


The 20th Anniversary of Pegaptanib (MacugenTM), the First Approved Aptamer Medicine: History, Recent Advances and Future Prospects of Aptamers in Therapy


Abstract

1. Introduction
2. Pegaptanib
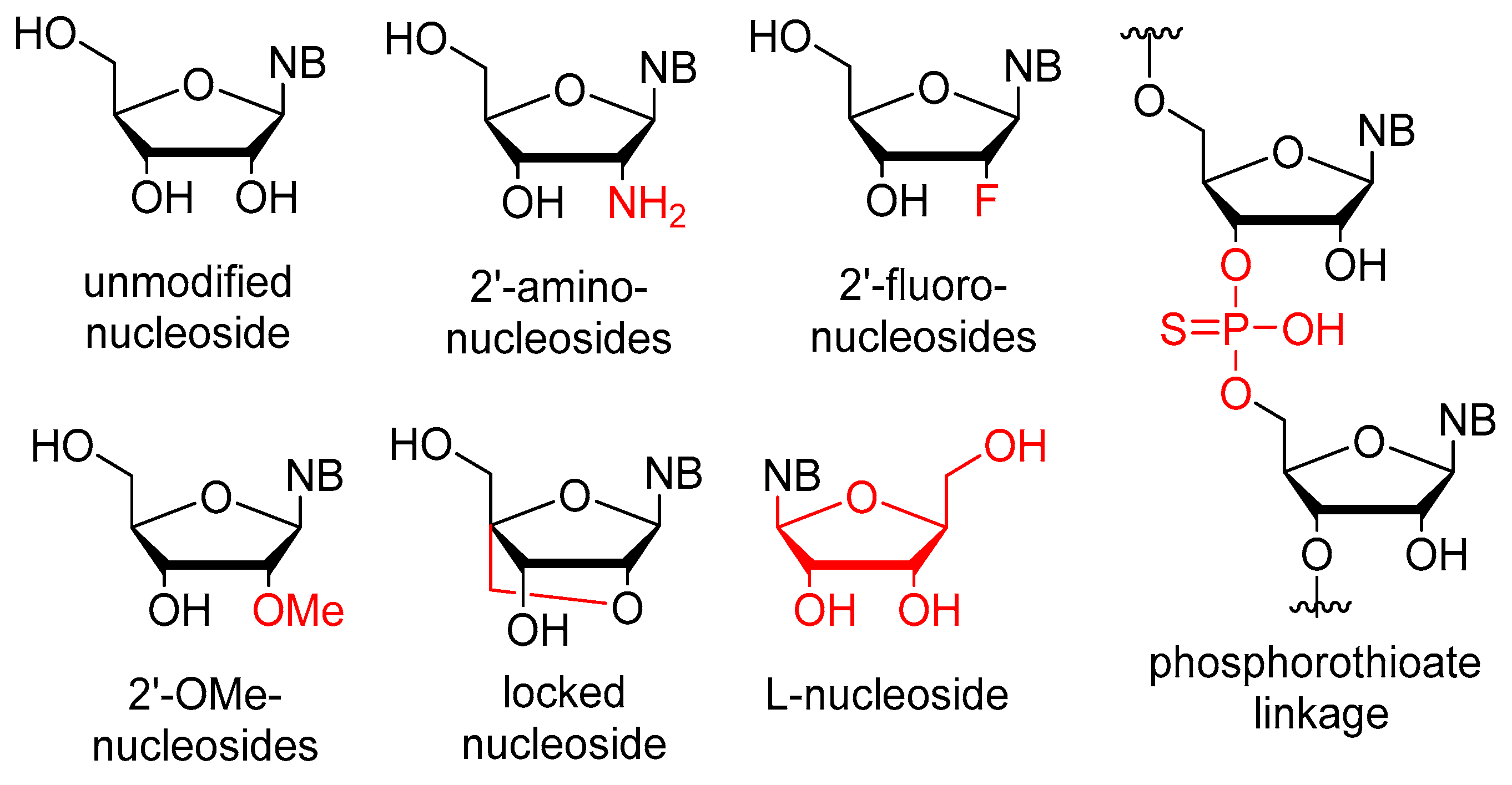
2.1. Chemistry and Mechanism of Action
2.2. Development and Approval
2.2.1. Chemical Development and In Vitro Assays
2.2.2. Preclinical Studies
2.2.3. Clinical Studies
2.3. After Approval
2.3.1. General Information
2.3.2. Efficacy
Age Related Macular Degeneration (AMD)
Diabetic Retinopathy (DR) and Diabetic Macular Edema (DME)
Coats Disease
Retinal Vein Occlusion
Choroidal Neovascularization in Different Diseases
Macular Edemas
Other Diseases
2.3.3. Safety and Adverse Events
General Safety
Endophtalmitis
Increased Intraocular Pressure (IOP)
Retinal Pigment Epithelial Tear
Silicon Oil Droplets
Other Studies on Potential Side Effects and Drug Interactions
2.3.4. Cost-Effectiveness
2.3.5. Comparing with Antibodies
Efficacy
Safety
Cost-Effectiveness
2.3.6. Other Results
Efficacy and Safety in Nonclinical Trials
Strategies for Maximizing the Potential of Pegaptanib
VEGF Isoforms-, Cell-, and Site-Specific Effects of Pegaptanib
Higher Order Structure of Pegaptanib
3. Aptamers Beyond Pegaptanib—The Latest Breakthrough in AMD and Emerging New Applications
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Ireson, C.R.; Kelland, L.R. Discovery and development of anticancer aptamers. Mol. Cancer Ther. 2006, 5, 2957–2962. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, H.; Trujillo, C.A.; Nery, A.A.; Alves, J.M.; Majumder, P.; Resende, R.R.; Martins, A.H. DNA and RNA aptamers: From tools for basic research towards therapeutic applications. Comb. Chem. High Throughput Screen. 2006, 9, 619–632. [Google Scholar] [CrossRef] [PubMed]
- Nimjee, S.M.; White, R.R.; Becker, R.C.; Sullenger, B.A. Aptamers as therapeutics. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 61–79. [Google Scholar] [CrossRef] [PubMed]
- Ospina-Villa, J.D.; López-Camarillo, C.; Castañón-Sánchez, C.A.; Soto-Sánchez, J.; Ramírez-Moreno, E.; Marchat, L.A. Advances on aptamers against protozoan parasites. Genes 2018, 9, 584. [Google Scholar] [CrossRef]
- Aljohani, M.M.; Cialla-May, D.; Popp, J.; Chinnappan, R.; Al-Kattan, K.; Zourob, M. Aptamers: Potential diagnostic and therapeutic agents for blood diseases. Molecules 2022, 27, 383. [Google Scholar] [CrossRef]
- Nimjee, S.M.; Rusconi, C.P.; Sullenger, B.A. Aptamers: An emerging class of therapeutics. Annu. Rev. Med. 2005, 56, 555–583. [Google Scholar] [CrossRef]
- Kaur, G.; Roy, I. Therapeutic applications of aptamers. Expert Opin. Investig. Drugs 2008, 17, 43–60. [Google Scholar] [CrossRef]
- Dausse, E.; Gomes, S.D.R.; Toulmé, J.-J. Aptamers: A new class of oligonucleotides in the drug discovery pipeline? Curr. Opin. Pharmacol. 2009, 9, 602–607. [Google Scholar] [CrossRef]
- Fallah, A.; Fooladi, A.A.I.; Havaei, S.A.; Mahboobi, M.; Sedighian, H. Recent advances in aptamer discovery, modification and improving performance. Biochem. Biophys. Rep. 2024, 40, 101852. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, N.; Chan, C.-W.; Lu, A.; Yu, Y.; Zhang, G.; Ren, K. The application of microfluidic technologies in aptamer selection. Front. Cell Dev. Biol. 2021, 9, 730035. [Google Scholar] [CrossRef]
- Ang, A.; Ouellet, E.; Cheung, K.C.; Haynes, C. Highly Efficient and Reliable DNA Aptamer Selection Using the Partitioning Capabilities of Ddpcr: The Hi-Fi Selex Method. In Digital PCR: Methods in Molecular Biology; Humana Press: New York, NY, USA, 2018; pp. 531–554. [Google Scholar] [CrossRef]
- Guan, J.; He, K.; Gunasekaran, S. Selection of ssDNA aptamer using GO-SELEX and development of DNA nanostructure-based electrochemical aptasensor for penicillin. Biosens. Bioelectron. X 2022, 12, 100220. [Google Scholar] [CrossRef]
- Sattari, R.; Palizban, A.; Khanahmad, H. Single-strand DNA-like oligonucleotide aptamer against proprotein convertase subtilisin/kexin 9 using CE-SELEX: PCSK9 targeting selection. Cardiovasc. Drugs Ther. 2020, 34, 475–485. [Google Scholar] [CrossRef]
- Eaton, R.M.; Shallcross, J.A.; Mael, L.E.; Mears, K.S.; Minkoff, L.; Scoville, D.J.; Whelan, R.J. Selection of DNA aptamers for ovarian cancer biomarker HE4 using CE-SELEX and high-throughput sequencing. Anal. Bioanal. Chem. 2015, 407, 6965–6973. [Google Scholar] [CrossRef] [PubMed]
- Lyu, C.; Khan, I.M.; Wang, Z. Capture-SELEX for aptamer selection: A short review. Talanta 2021, 229, 122274. [Google Scholar] [CrossRef] [PubMed]
- Niu, C.; Zhang, C.; Liu, J. Capture-SELEX of DNA aptamers for estradiol specifically and estrogenic compounds collectively. Environ. Sci. Technol. 2022, 56, 17702–17711. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Liu, C.; Zhou, Q.; Chen, X.; Li, H.; Wang, S.; Guo, Y. Selecting specific aptamers that bind to ovine pregnancy-associated glycoprotein 7 using real serum sample–assisted FluMag-SELEX to develop magnetic microparticle–based colorimetric aptasensor. Anal. Chim. Acta 2022, 1191, 339291. [Google Scholar] [CrossRef]
- Birch, C.M.; Hou, H.W.; Han, J.; Niles, J.C. Identification of malaria parasite-infected red blood cell surface aptamers by inertial microfluidic SELEX (I-SELEX). Sci. Rep. 2015, 5, 11347. [Google Scholar] [CrossRef]
- Wang, L.; Alkhamis, O.; Canoura, J.; Yu, H.; Xiao, Y. Rapid Nuclease-Assisted Selection of High-Affinity Small-Molecule Aptamers. J. Am. Chem. Soc. 2024, 146, 21296–21307. [Google Scholar] [CrossRef]
- Sedighian, H.; Halabian, R.; Amani, J.; Heiat, M.; Amin, M.; Fooladi, A.A.I. Staggered Target SELEX, a novel approach to isolate non-cross-reactive aptamer for detection of SEA by apta-qPCR. J. Biotechnol. 2018, 286, 45–55. [Google Scholar] [CrossRef]
- Duan, Y.; Zhang, C.; Wang, Y.; Chen, G. Research progress of whole-cell-SELEX selection and the application of cell-targeting aptamer. Mol. Biol. Rep. 2022, 49, 7979–7993. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, Y.; Chen, Y.; Hong, S.; Sun, Y.; Sun, N.; Pei, R. In vitro selection of DNA aptamers against renal cell carcinoma using living cell-SELEX. Talanta 2017, 175, 235–242. [Google Scholar] [CrossRef]
- Ning, Y.; Hu, J.; Lu, F. Aptamers used for biosensors and targeted therapy. Biomed. Pharmacother. 2020, 132, 110902. [Google Scholar] [CrossRef]
- Ruscito, A.; DeRosa, M.C. Small-molecule binding aptamers: Selection strategies, characterization, and applications. Front. Chem. 2016, 4, 14. [Google Scholar] [CrossRef]
- Armutcu, C.; Karasu, T.; Pişkin, S.; Özgür, E.; Uzun, L. Selective aptasensor for trinitrotoluene detection: Comparison of the detecting performances from liquid and vapor phases. Colloids Surf. A Physicochem. Eng. Asp. 2023, 676, 132258. [Google Scholar] [CrossRef]
- Kelvin, D.; Suess, B. RNA aptamers: Promising tools in synthetic biology. at-Automatisierungstechnik 2024, 72, 666–671. [Google Scholar] [CrossRef]
- Morita, Y.; Leslie, M.; Kameyama, H.; Volk, D.E.; Tanaka, T. Aptamer therapeutics in cancer: Current and future. Cancers 2018, 10, 80. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Xiang, J. Aptamers, the nucleic acid antibodies, in cancer therapy. Adv. Med. Biochem. Genom. Physiol. Pathol. 2021, 21, 2793. [Google Scholar] [CrossRef]
- Liu, S.; Xu, Y.; Jiang, X.; Tan, H.; Ying, B. Translation of aptamers toward clinical diagnosis and commercialization. Biosens. Bioelectron. 2022, 208, 114168. [Google Scholar] [CrossRef]
- Bunka, D.H.; Platonova, O.; Stockley, P.G. Development of aptamer therapeutics. Curr. Opin. Pharmacol. 2010, 10, 557–562. [Google Scholar] [CrossRef]
- Traber, G.M.; Yu, A.-M. RNAi-based therapeutics and novel RNA bioengineering technologies. J. Pharmacol. Exp. Ther. 2023, 384, 133–154. [Google Scholar] [CrossRef]
- Adachi, T.; Nakamura, Y. Aptamers: A review of their chemical properties and modifications for therapeutic application. Molecules 2019, 24, 4229. [Google Scholar] [CrossRef] [PubMed]
- Drolet, D.W.; Green, L.S.; Gold, L.; Janjic, N. Fit for the eye: Aptamers in ocular disorders. Nucleic Acid Ther. 2016, 26, 127–146. [Google Scholar] [CrossRef]
