
Safety Profile, Toxicokinetic, and Intestinal Absorption Differences of a Naturally-Derived Anti-Rheumatic Drug, Sinomenine Hydrochloride, in Normal and Arthritic Rats
 and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Animals
2.3. Induction and Evaluation of AIA
2.4. Assessment of the Acute Toxicity
2.5. TK Studies
2.6. Preparation of Everted Intestinal Sacs
2.7. Plasma Protein Binding
2.8. Sample Processing
2.9. Analytical Method
2.10. Method Validation
2.10.1. Selectivity
2.10.2. Calibration Curves
2.10.3. Precision and Accuracy
2.10.4. Recovery and Matrix Effect
2.10.5. Stability
2.11. Statistical Analysis
3. Results
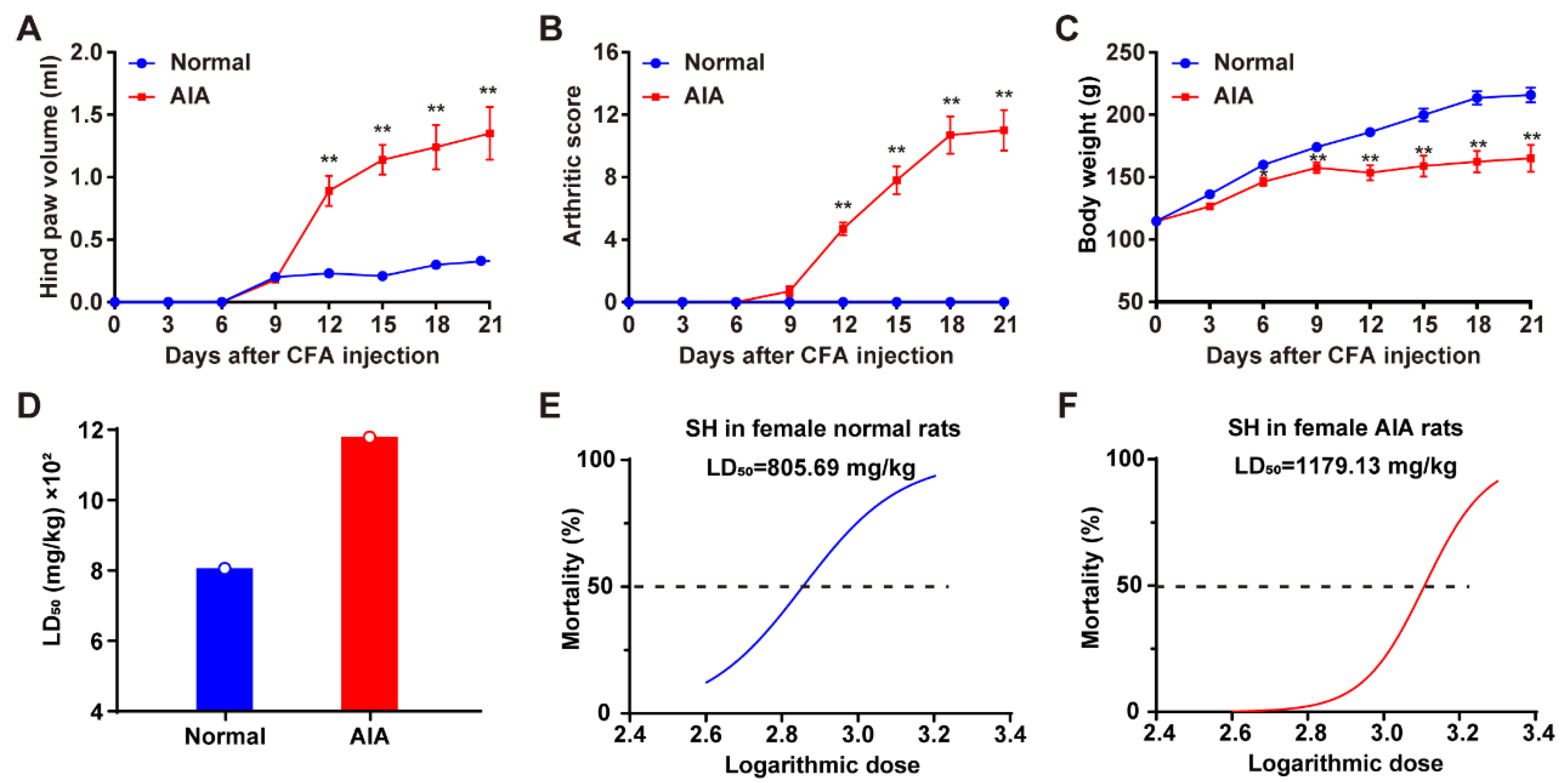
3.1. Induction of AIA and Assessment of the Acute Toxicity of SH
3.2. Method Validation
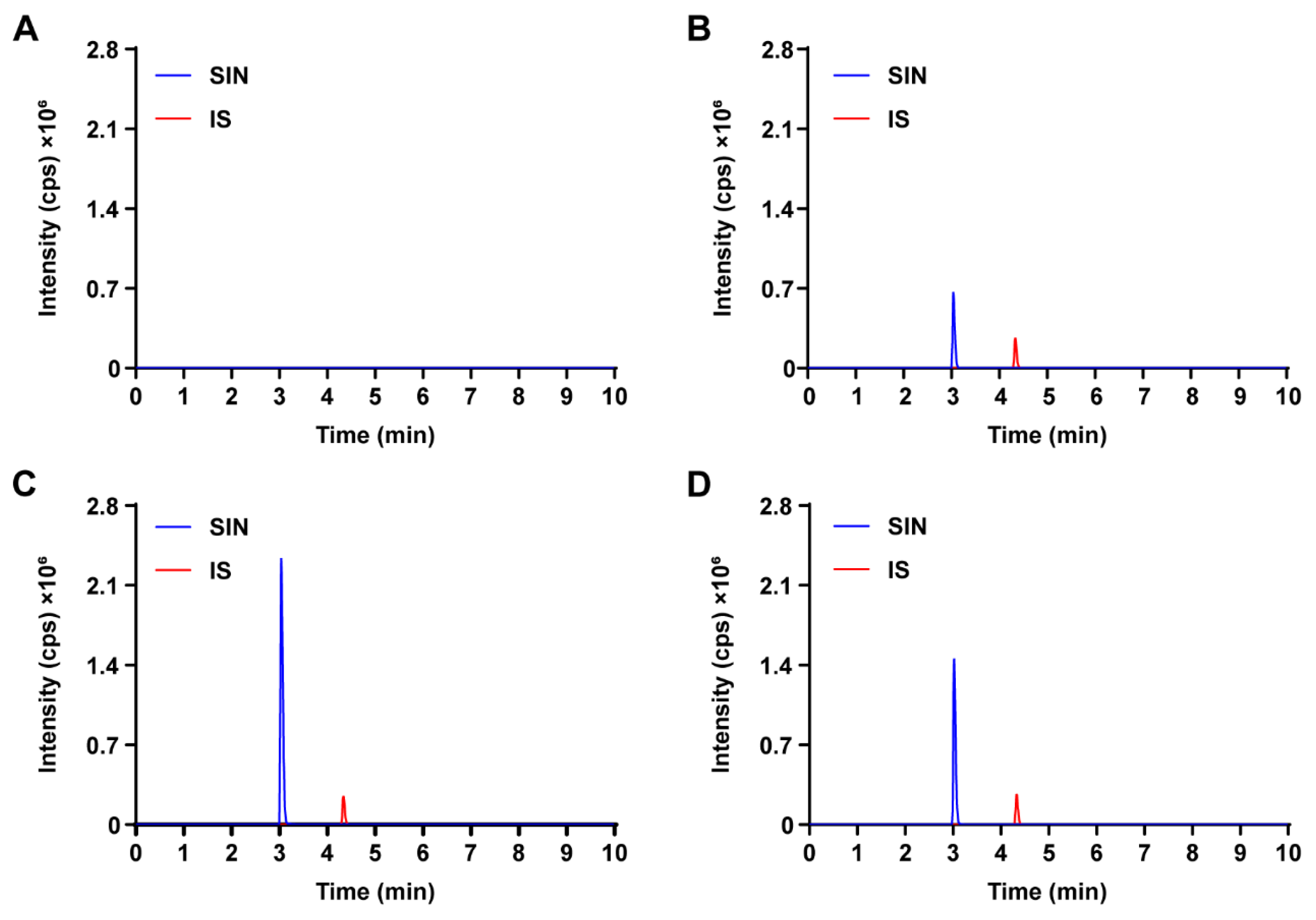
3.2.1. Selectivity
3.2.2. Linearity and Sensitivity
3.2.3. Accuracy and Precision
3.2.4. Matrix Effect and Recovery
3.2.5. Stability
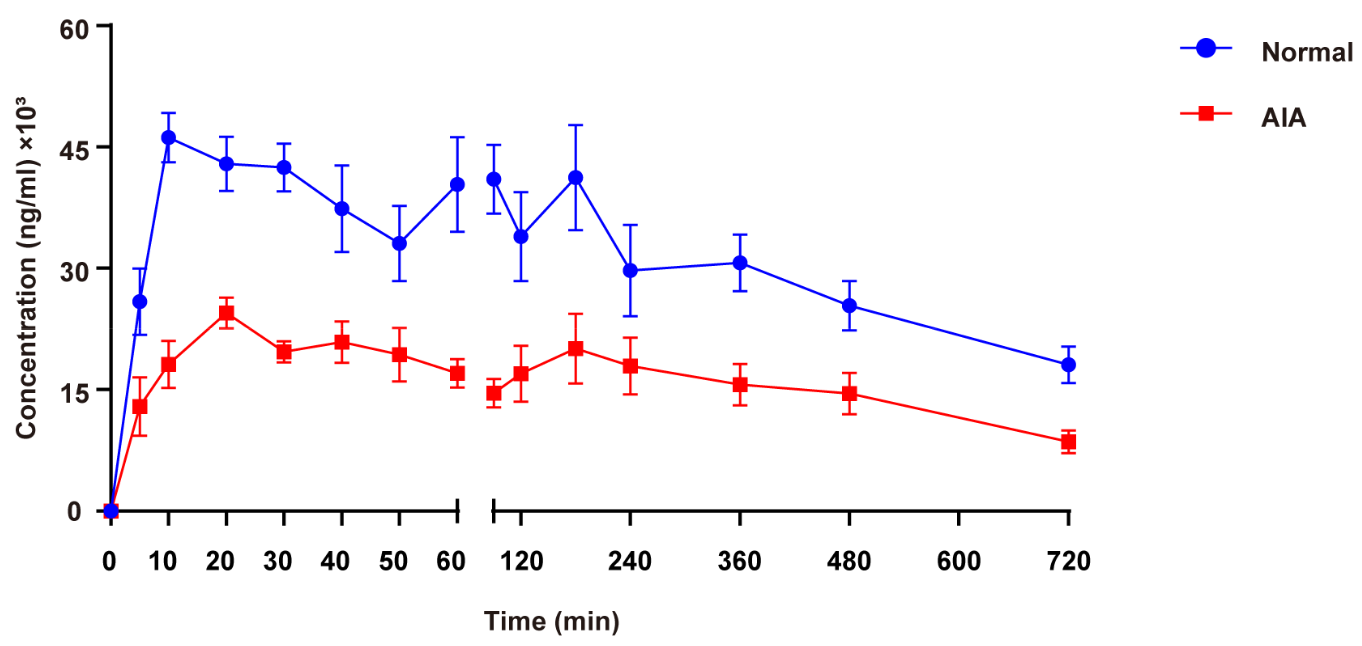
3.3. TK Study
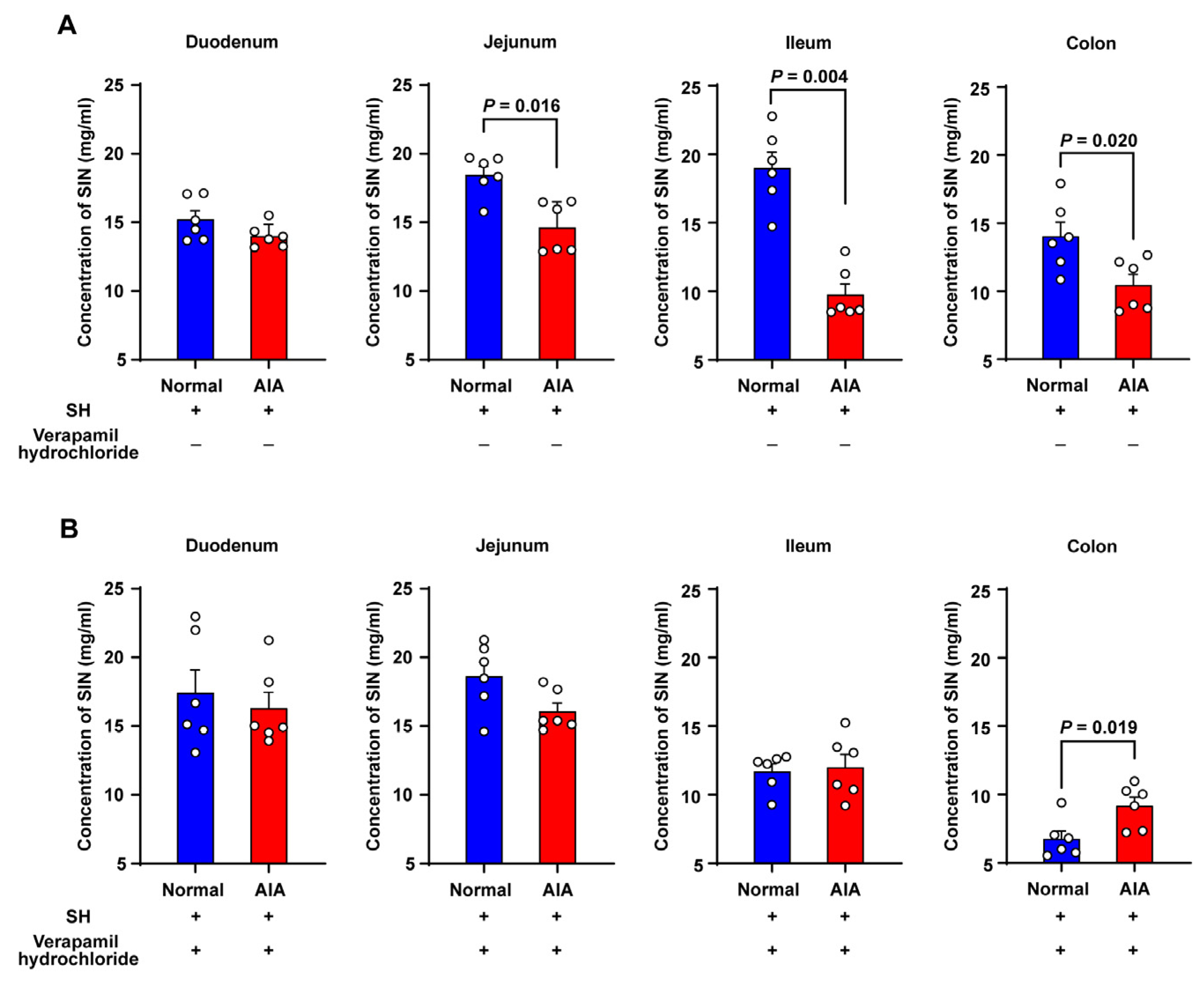
3.4. Absorption Studies Using Everted Gut Sac Model
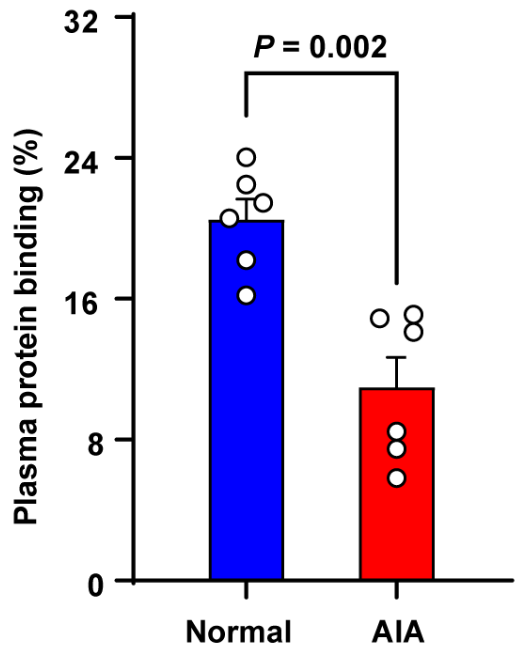
3.5. Plasma Protein Binding
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, Y.S.; Han, J.Y.; Iqbal, O.; Liang, A.H. Research advances and prospects on mechanism of sinomenin on histamine release and the binding to histamine receptors. Int. J. Mol. Sci. 2018, 20, 70. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Zhong, Z.; Ko, C.N.; Tian, T.; Yang, C. From mundane to classic: Sinomenine as a multi-therapeutic agent. Br. J. Pharmacol. 2023. [Google Scholar] [CrossRef]
- Xia, B.; Li, Q.; Wu, J.; Yuan, X.; Wang, F.; Lu, X.; Huang, C.; Zheng, K.; Yang, R.; Yin, L.; et al. Sinomenine Confers Protection Against Myocardial Ischemia Reperfusion Injury by Preventing Oxidative Stress, Cellular Apoptosis, and Inflammation. Front. Pharmacol. 2022, 13, 922484. [Google Scholar]
