Vitamin E TPGS-Based Nanomedicine, Nanotheranostics, and Targeted Drug Delivery: Past, Present, and Future. Pharmaceutics 2023, 15, 722. [Google Scholar] [CrossRef]
- Saboo, S.; Bapat, P.; Moseson, D.E.; Kestur, U.S.; Taylor, L.S. Exploring the Role of Surfactants in Enhancing Drug Release from Amorphous Solid Dispersions at Higher Drug Loadings. Pharmaceutics 2021, 13, 735. [Google Scholar] [CrossRef]
- European Medicines Agency. Chmp Assessment Report for Vedrop. Available online: https://s3-us-west-2.amazonaws.com/drugbank/cite_this/attachments/files/000/000/047/original/WC500047922.pdf?1525727854 (accessed on 30 January 2025).
- Yan, A.; Von Dem Bussche, A.; Kane, A.B.; Hurt, R.H. Tocopheryl Polyethylene Glycol Succinate as a Safe, Antioxidant Surfactant for Processing Carbon Nanotubes and Fullerenes. Carbon 2007, 45, 2463–2470. [Google Scholar] [CrossRef]
- Roszkowski, S.; Durczyńska, Z. Advantages and limitations of nanostructures for biomedical applications. Adv. Clin. Exp. Med. Off. Organ. Wroc. Med. Univ. 2024, 34, 447–456. [Google Scholar]
- George, A.; Shah, P.A.; Shrivastav, P.S. Natural biodegradable polymers based nano-formulations for drug delivery: A review. Int. J. Pharm. 2019, 561, 244–264. [Google Scholar] [CrossRef]
- Wang, X.; Zheng, Y.; Qiu, L.; Ouyang, H.; Xu, X.; Xu, W.; Zhang, Y.; Xu, W. Evaluation and antitumor mechanism of functionalized chitosan-based polymeric micelles for oral delivery of paclitaxel. Int. J. Pharm. 2022, 625, 122138. [Google Scholar] [CrossRef]
- Sampathi, S.; Rakesh, A.; Dodoala, S.; Vijaya, K. Biodegradable polymeric nanocarriers for oral delivery of antiretroviral drug: Pharmacokinetic and in vitro permeability studies. J. Appl. Pharm. Sci. 2021, 2021, 110405. [Google Scholar] [CrossRef]
- Sun, S.; Du, X.; Fu, M.; Khan, A.R.; Ji, J.; Liu, W.; Zhai, G. Galactosamine-modified PEG-PLA/TPGS micelles for the oral delivery of curcumin. Int. J. Pharm. 2021, 595, 120227. [Google Scholar] [CrossRef] [PubMed]
- Ge, Z.; Wang, Q.; Zhu, Q.; Yusif, M.; Yu, J.; Xu, X. Improved oral bioavailability, cellular uptake, and cytotoxic activity of zingerone via nano-micelles drug delivery system. J. Microencapsul. 2021, 38, 394–404. [Google Scholar] [CrossRef]
- Mahajan, H.S.; Patil, P.H. Central composite design-based optimization of lopinavir vitamin E-TPGS micelle: In vitro characterization and in vivo pharmacokinetic study. Colloids Surf. B Biointerfaces 2020, 194, 111149. [Google Scholar] [CrossRef]
- Shen, C.; Zhu, J.; Song, J.; Wang, J.; Shen, B.; Yuan, H.; Li, X. Formulation of pluronic F127/TPGS mixed micelles to improve the oral absorption of glycyrrhizic acid. Drug Dev. Ind. Pharm. 2020, 46, 1100–1107. [Google Scholar] [CrossRef]
- Piazzini, V.; Landucci, E.; Urru, M.; Chiarugi, A.; Pellegrini-Giampietro, D.E.; Bilia, A.R.; Bergonzi, M.C. Enhanced dissolution, permeation and oral bioavailability of aripiprazole mixed micelles: In vitro and in vivo evaluation. Int. J. Pharm. 2020, 583, 119361. [Google Scholar] [CrossRef]
- Chen, T.E.; Tu, L.; Wang, G.; Qi, N.; Wu, W.; Zhang, W.; Feng, J. Multi-functional chitosan polymeric micelles as oral paclitaxel delivery systems for enhanced bioavailability and anti-tumor efficacy. Int. J. Pharm. 2020, 578, 119105. [Google Scholar] [CrossRef]
- Zhen, L.; Wei, Q.; Wang, Q.; Zhang, H.; Adu-Frimpong, M.; Kesse Firempong, C.; Xu, X.; Yu, J. Preparation and in vitro/in vivo evaluation of 6-Gingerol TPGS/PEG-PCL polymeric micelles. Pharm. Dev. Technol. 2020, 25, 1–8. [Google Scholar] [CrossRef]
- Li, H.; Yan, L.; Tang, E.K.Y.; Zhang, Z.; Chen, W.; Liu, G.; Mo, J. Synthesis of TPGS/Curcumin Nanoparticles by Thin-Film Hydration and Evaluation of Their Anti-Colon Cancer Efficacy In Vitro and In Vivo. Front. Pharmacol. 2019, 10, 447207. [Google Scholar] [CrossRef]
- Wei, C.; Wang, Q.; Weng, W.; Wei, Q.; Xie, Y.; Adu-Frimpong, M.; Toreniyazov, E.; Ji, H.; Xu, X.; Yu, J. The characterisation, pharmacokinetic and tissue distribution studies of TPGS modified myricetrin mixed micelles in rats. J. Microencapsul. 2019, 36, 278–290. [Google Scholar] [CrossRef]
- Gu, S.-F.; Wang, L.-Y.; Tian, Y.-J.; Zhou, Z.-X.; Tang, J.-B.; Liu, X.-R.; Jiang, H.-P.; Shen, Y.-Q. Enhanced water solubility, antioxidant activity, and oral absorption of hesperetin by D-α-tocopheryl polyethylene glycol 1000 succinate and phosphatidylcholine. J. Zhejiang Univ. -Sci. B 2019, 20, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.Q.; Gong, Y.C.; Li, Z.L.; Li, Y.P.; Xiong, X.Y. Folate-conjugated pluronic/polylactic acid polymersomes for oral delivery of paclitaxel. Int. J. Biol. Macromol. 2019, 139, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Xi, Z.; Fei, Y.; Wang, Y.; Lin, Q.; Ke, Q.; Feng, G.; Xu, L. Solubility improvement of curcumin by crystallization inhibition from polymeric surfactants in amorphous solid dispersions. J. Drug Deliv. Sci. Technol. 2023, 83, 104351. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, J.; Liu, J.; Thant, Y.; Weng, W.; Wei, C.; Bao, R.; Adu-Frimpong, M.; Yu, Q.; Deng, W.; et al. Bisdemethoxycurcumin-conjugated vitamin E TPGS liposomes ameliorate poor bioavailability of free form and evaluation of its analgesic and hypouricemic activity in oxonate-treated rats. J. Nanoparticle Res. 2021, 23, 122. [Google Scholar] [CrossRef]
