
New and Effective Inhibitor of Class I HDACs, Eimbinostat, Reduces the Growth of Hematologic Cancer Cells and Triggers Apoptosis

 , , , , , , , , ,
, , , , , , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemistry
2.1.1. Reagents and Technical Equipment
2.1.2. General Procedure for the Preparation of Eimbinostat
- (i)
- Schiff base preparation—Belinostat hydrazide (HB) (900 mg, 2.84 mmol), prepared as described earlier [33], was suspended in i-PrOH (20 mL), then butyraldehyde (410 mg, 5.68 mmol) and AcOH in a catalytic amount (20 mg) were added, and the mixture was stirred for 18 h and used as-is in the next step.
- (ii)
- Eimbinostat—NaBH4 (500 mg, 13.2 mmol) was added to obtain the above suspension and the mixture was boiled for 1.25 h. After cooling to an ambient temperature, the reaction mixture was diluted with water (20 mL), neutralized with AcOH to pH~6.0, and evaporated to the final volume 15 mL. The supernatant was decanted, the gummy residue was solidified by twice evaporation with H2O (from 10 to 20 mL), with subsequent cooling in ice for 30 min and rubbing. The crude product was purified by silica gel chromatography using a mixture of two eluents: (a) n-hexane–EtOAc (1:2) and (b) CHCl3-EtOH (10:1) in equal proportions, affording eimbinostat in the yield of 756 mg (71.4%). 1H NMR (300 MHz, DMSO) δ 10.31 (s, 1H), 9.65 (s, 1H), 7.92 (s, 1H), 7.77 (d, J = 7.7 Hz, 1H), 7.71 (d, J = 8.1 Hz, 1H), 7.58 (t, J = 7.8 Hz, 1H), 7.45 (d, J = 15.9 Hz, 1H), 7.24 (t, J = 7.8 Hz, 2H), 7.10 (d, J = 7.5 Hz, 2H), 7.03 (t, J = 7.3 Hz, 1H), 6.59 (d, J = 15.8 Hz, 1H), 5.06 (s, 1H), 2.73 (t, J = 6.8 Hz, 2H), 1.36 (qt, J = 13.8, 6.7 Hz, 5H), 0.89 (t, J = 7.1 Hz, 3H). 13C NMR (75 MHz, DMSO) δ 163.65 (s), 140.76 (s), 137.95 (s), 137.15 (s), 136.39 (s), 132.41 (s), 130.47 (s), 129.65 (s), 127.52 (s), 125.26 (s), 124.77 (s), 122.95 (s), 120.81 (s), 51.31 (s), 30.17 (s), 20.19 (s), 14.33 (s). ESI-HRMS (m/z): Calcd for C19H23N3O3S [M + H]+ 374.1533, found 374.1514
2.2. Bioinformatics Analysis of Gene Expression and Gene Fitness Data
2.3. Biology
2.3.1. Cell Cultures
2.3.2. Cell Viability Analysis
2.3.3. Gene Expression Analysis
2.3.4. Western Blot Analysis and Acetylation Efficiency Assay