- Yang, W.; Feng, Q.; Li, M.; Su, J.; Wang, P.; Wang, X.; Yin, Y.; Wang, X.; Zhao, M. Sinomenine suppresses development of hepatocellular carcinoma cells via inhibiting MARCH1 and AMPK/STAT3 signaling pathway. Front. Mol. Biosci. 2021, 8, 684262. [Google Scholar]
- Gao, W.J.; Liu, J.X.; Xie, Y.; Luo, P.; Liu, Z.Q.; Liu, L.; Zhou, H. Suppression of macrophage migration by down-regulating Src/FAK/P130Cas activation contributed to the anti-inflammatory activity of sinomenine. Pharmacol. Res. 2021, 167, 105513. [Google Scholar]
- Xie, J.; Li, M.; Ye, W.; Shan, J.; Zhao, X.; Duan, Y.; Liu, Y.; Unger, B.H.; Cheng, Y.; Zhang, W.; et al. Sinomenine hydrochloride ameliorates fish foodborne enteritis via α7nAchR-mediated anti-inflammatory effect whilst altering microbiota composition. Front. Immunol. 2021, 12, 766845. [Google Scholar]
- Li, J.M.; Deng, H.S.; Yao, Y.D.; Wang, W.T.; Hu, J.Q.; Dong, Y.; Wang, P.X.; Liu, L.; Liu, Z.Q.; Xie, Y.; et al. Sinomenine ameliorates collagen-induced arthritis in mice by targeting GBP5 and regulating the P2X7 receptor to suppress NLRP3-related signaling pathways. Acta Pharmacol. Sin. 2023, 44, 2504–2524. [Google Scholar]
- Hong, H.; Lu, X.; Lu, Q.; Huang, C.; Cui, Z. Potential therapeutic effects and pharmacological evidence of sinomenine in central nervous system disorders. Front. Pharmacol. 2022, 13, 1015035. [Google Scholar]
- Chen, J.; Guo, P.; Han, M.; Chen, K.; Qin, J.; Yang, F. Cognitive protection of sinomenine in type 2 diabetes mellitus through regulating the EGF/Nrf2/HO-1 signaling, the microbiota-gut-brain axis, and hippocampal neuron ferroptosis. Phytother. Res. 2023, 37, 3323–3341. [Google Scholar]
- Chen, X.M.; Huang, R.Y.; Huang, Q.C.; Chu, Y.L.; Yan, J.Y. Systemic review and meta-analysis of the clinical efficacy and adverse Effects of Zhengqing Fengtongning combined with methotrexate in rheumatoid arthritis. Evid. Based Complement. Alternat. Med. 2015, 2015, 910376. [Google Scholar]
- Dumais, G.; Iovu, M.; du Souich, P. Inflammatory reactions and drug response: Importance of cytochrome P450 and membrane transporters. Expert Rev. Clin. Pharmacol. 2008, 1, 627–647. [Google Scholar] [PubMed]