- Bao, R.; Wang, Q.-L.; Li, R.; Adu-Frimpong, M.; Toreniyazov, E.; Ji, H.; Xu, X.-M.; Yu, J.-N. Improved oral bioavailability and target delivery of 6-shogaol via vitamin E TPGS-modified liposomes: Preparation, in-vitro and in-vivo characterizations. J. Drug Deliv. Sci. Technol. 2020, 59, 101842. [Google Scholar] [CrossRef]
- Xie, Z.; Zhang, Z.; Lv, H. Rapamycin loaded TPGS-Lecithins-Zein nanoparticles based on core-shell structure for oral drug administration. Int. J. Pharm. 2019, 568, 118529. [Google Scholar] [CrossRef]
- Liu, Y.; Sun, C.; Li, W.; Adu-Frimpong, M.; Wang, Q.; Yu, J.; Xu, X. Preparation and Characterization of Syringic Acid–Loaded TPGS Liposome with Enhanced Oral Bioavailability and In Vivo Antioxidant Efficiency. AAPS PharmSciTech 2019, 20, 98. [Google Scholar] [CrossRef]
- Rizwanullah, M.; Perwez, A.; Alam, M.; Ahmad, S.; Mir, S.R.; Rizvi, M.M.A.; Amin, S. Polymer-lipid hybrid nanoparticles of exemestane for improved oral bioavailability and anti-tumor efficacy: An extensive preclinical investigation. Int. J. Pharm. 2023, 642, 123136. [Google Scholar] [CrossRef]
- Ren, T.; Zheng, X.; Bai, R.; Yang, Y.; Jian, L. Bioadhesive poly(methyl vinyl ether-co-maleic anhydride)-TPGS copolymer modified PLGA/lipid hybrid nanoparticles for improving intestinal absorption of cabazitaxel. Int. J. Pharm. 2022, 611, 121301. [Google Scholar] [CrossRef]
- Anwer, K.E.; El-Sattar, N.E.A.A.; Shamaa, M.M.; Zakaria, M.Y.; Beshay, B.Y. Design, Green Synthesis and Tailoring of Vitamin E TPGS Augmented Niosomal Nano-Carrier of Pyrazolopyrimidines as Potential Anti-Liver and Breast Cancer Agents with Accentuated Oral Bioavailability. Pharmaceuticals 2022, 15, 330. [Google Scholar] [CrossRef]
- Yang, F.; Hu, S.; Sheng, X.; Liu, Y. Naringenin loaded multifunctional nanoparticles to enhance the chemotherapeutic efficacy in hepatic fibrosis. Biomed. Microdevices 2020, 22, 68. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.; Mundada, V.; Sawant, K. Enhanced intestinal absorption of asenapine maleate by fabricating solid lipid nanoparticles using TPGS: Elucidation of transport mechanism, permeability across Caco-2 cell line and in vivo pharmacokinetic studies. Artif. Cells Nanomed. Biotechnol. 2019, 47, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Bapat, P.; Ghadi, R.; Chaudhari, D.; Katiyar, S.S.; Jain, S. Tocophersolan stabilized lipid nanocapsules with high drug loading to improve the permeability and oral bioavailability of curcumin. Int. J. Pharm. 2019, 560, 219–227. [Google Scholar] [CrossRef]
- Ahmed, O.A.A.; El-Bassossy, H.M.; El-Sayed, H.M.; El-Hay, S.S.A. Rp-HPLC Determination of Quercetin in a Novel D-α-Tocopherol Polyethylene Glycol 1000 Succinate Based SNEDDS Formulation: Pharmacokinetics in Rat Plasma. Molecules 2021, 26, 1435. [Google Scholar] [CrossRef]
- Choi, M.-J.; Kim, J.S.; Yu, H.; Woo, M.R.; Choi, J.E.; Baek, K.; Kim, J.O.; Choi, Y.S.; Choi, H.-G.; Jin, S.G. Comparison of the physicochemical properties, aqueous solubility, and oral bioavailability of rivaroxaban-loaded high-pressure homogenised and Shirasu porous glass membrane emulsified solid self-nanoemulsifying drug delivery systems. J. Mol. Liq. 2022, 346, 117057. [Google Scholar] [CrossRef]
- Meher, J.G.; Dixit, S.; Singh, Y.; Pawar, V.K.; Konwar, R.; Saklani, R.; Chourasia, M.K. Paclitaxel-Loaded Colloidal Silica and TPGS-Based Solid Self-Emulsifying System Interferes Akt/mTOR Pathway in MDA-MB-231 and Demonstrates Anti-tumor Effect in Syngeneic Mammary Tumors. AAPS PharmSciTech 2020, 21, 313. [Google Scholar] [CrossRef]
- Liu, W.; Cheng, M.; Yuan, F.; He, J.; Feng, Y.; Jin, Y.; Feng, J.; Yang, S.; Tu, L. Enhancing oral bioavailability of andrographolide via sodium dodecyl sulfate and D-α-Tocopherol polyethylene glycol 1000 succinate copolymer modified nanocrystals. J. Drug Deliv. Sci. Technol. 2023, 79, 104006. [Google Scholar] [CrossRef]
- Liu, W.; Cheng, M.; Lu, Z.; Li, H.; Feng, Y.; Jin, Y.; Yang, S.; Feng, J.; Tu, L. Multi-functional chitosan copolymer modified nanocrystals as oral andrographolide delivery systems for enhanced bioavailability and anti-inflammatory efficacy. Drug Deliv. 2022, 29, 3432–3442. [Google Scholar] [CrossRef]
- Li, H.; Li, M.; Fu, J.; Ao, H.; Wang, W.; Wang, X. Enhancement of oral bioavailability of quercetin by metabolic inhibitory nanosuspensions compared to conventional nanosuspensions. Drug Deliv. 2021, 28, 1226–1236. [Google Scholar] [CrossRef]