2.3.5. HDAC Selectivity Assay Using the Cell-Test System (s3CTS)
2.3.6. Analysis of Apoptosis and Cell Cycle
2.3.7. Molecular Modeling and Docking
2.3.8. Data and Statistical Analysis
3. Results
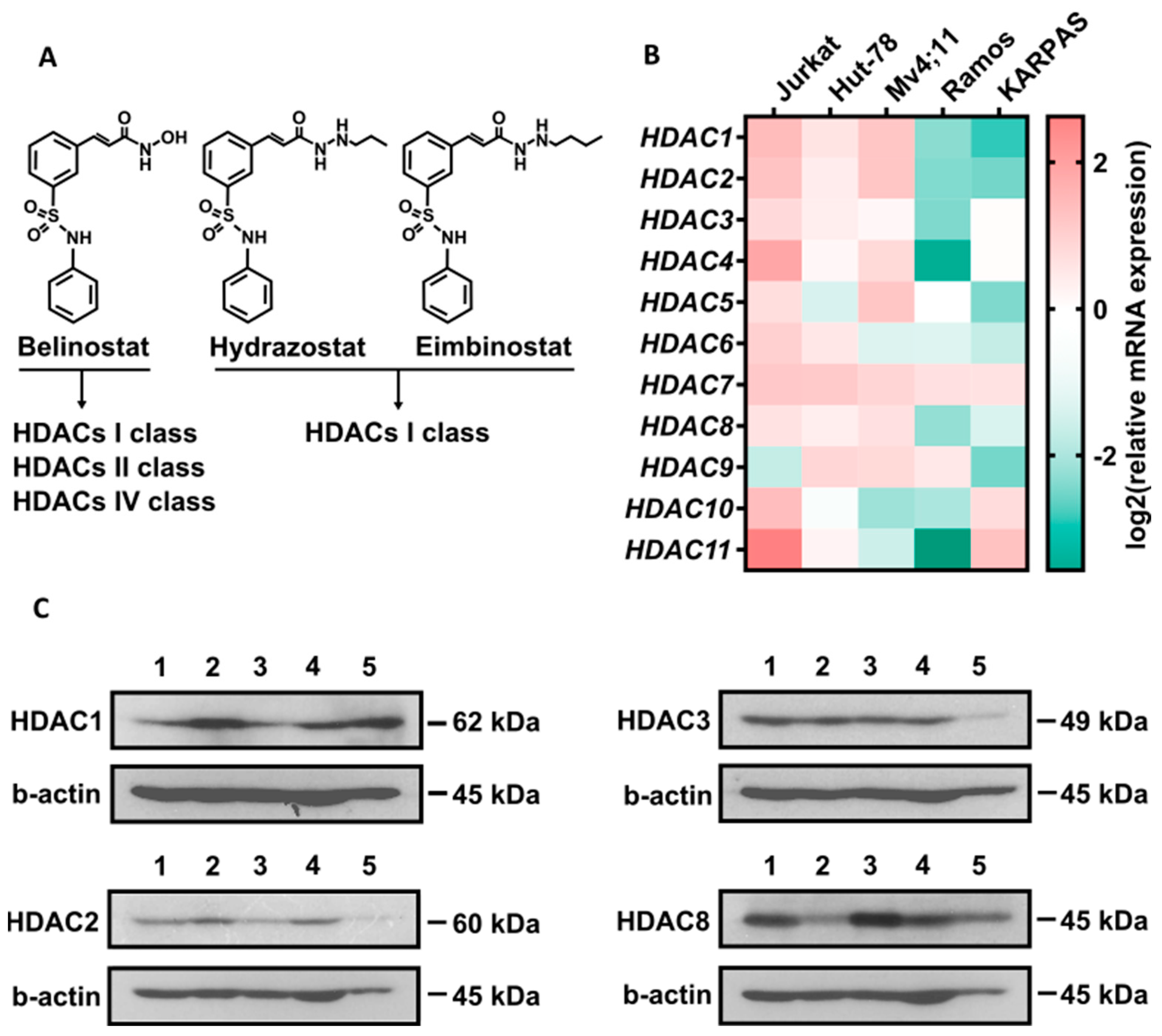
3.1. Synthesis and Characterization of Eimbinostat
3.2. Bioinformatics Assessment of the Effects of HDAC on Cell Viability
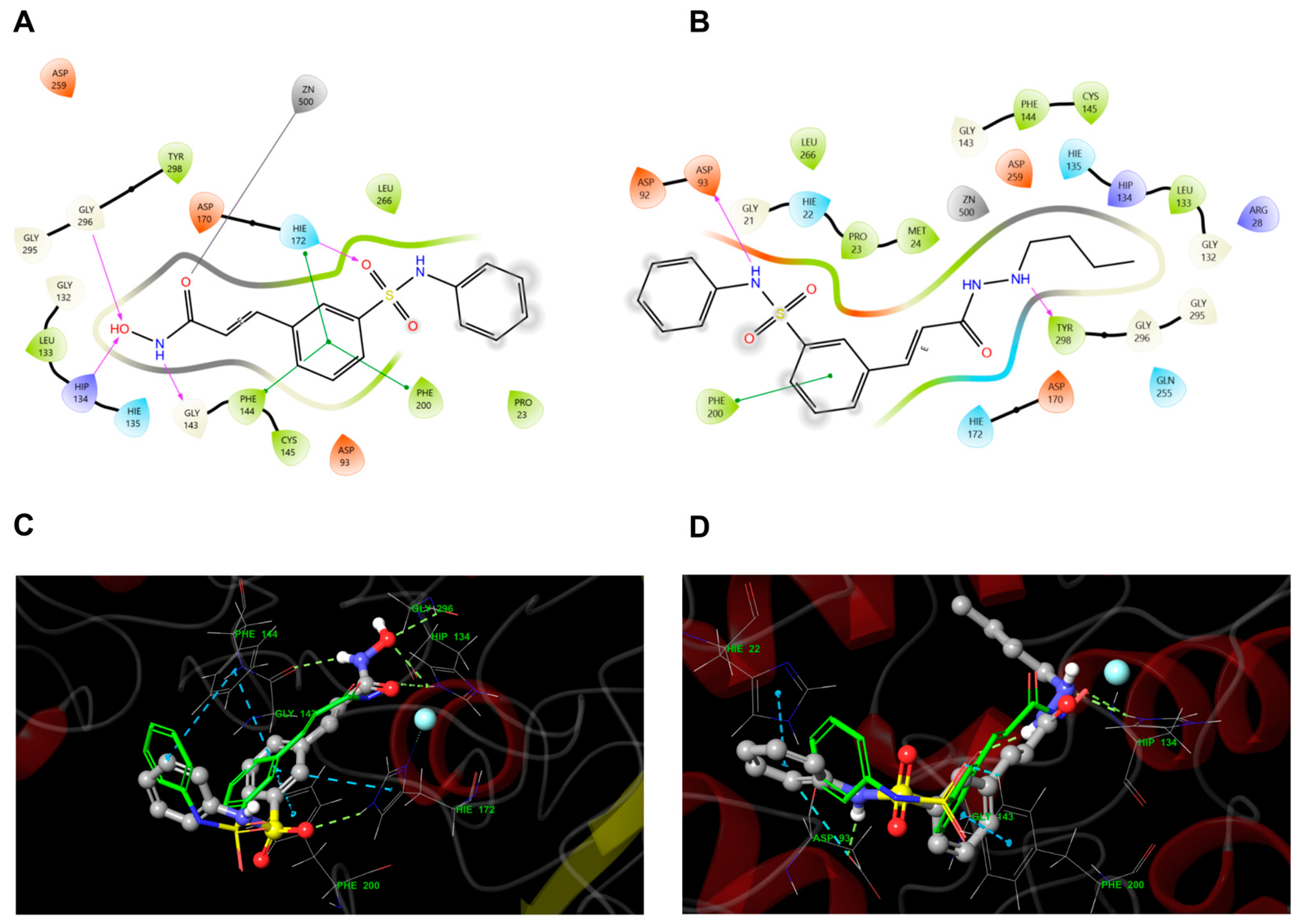
3.3. Molecular Docking
3.4. Eimbinostat Significantly Suppresses Growth of Lymphoma Cells and Selectively Inhibits Class I HDAC Deacetylation Activity
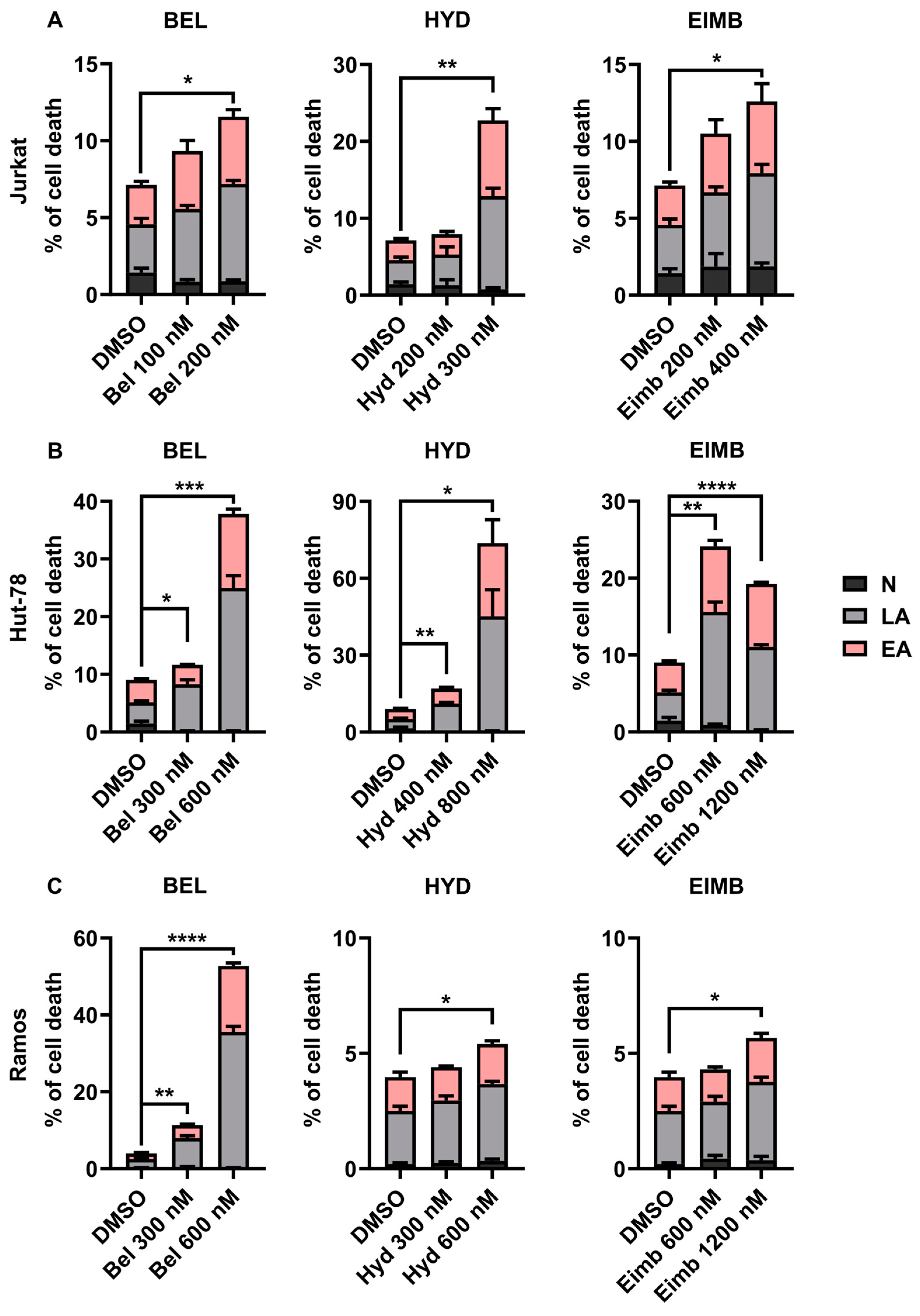
3.5. Eimbinostat Induces Apoptosis
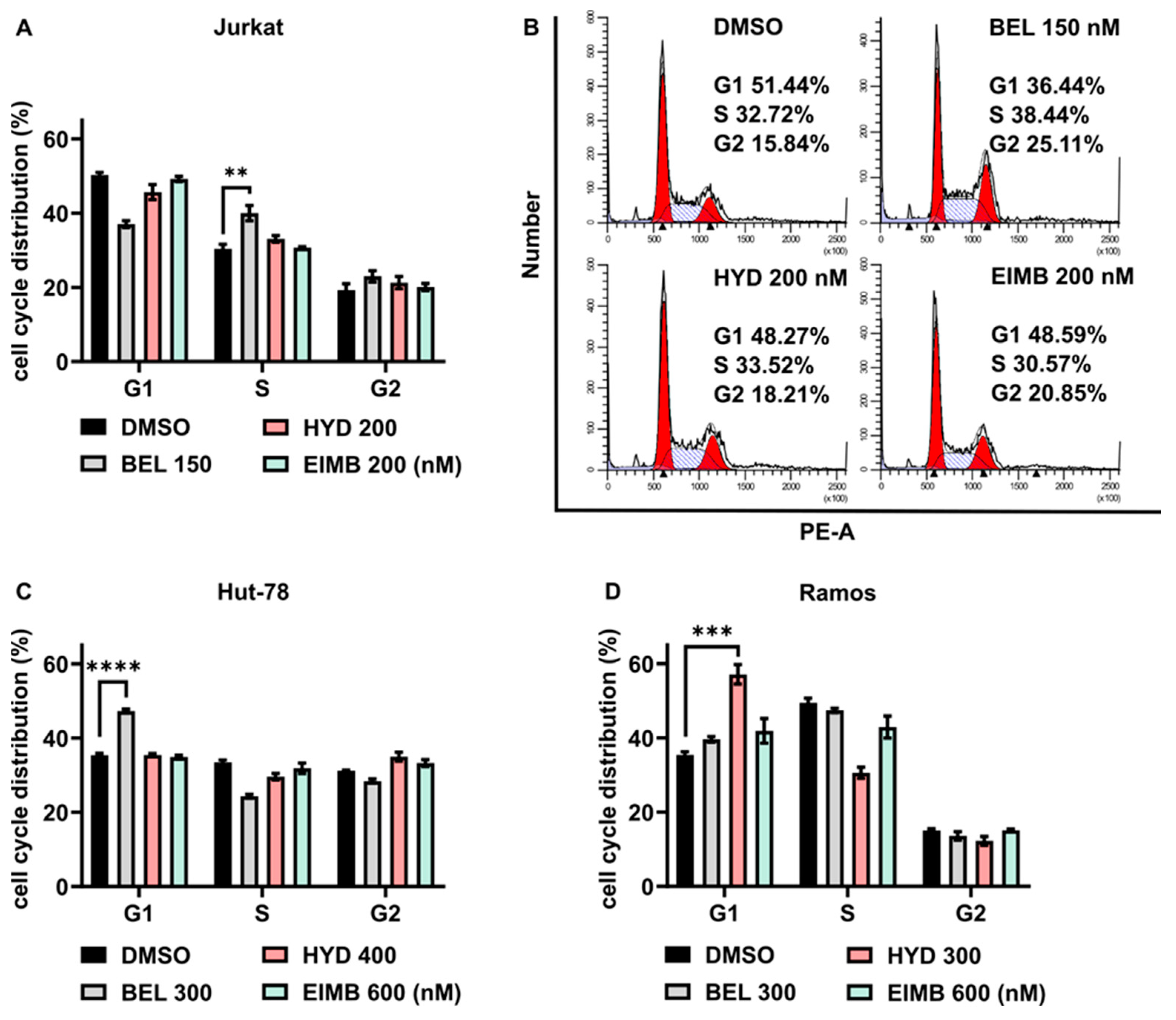
3.6. Eimbinostat Does Not Affect Cell Cycle
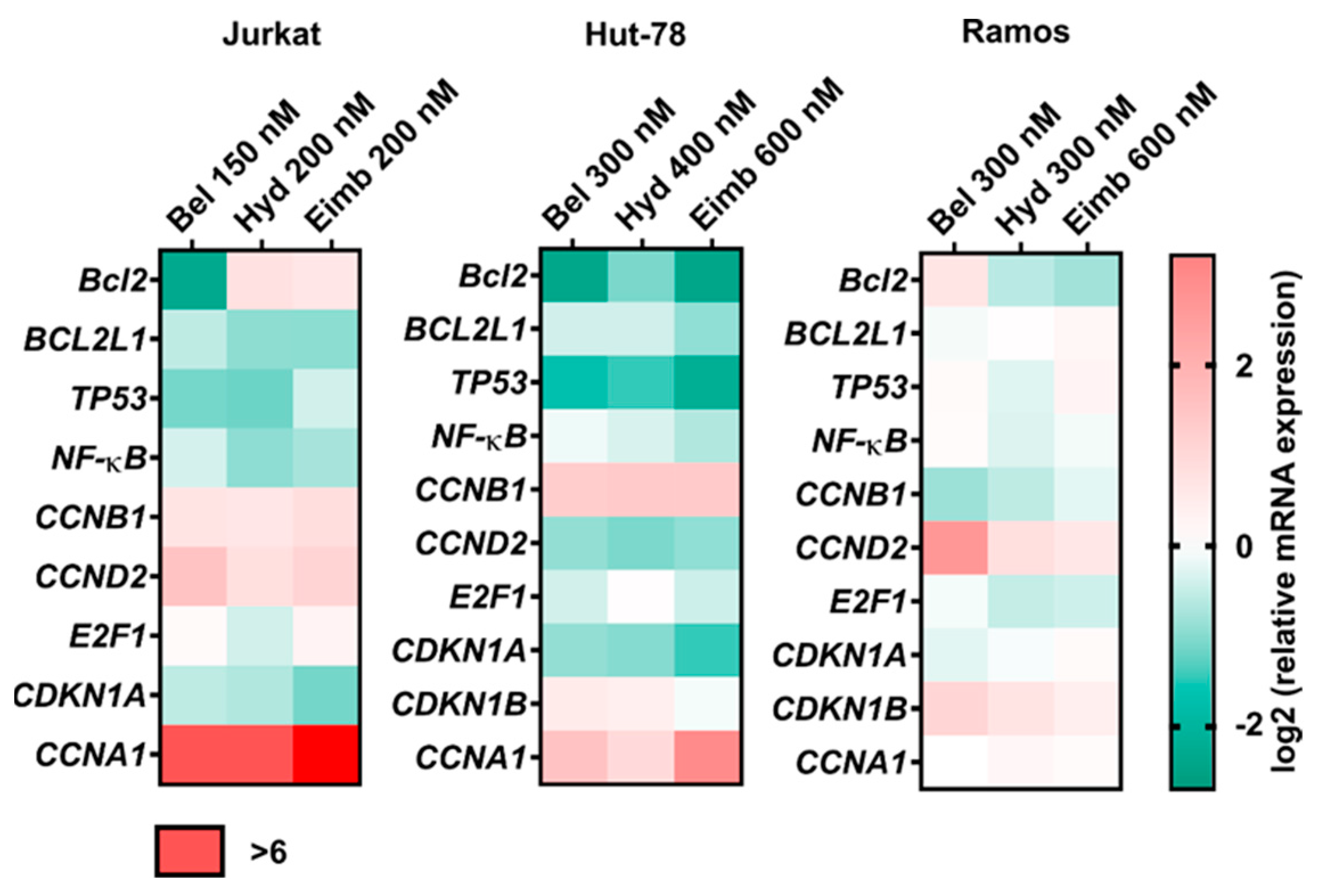
3.7. Treatment with Eimbinostat Alters Expression of Genes Associated with Leukemia/Lymphoma
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, N.; Wu, J.; Wang, Q.; Liang, Y.; Li, X.; Chen, G.; Ma, L.; Liu, X.; Zhou, F. Global Burden of Hematologic Malignancies and Evolution Patterns over the Past 30 Years. Blood Cancer J. 2023, 13, 82. [Google Scholar] [CrossRef] [PubMed]
- Forsberg, M.; Konopleva, M. AML Treatment: Conventional Chemotherapy and Emerging Novel Agents. Trends Pharmacol. Sci. 2024, 45, 430–448. [Google Scholar] [CrossRef]
- Takami, A. Hematopoietic Stem Cell Transplantation for Acute Myeloid Leukemia. Int. J. Hematol. 2018, 107, 513–518. [Google Scholar] [PubMed]
- Waldstein, C. Radiotherapy Update: Current Role of Radiotherapy in the Treatment of Lymphomas. Memo—Mag. Eur. Med. Oncol. 2023, 16, 62–66. [Google Scholar] [CrossRef]
- Blank, O.; von Tresckow, B.; Monsef, I.; Specht, L.; Engert, A.; Skoetz, N. Chemotherapy Alone versus Chemotherapy plus Radiotherapy for Adults with Early Stage Hodgkin Lymphoma. Cochrane Database Syst. Rev. 2017, 2017, CD007110. [Google Scholar] [CrossRef]
- Witt, O.; Deubzer, H.E.; Milde, T.; Oehme, I. HDAC Family: What Are the Cancer Relevant Targets? Cancer Lett. 2009, 277, 8–21. [Google Scholar] [CrossRef]
- Greer, C.B.; Tanaka, Y.; Kim, Y.J.; Xie, P.; Zhang, M.Q.; Park, I.H.; Kim, T.H. Histone Deacetylases Positively Regulate Transcription through the Elongation Machinery. Cell Rep. 2015, 13, 1444–1455. [Google Scholar] [CrossRef]