- Gao, S.; Zheng, X.; Jiao, B.; Wang, L. Post-SELEX optimization of aptamers. Anal. Bioanal. Chem. 2016, 408, 4567–4573. [Google Scholar] [CrossRef] [PubMed]
- Bege, M.; Borbás, A. The medicinal chemistry of artificial nucleic acids and therapeutic oligonucleotides. Pharmaceuticals 2022, 15, 909. [Google Scholar] [CrossRef] [PubMed]
- Dinis Ano Bom, A.P.; da Costa Neves, P.C.; Bonacossa de Almeida, C.E.; Silva, D.; Missailidis, S. Aptamers as delivery agents of siRNA and chimeric formulations for the treatment of cancer. Pharmaceutics 2019, 11, 684. [Google Scholar] [CrossRef]
- Wieleba, I.; Wojas-Krawczyk, K.; Krawczyk, P. Aptamers in non-small cell lung cancer treatment. Molecules 2020, 25, 3138. [Google Scholar] [CrossRef]
- Pugazhendhi, A.; Hubbell, M.; Jairam, P.; Ambati, B. Neovascular macular degeneration: A review of etiology, risk factors, and recent advances in research and therapy. Int. J. Mol. Sci. 2021, 22, 1170. [Google Scholar] [CrossRef]
- Wong, W.L.; Su, X.; Li, X.; Cheung, C.M.G.; Klein, R.; Cheng, C.-Y.; Wong, T.Y. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: A systematic review and meta-analysis. Lancet Glob. Health 2014, 2, e106–e116. [Google Scholar] [CrossRef]
- Pauleikhoff, D. Neovascular age-related macular degeneration: Natural history and treatment outcomes. Retina 2005, 25, 1065–1084. [Google Scholar] [CrossRef]
- Arrigo, A.; Bandello, F. Molecular features of classic retinal drugs, retinal therapeutic targets and emerging treatments. Pharmaceutics 2021, 13, 1102. [Google Scholar] [CrossRef]
- Egli, M.; Manoharan, M. Chemistry, structure and function of approved oligonucleotide therapeutics. Nucleic Acids Res. 2023, 51, 2529–2573. [Google Scholar] [CrossRef]
- Cao, J.; Zhang, F.; Xiong, W. Discovery of aptamers and the acceleration of the development of targeting research in ophthalmology. Int. J. Nanomed. 2023, 18, 4421–4430. [Google Scholar] [CrossRef]
- Ishida, S.; Usui, T.; Yamashiro, K.; Kaji, Y.; Amano, S.; Ogura, Y.; Hida, T.; Oguchi, Y.; Ambati, J.; Miller, J.W. VEGF164-mediated inflammation is required for pathological, but not physiological, ischemia-induced retinal neovascularization. J. Exp. Med. 2003, 198, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Nagineni, C.N.; Kommineni, V.K.; William, A.; Detrick, B.; Hooks, J.J. Regulation of VEGF expression in human retinal cells by cytokines: Implications for the role of inflammation in age-related macular degeneration. J. Cell. Physiol. 2012, 227, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Tucker, C.E.; Chen, L.-S.; Judkins, M.B.; Farmer, J.A.; Gill, S.C.; Drolet, D.W. Detection and plasma pharmacokinetics of an anti-vascular endothelial growth factor oligonucleotide-aptamer (NX1838) in rhesus monkeys. J. Chromatogr. B Biomed. Sci. Appl. 1999, 732, 203–212. [Google Scholar] [CrossRef]
- Lee, J.-H.; Canny, M.D.; De Erkenez, A.; Krilleke, D.; Ng, Y.-S.; Shima, D.T.; Pardi, A.; Jucker, F. A therapeutic aptamer inhibits angiogenesis by specifically targeting the heparin binding domain of VEGF165. Proc. Natl. Acad. Sci. USA 2005, 102, 18902–18907. [Google Scholar] [CrossRef]
- Amadio, M.; Govoni, S.; Pascale, A. Targeting VEGF in eye neovascularization: What’s new?: A comprehensive review on current therapies and oligonucleotide-based interventions under development. Pharmacol. Res. 2016, 103, 253–269. [Google Scholar] [CrossRef]
- Vinores, S.A. Pegaptanib in the treatment of wet, age-related macular degeneration. Int. J. Nanomed. 2006, 1, 263–268. [Google Scholar]
- Ruckman, J.; Green, L.S.; Beeson, J.; Waugh, S.; Gillette, W.L.; Henninger, D.D.; Claesson-Welsh, L.; Janjic, N. 2′-Fluoropyrimidine RNA-based aptamers to the 165-amino acid form of vascular endothelial growth factor (VEGF165): Inhibition of receptor binding and VEGF-induced vascular permeability through interactions requiring the exon 7-encoded domain. J. Biol. Chem. 1998, 273, 20556–20567. [Google Scholar] [CrossRef] [PubMed]
- Bell, C.; Lynam, E.; Landfair, D.J.; Janjic, N.; Wiles, M.E. Oligonucleotide NX1838 inhibits VEGF 165-mediated cellular responses in vitro. Vitr. Cell. Dev. Biol.-Anim. 1999, 35, 533–542. [Google Scholar] [CrossRef]
- Ng, E.W.; Shima, D.T.; Calias, P.; Cunningham Jr, E.T.; Guyer, D.R.; Adamis, A.P. Pegaptanib, a targeted anti-VEGF aptamer for ocular vascular disease. Nat. Rev. Drug Discov. 2006, 5, 123–132. [Google Scholar] [CrossRef]
- Jellinek, D.; Green, L.S.; Bell, C.; Janjic, N. Inhibition of receptor binding by high-affinity RNA ligands to vascular endothelial growth factor. Biochemistry 1994, 33, 10450–10456. [Google Scholar] [CrossRef] [PubMed]
- Green, L.S.; Jellinek, D.; Bell, C.; Beebe, L.A.; Feistner, B.D.; Gill, S.C.; Jucker, F.M.; Janjić, N. Nuclease-resistant nucleic acid ligands to vascular permeability factor/vascular endothelial growth factor. Chem. Biol. 1995, 2, 683–695. [Google Scholar] [CrossRef] [PubMed]
- Drolet, D.W.; Nelson, J.; Tucker, C.E.; Zack, P.M.; Nixon, K.; Bolin, R.; Judkins, M.B.; Farmer, J.A.; Wolf, J.L.; Gill, S.C. Pharmacokinetics and safety of an anti-vascular endothelial growth factor aptamer (NX1838) following injection into the vitreous humor of rhesus monkeys. Pharm. Res. 2000, 17, 1503–1510. [Google Scholar] [CrossRef]
- Eyetech Study Group. Preclinical and phase 1A clinical evaluation of an anti-VEGF pegylated aptamer (EYE001) for the treatment of exudative age-related macular degeneration. Retina 2002, 22, 143–152. [Google Scholar] [CrossRef]
- Ishida, S.; Usui, T.; Yamashiro, K.; Kaji, Y.; Ahmed, E.; Carrasquillo, K.G.; Amano, S.; Hida, T.; Oguchi, Y.; Adamis, A.P. VEGF164 is proinflammatory in the diabetic retina. Investig. Ophthalmol. Vis. Sci. 2003, 44, 2155–2162. [Google Scholar] [CrossRef]
- Carrasquillo, K.G.; Ricker, J.A.; Rigas, I.K.; Miller, J.W.; Gragoudas, E.S.; Adamis, A.P. Controlled delivery of the anti-VEGF aptamer EYE001 with poly (lactic-co-glycolic) acid microspheres. Investig. Ophthalmol. Vis. Sci. 2003, 44, 290–299. [Google Scholar] [CrossRef]
- Eyetech Study Group. Anti-vascular endothelial growth factor therapy for subfoveal choroidal neovascularization secondary to age-related macular degeneration: Phase II study results. Ophthalmology 2003, 110, 979–986. [Google Scholar] [CrossRef]
- Csaky, K. Anti-vascular endothelial growth factor therapy for neovascular age-related macular degeneration: Promises and pitfalls. Ophthalmology 2003, 110, 879–881. [Google Scholar] [CrossRef]
- Gragoudas, E.S.; Adamis, A.P.; Cunningham, E.T., Jr.; Feinsod, M.; Guyer, D.R. Pegaptanib for neovascular age-related macular degeneration. N. Engl. J. Med. 2004, 351, 2805–2816. [Google Scholar] [CrossRef]
- Rakic, J.-M.; Blaise, P.; Foidart, J.-M. Pegaptanib and age-related macular degeneration. N. Engl. J. Med. 2005, 352, 1720–1721. [Google Scholar] [CrossRef] [PubMed]
- Schachat, A.P. New treatments for age-related macular degeneration. Ophthalmology 2005, 112, 531–532. [Google Scholar] [CrossRef]
- Spaide, R. New treatments for AMD. Ophthalmology 2006, 113, 160–161. [Google Scholar] [CrossRef] [PubMed]
- Moshfeghi, A.A.; Puliafito, C.A. Pegaptanib sodium for the treatment of neovascular age-related macular degeneration. Expert Opin. Investig. Drugs 2005, 14, 671–682. [Google Scholar] [CrossRef]
- Doggrell, S.A. Pegaptanib: The first antiangiogenic agent approved for neovascular macular degeneration. Expert Opin. Pharmacother. 2005, 6, 1421–1423. [Google Scholar] [CrossRef] [PubMed]
- Sivaprasad, S.; Acharya, N.; Hykin, P. Pegaptanib sodium for neovascular age-related macular degeneration: Clinical experience in the UK. Clin. Ophthalmol. 2008, 2, 347–354. [Google Scholar] [CrossRef]
- VEGF Inhibition Study in Ocular Neovascularization Clinical Trial Group. Year 2 efficacy results of 2 randomized controlled clinical trials of pegaptanib for neovascular age-related macular degeneration. Ophthalmology 2006, 113, 1508.e1–1508.e25. [Google Scholar] [CrossRef]