- Na, Y.-G.; Pham, T.M.A.; Byeon, J.-J.; Kim, M.-K.; Han, M.-G.; Baek, J.-S.; Lee, H.-K.; Cho, C.-W. Development and evaluation of TPGS/PVA-based nanosuspension for enhancing dissolution and oral bioavailability of ticagrelor. Int. J. Pharm. 2020, 581, 119287. [Google Scholar] [CrossRef]
- Koopaie, M. 22-Nanoparticulate Systems for Dental Drug Delivery. In Nanoengineered Biomaterials for Advanced Drug Delivery; Mozafari, M., Ed.; Elsevier: Amsterdam, The Netherlands, 2020; pp. 525–559. [Google Scholar]
- Perumal, S.; Atchudan, R.; Lee, W. A Review of Polymeric Micelles and Their Applications. Polymers 2022, 14, 2510. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Liu, J.; Yu, X.; Li, J.; Lu, X.; Shen, T. Pluronic F127 and D-α-Tocopheryl Polyethylene Glycol Succinate (TPGS) Mixed Micelles for Targeting Drug Delivery across The Blood Brain Barrier. Sci. Rep. 2017, 7, 2964. [Google Scholar] [CrossRef]
- Ali, R.; Qamar, W.; Kalam, M.A.; Binkhathlan, Z. Soluplus–TPGS Mixed Micelles as a Delivery System for Brigatinib: Characterization and In Vitro Evaluation. ACS Omega 2024, 9, 41830–41840. [Google Scholar] [CrossRef] [PubMed]
- Konno, T.; Watanabe, J.; Ishihara, K. Enhanced solubility of paclitaxel using water-soluble and biocompatible 2-methacryloyloxyethyl phosphorylcholine polymers. J. Biomed. Mater. Res. A 2003, 65, 209–214. [Google Scholar] [CrossRef]
- Torne, S.J.; Ansari, K.A.; Vavia, P.R.; Trotta, F.; Cavalli, R. Enhanced oral paclitaxel bioavailability after administration of paclitaxel-loaded nanosponges. Drug Deliv. 2010, 17, 419–425. [Google Scholar] [CrossRef]
- Hu, Y.; Qiu, L. Polymersomes: Preparation and Characterization. In Pharmaceutical Nanotechnology: Basic Protocols; Weissig, V., Elbayoumi, T., Eds.; Springer: New York, NY, USA, 2019; pp. 247–265. [Google Scholar]
- Fonseca, M.; Jarak, I.; Victor, F.; Domingues, C.; Veiga, F.; Figueiras, A. Polymersomes as the Next Attractive Generation of Drug Delivery Systems: Definition, Synthesis and Applications. Materials 2024, 17, 319. [Google Scholar] [CrossRef]
- Zhang, J.; Guo, M.; Luo, M.; Cai, T. Advances in the development of amorphous solid dispersions: The role of polymeric carriers. Asian J. Pharm. Sci. 2023, 18, 100834. [Google Scholar] [CrossRef]
- Tambe, S.M.; Jain, D.D.; Meruva, S.; Rongala, G.; Juluri, A.; Nihalani, G.; Mamidi, H.K.; Nukala, P.K.; Bolla, P.K. Recent Advances in Amorphous Solid Dispersions: Preformulation, Formulation Strategies, Technological Advancements and Characterization. Pharmaceutics 2022, 14, 2203. [Google Scholar] [CrossRef]
- Sturm, L.; Poklar Ulrih, N. Basic Methods for Preparation of Liposomes and Studying Their Interactions with Different Compounds, with the Emphasis on Polyphenols. Int. J. Mol. Sci. 2021, 22, 6547. [Google Scholar] [CrossRef]
- Dymek, M.; Sikora, E. Liposomes as biocompatible and smart delivery systems–the current state. Adv. Colloid. Interface Sci. 2022, 309, 102757. [Google Scholar] [CrossRef]
- Nsairat, H.; Khater, D.; Sayed, U.; Odeh, F.; Al Bawab, A.; Alshaer, W. Liposomes: Structure, composition, types, and clinical applications. Heliyon 2022, 8, e09394. [Google Scholar] [CrossRef] [PubMed]
- Woodle, M. Sterically stabilized liposome therapeutics. Adv. Drug Deliv. Rev. 1995, 16, 249–265. [Google Scholar] [CrossRef]
- Karabasz, A.; Szuwarzyński, M.; Nowakowska, M.; Bzowska, M.; Lewandowska-Łańcucka, J. Stabilization of liposomes with silicone layer improves their elastomechanical properties while not compromising biological features. Colloids Surf. B Biointerfaces 2020, 195, 111272. [Google Scholar] [CrossRef]
- Zhang, L.; Granick, S. How to stabilize phospholipid liposomes (using nanoparticles). Nano Lett. 2006, 6, 694–698. [Google Scholar]
- Mukherjee, A.; Waters, A.K.; Kalyan, P.; Achrol, A.S.; Kesari, S.; Yenugonda, V.M. Lipid-polymer hybrid nanoparticles as a next-generation drug delivery platform: State of the art, emerging technologies, and perspectives. Int. J. Nanomed. 2019, 14, 1937–1952. [Google Scholar] [CrossRef]
- Dave, V.; Tak, K.; Sohgaura, A.; Gupta, A.; Sadhu, V.; Reddy, K.R. Lipid-polymer hybrid nanoparticles: Synthesis strategies and biomedical applications. J. Microbiol. Methods 2019, 160, 130–142. [Google Scholar] [CrossRef]
- Bashkeran, T.; Kamaruddin, A.H.; Ngo, T.X.; Suda, K.; Umakoshi, H.; Watanabe, N.; Nadzir, M.M. Niosomes in cancer treatment: A focus on curcumin encapsulation. Heliyon 2023, 9, e18710. [Google Scholar] [CrossRef]
- Ge, X.; Wei, M.; He, S.; Yuan, W.E. Advances of Non-Ionic Surfactant Vesicles (Niosomes) and Their Application in Drug Delivery. Pharmaceutics 2019, 11, 55. [Google Scholar] [CrossRef]
- Mawazi, S.M.; Ge, Y.; Widodo, R.T. Niosome Preparation Techniques and Structure—An Illustrated Review. Pharmaceutics 2025, 17, 67. [Google Scholar] [CrossRef]
- Duong, V.A.; Nguyen, T.T.; Maeng, H.J. Preparation of Solid Lipid Nanoparticles and Nanostructured Lipid Carriers for Drug Delivery and the Effects of Preparation Parameters of Solvent Injection Method. Molecules 2020, 25, 4781. [Google Scholar] [CrossRef]