- Contreras-Sanzón, E.; Prado-Garcia, H.; Romero-Garcia, S.; Nuñez-Corona, D.; Ortiz-Quintero, B.; Luna-Rivero, C.; Martínez-Cruz, V.; Carlos-Reyes, Á. Histone Deacetylases Modulate Resistance to the Therapy in Lung Cancer. Front. Genet. 2022, 13, 960263. [Google Scholar] [CrossRef]
- José-Enériz, E.S.; Gimenez-Camino, N.; Agirre, X.; Prosper, F. HDAC Inhibitors in Acute Myeloid Leukemia. Cancers 2019, 11, 1794. [Google Scholar] [CrossRef]
- Zhang, J.; Gao, X.; Yu, L. Roles of Histone Deacetylases in Acute Myeloid Leukemia with Fusion Proteins. Front. Oncol. 2021, 11, 741746. [Google Scholar]
- Brancolini, C.; Gagliano, T.; Minisini, M. HDACs and the Epigenetic Plasticity of Cancer Cells: Target the Complexity. Pharmacol. Ther. 2022, 238, 108190. [Google Scholar]
- Maiques-Diaz, A.; Spencer, G.J.; Lynch, J.T.; Ciceri, F.; Williams, E.L.; Amaral, F.M.R.; Wiseman, D.H.; Harris, W.J.; Li, Y.; Sahoo, S.; et al. Enhancer Activation by Pharmacologic Displacement of LSD1 from GFI1 Induces Differentiation in Acute Myeloid Leukemia. Cell Rep. 2018, 22, 3641–3659. [Google Scholar] [CrossRef]
- van Bergen, M.G.J.M.; van der Reijden, B.A. Targeting the GFI1/1B—CoREST Complex in Acute Myeloid Leukemia. Front. Oncol. 2019, 9, 1027. [Google Scholar]
- Simeoni, F.; Romero-Camarero, I.; Camera, F.; Amaral, F.M.R.; Sinclair, O.J.; Papachristou, E.K.; Spencer, G.J.; Lie-A-Ling, M.; Lacaud, G.; Wiseman, D.H.; et al. Enhancer Recruitment of Transcription Repressors RUNX1 and TLE3 by Mis-Expressed FOXC1 Blocks Differentiation in Acute Myeloid Leukemia. Cell Rep. 2021, 36, 109725. [Google Scholar] [CrossRef] [PubMed]
- Cantor, D.J.; David, G. The Potential of Targeting Sin3b and Its Associated Complexes for Cancer Therapy. Expert Opin. Ther. Targets 2017, 21, 1051–1061. [Google Scholar] [CrossRef] [PubMed]
- Turnbull, R.E.; Fairall, L.; Saleh, A.; Kelsall, E.; Morris, K.L.; Ragan, T.J.; Savva, C.G.; Chandru, A.; Millard, C.J.; Makarova, O.V.; et al. The MiDAC Histone Deacetylase Complex Is Essential for Embryonic Development and Has a Unique Multivalent Structure. Nat. Commun. 2020, 11, 3252. [Google Scholar] [CrossRef] [PubMed]
- Lai, A.Y.; Wade, P.A. Cancer Biology and NuRD: A Multifaceted Chromatin Remodelling Complex. Nat. Rev. Cancer 2011, 11, 588–596. [Google Scholar]