- Morgan, E.T.; Goralski, K.B.; Piquette-Miller, M.; Renton, K.W.; Robertson, G.R.; Chaluvadi, M.R.; Charles, K.A.; Clarke, S.J.; Kacevska, M.; Liddle, C.; et al. Regulation of drug-metabolizing enzymes and transporters in infection, inflammation, and cancer. Drug Metab. Dispos. 2008, 36, 205–216. [Google Scholar] [PubMed]
- Morgan, E.T. Impact of infectious and inflammatory disease on cytochrome P450-mediated drug metabolism and pharmacokinetics. Clin. Pharmacol. Ther. 2009, 85, 434–438. [Google Scholar]
- Murakami, T.; Bodor, E.; Bodor, N. Modulation of expression/function of intestinal P-glycoprotein under disease states. Expert Opin. Drug Metab. Toxicol. 2020, 16, 59–78. [Google Scholar] [PubMed]
- Saib, S.; Delavenne, X. Inflammation induces changes in the functional expression of P-gp, BCRP, and MRP2: An overview of different models and consequences for drug disposition. Pharmaceutics 2021, 13, 1544. [Google Scholar] [CrossRef]
- Schmith, V.D.; Foss, J.F. Inflammation: Planning for a source of pharmacokinetic/pharmacodynamic variability in translational studies. Clin. Pharmacol. Ther. 2010, 87, 488–491. [Google Scholar]
- Shah, R.R.; Smith, R.L. Inflammation-induced phenoconversion of polymorphic drug metabolizing enzymes: Hypothesis with implications for personalized medicine. Drug Metab. Dispos. 2015, 43, 400–410. [Google Scholar]
- García-Carrasco, M.; Mendoza-Pinto, C.; Macias Díaz, S.; Vera-Recabarren, M.; Vázquez de Lara, L.; Méndez Martínez, S.; Soto-Santillán, P.; González-Ramírez, R.; Ruiz-Arguelles, A. P-glycoprotein in autoimmune rheumatic diseases. Autoimmun. Rev. 2015, 14, 594–600. [Google Scholar]
- Suzuki, T.; Zhao, Y.L.; Nadai, M.; Naruhashi, K.; Shimizu, A.; Takagi, K.; Takagi, K.; Hasegawa, T. Gender-related differences in expression and function of hepatic P-glycoprotein and multidrug resistance-associated protein (Mrp2) in rats. Life Sci. 2006, 79, 455–461. [Google Scholar]