- Mukherjee, S.; Ray, S.; Thakur, R.S. Solid lipid nanoparticles: A modern formulation approach in drug delivery system. Indian. J. Pharm. Sci. 2009, 71, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Beussink-Nelson, L.; Baldridge, A.S.; Hibler, E.; Bello, N.A.; Epps, K.; Cameron, K.A.; Lloyd-Jones, D.M.; Gooding, H.C.; Catov, J.M.; Rich-Edwards, J.W.; et al. Knowledge and perception of cardiovascular disease risk in women of reproductive age. Am. J. Prev. Cardiol. 2022, 11, 100364. [Google Scholar] [CrossRef] [PubMed]
- Urimi, D.; Hellsing, M.; Mahmoudi, N.; Söderberg, C.; Widenbring, R.; Gedda, L.; Edwards, K.; Loftsson, T.; Schipper, N. Structural Characterization Study of a Lipid Nanocapsule Formulation Intended for Drug Delivery Applications Using Small-Angle Scattering Techniques. Mol. Pharm. 2022, 19, 1068–1077. [Google Scholar] [CrossRef] [PubMed]
- Huynh, N.T.; Passirani, C.; Saulnier, P.; Benoit, J.P. Lipid nanocapsules: A new platform for nanomedicine. Int. J. Pharm. 2009, 379, 201–209. [Google Scholar] [CrossRef]
- Raman Kallakunta, V.; Dudhipala, N.; Nyavanandi, D.; Sarabu, S.; Yadav Janga, K.; Ajjarapu, S.; Bandari, S.; Repka, M.A. Formulation and processing of solid self-emulsifying drug delivery systems (HME S-SEDDS): A single-step manufacturing process via hot-melt extrusion technology through response surface methodology. Int. J. Pharm. 2023, 641, 123055. [Google Scholar] [CrossRef]
- Rani, E.R.; Radha, G.V. Insights into Novel Excipients of Self-Emulsifying Drug Delivery Systems and Their Significance: An Updated Review. Crit. Rev. Ther. Drug Carr. Syst. 2021, 38, 27–74. [Google Scholar] [CrossRef]
- Salawi, A. Self-emulsifying drug delivery systems: A novel approach to deliver drugs. Drug Deliv. 2022, 29, 1811–1823. [Google Scholar] [CrossRef]
- Maji, I.; Mahajan, S.; Sriram, A.; Medtiya, P.; Vasave, R.; Khatri, D.K.; Kumar, R.; Singh, S.B.; Madan, J.; Singh, P.K. Solid self emulsifying drug delivery system: Superior mode for oral delivery of hydrophobic cargos. J. Control. Release 2021, 337, 646–660. [Google Scholar] [CrossRef]
- Lu, Y.; Li, Y.; Wu, W. Injected nanocrystals for targeted drug delivery. Acta Pharm. Sin. B 2016, 6, 106–113. [Google Scholar] [CrossRef]
- Lhaglham, P.; Jiramonai, L.; Jia, Y.; Huang, B.; Huang, Y.; Gao, X.; Zhang, J.; Liang, X.-J.; Zhu, M. Drug nanocrystals: Surface engineering and its applications in targeted delivery. iScience 2024, 27, 111185. [Google Scholar] [CrossRef]
- Patel, V.R.; Agrawal, Y.K. Nanosuspension: An approach to enhance solubility of drugs. J. Adv. Pharm. Technol. Res. 2011, 2, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Jacob, S.; Nair, A.B.; Shah, J. Emerging role of nanosuspensions in drug delivery systems. Biomater. Res. 2020, 24, 3. [Google Scholar] [CrossRef]
- Liu, D.; Qiao, S.; Cheng, B.; Li, D.; Chen, J.; Wu, Q.; Pan, H.; Pan, W. Enhanced Oral Delivery of Curcumin via Vitamin E TPGS Modified Nanodiamonds: A Comparative Study on the Efficacy of Non-covalent and Covalent Conjugated Strategies. AAPS PharmSciTech 2020, 21, 187. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Tan, S.; Feng, S.S. Vitamin E TPGS as a molecular biomaterial for drug delivery. Biomaterials 2012, 33, 4889–4906. [Google Scholar] [CrossRef]
- Price, G.; Patel, D.A. Drug Bioavailability; StatPearls: Treasure Island, FL, USA, 2023. [Google Scholar]
- Danaei, M.; Dehghankhold, M.; Ataei, S.; Hasanzadeh Davarani, F.; Javanmard, R.; Dokhani, A.; Khorasani, S.; Mozafari, M.R. Impact of Particle Size and Polydispersity Index on the Clinical Applications of Lipidic Nanocarrier Systems. Pharmaceutics 2018, 10, 57. [Google Scholar] [CrossRef]
- Cohen, T.; Zemmour, C.; Cohen, O.T.; Benny, O. Elongated Particles Show a Preferential Uptake in Invasive Cancer Cells. Nanomaterials 2024, 14, 1891. [Google Scholar] [CrossRef]
- Sousa de Almeida, M.; Susnik, E.; Drasler, B.; Taladriz-Blanco, P.; Petri-Fink, A.; Rothen-Rutishauser, B. Understanding nanoparticle endocytosis to improve targeting strategies in nanomedicine. Chem. Soc. Rev. 2021, 50, 5397–5434. [Google Scholar] [CrossRef]
- Wu, J. The Enhanced Permeability and Retention (EPR) Effect: The Significance of the Concept and Methods to Enhance Its Application. J. Pers. Med. 2021, 11, 771. [Google Scholar] [CrossRef]
- Wang, D.; Jiang, Q.; Dong, Z.; Meng, T.; Hu, F.; Wang, J.; Yuan, H. Nanocarriers transport across the gastrointestinal barriers: The contribution to oral bioavailability via blood circulation and lymphatic pathway. Adv. Drug Deliv. Rev. 2023, 203, 115130. [Google Scholar] [CrossRef]
- Fatfat, Z.; Karam, M.; Maatouk, B.; Fahs, D.; Gali-Muhtasib, H. Chapter 7-Nanoliposomes as Safe and Efficient Drug Delivery Nanovesicles. In Advanced and Modern Approaches for Drug Delivery; Nayak, A.K., Hasnain, M.S., Laha, B., Maiti, S., Eds.; Academic Press: New York, NY, USA, 2023; pp. 159–197. [Google Scholar]