- Gryziewicz, L. Regulatory aspects of drug approval for macular degeneration. Adv. Drug Deliv. Rev. 2005, 57, 2092–2098. [Google Scholar] [CrossRef]
- Hussar, D.A. New drugs: Entecavir, ibandronate sodium, and pegaptanib sodium. J. Am. Pharm. Assoc. 2005, 45, 412–415. [Google Scholar] [CrossRef]
- Mills, E.; Heels-Ansdell, D.; Kelly, S.; Guyatt, G. A randomized trial of Pegaptanib sodium for age-related macular degeneration used an innovative design to explore disease-modifying effects. J. Clin. Epidemiol. 2007, 60, 456–460. [Google Scholar] [CrossRef]
- Ng, E.W.; Adamis, A.P. Targeting angiogenesis, the underlying disorder in neovascular age-related macular degeneration. Can. J. Ophthalmol. 2005, 40, 352–368. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, M.A.A.; Keating, G.M. Pegaptanib: In exudative age-related macular degeneration. Drugs 2005, 65, 1571–1577. [Google Scholar] [CrossRef] [PubMed]
- Leys, A.; Zlateva, G.; Shah, S.; Patel, M. Quality of life in patients with age-related macular degeneration: Results from the VISION study. Eye 2008, 22, 792–798. [Google Scholar] [CrossRef] [PubMed]
- VEGF Inhibition Study in Ocular Neovascularization Clinical Trial Group. Enhanced efficacy associated with early treatment of neovascular age-related macular degeneration with pegaptanib sodium: An exploratory analysis. Retina 2005, 25, 815–827. [Google Scholar] [CrossRef]
- Quiram, P.A.; Hassan, T.S.; Williams, G.A. Treatment of naïve lesions in neovascular age-related macular degeneration with pegaptanib. Retina 2007, 27, 851–856. [Google Scholar] [CrossRef]
- Williams, A.J.; Fekrat, S. Progression of choroidal neovascularization following injection of pegaptanib sodium (Macugen) in two eyes with neovascular age-related macular degeneration. Am. J. Ophthalmol. 2006, 142, 683–685.e681. [Google Scholar] [CrossRef]
- Schuman, S.; Rogers, A.H.; Duker, J.S.; Reichel, E.; Baumal, C.R. Six-week outcomes after pegaptanib. Ophthalmology 2006, 113, 501. [Google Scholar] [CrossRef]
- Emerson, G.G.; Flaxel, C.J.; Lauer, A.K.; Stout, J.T.; Emerson, M.V.; Nolte, S.; Wilson, D.J.; Klein, M.L. Optical coherence tomography findings during pegaptanib therapy for neovascular age-related macular degeneration. Retina 2007, 27, 724–729. [Google Scholar] [CrossRef]
- Lipski, A.; Bornfeld, N.; Jurklies, B. Multifocal electroretinography in patients with exudative amd and intravitreal treatment with pegaptanib sodium. Retina 2007, 27, 864–872. [Google Scholar] [CrossRef]
- Feucht, N.; Matthias, H.; Lohmann, C.P.; Maier, M. Pegaptanib sodium treatment in neovascular age-related macular degeneration: Clinical experience in Germany. Clin. Ophthalmol. 2008, 2, 253–259. [Google Scholar] [CrossRef]
- Thelen, U. Clinical experience with pegaptanib in the treatment of age-related macular degeneration (AMD). Klin. Monatsblatter Augenheilkd. 2009, 227, 67–72. [Google Scholar] [CrossRef]
- Forte, R.; Cennamo, G.; Finelli, M.; Cesarano, I.; D’Amico, G.; De Crecchio, G.; Cennamo, G. Intravitreal triamcinolone, bevacizumab and pegaptanib for occult choroidal neovascularization. Acta Ophthalmol. 2010, 88, e305–e310. [Google Scholar] [CrossRef] [PubMed]
- Weber, P.A.; Wirostko, B.M.; Xu, X.; Goss, T.F.; Zlateva, G. Newly diagnosed exudative age-related macular degeneration treated with pegaptanib sodium monotherapy in US community-based practices: Medical chart review study. BMC Ophthalmol. 2010, 10, 2. [Google Scholar] [CrossRef]
- Lesniak, S.P.; Fine, H.F.; Prenner, J.L.; Roth, D.B. Long-term follow-up of spontaneous retinal pigment epithelium tears in age-related macular degeneration treated with anti-VEGF therapy. Eur. J. Ophthalmol. 2011, 21, 73–76. [Google Scholar] [CrossRef]
- Sivaprasad, S.; Hykin, P.; Saeed, A.; Beatty, S.; Grisanti, S.; Staurenghi, G.; Olea, J.L.; Campos, A.; Barbosa, A.; Rito, L. Intravitreal pegaptanib sodium for choroidal neovascularisation secondary to age-related macular degeneration: Pan-European experience. Eye 2010, 24, 793–798. [Google Scholar] [CrossRef]
- Ricci, F.; Missiroli, F.; Cedrone, C.; Grossi, M.; Regine, F. Compassionate use of intravitreal pegaptanib in patients with age-related macular degeneration. Semin.Ophthalmol. 2010, 25, 16–20. [Google Scholar] [CrossRef]
- Battaglia Parodi, M.; Di Bartolo, E.; Brue, C.; Cappello, E.; Furino, C.; Giuffrida, S.; Imparato, M.; Reibaldi, M. Pegaptanib: Choroidal neovascularization in patients with age-related macular degeneration and previous arterial thromboembolic events. Eur. J. Ophthalmol. 2018, 28, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Atmani, K.; Coscas, F.; Coscas, G.; Soubrane, G. Pegaptanib sodium for occult choroidal neovascularization in neovascular age-related macular degeneration: A prospective case series. Eye 2009, 23, 1150–1154. [Google Scholar] [CrossRef]
- Kaiser, P. Verteporfin photodynamic therapy and anti-angiogenic drugs: Potential for combination therapy in exudative age-related macular degeneration. Curr. Med. Res. Opin. 2007, 23, 477–487. [Google Scholar] [CrossRef]
- Liggett, P.E.; Colina, J.; Chaudhry, N.A.; Tom, D.; Haffner, G. Triple therapy of intravitreal triamcinolone, photodynamic therapy, and pegaptanib sodium for choroidal neovascularization. Am. J. Ophthalmol. 2006, 142, 1072–1074. [Google Scholar] [CrossRef]
- Calvo-González, C.; Reche-Frutos, J.; Donate-López, J.; García-Feijoó, J.; Leila, M.; Fernández-Pérez, C.; Garcia-Sánchez, J. Combined Pegaptanib sodium (Macugen) and photodynamic therapy in predominantly classic juxtafoveal choroidal neovascularisation in age related macular degeneration. Br. J. Ophthalmol. 2008, 92, 74–75. [Google Scholar] [CrossRef] [PubMed]
- Ozturk, T.; Oner, H.; Saatci, A.O.; Kaynak, S. Low-fluence photodynamic therapy combinations in the treatment of exudative age-related macular degeneration. Int. J. Ophthalmol. 2012, 5, 377. [Google Scholar] [CrossRef]
- Schmid-Kubista, K.E.; Krebs, I.; Ansari-Shahrezaei, S.; Haas, P.; Hagen, S.; Binder, S. Comparing treatment of neovascular age-related macular degeneration with sequential intravitreal Avastin and Macugen versus intravitreal mono-therapy—A pilot study. Curr. Eye Res. 2011, 36, 958–963. [Google Scholar] [CrossRef]
- Friberg, T.R.; Tolentino, M.; Group, L.S. Pegaptanib sodium as maintenance therapy in neovascular age-related macular degeneration: The LEVEL study. Br. J. Ophthalmol. 2010, 94, 1611–1617. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, T.; Group, L.-J.S. Maintenance therapy with pegaptanib sodium for neovascular age-related macular degeneration: An exploratory study in Japanese patients (LEVEL-J study). Jpn. J. Ophthalmol. 2013, 57, 417–423. [Google Scholar] [CrossRef]
- Inoue, M.; Kadonosono, K.; Arakawa, A.; Yamane, S.; Ishibashi, T. Long-term outcome of intravitreal pegaptanib sodium as maintenance therapy in Japanese patients with neovascular age-related macular degeneration. Jpn. J. Ophthalmol. 2015, 59, 173–178. [Google Scholar] [CrossRef]
- Bennett, M.D.; Yee, W.; Bryan, J.S. Pegaptanib combined with intravitreal injection of moxifloxacin as treatment of wet macular degeneration. Retina 2008, 28, 976–980. [Google Scholar] [CrossRef]
- Stewart, M.W. Anti-vascular endothelial growth factor drug treatment of diabetic macular edema: The evolution continues. Curr. Diabetes Rev. 2012, 8, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Macugen Diabetic Retinopathy Study Group. A phase II randomized double-masked trial of pegaptanib, an Anti–Vascular endothelial growth factor aptamer, for diabetic macular edema. Ophthalmology 2005, 112, 1747–1757. [Google Scholar] [CrossRef]
- Macugen Diabetic Retinopathy Study Group. Changes in retinal neovascularization after pegaptanib (Macugen) therapy in diabetic individuals. Ophthalmology 2006, 113, 23–28. [Google Scholar] [CrossRef]
- Bansal, A.G.; Narayanan, R.; Majji, A.B.; Thomas, R. Neovascular changes after pegaptanib in diabetics. Ophthalmology 2007, 114, 615–616. [Google Scholar] [CrossRef] [PubMed]