- Liu, Z.Q.; Chan, K.; Zhou, H.; Jiang, Z.H.; Wong, Y.F.; Xu, H.X.; Liu, L. The pharmacokinetics and tissue distribution of sinomenine in rats and its protein binding ability in vitro. Life Sci. 2005, 77, 3197–3209. [Google Scholar]
- Moshage, H.J.; Janssen, J.A.; Franssen, J.H.; Hafkenscheid, J.C.; Yap, S.H. Study of the molecular mechanism of decreased liver synthesis of albumin in inflammation. J. Clin. Investig. 1987, 79, 1635–1641. [Google Scholar] [PubMed]
- Hochepied, T.; Berger, F.G.; Baumann, H.; Libert, C. Alpha(1)-acid glycoprotein: An acute phase protein with inflammatory and immunomodulating properties. Cytokine Growth Factor Rev. 2003, 14, 25–34. [Google Scholar] [PubMed]
- Beales, I.L.; Calam, J. Interleukin 1 beta and tumour necrosis factor alpha inhibit acid secretion in cultured rabbit parietal cells by multiple pathways. Gut 1998, 42, 227–234. [Google Scholar]
- Mugford, C.A.; Kedderis, G.L. Sex-dependent metabolism of xenobiotics. Drug Metab. Rev. 1998, 30, 441–498. [Google Scholar]
- Cai, X.; Wong, Y.F.; Zhou, H.; Liu, Z.Q.; Xie, Y.; Jiang, Z.H.; Bian, Z.X.; Xu, H.X.; Liu, L. Manipulation of the induction of adjuvant arthritis in Sprague-Dawley rats. Inflamm. Res. 2006, 55, 368–377. [Google Scholar]
- Li, Y.; Liu, S.S.; Guo, Z.Y.; Yi, H.; Li, C.; Chen, L.M.; Gao, H.M.; Yan, L.H.; Zhang, W.W.; Feng, X.X.; et al. Discovery of potential pharmacodynamic ingredients of Dang-Gui-Si-Ni decoction based on absorbed ingredients and molecular docking. J. Ethnopharmacol. 2021, 275, 114045. [Google Scholar]
- Xie, B.; Jiang, S.Q.; Shen, X.L.; Wu, H.Q.; Hu, Y.J. Pharmacokinetics, plasma protein binding, and metabolism of a potential natural chemosensitizer from Marsdenia tenacissima in rats. J. Ethnopharmacol. 2021, 281, 114544. [Google Scholar]
- Huang, H.; Zhang, E.B.; Yi, O.Y.; Wu, H.; Deng, G.; Huang, Y.M.; Liu, W.L.; Yan, J.Y.; Cai, X. Sex-related differences in safety profiles, pharmacokinetics and tissue distribution of sinomenine hydrochloride in rats. Arch. Toxicol. 2022, 96, 3245–3255. [Google Scholar]