- Selvamani, V. Chapter 15-Stability Studies on Nanomaterials Used in Drugs. In Characterization and Biology of Nanomaterials for Drug Delivery; Mohapatra, S.S., Ranjan, S., Dasgupta, N., Mishra, R.K., Thomas, S., Eds.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 425–444. [Google Scholar]
- Piacentini, E. Encapsulation Efficiency. In Encyclopedia of Membranes; Drioli, E., Giorno, L., Eds.; Springer: Berlin/Heidelberg, Germany, 2016; pp. 706–707. [Google Scholar]
- Mirasol, F. Stability testing determines proper drug storage parameters. Pharm. Technol. Eur. 2018, 30, 34–37. [Google Scholar]
- Wu, L.; Zhang, J.; Watanabe, W. Physical and chemical stability of drug nanoparticles. Adv. Drug Deliv. Rev. 2011, 63, 456–469. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. ICH Harmonised Tripartite Guideline: Stability Testing of New Drug Substances and Products Q1A(R2) Current Step 4 Version. 2003, pp. 1–24. Available online: https://www.ema.europa.eu/en/ich-q1a-r2-stability-testing-new-drug-substances-drug-products-scientific-guideline (accessed on 30 March 2025).
- Parasuraman, S. Toxicological screening. J. Pharmacol. Pharmacother. 2011, 2, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Pognan, F.; Beilmann, M.; Boonen, H.C.M.; Czich, A.; Dear, G.; Hewitt, P.; Mow, T.; Oinonen, T.; Roth, A.; Steger-Hartmann, T.; et al. The evolving role of investigative toxicology in the pharmaceutical industry. Nat. Rev. Drug Discov. 2023, 22, 317–335. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Vayer, P.; Tanwar, S.; Poyet, J.-L.; Tsaioun, K.; Villoutreix, B.O. Drug discovery and development: Introduction to the general public and patient groups. Front. Drug Discov. 2023, 3, 1201419. [Google Scholar] [CrossRef]
- Kumar, R.; Islam, T.; Nurunnabi, M. Mucoadhesive carriers for oral drug delivery. J. Control Release 2022, 351, 504–559. [Google Scholar] [CrossRef]
- Neophytou, C.M.; Constantinou, A.I. Drug Delivery Innovations for Enhancing the Anticancer Potential of Vitamin E Isoforms and Their Derivatives. BioMed Res. Int. 2015, 2015, 584862. [Google Scholar] [CrossRef]
- Davis, S.; Davis, B.M.; Richens, J.L.; Vere, K.A.; Petrov, P.G.; Winlove, C.P.; O‘Shea, P. alpha-Tocopherols modify the membrane dipole potential leading to modulation of ligand binding by P-glycoprotein. J. Lipid Res. 2015, 56, 1543–1550. [Google Scholar] [CrossRef]
- Suk, J.S.; Xu, Q.; Kim, N.; Hanes, J.; Ensign, L.M. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv. Drug Deliv. Rev. 2016, 99, 28–51. [Google Scholar] [CrossRef]
- Lee, C.-C.; Su, Y.-C.; Ko, T.-P.; Lin, L.-L.; Yang, C.-Y.; Chang, S.S.-C.; Roffler, S.R.; Wang, A.H.J. Structural basis of polyethylene glycol recognition by antibody. J. Biomed. Sci. 2020, 27, 12. [Google Scholar] [CrossRef]
- Ramachandran, P.V.; Alawaed, A.A.; Hamann, H.J. Catalyst- and Stoichiometry-Dependent Deoxygenative Reduction of Esters to Ethers or Alcohols with Borane–Ammonia. Org. Lett. 2023, 25, 6902–6906. [Google Scholar] [CrossRef]
- Zhao, J.; Feng, S.-S. Effects of PEG tethering chain length of vitamin E TPGS with a Herceptin-functionalized nanoparticle formulation for targeted delivery of anticancer drugs. Biomaterials 2014, 35, 3340–3347. [Google Scholar] [CrossRef]
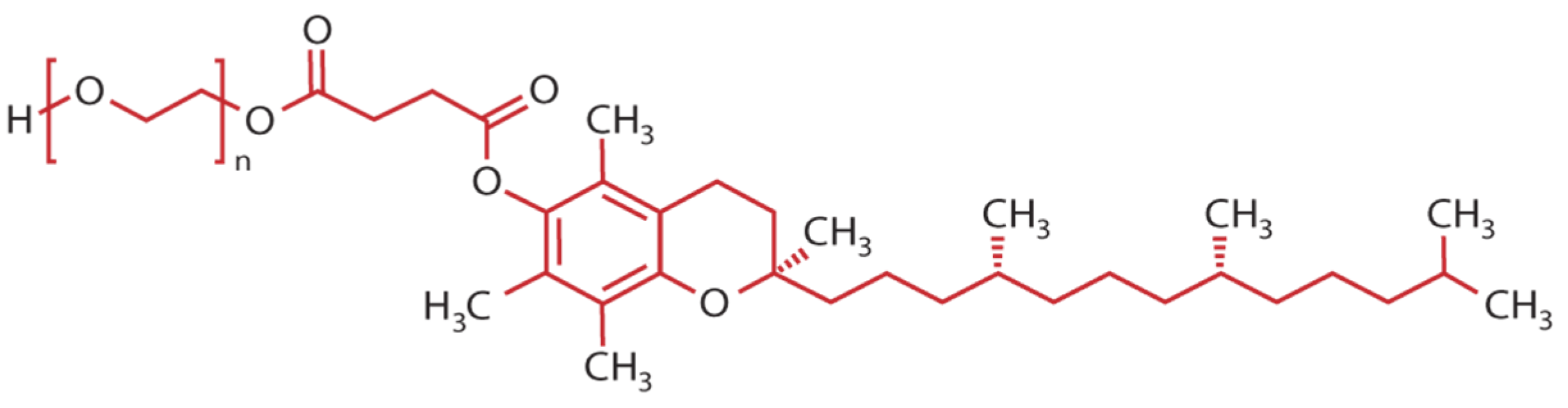
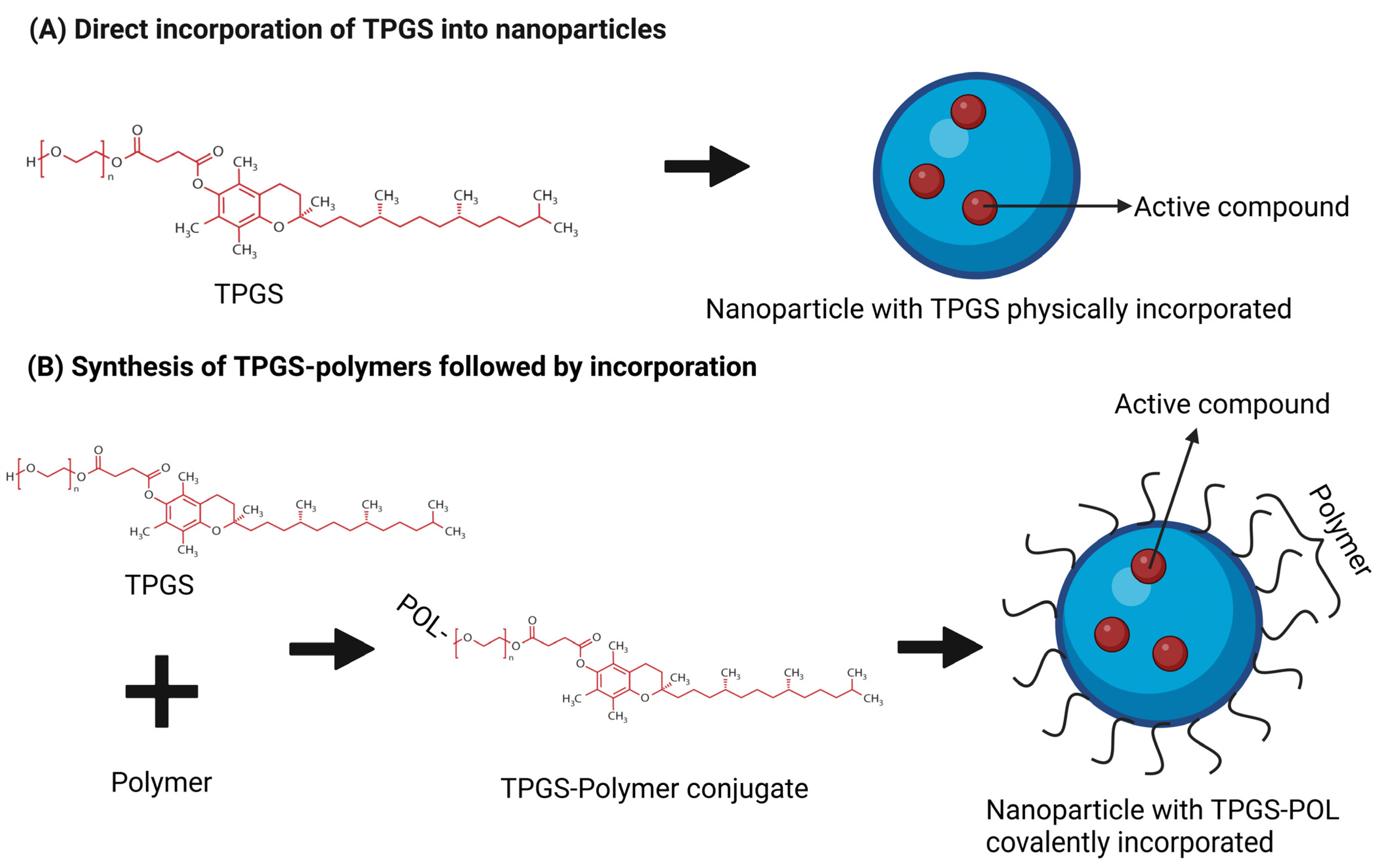

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Nanocarrier | Characteristics | ||
| Structure | Drug Loading | Stability | |
| Polymer-based | |||
| Micelle | Amphiphilic block copolymer self-assembled into a core–shell structure | Hydrophobic drug loaded into the core | Sensitive to dilution and environmental changes |
| Polymersome | Vesicle composed of amphiphilic block copolymers | Can entrap both hydrophilic and hydrophobic drugs | Enhanced stability compared to liposome |
| Amorphous solid dispersion | Drug dispersed in a polymer matrix in an amorphous state | Improve solubility of poorly water-soluble drugs | Thermodynamic instable and may crystallize over time |
| Lipid-based | |||
| Liposome | Phospholipid bilayer surrounding an aqueous core | Hydrophilic drug in core, hydrophobic drug in bilayer | Prone to fusion and leakage |
| Polymer lipid hybrid nanoparticle | Polymer core with a lipid shell | Can load both hydrophilic and hydrophobic drugs | Stable as it inherits advantages from both parental carriers |
| Niosome | Non-ionic surfactant vesicle with a bilayer structure | Hydrophilic drug within the core and hydrophobic drug between the bilayer | More stable than liposome |
| Solid lipid nanoparticle | Solid lipid core stabilized by surfactants | Can load both hydrophilic and hydrophobic drugs | Susceptible to lipid polymorphism-induced drug leakage |
| Lipid nanocapsule | Oily liquid core enclosed by a solid lipid-based shell | Lipophilic drug within the core | Highly stable |