- Wong, M.M.; Guo, C.; Zhang, J. Nuclear Receptor Corepressor Complexes in Cancer: Mechanism, Function and Regulation. Am. J. Clin. Exp. Urol. 2014, 2, 169–187. [Google Scholar]
- Zhang, Q.; Wang, S.; Chen, J.; Yu, Z. Histone Deacetylases (HDACs) Guided Novel Therapies for T-Cell Lymphomas. Int. J. Med. Sci. 2019, 16, 424–442. [Google Scholar]
- Li, G.; Tian, Y.; Zhu, W.G. The Roles of Histone Deacetylases and Their Inhibitors in Cancer Therapy. Front. Cell Dev. Biol. 2020, 8, 576946. [Google Scholar]
- Lee, H.Z.; Kwitkowski, V.E.; Del Valle, P.L.; Ricci, M.S.; Saber, H.; Habtemariam, B.A.; Bullock, J.; Bloomquist, E.; Shen, Y.L.; Chen, X.H.; et al. FDA Approval: Belinostat for the Treatment of Patients with Relapsed or Refractory Peripheral T-Cell Lymphoma. Clin. Cancer Res. 2015, 21, 2666–2670. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Cao, J.; Xu, W. Selective Histone Deacetylase Inhibitors. Anti-Cancer Agents Med. Chem. 2012, 12, 247–270. [Google Scholar] [CrossRef]
- Vagapova, E.; Kozlov, M.; Lebedev, T.; Ivanenko, K.; Leonova, O.; Popenko, V.; Spirin, P.; Kochetkov, S.; Prassolov, V. Selective Inhibition of Hdac Class i Sensitizes Leukemia and Neuroblastoma Cells to Anticancer Drugs. Biomedicines 2021, 9, 1846. [Google Scholar] [CrossRef]
- O’Connor, O.A.; Horwitz, S.; Masszi, T.; Van Hoof, A.; Brown, P.; Doorduijn, J.; Hess, G.; Jurczak, W.; Knoblauch, P.; Chawla, S.; et al. Belinostat in Patients with Relapsed or Refractory Peripheral T-Cell Lymphoma: Results of the Pivotal Phase II BELIEF (CLN-19) Study. J. Clin. Oncol. 2015, 33, 2492–2499. [Google Scholar] [CrossRef]
- Peter, R.M.; Sarwar, M.S.; Mostafa, S.Z.; Wang, Y.; Su, X.; Kong, A.N. Histone Deacetylase Inhibitor Belinostat Regulates Metabolic Reprogramming in Killing KRAS-Mutant Human Lung Cancer Cells. Mol. Carcinog. 2023, 62, 1136–1146. [Google Scholar] [CrossRef]
- Vandermolen, K.M.; McCulloch, W.; Pearce, C.J.; Oberlies, N.H. Romidepsin (Istodax, NSC 630176, FR901228, FK228, Depsipeptide): A Natural Product Recently Approved for Cutaneous T-Cell Lymphoma. J. Antibiot. 2011, 64, 525–531. [Google Scholar] [CrossRef]