- Querques, G.; Bux, A.; Fusco, A.; Iaculli, C.; Delle Noci, N. Pegaptanib sodium versus pegaptanib sodium combined with macular laser photocoagulation or laser alone for diabetic macular edema. J. Ophthalmol. 2009, 2009, 672178. [Google Scholar] [CrossRef]
- Montero, J.; Ruiz-Moreno, J.; De La Vega, C. Incomplete posterior hyaloid detachment after intravitreal pegaptanib injection in diabetic macular edema. Eur. J. Ophthalmol. 2018, 18, 469–472. [Google Scholar] [CrossRef]
- Mendrinos, E.; Donati, G.; Pournaras, C.J. Rapid and persistent regression of severe new vessels on the disc in proliferative diabetic retinopathy after a single intravitreal injection of pegaptanib. Acta Ophthalmol. 2009, 87, 683–684. [Google Scholar] [CrossRef] [PubMed]
- Querques, G.; Bux, A.V.; Martinelli, D.; Iaculli, C.; Noci, N.D. Intravitreal pegaptanib sodium (Macugen®) for diabetic macular oedema. Acta Ophthalmol. 2009, 87, 623–630. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, V.H.; Giuliari, G.P.; Banda, R.M.; Guel, D.A. Intravitreal injection of pegaptanib sodium for proliferative diabetic retinopathy. Br. J. Ophthalmol. 2009, 93, 1474–1478. [Google Scholar] [CrossRef]
- Hornan, D.; Edmeades, N.; Krishnan, R.; Khan, J.; Lochhead, J. Use of pegaptanib for recurrent and non-clearing vitreous haemorrhage in proliferative diabetic retinopathy. Eye 2010, 24, 1315–1319. [Google Scholar] [CrossRef]
- Sultan, M.B.; Zhou, D.; Loftus, J.; Dombi, T.; Ice, K.S.; Group, M.S. A phase 2/3, multicenter, randomized, double-masked, 2-year trial of pegaptanib sodium for the treatment of diabetic macular edema. Ophthalmology 2011, 118, 1107–1118. [Google Scholar] [CrossRef]
- Loftus, J.V.; Sultan, M.B.; Pleil, A.M.; Group, M.S. Changes in vision-and health-related quality of life in patients with diabetic macular edema treated with pegaptanib sodium or sham. Investig. Ophthalmol. Vis. Sci. 2011, 52, 7498–7505. [Google Scholar] [CrossRef]
- Kiire, C.A.; Morjaria, R.; Rudenko, A.; Fantato, A.; Smith, L.; Smith, A.; Chong, V. Intravitreal pegaptanib for the treatment of ischemic diabetic macular edema. Clin. Ophthalmol. 2015, 9, 2305–2311. [Google Scholar] [CrossRef]
- Rinaldi, M.; Chiosi, F.; Dell’Omo, R.; Romano, M.R.; Parmeggiani, F.; Semeraro, F.; Mastropasqua, R.; Costagliola, C. Intravitreal pegaptanib sodium (Macugen®) for treatment of diabetic macular oedema: A morphologic and functional study. Br. J. Clin. Pharmacol. 2012, 74, 940–946. [Google Scholar] [CrossRef] [PubMed]
- Hussain, R.M.; Harris, A.; Siesky, B.; Yung, C.-W.; Ehrlich, R.; Prall, R. The effect of pegaptanib (Macugen) injection on retinal and retrobulbar blood flow in retinal Ischaemic diseases. Acta Ophthalmol. 2015, 93, e399–e400. [Google Scholar] [CrossRef]
- Sen, M.; Shields, C.L.; Honavar, S.G.; Shields, J.A. Coats disease: An overview of classification, management and outcomes. Indian J. Ophthalmol. 2019, 67, 763–771. [Google Scholar] [CrossRef]
- Sun, Y.; Jain, A.; Moshfeghi, D.M. Elevated vascular endothelial growth factor levels in Coats disease: Rapid response to pegaptanib sodium. Graefes Arch. Clin. Exp. Ophthalmol. 2007, 245, 1387–1388. [Google Scholar] [CrossRef] [PubMed]
- Kaul, S.; Uparkar, M.; Mody, K.; Walinjkar, J.; Kothari, M.; Natarajan, S. Intravitreal anti-vascular endothelial growth factor agents as an adjunct in the management of Coats’ disease in children. Indian J. Ophthalmol. 2010, 58, 76–78. [Google Scholar] [CrossRef]
- Campochiaro, P.A. Anti-vascular endothelial growth factor treatment for retinal vein occlusions. Ophthalmologica 2012, 227, 30–35. [Google Scholar] [CrossRef]
- Wroblewski, J.J.; Wells, J.A.; Adamis, A.P.; Buggage, R.R.; Cunningham, E.T.; Goldbaum, M.; Guyer, D.R.; Katz, B.; Altaweel, M.M.; Pegaptanib in Central Retinal Vein Occlusion Study Group. Pegaptanib sodium for macular edema secondary to central retinal vein occlusion. Arch. Ophthalmol. 2009, 127, 374–380. [Google Scholar] [CrossRef]
- Wroblewski, J.J.; Wells III, J.A.; Gonzales, C.R. Pegaptanib sodium for macular edema secondary to branch retinal vein occlusion. Am. J. Ophthalmol. 2010, 149, 147–154. [Google Scholar] [CrossRef]
- Wong, I.; Koo, S.; Chan, C. Pegaptanib for branch retinal vein occlusion. Am. J. Ophthalmol. 2010, 150, 129. [Google Scholar] [CrossRef]
- Udaondo, P.; Garcia-Delpech, S.; Salom, D.; Garcia-Pous, M.; Diaz-Llopis, M. Intravitreal pegaptanib for refractory macular edema secondary to retinal vein occlusion. Clin. Ophthalmol. 2011, 5, 941–944. [Google Scholar] [CrossRef]
- Bennett, M.D.; Yee, W. Pegaptanib for myopic choroidal neovascularization in a young patient. Graefes Arch. Clin. Exp. Ophthalmol. 2007, 245, 903–905. [Google Scholar] [CrossRef]
- Rinaldi, M.; Chiosi, F.; Dell’Omo, R.; Romano, M.R.; Parmeggiani, F.; Semeraro, F.; Menzione, M.; Costagliola, C. Intravitreal pegaptanib sodium (Macugen) for treatment of myopic choroidal neovascularization: A morphologic and functional study. Retina 2013, 33, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Friberg, T.R. Ranibizumab-pegaptanib therapy for idiopathic choroidal neovascularization: Report of a case. Semin. Ophthalmol. 2007, 22, 137–139. [Google Scholar] [CrossRef]
- Molina Guilabert, I.; Calvo-González, C.; Reche-Frutos, J.; Donate-López, J.; García-Feijoó, J.; Leila, M.; García-Sánchez, J. Intravitreal pegabtanib sodium in choroidal neovascularization secondary to angioid streaks. Acta Ophthalmol. 2009, 87, 581–582. [Google Scholar] [CrossRef] [PubMed]
- Çekiç, O.; Göçmez, E.; Kocabora, M.S. Management of CNV in angioid streaks by intravitreal use of specific anti-VEGF165 aptamer (pegaptanib sodium): Long-term results. Curr. Eye Res. 2011, 36, 492–495. [Google Scholar] [CrossRef]
- Vinekar, A.; Sund, N.; Quiram, P.; Capone, A., Jr. Choroidal neovascular membrane in persistent fetal vasculature syndrome managed with intravitreal pegaptanib sodium in an infant. Retina 2010, 30, S41–S44. [Google Scholar] [CrossRef]
- Kohly, R.P.; Muni, R.H.; Kertes, P.J.; Lam, W.-C. Management of pediatric choroidal neovascular membranes with intravitreal anti-VEGF agents: A retrospective consecutive case series. Can. J. Ophthalmol. 2011, 46, 46–50. [Google Scholar] [CrossRef] [PubMed]
- Bourla, D.H.; Gonzales, C.R.; Mango, C.W.; Moral, J.N.; Wirthlin, R.S.; Schwartz, S.D. Intravitreous vascular endothelial growth factor (VEGF) inhibitor therapy for tamoxifen induced macular edema. Semin. Ophthalmol. 2007, 22, 87–88. [Google Scholar] [CrossRef]
- Cervera, E.; Diaz-Llopis, M.; Udaondo, P.; Garcia-Delpech, S. Intravitreal pegaptanib sodium for refractory pseudophakic macular oedema. Eye 2008, 22, 1180–1182. [Google Scholar] [CrossRef]
- Querques, G.; Prascina, F.; Iaculli, C.; Noci, N.D. Intravitreal pegaptanib sodium (Macugen) for refractory cystoid macular edema in pericentral retinitis pigmentosa. Int. Ophthalmol. 2009, 29, 103–107. [Google Scholar] [CrossRef]
- Amselem, L.; Diaz-Llopis, M.; Cervera, E.; Salom, D.; Huste, F.; Montero, J. Pegaptanib sodium for acute retinal necrosis-induced macular oedema. Acta Ophthalmol. 2010, 88, e42–e43. [Google Scholar] [CrossRef] [PubMed]
- Querques, G.; Iaculli, C.; Delle Noci, N. Intravitreal pegaptanib sodium for Irvine-Gass syndrome. Eur. J. Ophtalmol. 2008, 18, 138–141. [Google Scholar] [CrossRef]
- Dahr, S.S.; Cusick, M.; Rodriguez-Coleman, H.; Srivastava, S.K.; Thompson, D.J.; Linehan, W.M.; FERRIS III, F.L.; Chew, E.Y. Intravitreal anti-vascular endothelial growth factor therapy with pegaptanib for advanced von Hippel-Lindau disease of the retina. Retina 2007, 27, 150–158. [Google Scholar] [CrossRef]
- Vianna, R.N.; Squeri, G.; Turquetti, R.; Brasil, O.; Burnier, M., Jr. Intravitreal pegaptanib reduces fluorescein leakage in idiopathic parafoveal telangiectasis. Can. J. Ophthalmol. 2008, 43, 492–493. [Google Scholar] [CrossRef]
- Querques, G.; Prascina, F.; Iaculli, C.; Noci, N.D. Intravitreal pegaptanib sodium (Macugen®) for radiation retinopathy following episcleral plaque radiotherapy. Acta Ophthalmol. 2008, 86, 700–701. [Google Scholar] [CrossRef]