| Self-emulsifying drug delivery system | Oil in water emulsion comprising a mixture of active compound, liquid oil, surfactant, and co-surfactant | Able to incorporate both hydrophilic and hydrophobic drugs | Thermodynamically stable |
| Nanocrystal | |||
| Nanocrystal | Crystalline drug particles, stabilized by surfactants | Incorporate hydrophobic drug | Require stabilizer to enhance stability |
| Nanosuspension | |||
| Nanosuspension | Nanodrug particles dispersed within aqueous or organic medium | Improve solubility of hydrophobic drug | Thermodynamically unstable |
| Challenges in Oral Active Compound Formulation | Potential of TPGS as an Oral Active Compound Bioavailability Enhancer |
|---|---|
| Solubility: Many active compounds have poor water solubility, leading to low bioavailability when administered orally. | The amphiphilic nature of TPGS enables it to form hydrogen bonding with hydrophilic active compounds and engage in hydrophobic interaction with lipophilic active compounds, allowing it to effectively dissolve both types of active compounds, making it a potent solubility enhancer. |
| Stability: Active compounds can degrade due to factors such as moisture, heat, light, and pH variations in the gastrointestinal tract. | Encapsulation of active compound within TPGS coating effectively forms a protective shield around the active compound, blocking the access of degradative enzymes to the active pharmaceutical compound. |
| First-pass metabolism: The liver metabolizes many active compounds before they reach systemic circulation, significantly reducing their bioavailability. | TPGS-containing formulation strategies such as proactive compounds, enzyme inhibitors, etc., are used to bypass or reduce first-pass metabolism. |
| Permeability: Some active compounds have low permeability across the gastrointestinal membrane. | TPGS enhances active compound permeability by inhibiting P-gp through modulation of membrane fluidity and P-gp ATPase inhibition. |
| Bioavailability: The fraction of the administered active compound that reaches the systemic circulation in an active form can be very low for some active compounds. | Enhancing solubility, using permeation enhancers, and employing active compound delivery systems like nanoparticles can improve bioavailability. |
| Common Absorption Enhancer | Mechanism of Action | Advantage(s) | Disadvantage(s) |
|---|---|---|---|
| TPGS | Solubilizes drug and inhibit P-gp efflux | Tailorable chemical modifications to meet the specific needs of drug delivery systems | Non-specific P-gp inhibition |
| Chitosan | Increases permeability of intestinal wall | Non-toxic and biodegradable | pH-sensitive |
| Bile salt | Reduces surface tension and increases drug solubility | Biocompatible and readily metabolized by the body | High concentration can cause significant membrane damage and local irritation |
| Chelating agent such as ethylene glycol tetraacetic acid and ethylene diamine tetraacetic acid | Bind to calcium ions, disrupting cell–cell contacts | Synergistic with other enhancers |
|
| Fatty acid and derivatives such as sodium caprate and salcaprozate sodium | Disrupt cellular tight junctions | Biocompatible and protects drug against degradation | Dose-dependent irritation and less useful for lipophilic drugs |
| Type of Nanocarrier | Study | Co-Ingredient | Preparation Method | Active Compound | Particle Size (nm) | Zeta Potential (mV) | Polydispersity Index | Encapsulation Efficiency (%) | Experimental Model | Dosage (mg/kg) | AUC (Free Form in Bracket) | AUC Increment as Compared to Free Form (-Fold) | T1/2 (h) (Free Form in Bracket) | Tmax (h) (Free Form in Bracket) | Cmax (Free Form in Bracket) | Storage Stability (Month) | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Micelle | 1 | Carboxymethyl chitosan (CMCS) and rhein | Sonication and dialysis | Paclitaxel | 193.0 ± 1.0 | –32.1 ± 0.4 | 0.126 ± 0.004 | 85.53 ± 3.36 | Sprague Dawley rat | 20 | 10.50 ± 1.96 (0.57 ± 0.23) µg/mL × h | 18.420 | N/A | 3.20 ± 0.45 (1.60 ± 0.89) | 1.14 ± 0.61 (0.13 ± 0.06) µg/mL | N/A | [36] |
| 2 | Poly(ε-caprolactone) (PCL) | Solvent emulsification–evaporation | Darunavir (DRV) | 173.74 ± 8.01 | −21.5 ± 0.212 | 0.218 ± 0.01 | 82.32 ± 4.18 | Sprague Dawley rat | 20 | 22,3031.61 ± 11.4 (65,248.79 ± 8.19) ng/mL × h | 3.42 | N/A | 12.14 ± 1.45 (5.07 ± 0.30) | 15,645.65 ± 2.03 (8467.36 ± 4.78) ng/mL | N/A | [37] | |
| 3 | Galactosamine, polyethylene glycol (PEG) and polylactic acid (PLA) | Thin-film dispersion | Curcumin | 100.02 ± 0.55 | −8.77 ± 0.73 | 0.127 ± 0.01 | 84.31 ± 0.11 | Female Wistar rat | 50 | 2. 20 ± 0.40 µg/mL × h | N/A | N/A | 1.17 ± 0.65 | 245.87 ± 0.17 ng/mL | N/A | [38] | |
| 4 | Phospholipid, sodium cholate and polyvinylpyrrolidone K30 (PVP K30) | Thin-film dispersion | Zingerone | 50.62 ± 0.25 | −28.07 ± 0.33 | 0.168 ± 0.006 | 94.71 ± 2.02 | Male Sprague Dawley rat | 300 | 44.80 ± 0.78 (8.79 ± 0.21) µg/mL × h | 5.10 | 3.22 ± 0.24 (1.7660:09) | 2 (0.5) | 8.86 ± 0.03 (4.2860:07) µg/mL | 1 | [39] | |