- Di Matteo, A.; Bathon, J.M.; Emery, P. Rheumatoid arthritis. Lancet 2023, 402, 2019–2033. [Google Scholar]
- Alam, M.A.; Al-Jenoobi, F.I.; Al-Mohizea, A.M. Everted gut sac model as a tool in pharmaceutical research: Limitations and applications. J. Pharm. Pharmacol. 2012, 64, 326–336. [Google Scholar]
- Álvarez-Olguín, M.A.; Beltrán-Barrientos, L.M.; Hernandez-Mendoza, A.; González-Córdova, A.F.; Vallejo-Cordoba, B. Current trends and perspectives on bioaccessibility and bioavailability of food bioactive peptides: In vitro and ex vivo studies. J. Sci. Food Agric. 2022, 102, 6824–6834. [Google Scholar] [PubMed]
- Silva, V.; Gil-Martins, E.; Silva, B.; Rocha-Pereira, C.; Sousa, M.E.; Remião, F.; Silva, R. Xanthones as P-glycoprotein modulators and their impact on drug bioavailability. Expert Opin. Drug Metab. Toxicol. 2021, 17, 441–482. [Google Scholar] [PubMed]
- Tsai, T.H.; Wu, J.W. Regulation of hepatobiliary excretion of sinomenine by P-glycoprotein in Sprague-Dawley rats. Life Sci. 2003, 72, 2413–2426. [Google Scholar] [PubMed]
- Tang, H.; Wu, Y.J.; Xiao, F.; Wang, B.; Asenso, J.; Wang, Y.; Sun, W.; Wang, C.; Wei, W. Regulation of CP-25 on P-glycoprotein in synoviocytes of rats with adjuvant arthritis. Biomed. Pharmacother. 2019, 119, 109432. [Google Scholar]
- Slifman, N.R.; Gershon, S.K.; Lee, J.H.; Edwards, E.T.; Braun, M.M. Listeria monocytogenes infection as a complication of treatment with tumor necrosis factor alpha-neutralizing agents. Arthritis Rheum. 2003, 48, 319–324. [Google Scholar]
- Verhoef, L.M.; van den Bemt, B.J.; van der Maas, A.; Vriezekolk, J.E.; Hulscher, M.E.; van den Hoogen, F.H.; Jacobs, W.C.; van Herwaarden, N.; den Broeder, A.A. Down-titration and discontinuation strategies of tumour necrosis factor-blocking agents for rheumatoid arthritis in patients with low disease activity. Cochrane Database Syst. Rev. 2019, 5, CD010455. [Google Scholar]