- Quiram, P.A.; Drenser, K.A.; Lai, M.M.; Antonio Capone, J.; Trese, M.T. Treatment of vascularly active familial exudative vitreoretinopathy with pegaptanib sodium (Macugen). Retina 2008, 28, S8–S12. [Google Scholar] [CrossRef] [PubMed]
- Mitry, D.; Schmoll, C.; Hegde, V.; Borooah, S.; Singh, J.; Bennett, H. Use of pegaptanib in the treatment of vitreous haemorrhage in idiopathic retinal vasculitis. Eye 2008, 22, 1449–1450. [Google Scholar] [CrossRef]
- Mahmood, S.; Kumar, N.; Lenfestey, P.; Murjaneh, S.; Heimann, H.; Harding, S. Early response of retinal angiomatous proliferation treated with intravitreal pegaptanib: A retrospective review. Eye 2009, 23, 530–535. [Google Scholar] [CrossRef]
- Sánchez-Espino, L.F.; Ivars, M.; Antoñanzas, J.; Baselga, E. Sturge-Weber syndrome: A review of pathophysiology, genetics, clinical features, and current management approache. Appl. Clin. Genet. 2023, 16, 63–81. [Google Scholar] [CrossRef]
- Paulus, Y.M.; Jain, A.; Moshfeghi, D.M. Resolution of persistent exudative retinal detachment in a case of Sturge-Weber syndrome with anti-VEGF administration. Ocul. Immunol. Inflamm. 2009, 17, 292–294. [Google Scholar] [CrossRef]
- Bastion, M.; Then, K.; Faridah, H.; Mushawiahti, M.; Othmaliza, O.; Wong, H. The Adjunctive Use of Anti-vascular Endothelial Growth Factor Agents in the Management of Iris Neovascularisation in Malaysia. Med. J. Malays. 2011, 66, 10–14. [Google Scholar]
- Arevalo, J.F.; Espinoza, J.V. Single-session combined photodynamic therapy with verteporfin and intravitreal anti-vascular endothelial growth factor therapy for chronic central serous chorioretinopathy: A pilot study at 12-month follow-up. Graefes Arch. Clin. Exp. Ophthalmol. 2011, 249, 1159–1166. [Google Scholar] [CrossRef]
- Autrata, R.; Krejčířová, I.; Šenková, K.; Holoušová, M.; Doležel, Z.; Borek, I. Intravitreal pegaptanib combined with diode laser therapy for stage 3+ retinopathy of prematurity in zone I and posterior zone II. Eur. J. Ophthalmol. 2012, 22, 687–694. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Sun, X.; Niu, B. The importance of pegaptanib sodium treatment for patients with vascular active vitreoretinopathy. Exp. Ther. Med. 2017, 14, 6002–6006. [Google Scholar] [CrossRef]
- Avery, R.L.; Gordon, G.M. Systemic safety of prolonged monthly anti–vascular endothelial growth factor therapy for diabetic macular edema: A systematic review and meta-analysis. JAMA Ophthalmol. 2016, 134, 21–29. [Google Scholar] [CrossRef]
- Chong, V. Biological, preclinical and clinical characteristics of inhibitors of vascular endothelial growth factors. Ophthalmologica 2012, 227, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Morjaria, R.; Chong, N.V. Pharmacokinetic evaluation of pegaptanib octasodium for the treatment of diabetic edema. Expert Opin. Drug Metab. Toxicol. 2014, 10, 1185–1192. [Google Scholar] [CrossRef]
- D’Amico, D.J.; VEGF Inhibition Study in Ocular Neovascularization (VISION) Clinical Trial Group. Pegaptanib sodium for neovascular age-related macular degeneration: Two-year safety results of the two prospective, multicenter, controlled clinical trials. Ophthalmology 2006, 113, 992–1001.e6. [Google Scholar] [CrossRef]
- Tezel, T.H.; Kaplan, H.J. Are intravitreal anti-VEGF antibodies safe? Ocul. Immunol. Inflamm. 2007, 15, 1–2. [Google Scholar] [CrossRef]
- Singerman, L.J.; Masonson, H.; Patel, M.; Adamis, A.P.; Buggage, R.; Cunningham, E.; Goldbaum, M.; Katz, B.; Guyer, D. Pegaptanib sodium for neovascular age-related macular degeneration: Third-year safety results of the VEGF Inhibition Study in Ocular Neovascularisation (VISION) trial. Br. J. Ophthalmol. 2008, 92, 1606–1611. [Google Scholar] [CrossRef]
- Fung, A.E.; Bhisitkul, R.B. Safety monitoring with ocular anti-vascular endothelial growth factor therapies. Br. J. Ophthalmol. 2008, 92, 1573–1574. [Google Scholar] [CrossRef] [PubMed]
- Macugen AMD Study Group. Pegaptanib 1-year systemic safety results from a safety–pharmacokinetic trial in patients with neovascular age-related macular degeneration. Ophthalmology 2007, 114, 1702–1712.e2. [Google Scholar] [CrossRef]
- Dombi, T.; Kwok, K.K.; Sultan, M.B. A retrospective, pooled data analysis of the safety of pegaptanib sodium in the treatment of age-related macular degeneration in subjects with or without diabetes mellitus. BMC Ophthalmol. 2012, 12, 37. [Google Scholar] [CrossRef]
- Sivaprasad, S.; Browning, R.C.; Starita, C. An open-label, one-year, noncomparative study to evaluate the safety and tolerability of intravitreal pegaptanib sodium in patients with diabetic macular edema. Clin. Ophthalmol. 2014, 8, 1565–1571. [Google Scholar] [CrossRef]
- Nuzzi, R.; Tridico, F. Local and systemic complications after intravitreal administration of anti-vascular endothelial growth factor agents in the treatment of different ocular diseases: A five-year retrospective study. Semin. Ophthalmol. 2015, 30, 129–135. [Google Scholar] [CrossRef]
- Dalvin, L.A.; Starr, M.R.; AbouChehade, J.E.; Damento, G.M.; Garcia, M.; Shah, S.M.; Hodge, D.O.; Meissner, I.; Bakri, S.J.; Iezzi, R. Association of intravitreal anti–vascular endothelial growth factor therapy with risk of stroke, myocardial infarction, and death in patients with exudative age-related macular degeneration. JAMA Ophthalmol. 2019, 137, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Pilli, S.; Kotsolis, A.; Spaide, R.F.; Slakter, J.; Freund, K.B.; Sorenson, J.; Klancnik, J.; Cooney, M. Endophthalmitis associated with intravitreal anti-vascular endothelial growth factor therapy injections in an office setting. Am. J. Ophthalmol. 2008, 145, 879–882. [Google Scholar] [CrossRef]
- Klein, K.S.; Walsh, M.K.; Hassan, T.S.; Halperin, L.S.; Castellarin, A.A.; Roth, D.; Driscoll, S.; Prenner, J.L. Endophthalmitis after anti-VEGF injections. Ophthalmology 2009, 116, 1225–1225.e1. [Google Scholar] [CrossRef]
- Diago, T.; McCANNEL, C.A.; Bakri, S.J.; Pulido, J.S.; Edwards, A.O.; Pach, J.M. Infectious endophthalmitis after intravitreal injection of antiangiogenic agents. Retina 2009, 29, 601–605. [Google Scholar] [CrossRef]
- Inoue, M.; Kobayakawa, S.; Sotozono, C.; Komori, H.; Tanaka, K.; Suda, Y.; Matsushima, H.; Kinoshita, S.; Senoo, T.; Tochikubo, T. Evaluation of the incidence of endophthalmitis after intravitreal injection of anti-vascular endothelial growth factor. Ophthalmologica 2011, 226, 145–150. [Google Scholar] [CrossRef]
- Moshfeghi, A.A.; Rosenfeld, P.J.; Schwartz, S.G.; Flynn Jr, H.W.; Murray, T.G.; Smiddy, W.E.; Berrocal, A.M.; Dubovy, S.R.; Lee, W.-H.; Albini, T.A. Endophthalmitis After Intravitreal Anti-Vascular Endothelial Growth Factor Antagonists: A Six-Year Experience at a University Referral Center. Retina 2012, 32, 1228. [Google Scholar] [CrossRef]
- Chen, E.; Lin, M.Y.; Cox, J.; Brown, D.M. Endophthalmitis after intravitreal injection: The importance of viridans streptococci. Retina 2011, 31, 1525–1533. [Google Scholar] [CrossRef]
- Inman, Z.D.; Anderson, N.G. Incidence of endophthalmitis after intravitreal injection of antivascular endothelial growth factor medications using topical lidocaine gel anesthesia. Retina 2011, 31, 669–672. [Google Scholar] [CrossRef] [PubMed]
- Cheung, C.S.; Wong, A.W.; Lui, A.; Kertes, P.J.; Devenyi, R.G.; Lam, W.-C. Incidence of endophthalmitis and use of antibiotic prophylaxis after intravitreal injections. Ophthalmology 2012, 119, 1609–1614. [Google Scholar] [CrossRef]
- Flaxel, C.J.; Schlesinger, T.; Lauer, A. Culture-positive endophthalmitis 6 h after intravitreal antibiotics. Retin. Cases Brief Rep. 2011, 5, 14–15. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.; Ho, A.C.; Garg, S.J.; Brown, G.C.; Kaiser, R.S. Streptococcus mitis endophthalmitis presenting as frosted branch angiitis after intravitreal pegaptanib sodium injection. Ophthalmic Surg. Lasers Imaging Retin. 2009, 40, 192–194. [Google Scholar] [CrossRef]
- Shimada, H.; Hattori, T.; Mori, R.; Nakashizuka, H.; Fujita, K.; Yuzawa, M. Minimizing the endophthalmitis rate following intravitreal injections using 0.25% povidone–iodine irrigation and surgical mask. Graefes Arch. Clin. Exp. Ophthalmol. 2013, 251, 1885–1890. [Google Scholar] [CrossRef] [PubMed]