- Eleutherakis-Papaiakovou, E.; Kanellias, N.; Kastritis, E.; Gavriatopoulou, M.; Terpos, E.; Dimopoulos, M.A. Efficacy of Panobinostat for the Treatment of Multiple Myeloma. J. Oncol. 2020, 2020, 7131802. [Google Scholar] [PubMed]
- Gupta, V.G.; Hirst, J.; Petersen, S.; Roby, K.F.; Kusch, M.; Zhou, H.; Clive, M.L.; Jewell, A.; Pathak, H.B.; Godwin, A.K.; et al. Entinostat, a Selective HDAC1/2 Inhibitor, Potentiates the Effects of Olaparib in Homologous Recombination Proficient Ovarian Cancer. Gynecol. Oncol. 2021, 162, 163–172. [Google Scholar] [CrossRef]
- Wu, C.; Song, Q.; Gao, S.; Wu, S. Targeting HDACs for Diffuse Large B-Cell Lymphoma Therapy. Sci. Rep. 2024, 14, 289. [Google Scholar] [CrossRef]
- Smalley, J.P.; Baker, I.M.; Pytel, W.A.; Lin, L.Y.; Bowman, K.J.; Schwabe, J.W.R.; Cowley, S.M.; Hodgkinson, J.T. Optimization of Class i Histone Deacetylase PROTACs Reveals That HDAC1/2 Degradation Is Critical to Induce Apoptosis and Cell Arrest in Cancer Cells. J. Med. Chem. 2022, 65, 5642–5659. [Google Scholar] [CrossRef]
- Lin, C.L.; Tsai, M.L.; Lin, C.Y.; Hsu, K.W.; Hsieh, W.S.; Chi, W.M.; Huang, L.C.; Lee, C.H. HDAC1 and HDAC2 Double Knockout Triggers Cell Apoptosis in Advanced Thyroid Cancer. Int. J. Mol. Sci. 2019, 20, 454. [Google Scholar] [CrossRef]
- Li, X.; Jiang, Y.; Peterson, Y.K.; Xu, T.; Himes, R.A.; Luo, X.; Yin, G.; Inks, E.S.; Dolloff, N.; Halene, S.; et al. Design of Hydrazide-Bearing HDACIs Based on Panobinostat and Their P53 and FLT3-ITD Dependency in Antileukemia Activity. J. Med. Chem. 2020, 63, 5501–5525. [Google Scholar] [CrossRef]
- Kozlov, M.V.; Konduktorov, K.A.; Shcherbakova, A.S.; Kochetkov, S.N. Synthesis of N′-Propylhydrazide Analogs of Hydroxamic Inhibitors of Histone Deacetylases (HDACs) and Evaluation of Their Impact on Activities of HDACs and Replication of Hepatitis C Virus (HCV). Bioorganic Med. Chem. Lett. 2019, 29, 2369–2374. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Stowe, R.L.; Pinello, C.E.; Tian, G.; Madoux, F.; Li, D.; Zhao, L.Y.; Li, J.L.; Wang, Y.; Wang, Y.; et al. Identification of Histone Deacetylase Inhibitors with Benzoylhydrazide Scaffold That Selectively Inhibit Class i Histone Deacetylases. Chem. Biol. 2015, 22, 273–284. [Google Scholar] [CrossRef]