- Liu, J.; Zhou, F.; Chen, Q.; Kang, A.; Lu, M.; Liu, W.; Zang, X.; Wang, G.; Zhang, J. Chronic inflammation up-regulates P-gp in peripheral mononuclear blood cells via the STAT3/Nf-κb pathway in 2,4,6-trinitrobenzene sulfonic acid-induced colitis mice. Sci. Rep. 2015, 5, 13558. [Google Scholar]
- Bailey, D.G.; Dresser, G.K.; Leake, B.F.; Kim, R.B. Naringin is a major and selective clinical inhibitor of organic anion-transporting polypeptide 1A2 (OATP1A2) in grapefruit juice. Clin. Pharmacol. Ther. 2007, 81, 495–502. [Google Scholar]
- Ahlin, G.; Chen, L.; Lazorova, L.; Chen, Y.; Ianculescu, A.G.; Davis, R.L.; Giacomini, K.M.; Artursson, P. Genotype-dependent effects of inhibitors of the organic cation transporter, OCT1: Predictions of metformin interactions. Pharmacogenom. J. 2011, 11, 400–411. [Google Scholar]
- Grigat, S.; Harlfinger, S.; Pal, S.; Striebinger, R.; Golz, S.; Geerts, A.; Lazar, A.; Schömig, E.; Gründemann, D. Probing the substrate specificity of the ergothioneine transporter with methimazole, hercynine, and organic cations. Biochem. Pharmacol. 2007, 74, 309–316. [Google Scholar]
- Smith, D.A.; Di, L.; Kerns, E.H. The effect of plasma protein binding on in vivo efficacy: Misconceptions in drug discovery. Nat. Rev. Drug Discov. 2010, 9, 929–939. [Google Scholar] [PubMed]
- Yun, Y.E.; Edginton, A.N. Evaluation of models for predicting pediatric fraction unbound in plasma for human health risk assessment. J. Toxicol. Environ. Health A 2021, 84, 67–83. [Google Scholar] [PubMed]
- Yu, X.; Jiao, Q.; Jiang, Y.; Guo, S.; Zhang, W.; Liu, B. Study on the Plasma protein binding rate and compatibility regularity of the constituents migrating to blood of Simiao Yong’an Decoction. Curr. Drug Metab. 2020, 21, 979–993. [Google Scholar]
- Miyauchi, S.; Kim, S.J.; Lee, W.; Sugiyama, Y. Consideration of albumin-mediated hepatic uptake for highly protein-bound anionic drugs: Bridging the gap of hepatic uptake clearance between in vitro and in vivo. Pharmacol. Ther. 2022, 229, 107938. [Google Scholar] [CrossRef]