- Boyer, D.S.; Goldbaum, M.; Leys, A.M.; Starita, C.; Group, V.S. Effect of pegaptanib sodium 0.3 mg intravitreal injections (Macugen) in intraocular pressure: Posthoc analysis from VISION study. Br. J. Ophthalmol. 2014, 98, 1543–1546. [Google Scholar] [CrossRef]
- Hariprasad, S.M.; Shah, G.K.; Blinder, K.J. Short-term intraocular pressure trends following intravitreal pegaptanib (Macugen) injection. Am. J. Ophthalmol. 2006, 141, 200–201. [Google Scholar] [CrossRef]
- Bakri, S.J.; Pulido, J.; McCannel, C.; Hodge, D.; Diehl, N.; Hillemeier, J. Immediate intraocular pressure changes following intravitreal injections of triamcinolone, pegaptanib, and bevacizumab. Eye 2009, 23, 181–185. [Google Scholar] [CrossRef]
- Kim, J.E.; Mantravadi, A.V.; Hur, E.Y.; Covert, D.J. Short-term intraocular pressure changes immediately after intravitreal injections of anti–vascular endothelial growth factor agents. Am. J. Ophthalmol. 2008, 146, 930–934.e1. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.Y.; Ortube, M.C.; Mccannel, C.A.; Sarraf, D.; Hubschman, J.-P.; Mccannel, T.A.; Gorin, M.B. Sustained elevated intraocular pressures after intravitreal injection of bevacizumab, ranibizumab, and pegaptanib. Retina 2011, 31, 1028–1035. [Google Scholar] [CrossRef] [PubMed]
- Frenkel, R.E.; Mani, L.; Toler, A.R.; Frenkel, M.P. Intraocular pressure effects of pegaptanib (Macugen) injections in patients with and without glaucoma. Am. J. Ophthalmol. 2007, 143, 1034–1035. [Google Scholar] [CrossRef]
- Knip, M.M.; Välimäki, J. Effects of pegaptanib injections on intraocular pressure with and without anterior chamber paracentesis: A prospective study. Acta Ophthalmol. 2012, 90, 254–258. [Google Scholar] [CrossRef]
- Frenkel, M.P.; Haji, S.A.; Frenkel, R.E. Effect of prophylactic intraocular Pressure–Lowering medication on intraocular pressure spikes after intravitreal injections. Arch. Ophthalmol. 2010, 128, 1523–1527. [Google Scholar] [CrossRef]
- Dhalla, M.S.; Blinder, K.J.; Tewari, A.; Hariprasad, S.M.; Apte, R.S. Retinal pigment epithelial tear following intravitreal pegaptanib sodium. Am. J. Ophthalmol. 2006, 141, 752–754. [Google Scholar] [CrossRef]
- Singh, R.P.; Sears, J.E. Retinal pigment epithelial tears after pegaptanib injection for exudative age-related macular degeneration. Am. J. Ophthalmol. 2006, 142, 160–162. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.K.; Flaxel, C.J.; Lauer, A.K.; Sarraf, D. RPE tears after pegaptanib treatment in age-related macular degeneration. Retina 2007, 27, 857–863. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.K.; Sarraf, D. Tears of the retinal pigment epithelium: An old problem in a new era. Retina 2007, 27, 523–534. [Google Scholar] [CrossRef]
- Chiang, A.; Chang, L.K.; Yu, F.; Sarraf, D. Predictors of anti-VEGF-associated retinal pigment epithelial tear using FA and OCT analysis. Retina 2008, 28, 1265–1269. [Google Scholar] [CrossRef]
- Gutfleisch, M.; Heimes, B.; Schumacher, M.; Dietzel, M.; Lommatzsch, A.; Bird, A.; Pauleikhoff, D. Long-term visual outcome of pigment epithelial tears in association with anti-VEGF therapy of pigment epithelial detachment in AMD. Eye 2011, 25, 1181–1186. [Google Scholar] [CrossRef] [PubMed]
- Freund, K.B.; Laud, K.; Eandi, C.M.; Spaide, R.F. Silicone oil droplets following intravitreal injection. Retina 2006, 26, 701–703. [Google Scholar] [CrossRef]
- Bakri, S.J.; Ekdawi, N.S. Intravitreal silicone oil droplets after intravitreal drug injections. Retina 2008, 28, 996–1001. [Google Scholar] [CrossRef] [PubMed]
- Kocabora, M.S.; Ozbilen, K.T.; Serefoglu, K. Intravitreal silicone oil droplets following pegaptanib injection. Acta Ophthalmol. 2010, 88, e44–e45. [Google Scholar] [CrossRef] [PubMed]
- Steffensmeier, A.C.; Azar, A.E.; Fuller, J.J.; Muller, B.A.; Russell, S.R. Vitreous injections of pegaptanib sodium triggering allergic reactions. Am. J. Ophthalmol. 2007, 143, 512–513. [Google Scholar] [CrossRef]
- Dayani, P.N.; Siddiqi, O.K.; Holekamp, N.M. Safety of intravitreal injections in patients receiving warfarin anticoagulation. Am. J. Ophthalmol. 2007, 144, 451–453. [Google Scholar] [CrossRef]
- Querques, G.; Bux, A.V.; Delle Noci, N. Foveal geographic atrophy following intravitreal pegaptanib sodium (Macugen) for drusenoid pigment epithelium detachment. Eur. J. Ophthalmol. 2009, 19, 890–893. [Google Scholar] [CrossRef]
- Krishnan, R.; Goverdhan, S.; Lochhead, J. Intravitreal pegaptanib in severe proliferative diabetic retinopathy leading to the progression of tractional retinal detachment. Eye 2009, 23, 1238–1239. [Google Scholar] [CrossRef]
- Hamrah, P.; Singh, P.; Hoesl, L.; Tezel, T. Development of crystalline keratopathy after intravitreal injections of pegaptanib. Eye 2010, 24, 1527–1528. [Google Scholar] [CrossRef]
- Querques, G.; Bux, A.V.; Iaculli, C.; Noci, N.D. Lamellar macular hole following intravitreal pegaptanib sodium (Macugen) injection for diabetic macular edema. Int. Ophthalmol. 2011, 31, 525–527. [Google Scholar] [CrossRef]
- Seth, R.K.; Salim, S.; Shields, M.B.; Adelman, R.A. Assessment of optic nerve cup-to-disk ratio changes in patients receiving multiple intravitreal injections of antivascular endothelial growth factor agents. Retina 2009, 29, 956–959. [Google Scholar] [CrossRef] [PubMed]
- Horsley, M.B.; Mandava, N.; Maycotte, M.A.; Kahook, M.Y. Retinal nerve fiber layer thickness in patients receiving chronic anti–vascular endothelial growth factor therapy. Am. J. Ophthalmol. 2010, 150, 558–561.e1. [Google Scholar] [CrossRef] [PubMed]
- Sultana, J.; Scondotto, G.; Cutroneo, P.M.; Morgante, F.; Trifirò, G. Intravitreal anti-VEGF drugs and signals of dementia and Parkinson-like events: Analysis of the VigiBase database of spontaneous reports. Front. Pharmacol. 2020, 11, 315. [Google Scholar] [CrossRef]
- Van Wijngaarden, P.; Coster, D.J.; Williams, K.A. Inhibitors of ocular neovascularization: Promises and potential problems. JAMA 2005, 293, 1509–1513. [Google Scholar] [CrossRef]
- From the BMJ Group A view on new drugs for macular degeneration. Drug Ther. Bull. 2007, 45, 49–52. [CrossRef]
- Wolowacz, S.E.; Roskell, N.; Kelly, S.; Maciver, F.M.; Brand, C.S. Cost effectiveness of pegaptanib for the treatment of age-related macular degeneration in the UK. Pharmacoeconomics 2007, 25, 863–879. [Google Scholar] [CrossRef]
- Earnshaw, S.R.; Moride, Y.; Rochon, S. Cost-effectiveness of pegaptanib compared to photodynamic therapy with verteporfin and to standard care in the treatment of subfoveal wet age-related macular degeneration in Canada. Clin. Ther. 2007, 29, 2096–2106. [Google Scholar] [CrossRef] [PubMed]
- Smiddy, W.E. Relative cost of a line of vision in age-related macular degeneration. Ophthalmology 2007, 114, 847–854. [Google Scholar] [CrossRef]
- Azad, R.; Chandra, P.; Gupta, R. The economic implications of the use of anti-vascular endothelial growth factor drugs in age-related macular degeneration. Indian J. Ophthalmol. 2007, 55, 441–443. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.C.; Brown, M.M.; Brown, H.C.; Kindermann, S.; Sharma, S. A value-based medicine comparison of interventions for subfoveal neovascular macular degeneration. Ophthalmology 2007, 114, 1170–1178. [Google Scholar] [CrossRef]
- Javitt, J.C.; Zlateva, G.P.; Earnshaw, S.R.; Pleil, A.M.; Graham, C.N.; Brogan, A.J.; Shah, S.N.; Adamis, A.P. Cost-effectiveness model for neovascular age-related macular degeneration: Comparing early and late treatment with pegaptanib sodium based on visual acuity. Value Health 2008, 11, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Colquitt, J.; Jones, J.; Tan, S.; Takeda, A.; Clegg, A.; Price, A. Ranibizumab and pegaptanib for the treatment of age-related macular degeneration: A systematic review and economic evaluation. Health Technol. Assess. 2008, 12, 1–222. [Google Scholar] [CrossRef]