- Seashore-Ludlow, B.; Rees, M.G.; Cheah, J.H.; Coko, M.; Price, E.V.; Coletti, M.E.; Jones, V.; Bodycombe, N.E.; Soule, C.K.; Gould, J.; et al. Harnessing Connectivity in a Large-Scale Small-Molecule Sensitivity Dataset. Cancer Discov. 2015, 5, 1210–1223. [Google Scholar] [CrossRef] [PubMed]
- Lebedev, T.; Kousar, R.; Patrick, B.; Usama, M.; Lee, M.K.; Tan, M.; Li, X.G. Targeting ARID1A-Deficient Cancers: An Immune-Metabolic Perspective. Cells 2023, 12, 952. [Google Scholar] [CrossRef] [PubMed]
- Lebedev, T.; Buzdin, A.; Khabusheva, E.; Spirin, P.; Suntsova, M.; Sorokin, M.; Popenko, V.; Rubtsov, P.; Prassolov, V. Subtype of Neuroblastoma Cells with High KIT Expression Are Dependent on KIT and Its Knockdown Induces Compensatory Activation of Pro-Survival Signaling. Int. J. Mol. Sci. 2022, 23, 7724. [Google Scholar] [CrossRef]
- McDonald, E.R.; de Weck, A.; Schlabach, M.R.; Billy, E.; Mavrakis, K.J.; Hoffman, G.R.; Belur, D.; Castelletti, D.; Frias, E.; Gampa, K.; et al. Project DRIVE: A Compendium of Cancer Dependencies and Synthetic Lethal Relationships Uncovered by Large-Scale, Deep RNAi Screening. Cell 2017, 170, 577–592. [Google Scholar] [CrossRef]
- McFarland, J.M.; Ho, Z.V.; Kugener, G.; Dempster, J.M.; Montgomery, P.G.; Bryan, J.G.; Krill-Burger, J.M.; Green, T.M.; Vazquez, F.; Boehm, J.S.; et al. Improved Estimation of Cancer Dependencies from Large-Scale RNAi Screens Using Model-Based Normalization and Data Integration. Nat. Commun. 2018, 9, 4610. [Google Scholar] [CrossRef]
- Behan, F.M.; Iorio, F.; Picco, G.; Gonçalves, E.; Beaver, C.M.; Migliardi, G.; Santos, R.; Rao, Y.; Sassi, F.; Pinnelli, M.; et al. Prioritization of Cancer Therapeutic Targets Using CRISPR–Cas9 Screens. Nature 2019, 568, 511–516. [Google Scholar] [CrossRef]
- Gu, Z.; Eils, R.; Schlesner, M. Complex Heatmaps Reveal Patterns and Correlations in Multidimensional Genomic Data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [PubMed]
- Kozlov, M.V.; Malikova, A.Z.; Kamarova, K.A.; Konduktorov, K.A.; Kochetkov, S.N. Synthesis of Pyridyl-4-Oxy-Substituted N-Hydroxy Amides of Cinnamic Acid as New Inhibitors of Histone Deacetylase Activity and Hepatitis C Virus Replication. Russ. J. Bioorganic Chem. 2018, 44, 453–460. [Google Scholar] [CrossRef]