- Di, L. An update on the importance of plasma protein binding in drug discovery and development. Exp. Opin Drug Discov. 2021, 16, 1453–1465. [Google Scholar]
- Lambrinidis, G.; Vallianatou, T.; Tsantili-Kakoulidou, A. In vitro, in silico and integrated strategies for the estimation of plasma protein binding. A review. Adv. Drug Del. Rev. 2015, 86, 27–45. [Google Scholar]
- Garg, U.; Jain, N.; Kaul, S.; Nagaich, U. Role of albumin as a targeted drug carrier in the management of rheumatoid arthritis: A comprehensive Review. Mol. Pharm. 2023, 20, 5345–5358. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tissue | Female Normal Rats | |
|---|---|---|
| Standard Curve | Linearity | |
| Plasma | Y = 0.00750X + 0.1500 | r = 0.9916 |
| Intestinal serosal fluid | Y = 0.00920X + 0.0314 | r = 0.9974 |
| Plasma protein binding | Y = 0.00879X + 0.0016 | r = 0.9995 |
| Matrix | Concentration (ng/mL) | Intra–Day (n = 6) | Inter–Day (n = 18) | Extraction Recovery (%) (Mean ± SD, n = 6) | Matrix Effect (%) (Mean ± SD, n = 6) | ||
|---|---|---|---|---|---|---|---|
| Accuracy (%) | Precision RSD (%) | Accuracy (%) | Precision RSD (%) | ||||
| Intestinal serosal fluid | 200.00 | 99.50 | 4.63 | 106.03 | 6.10 | 97.03 ± 2.39 | 98.15 ± 2.80 |
| 350.00 | 99.50 | 1.68 | 105.40 | 4.43 | 104.05 ± 2.49 | 104.26 ± 8.76 | |
| 700.00 | 102.62 | 2.11 | 100.20 | 2.95 | 98.83 ± 3.33 | 101.01 ± 1.43 | |
| Matrix | Condition | Nominal Concentration (ng/mL) | Measured Concentration (mean ± SD, ng/mL) | RSD Precision (%) |
|---|---|---|---|---|
| Intestinal serosal fluid | Room temperature stability | 200.00 | 222.17 ± 1.94 | 0.87 |
| 350.00 | 356.17 ± 6.62 | 1.86 | ||
| 700.00 | 649.50 ± 14.61 | 2.25 | ||
| Freeze–thaw stability | 200.00 | 204.50 ± 6.47 | 3.17 | |
| 350.00 | 371.17 ± 4.58 | 1.23 | ||
| 700.00 | 702.83 ± 10.48 | 1.49 | ||
| Long term freezing stability | 200.00 | 229.67 ± 4.76 | 2.07 | |
| 350.00 | 386.33 ± 7.00 | 1.81 | ||
| 700.00 | 689.50 ± 11.50 | 1.67 |
| TK Parameter | Female Normal Rats | Female AIA Rats | p-Value |
|---|---|---|---|