- Gower, E.W.; Cassard, S.D.; Bass, E.B.; Schein, O.D.; Bressler, N.M. A cost-effectiveness analysis of three treatments for age-related macular degeneration. Retina 2010, 30, 212–221. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.; Hurwitz, H.I.; Sandler, A.B.; Miles, D.; Coleman, R.L.; Deurloo, R.; Chinot, O.L. Bevacizumab (Avastin®) in cancer treatment: A review of 15 years of clinical experience and future outlook. Cancer Treat. Rev. 2020, 86, 102017. [Google Scholar] [CrossRef] [PubMed]
- Stewart, M.W. A review of ranibizumab for the treatment of diabetic retinopathy. Ophthalmol. Ther. 2017, 6, 33–47. [Google Scholar] [CrossRef]
- Nagpal, M.; Nagpal, K.; Nagpal, P. A comparative debate on the various anti-vascular endothelial growth factor drugs: Pegaptanib sodium (Macugen), ranibizumab (Lucentis) and bevacizumab (Avastin). Indian J. Ophthalmol. 2007, 55, 437–439. [Google Scholar] [CrossRef]
- Brown, D.M.; Kaiser, P.K.; Michels, M.; Soubrane, G.; Heier, J.S.; Kim, R.Y.; Sy, J.P.; Schneider, S. Ranibizumab versus verteporfin for neovascular age-related macular degeneration. N. Engl. J. Med. 2006, 355, 1432–1444. [Google Scholar] [CrossRef]
- Rosenfeld, P.J.; Brown, D.M.; Heier, J.S.; Boyer, D.S.; Kaiser, P.K.; Chung, C.Y.; Kim, R.Y. Ranibizumab for neovascular age-related macular degeneration. N. Engl. J. Med. 2006, 355, 1419–1431. [Google Scholar] [CrossRef]
- Heier, J.S.; Boyer, D.S.; Ciulla, T.A.; Ferrone, P.J.; Jumper, J.M.; Gentile, R.C.; Kotlovker, D.; Chung, C.Y.; Kim, R.Y.; Group, F.S. Ranibizumab combined with verteporfin photodynamic therapy in neovascular age-related macular degeneration: Year 1 results of the FOCUS Study. Arch. Ophthalmol. 2006, 124, 1532–1542. [Google Scholar] [CrossRef]
- Antoszyk, A.N.; Tuomi, L.; Chung, C.Y.; Singh, A.; FOCUS Study Group. Ranibizumab combined with verteporfin photodynamic therapy in neovascular age-related macular degeneration (FOCUS): Year 2 results. Am. J. Ophthalmol. 2008, 145, 862–874.e3. [Google Scholar] [CrossRef]
- Wong, T.Y.; Liew, G.; Mitchell, P. Clinical update: New treatments for age-related macular degeneration. Lancet 2007, 370, 204–206. [Google Scholar] [CrossRef] [PubMed]
- Takeda, A.L.; Colquitt, J.; Clegg, A.J.; Jones, J. Pegaptanib and ranibizumab for neovascular age-related macular degeneration: A systematic review. Br. J. Ophthalmol. 2007, 91, 1177–1182. [Google Scholar] [CrossRef] [PubMed]
- Avery, R.L.; Pieramici, D.J.; Rabena, M.D.; Castellarin, A.A.; Ma’an, A.N.; Giust, M.J. Intravitreal bevacizumab (Avastin) for neovascular age-related macular degeneration. Ophthalmology 2006, 113, 363–372.e5. [Google Scholar] [CrossRef] [PubMed]
- Yoganathan, P.; Deramo, V.A.; Lai, J.C.; Tibrewala, R.K.; Fastenberg, D.M. Visual improvement following intravitreal bevacizumab (Avastin) in exudative age-related macular degeneration. Retina 2006, 26, 994–998. [Google Scholar] [CrossRef]
- Tufail, A.; Patel, P.J.; Egan, C.; Hykin, P.; da Cruz, L.; Gregor, Z.; Dowler, J.; Majid, M.A.; Bailey, C.; Mohamed, Q. Bevacizumab for neovascular age related macular degeneration (ABC Trial): Multicentre randomised double masked study. BMJ 2010, 340, c2459. [Google Scholar] [CrossRef]
- Keane, P.A.; Heussen, F.M.; Ouyang, Y.; Mokwa, N.; Walsh, A.C.; Tufail, A.; Sadda, S.R.; Patel, P.J. Assessment of differential pharmacodynamic effects using optical coherence tomography in neovascular age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2012, 53, 1152–1161. [Google Scholar] [CrossRef]
- Joeres, S.; Kaplowitz, K.; Brubaker, J.W.; Updike, P.G.; Collins, A.T.; Walsh, A.C.; Romano, P.W.; Sadda, S.R. Quantitative comparison of optical coherence tomography after pegaptanib or bevacizumab in neovascular age-related macular degeneration. Ophthalmology 2008, 115, 347–354.e2. [Google Scholar] [CrossRef]
- Arias, L. Treatment of retinal pigment epithelial detachment with antiangiogenic therapy. Clin. Ophthalmol. 2010, 4, 369–374. [Google Scholar] [CrossRef]
- Lommatzsch, A.; Heimes, B.; Gutfleisch, M.; Spital, G.; Zeimer, M.; Pauleikhoff, D. Serous pigment epithelial detachment in age-related macular degeneration: Comparison of different treatments. Eye 2009, 23, 2163–2168. [Google Scholar] [CrossRef]
- Nishimura, Y.; Taguchi, M.; Nagai, T.; Fujihara, M.; Honda, S.; Uenishi, M. Comparison of the effect between pegaptanib and ranibizumab on exudative age-related macular degeneration with small lesion size. Clin. Ophthalmol. 2012, 6, 365–368. [Google Scholar] [CrossRef]
- Ollendorf, D.A.; Colby, J.A.; Pearson, S.D. Comparative effectiveness of anti-VEGF agents for diabetic macular edema. Int. J. Technol. Assess. Health Care 2013, 29, 392–401. [Google Scholar] [CrossRef]
- Shiragami, C.; Ono, A.; Kobayashi, M.; Manabe, S.; Yamashita, A.; Shiraga, F. Effect of switching therapy to pegaptanib in eyes with the persistent cases of exudative age-related macular degeneration. Medicine 2014, 93, e116. [Google Scholar] [CrossRef]
- Khalili, M.R.; Hosseini, H. Debate on the various anti-vascular endothelial growth factor drugs. Indian J. Ophthalmol. 2008, 56, 255–256. [Google Scholar] [CrossRef] [PubMed]
- Curtis, L.H.; Hammill, B.G.; Schulman, K.A.; Cousins, S.W. Risks of mortality, myocardial infarction, bleeding, and stroke associated with therapies for age-related macular degeneration. Arch. Ophthalmol. 2010, 128, 1273–1279. [Google Scholar] [CrossRef] [PubMed]
- Biagi, C.; Conti, V.; Montanaro, N.; Melis, M.; Buccellato, E.; Donati, M.; Covezzoli, A.; Amato, R.; Pazzi, L.; Venegoni, M. Comparative safety profiles of intravitreal bevacizumab, ranibizumab and pegaptanib: The analysis of the WHO database of adverse drug reactions. Eur. J. Clin. Pharmacol. 2014, 70, 1505–1512. [Google Scholar] [CrossRef]
- Mikačić, I.; Bosnar, D. Intravitreal bevacizumab and cardiovascular risk in patients with age-related macular degeneration: Systematic review and meta-analysis of randomized controlled trials and observational studies. Drug Saf. 2016, 39, 517–541. [Google Scholar] [CrossRef] [PubMed]
- Meredith, G.G.; Schkade, P.A.; Joondeph, B.C. Allergic reactions upon intravitreal administration of anti-vascular endothelial growth factor agents. Retin. Cases Brief Rep. 2019, 13, 287–289. [Google Scholar] [CrossRef]
- Hernández-Pastor, L.J.; Ortega, A.; García-Layana, A.; Giráldez, J. Cost-effectiveness of ranibizumab compared with pegaptanib in neovascular age-related macular degeneration. Graefes Arch. Clin. Exp. Ophthalmol. 2010, 248, 467–476. [Google Scholar] [CrossRef]
- Wiwanitkit, V. Ranibizumab vs. pegaptanib: A cost-effectiveness study? Graefes Arch. Clin. Exp. Ophthalmol. 2010, 248, 1675. [Google Scholar] [CrossRef]
- Hodge, W.; Brown, A.; Kymes, S.; Cruess, A.; Blackhouse, G.; Hopkins, R.; McGahan, L.; Sharma, S.; Pan, I.; Blair, J. Pharmacologic management of neovascular age-related macular degeneration: Systematic review of economic evidence and primary economic evaluation. Can. J. Ophthalmol. 2010, 45, 223–230. [Google Scholar] [CrossRef]
- Athanasakis, K.; Fragoulakis, V.; Tsiantou, V.; Masaoutis, P.; Maniadakis, N.; Kyriopoulos, J. Cost-effectiveness analysis of ranibizumab versus verteporfin photodynamic therapy, pegaptanib sodium, and best supportive care for the treatment of age-related macular degeneration in Greece. Clin. Ther. 2012, 34, 446–456. [Google Scholar] [CrossRef]
- Studnička, J.; Říhová, B.; Rencová, E.; Rozsíval, P.; Dubská, Z.; Chrapek, O.; Kolář, P.; Kandrnal, V.; Demlová, R.; Pitrová, Š. Cost and effectiveness of therapy for wet age-related macular degeneration in routine clinical practice. Ophthalmologica 2013, 230, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Akar, E.E.; Öner, V.; Küçükerdönmez, C.; Akova, Y.A. Comparison of subconjunctivally injected bevacizumab, ranibizumab, and pegaptanib for inhibition of corneal neovascularization in a rat model. Int. J. Ophthalmol. 2013, 6, 136. [Google Scholar] [CrossRef]
- Sener, E.; Yuksel, N.; Yildiz, D.K.; Yilmaz, B.; Ozdemir, O.; Caglar, Y.; Degirmenci, E. The impact of subconjuctivally injected EGF and VEGF inhibitors on experimental corneal neovascularization in rat model. Curr. Eye Res. 2011, 36, 1005–1013. [Google Scholar] [CrossRef] [PubMed]