- Kleymenova, A.; Zemskaya, A.; Kochetkov, S.; Kozlov, M. In-Cell Testing of Zinc-Dependent Histone Deacetylase Inhibitors in the Presence of Class-Selective Fluorogenic Substrates: Potential and Limitations of the Method. Biomedicines 2024, 12, 1203. [Google Scholar] [CrossRef] [PubMed]
- Hai, Y.; Christianson, D.W. Histone Deacetylase 6 Structure and Molecular Basis of Catalysis and Inhibition. Nat. Chem. Biol. 2016, 12, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Watson, P.J.; Fairall, L.; Santos, G.M.; Schwabe, J.W.R. Structure of HDAC3 Bound to Co-Repressor and Inositol Tetraphosphate. Nature 2012, 481, 335–340. [Google Scholar] [CrossRef]
- Lu, C.; Wu, C.; Ghoreishi, D.; Chen, W.; Wang, L.; Damm, W.; Ross, G.A.; Dahlgren, M.K.; Russell, E.; Von Bargen, C.D.; et al. OPLS4: Improving Force Field Accuracy on Challenging Regimes of Chemical Space. J. Chem. Theory Comput. 2021, 17, 4291–4300. [Google Scholar] [CrossRef]
- Bülbül, E.F.; Robaa, D.; Sun, P.; Mahmoudi, F.; Melesina, J.; Zessin, M.; Schutkowski, M.; Sippl, W. Application of Ligand- and Structure-Based Prediction Models for the Design of Alkylhydrazide-Based HDAC3 Inhibitors as Novel Anti-Cancer Compounds. Pharmaceuticals 2023, 16, 968. [Google Scholar] [CrossRef]
- Miller, E.B.; Murphy, R.B.; Sindhikara, D.; Borrelli, K.W.; Grisewood, M.J.; Ranalli, F.; Dixon, S.L.; Jerome, S.; Boyles, N.A.; Day, T.; et al. Reliable and Accurate Solution to the Induced Fit Docking Problem for Protein-Ligand Binding. J. Chem. Theory Comput. 2021, 17, 2630–2639. [Google Scholar] [CrossRef]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef]
- Li, Y.; Seto, E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb. Perspect. Med. 2016, 6, a026831. [Google Scholar] [CrossRef]
- Liang, T.; Wang, F.; Elhassan, R.M.; Cheng, Y.; Tang, X.; Chen, W.; Fang, H.; Hou, X. Targeting Histone Deacetylases for Cancer Therapy: Trends and Challenges. Acta Pharm. Sin. B 2023, 13, 2425–2463. [Google Scholar] [PubMed]
- Sun, P.; Wang, J.; Khan, K.S.; Yang, W.; Ng, B.W.L.; Ilment, N.; Zessin, M.; Bülbül, E.F.; Robaa, D.; Erdmann, F.; et al. Development of Alkylated Hydrazides as Highly Potent and Selective Class i Histone Deacetylase Inhibitors with T Cell Modulatory Properties. J. Med. Chem. 2022, 65, 16313–16337. [Google Scholar] [CrossRef] [PubMed]