| 5 | NO | Thin-film hydration | Lopinavir | 91.71 | −24.8 | 0.129 | 99.36 ± 1.06 | New Zealand rabbit | 20 | 639.22 ± 28.39 (201.33 ± 17.68) ng/mL × h | 3.17 | 15.57 ± 4.80 (4.66 ± 0.27) | 1.54 ± 0.033 (1.55 ± 0.03) | 165.573 ± 3.05 (49.05 ± 2.53) ng/mL | 6 | [40] | |
| 6 | Pluronic F127 | Thin-film hydration | Glycyrrhizic acid (GL) | 27.41 ± 4.90 | −5.92 ± 0.68 | 0.19 ± 0.07 | 95.38 ± 3.22 | Male Sprague Dawley rat | 50 | 79.66 ± 10.83 (44.20 ± 15.25) mg/L × h | 1.80 | 4.83 ± 2.06 (4.58 ± 1.79) | 6.33 ± 0.82 (8.00 ± 0.00) | 8.16 ± 2.19 (4.83 ± 1.41) mg/L | N/A | [41] | |
| 7 | Soluplus, borneol | Thin-film hydration | Aripiprazole (ARP) | 52.37 ± 1.35 | −4.44 ± 0.32 | 0.07 ± 0.02 | 98.48 ± 2.39 | Male CD1 mice | 0.2 | 14,181 ± 551 (8463 ± 503) ng/mL × h | 1.68 | 17.50 ± 0.83 (13.40 ± 0.75) | 4 (4) | 738 ± 28.7 (437 ± 26.4) ng/mL | 3 | [42] | |
| 8 | Gallic acid and chitosan | Ultrasonic emulsification | Paclitaxel | 134.9 ± 10.2 | 34.8 ± 1.3 | 0.172 ± 0.130 | 80 ± 3 | Male Sprague Dawley rat | 11 | 2528 ± 294 (665 ± 129) ng/mL × h | 3.80 | 19.8 ± 10.4 (12.7 ± 3.4) | 3.0 ± 0 (1.5 ± 0) | 308 ± 103 (78 ± 34) ng/mL | N/A | [43] | |
| 9 | Poly (ethylene glycol)-poly (ε-caprolactone) (PEG-PCL) | Solvent injection | 6-gingerol | 73.24 ± 2.84 | −2.74 ± 0.92 | 0.129 ± 0.03 | 79.68 | Male Sprague Dawley rat | 250 | 497.36 ± 48.11 (89.88 ± 22.84) µg/mL × min | 5.53 | N/A | 0.25 (0.083) | 9.55 ± 1.12 (2.66 ± 0.19) µg/mL | N/A | [44] | |
| 10 | NO | Thin-film hydration | Curcumin | 12.3 ± 0.1 | N/A | 0.17 | N/A | Male Wistar rat | 150 | 3461.48 ± 102.47 (529.49 ± 22.32) ng/mL × h | 6.54 | 3.16 ± 0.78 (0.35 ± 0.07) | 2 (0) | 794.97 ± 43.94 (311.42 ± 15.51) ng/mL | 0.25 | [45] | |
| 11 | Phospholipids, sodium cholate and polyvinylpyrrolidone K30 (PVP K30) | Thin-film dispersion | Myricetin (MRC) | 26.42 ± 0.89 | −23.23 ± 0.79 | 0.135 ± 0.017 | 93.8 ± 1.18 | Male Sprague Dawley rat | 300 | 19.648 ± 2.779 (5.535 ± 0.729) µg/mL × h | 3.55 | 9.208 ± 0.233 (8.101 ± 0.0534) | 7 ± 0 (7 ± 0) | 1.630 ± 0.112 (0.584 ± 0.052) µg/mL | N/A | [46] | |
| 12 | NO | Solvent dispersion | Hesperetin | 26.19 ± 0.05 | N/A | 0.257 ± 0.024 | N/A | Female Sprague Dawley rat | 100 | 53.01 ± 4.39 (3.28 ± 0.68) µg/mL × h | 16.16 | 18.45 ± 17.86 (0.39 ± 0.00) | 0.39 ± 0.10 (0.61 ± 0.24) | 20.67 ± 8.27 (2.64 ± 0.76) µg/mL | N/A | [47] | |
| Polymersome | 13 | Folic acid (FA), Pluronic F127 and polylactic acid (PLA) | Direct injection and dialysis | Paclitaxel | 108.53 | N/A | 0.34 | 15.53 | Sprague Dawley rat | 0.15 | 3737.14 ± 631.58 (559.18 ± 113.90) ng/mL × h | 6.68 | N/A | 3.20 ± 1.34 (1.40 ± 0.55) | 228.31 ± 59.46 (51.72 ± 17.52) ng/mL | N/A | [48] |
| Amorphous solid dispersion | 14 | Kollidon CLSF | Solvent Evaporation | Curcumin | N/A | N/A | N/A | N/A | Sprague Dawley rat | 100 | 1186.0 ± 59.7 (724.3 ± 11.0) ng/mL × h | 1.64 | N/A | 0.33 (0.50) | 625.20 ± 66.98 (227.28 ± 22.28) ng/mL | 2 | [49] |
| Liposome | 15 | Lecithin and cholesterol | Thin-film hydration | Bisdemethoxycurcumin (BDMC) | 75.98 ± 5.46 | − 38.21 ± 0.29 | 0.35 ± 0.016 | 96.98 ± 0.17 | Male Sprague Dawley rat | 100 | 6.06 ± 1.18 (0.58 ± 0.18) mg/L × h | 10.45 | 5.7 ± 0.98 (1.512 ± 0.34) | 0.5 ± 0 (0.17 ± 0) | 1.38 ± 0.37 (0.4 ± 0.08) mg/L | N/A | [50] |
| 16 | Phospholipid, cholesterol, sodium cholate, isopropyl myristate (IPM) | Thin-film dispersion | 6-shogaol | 23.50 ± 0.09 | −45.40 ± 2.2 | 0.140 ± 0.003 | 95.18 ± 0.28 | Male Sprague Dawley rat | 200 | 2220.41 ± 24.21 (382.80 ± 47.24) µg/mL × min | 5.80 | 6.08 (3.25) | 1 (0.5) | 5.09 ± 0.24 (2.23 ± 0.16) µg/mL | N/A | [51] | |
| 17 | Zein and lecithin | Phase separation | Rapamycin | 190.31 ± 9.02 | −19.71 ± 1.125 | 0.256 ± 0.029 | 86.64 ± 2.43 | Male Sprague Dawley rat | 20 | 18,021.44 ± 1300.29 (7313.65 ± 934.83) ng/mL × h | 2.46 | 55.95 ± 3.39 (34.12 ± 2.65) | 2.00 ± 0.64 (4.00 ± 0.36) | 1052.05 ± 173.11 (516.80 ± 33.05) ng/mL | N/A | [52] | |
| 18 | Lecithin and cholesterol | Thin-film dispersion | Syringic acid (SA) | 40.01 ± 0.48 | − 38.50 ± 0.05 | 0.22 ± 0.02 | 96.48 ± 0.76 | Male Sprague Dawley rat | 25 | 338.08 ± 3.65 (120.58 ± 2.92) µg/mL × min | 2.80 | 0.83 (0.29) | 0.133 (0.133) | 4.50 ± 0.04 (4.50 ± 0.04) µg/mL | 1 | [53] | |
| Polymer Lipid Hybrid Nanoparticle | 19 | Polycaprolactone, phospholipon 90 G, poloxamer | Single-step nanoprecipitation | Exemestane | 136.37 ± 3.27 | N/A | 0.110 ± 0.013 | 88.56 ± 2.15 | Female Wistar rat | 20 | 2444.33 ± 204.66 (520.29 ± 122.29) ng/mL × h | 4.70 | 22.211 ± 0.754 (11.898 ± 1.025) h | 4 (2) h | 312.8 ± 18.21 (76.81 ± 9.95) ng/mL | N/A | [54] |
| 20 | Poly(lactic-co-glycolic acid) (PLGA), glyceryl monostearate, soybean lecithin, poly(methyl vinyl ether-co-maleic anhydride) (PVMMA) and poloxamer 188 | Emulsification-solvent evaporation | Cabazitaxel (CTX) | 192.2 ± 4.0 | −35.65 ± 2.46 | 0.241 ± 0.015 | 92.1 ± 3.7 | Male Sprague Dawley rat | 10 | 615.77 ± 296.87 (127.78 ± 76.77) µg/L × h | 4.82 | N/A | 1.08 ± 0.22 (0.30 ± 0.11) | 209.94 ± 76.18 (53.53 ± 34.97) µg/L | N/A | [55] | |