| Tmax (min) | 73.3 ± 58.5 | 86.7 ± 97.7 | 0.746 |
| Cmax (ng/mL) | 54,500.0 ± 5950.8 | 27,105.0 ± 6689.2 | 0.000 |
| T1/2 (min) | 476.9 ± 259.1 | 425.6 ± 129.4 | 0.070 |
| AUC(0-t) (ng·min/mL) | 21,157,950.0 ± 5,011,214.1 | 10,927,737.5 ± 4,038,606.3 | 0.003 |
| AUC(0-∞) (ng·min/mL) | 34,574,304.2 ± 8,516,605.5 | 16,168,486.3 ± 5,832,812.7 | 0.001 |
| CL/F (mL/min/kg) | 18.4 ± 5.4 | 41.3 ± 14.8 | 0.005 |
| V/F (mL/kg) | 11,537.8 ± 4686.3 | 25,788.8 ± 13,299.8 | 0.047 |
| ka (h−1) | 1.32 ± 1.44 | 1.38 ± 0.92 | 0.749 |
| ke (h−1) | 0.11 ± 0.06 | 0.10 ± 0.03 | 0.798 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, Y.; Huang, H.; Li, G.; Zhang, Y.; He, J.; Lin, Y.; Wu, F.; Yan, J.; Cai, X.; Liu, L. Safety Profile, Toxicokinetic, and Intestinal Absorption Differences of a Naturally-Derived Anti-Rheumatic Drug, Sinomenine Hydrochloride, in Normal and Arthritic Rats. Pharmaceutics 2025, 17, 484. https://doi.org/10.3390/pharmaceutics17040484
He Y, Huang H, Li G, Zhang Y, He J, Lin Y, Wu F, Yan J, Cai X, Liu L. Safety Profile, Toxicokinetic, and Intestinal Absorption Differences of a Naturally-Derived Anti-Rheumatic Drug, Sinomenine Hydrochloride, in Normal and Arthritic Rats. Pharmaceutics. 2025; 17(4):484. https://doi.org/10.3390/pharmaceutics17040484
Chicago/Turabian StyleHe, Yini, Hong Huang, Gejing Li, Ye Zhang, Junjie He, Ye Lin, Feichi Wu, Jianye Yan, Xiong Cai, and Liang Liu. 2025. "Safety Profile, Toxicokinetic, and Intestinal Absorption Differences of a Naturally-Derived Anti-Rheumatic Drug, Sinomenine Hydrochloride, in Normal and Arthritic Rats" Pharmaceutics 17, no. 4: 484. https://doi.org/10.3390/pharmaceutics17040484
APA StyleHe, Y., Huang, H., Li, G., Zhang, Y., He, J., Lin, Y., Wu, F., Yan, J., Cai, X., & Liu, L. (2025). Safety Profile, Toxicokinetic, and Intestinal Absorption Differences of a Naturally-Derived Anti-Rheumatic Drug, Sinomenine Hydrochloride, in Normal and Arthritic Rats. Pharmaceutics, 17(4), 484. https://doi.org/10.3390/pharmaceutics17040484