- Melesina, J.; Simoben, C.V.; Praetorius, L.; Bülbül, E.F.; Robaa, D.; Sippl, W. Strategies To Design Selective Histone Deacetylase Inhibitors. ChemMedChem 2021, 16, 1336–1359. [Google Scholar]
- Havas, A.P.; Tula-Sanchez, A.A.; Steenhoek, H.M.; Bhakta, A.; Wingfield, T.; Huntley, M.J.; Nofal, A.S.; Ahmed, T.; Jaime-Frias, R.; Smith, C.L. Defining Cellular Responses to HDAC-Selective Inhibitors Reveals That Efficient Targeting of HDAC3 Is Required to Elicit Cytotoxicity and Overcome Naïve Resistance to Pan-HDACi in Diffuse Large B Cell Lymphoma. Transl. Oncol. 2024, 39, 101779. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spirin, P.; Vedernikova, V.; Volkava, T.; Morozov, A.; Kleymenova, A.; Zemskaya, A.; Shyrokova, L.; Porozov, Y.; Glumakova, K.; Lebedev, T.; et al. New and Effective Inhibitor of Class I HDACs, Eimbinostat, Reduces the Growth of Hematologic Cancer Cells and Triggers Apoptosis. Pharmaceutics 2025, 17, 416. https://doi.org/10.3390/pharmaceutics17040416
Spirin P, Vedernikova V, Volkava T, Morozov A, Kleymenova A, Zemskaya A, Shyrokova L, Porozov Y, Glumakova K, Lebedev T, et al. New and Effective Inhibitor of Class I HDACs, Eimbinostat, Reduces the Growth of Hematologic Cancer Cells and Triggers Apoptosis. Pharmaceutics. 2025; 17(4):416. https://doi.org/10.3390/pharmaceutics17040416
Chicago/Turabian StyleSpirin, Pavel, Valeria Vedernikova, Tatsiana Volkava, Alexey Morozov, Alla Kleymenova, Anastasia Zemskaya, Lena Shyrokova, Yuri Porozov, Ksenia Glumakova, Timofey Lebedev, and et al. 2025. "New and Effective Inhibitor of Class I HDACs, Eimbinostat, Reduces the Growth of Hematologic Cancer Cells and Triggers Apoptosis" Pharmaceutics 17, no. 4: 416. https://doi.org/10.3390/pharmaceutics17040416
APA StyleSpirin, P., Vedernikova, V., Volkava, T., Morozov, A., Kleymenova, A., Zemskaya, A., Shyrokova, L., Porozov, Y., Glumakova, K., Lebedev, T., Kozlov, M., & Prassolov, V. (2025). New and Effective Inhibitor of Class I HDACs, Eimbinostat, Reduces the Growth of Hematologic Cancer Cells and Triggers Apoptosis. Pharmaceutics, 17(4), 416. https://doi.org/10.3390/pharmaceutics17040416