| Niosome | 21 | Span 60 or Span 40 and cholesterol | Thin-film hydration | Pyrazolopyrimidine | 138.9 ± 16.8 | −45.1 ± 8.9 | 0.21 ± 0.03 | 90.6 ± 5.1 | Male Wistar albino rat | 20 | 126.6 ± 16.3 (28.1 ± 8.2) mcg/mL × h | 4.51 | N/A | N/A | 9.2 ± 2.1 (2.52 ± 0.71) mcg/ml | N/A | [56] |
| Solid Lipid Nanoparticle | 22 | Glycerylmonooleate (GMO) | Emulsification and homogenization | Naringenin | 256 | −10.6 ± 2.1 | N/A | 72.70 ± 0.80 | Male Wistar rat | 20 | 15.0 ± 4.0 (2.0 ± 0.5) µg/mL × h | 7.5 | 2.3 (2.3) | N/A | 4.3 ± 1.2 (0.3 ± 0.1) µg/mL | N/A | [57] |
| 23 | Glyceryl monostearate (GMS) and Poloxamer 188 | High-speed homogenization followed by ultrasonication | Asenapine maleate (AM) | 114.3 ± 3.5 | −12.9 ± 3.8 | 0.188 ± 0.010 | 84.10 ± 2.90 | Female Sprague Dawley rat | 1.033 | 27,460.50 ± 151.90 (547.12 ± 28.47) ng/mL × h | 50.19 | 7.61 ± 1.19 (4.13 ± 0.58) | 8.00 ± 0.21 (1.00 ± 0.31) | 1396.44 ± 116.81 (67.19 ± 8.40) ng/mL | 3 | [58] | |
| Lipid nanocapsule | 24 | Maisine™ 35-1 | Antisolvent precipitation | Curcumin | 190 | N/A | 0.24 | 51.06 ± 7.27 | Male Sprague Dawley rat | 100 | 1174.42 ± 567.99 (95.64 ± 34.77) ng/mL × h | 12.28 | N/A | 2.0 ± 0.1 (0.5 ± 0.1) | 171.23 ± 67.88 (30.56 ± 10.22) ng/mL | 3 | [59] |
| Self-emulsifying drug delivery system (SEDDS) | 25 | Polyethylene glycol 200 (PEG 200) and pumpkin seed oil (PSO) | Probe sonication | Quercetin (QU) | 320 ± 34.3 | −28.6 ± 4.1 | 0.37 ± 0.07 | N/A | Male Wistar rat | 25 | 2286 ± 500.1 (1525.7 ± 378.8) µg/L × h | 1.50 | N/A | 0.5 ± 0.0 (0.83 ± 0.26) | 491.3 ± 172.2 (163.2 ± 74) µg/L | 0.5 | [60] |
| 26 | Peceol, Solutol HS15 (polyethylene glycol-15-hydroxystearate) and calcium silicate | High-pressure homogenization and spray drying | Rivaroxaban | 241.2 ± 26.0 | N/A | 0.184 ± 0.079 | N/A | Male Sprague Dawley rat | 0.5 | 554.11 ± 130.83 (156.49 ± 76.67) ng/mL × h | 3.54 | 1.32 ± 0.45 (2.88 ± 3.83) | 0.63 ± 0.14 (0.46 ± 0.19) | 320.92 ± 96.91 (80.45 ± 14.85) ng/mL | N/A | [61] | |
| 27 | Vitamin E, Labrafa, Capryol® 90 and Gelucire® | Mixing and Spray-drying | Paclitaxel | 30.00 ± 2.00 | 17.38 ± 2.88 | 0.198 ± 0.050 | 95.63 ± 3.36 | Female Sprague Dawley rat | 20 | 16,071.00 ± 2580.00 (2657.00 ± 208.80) ng/mL × h | 6.05 | N/A | 4.46 ± 0.59 (1.14 ± 0.16) | 1627.00 ± 281.50 (403.90 ± 78.02) ng/mL | 6 | [62] | |
| Nanocrystal | 28 | Sodium dodecyl sulfate (SDS) | High-pressure homogenization | Andrographolide (ADR) | 604.6 ± 5.7 | N/A | 0.198 ± 0.038 | N/A | Male Sprague Dawley rat | 50 | 3.428 ± 0.789 (1.286 ± 0.218) mg/L × h | 2.67 | 8.246 ± 2.915 (2.809 ± 0.232) h | 0.517 ± 0.32 (0.717 ± 0.415)5 h | 0.602 ± 0.146 (0.229 ± 0.082) mg/L | N/A | [63] |
| 29 | NO | 573.2 ± 3.9 | 46.27 ± 0.25 | 0.223 ± 0.024 | N/A | 4.602 ± 0.969 (1.286 ± 0.218) mg/L × h | 3.58 | 8.145 ± 1.227 (2.809 ± 0.232) h | 0.850 ± 0.224 (0.717 ± 0.415) h | 0.791 ± 0.330 (0.229 ± 0.082) mg/L | N/A | [64] | |||||
| Nanosuspension | 30 | NO | Tetragonal zirconia polycrystal (TZP) grinding method | Quercetin (QU) | 182.1 ± 1.5 | N/A | 0.21 ± 0.02 | N/A | Male Sprague Dawley rat | 50 | 387.09 ± 60.28 (89.93 ± 38.42) ng/mL × h | 4.30 | N/A | 0.61 ± 0.71 (0.53 ± 0.34) | 52.68 ± 16.87 (16.73 ± 6.33) ng/mL | 1 | [65] |
| 31 | Polyvinyl alcohol (PVA) | Antisolvent precipitation | Ticagrelor (TCG) | 233 ± 2 | –8.9 ± 0.5 | 0.173 ± 0.022 | N/A | Male Sprague Dawley rat | 10 | 2112.2 ± 268.1 (1026.5 ± 463.7) ng/mL × h | 2.06 | 4.4 ± 2.3 (4.3 ± 1.8) | 0.7 ± 0.5 (1.8 ± 1.3) | 571.3 ± 144.9 (189.0 ± 54.7) ng/mL | N/A | [66] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wong, C.N.; Lee, S.-K.; Lim, Y.M.; Yang, S.-B.; Chew, Y.-L.; Chua, A.-L.; Liew, K.B. Recent Advances in Vitamin E TPGS-Based Organic Nanocarriers for Enhancing the Oral Bioavailability of Active Compounds: A Systematic Review. Pharmaceutics 2025, 17, 485. https://doi.org/10.3390/pharmaceutics17040485
Wong CN, Lee S-K, Lim YM, Yang S-B, Chew Y-L, Chua A-L, Liew KB. Recent Advances in Vitamin E TPGS-Based Organic Nanocarriers for Enhancing the Oral Bioavailability of Active Compounds: A Systematic Review. Pharmaceutics. 2025; 17(4):485. https://doi.org/10.3390/pharmaceutics17040485
Chicago/Turabian StyleWong, Chee Ning, Siew-Keah Lee, Yang Mooi Lim, Shi-Bing Yang, Yik-Ling Chew, Ang-Lim Chua, and Kai Bin Liew. 2025. "Recent Advances in Vitamin E TPGS-Based Organic Nanocarriers for Enhancing the Oral Bioavailability of Active Compounds: A Systematic Review" Pharmaceutics 17, no. 4: 485. https://doi.org/10.3390/pharmaceutics17040485
APA StyleWong, C. N., Lee, S.-K., Lim, Y. M., Yang, S.-B., Chew, Y.-L., Chua, A.-L., & Liew, K. B. (2025). Recent Advances in Vitamin E TPGS-Based Organic Nanocarriers for Enhancing the Oral Bioavailability of Active Compounds: A Systematic Review. Pharmaceutics, 17(4), 485. https://doi.org/10.3390/pharmaceutics17040485