- Ju, M.; Mailhos, C.; Bradley, J.; Dowie, T.; Ganley, M.; Cook, G.; Calias, P.; Lange, N.; Adamis, A.P.; Shima, D.T. Simultaneous but not prior inhibition of VEGF165 enhances the efficacy of photodynamic therapy in multiple models of ocular neovascularization. Investig. Ophthalmol. Vis. Sci. 2008, 49, 662–670. [Google Scholar] [CrossRef]
- Verhoeff, J.J.; Stalpers, L.J.; Claes, A.; Hovinga, K.E.; Musters, G.D.; Vandertop, W.P.; Richel, D.J.; Leenders, W.P.; Van Furth, W.R. Tumour control by whole brain irradiation of anti-VEGF-treated mice bearing intracerebral glioma. Eur. J. Cancer 2009, 45, 3074–3080. [Google Scholar] [CrossRef] [PubMed]
- Lytvynchuk, L.; Sergienko, A.; Lavrenchuk, G.; Petrovski, G. Antiproliferative, apoptotic, and autophagic activity of ranibizumab, bevacizumab, pegaptanib, and aflibercept on fibroblasts: Implication for choroidal neovascularization. J. Ophthalmol. 2015, 2015, 934963. [Google Scholar] [CrossRef]
- Carneiro, Â.; Falcão, M.; Pirraco, A.; Milheiro-Oliveira, P.; Falcão-Reis, F.; Soares, R. Comparative effects of bevacizumab, ranibizumab and pegaptanib at intravitreal dose range on endothelial cells. Exp. Eye Res. 2009, 88, 522–527. [Google Scholar] [CrossRef]
- Criswell, M.H.; Hu, W.-Z.; Steffens, T.J.; Li, R.; Margaron, P. Comparing pegaptanib and triamcinolone efficacy in the rat choroidal neovascularization model. Arch. Ophthalmol. 2008, 126, 946–952. [Google Scholar] [CrossRef]
- Lu, F.; Adelman, R.A. Are intravitreal bevacizumab and ranibizumab effective in a rat model of choroidal neovascularization? Graefes Arch. Clin. Exp. Ophthalmol. 2009, 247, 171–177. [Google Scholar] [CrossRef]
- Sertbas, I.; Yilmaz, A.; Yildirim, T.; Karatay, M.; Celik, H.; Bayar, M. The role of pegaptanib sodium in the suppression of epidural fibrosis in a postlaminectomy rat model. Bratisl. Lek. Listy 2017, 118, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Sato, J.; Inage, K.; Miyagi, M.; Sakuma, Y.; Yamauchi, K.; Koda, M.; Furuya, T.; Nakamura, J.; Suzuki, M.; Kubota, G. Inhibiting vascular endothelial growth factor in injured intervertebral discs attenuates pain-related neuropeptide expression in dorsal root ganglia in rats. Asian Spine J. 2017, 11, 556. [Google Scholar] [CrossRef]
- Dorrell, M.I.; Aguilar, E.; Jacobson, R.; Yanes, O.; Gariano, R.; Heckenlively, J.; Banin, E.; Ramirez, G.A.; Gasmi, M.; Bird, A. Antioxidant or neurotrophic factor treatment preserves function in a mouse model of neovascularization-associated oxidative stress. J. Clin. Investig. 2009, 119, 611–623. [Google Scholar] [CrossRef]
- Klettner, A.; Roider, J. Comparison of bevacizumab, ranibizumab, and pegaptanib in vitro: Efficiency and possible additional pathways. Investig. Ophthalmol. Vis. Sci. 2008, 49, 4523–4527. [Google Scholar] [CrossRef]
- Mitchell, T.S.; Bradley, J.; Robinson, G.S.; Shima, D.T.; Ng, Y.-S. RGS5 expression is a quantitative measure of pericyte coverage of blood vessels. Angiogenesis 2008, 11, 141–151. [Google Scholar] [CrossRef]
- Foy, J.W.-D.; Rittenhouse, K.; Modi, M.; Patel, M. Local tolerance and systemic safety of pegaptanib sodium in the dog and rabbit. J. Ocul. Pharmacol. Ther. 2007, 23, 452–466. [Google Scholar] [CrossRef]
- Avci, B.; Avci, R.; Inan, U.U.; Kaderli, B. Comparative evaluation of apoptotic activity in photoreceptor cells after intravitreal injection of bevacizumab and pegaptanib sodium in rabbits. Investig. Ophthalmol. Vis. Sci. 2009, 50, 3438–3446. [Google Scholar] [CrossRef] [PubMed]
- Myers, A.C.; Lövestam Adrian, M.; Bruun, A.; Ghosh, F.; Andréasson, S.; Ponjavic, V. Retinal function and morphology in rabbit after intravitreal injection of VEGF inhibitors. Curr. Eye Res. 2012, 37, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Thaler, S.; Fiedorowicz, M.; Choragiewicz, T.J.; Bolz, S.; Tura, A.; Henke-Fahle, S.; Yoeruek, E.; Zrenner, E.; Bartz-Schmidt, K.U.; Ziemssen, F. Toxicity testing of the VEGF inhibitors bevacizumab, ranibizumab and pegaptanib in rats both with and without prior retinal ganglion cell damage. Acta Ophthalmol. 2010, 88, e170–e176. [Google Scholar] [CrossRef]
- Lüke, M.; Januschowski, K.; Tura, A.; Lüke, J.; Nassar, K.; Lüke, C.; Schneider, T.; Szurman, P.; Grisanti, S.; Bartz-Schmidt, K.U. Effects of pegaptanib sodium on retinal function in isolated perfused vertebrate retina. Curr. Eye Res. 2010, 35, 248–254. [Google Scholar] [CrossRef]
- Christoforidis, J.; Ricketts, R.; Pratt, C.; Pierce, J.; Bean, S.; Wells, M.; Zhang, X.; La Perle, K. The effect of intravitreal anti-VEGF agents on peripheral wound healing in a rabbit model. Clin. Ophthalmol. 2012, 6, 61–69. [Google Scholar] [CrossRef]
- Sawada, O.; Miyake, T.; Kakinoki, M.; Sawada, T.; Kawamura, H.; Ohji, M. Aqueous vascular endothelial growth factor after intravitreal injection of pegaptanib or ranibizumab in patients with age-related macular degeneration. Retina 2010, 30, 1034–1038. [Google Scholar] [CrossRef] [PubMed]
- Manresa, N.; Mulero, J.; Losada, M.; Zafrilla, P. Influence of anti-VEGF about cardiovascular biomarkers in age related macular degeneration. J. Nutr. Health Aging 2015, 19, 228–231. [Google Scholar] [CrossRef] [PubMed]
- Zehetner, C.; Kirchmair, R.; Huber, S.; Kralinger, M.T.; Kieselbach, G.F. Plasma levels of vascular endothelial growth factor before and after intravitreal injection of bevacizumab, ranibizumab and pegaptanib in patients with age-related macular degeneration, and in patients with diabetic macular oedema. Br. J. Ophthalmol. 2013, 97, 454–459. [Google Scholar] [CrossRef]
- Takahashi, H.; Nomura, Y.; Nishida, J.; Fujino, Y.; Yanagi, Y.; Kawashima, H. Vascular endothelial growth factor (VEGF) concentration is underestimated by enzyme-linked immunosorbent assay in the presence of anti-VEGF drugs. Investig. Ophthalmol. Vis. Sci. 2016, 57, 462–466. [Google Scholar] [CrossRef]
- Manresa, N.; Mulero, J.; Losada, M.; Zafrilla, P. Effect of pegaptanib and ranibizumab on plasma and vitreous homocysteine in patients with exudative age-related macular degeneration. Retina 2015, 35, 1765–1771. [Google Scholar] [CrossRef]
- Basile, A.S.; Hutmacher, M.; Nickens, D.; Nielsen, J.; Kowalski, K.; Whitfield, L.; Masayo, O.; Nakane, M. Population pharmacokinetics of pegaptanib in patients with neovascular, age-related macular degeneration. J. Clin. Pharmacol. 2012, 52, 1186–1199. [Google Scholar] [CrossRef]
- Basile, A.S.; Hutmacher, M.M.; Kowalski, K.G.; Gandelman, K.Y.; Nickens, D.J. Population pharmacokinetics of pegaptanib sodium (Macugen®) in patients with diabetic macular edema. Clin. Ophthalmol. 2015, 9, 323–335. [Google Scholar] [CrossRef]
- Chen, S.; Zhang, L.; Song, Y.; Xie, K.; Wang, Y.; Liang, Y. A Comprehensive Analysis of NRP1 in Malignancies Provide Therapeutic Implication for Treating Cancer Patients Infected with SARS-CoV-2. Biochem. Genet. 2024, 62, 2399–2417. [Google Scholar] [CrossRef]
- Lopez-Guajardo, L.; Del Valle, F.; Moreno, J.; Teus, M. Reduction of pegaptanib loss during intravitreal delivery using an oblique injection technique. Eye 2008, 22, 430–433. [Google Scholar] [CrossRef] [PubMed]
- Turgut, B.; Demir, T.; Celiker, U. The effects of injection site on the reflux following intravitreal injections. J. Clin. Med. Res. 2009, 1, 280. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Hwang, D.; Chung, J. Cotinine-conjugated aptamer/anti-cotinine antibody complexes as a novel affinity unit for use in biological assays. Exp. Mol. Med. 2012, 44, 554–561. [Google Scholar] [CrossRef] [PubMed]
- Heo, K.; Min, S.-W.; Sung, H.J.; Kim, H.G.; Kim, H.J.; Kim, Y.H.; Choi, B.K.; Han, S.; Chung, S.; Lee, E.S. An aptamer-antibody complex (oligobody) as a novel delivery platform for targeted cancer therapies. J. Control. Release 2016, 229, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Zhang, Y.; Ma, W.; Shao, X.; Zhan, Y.; Mao, C.; Zhu, B.; Zhou, Y.; Zhao, H.; Cai, X. Potent anti-angiogenesis and anti-tumour activity of pegaptanib-loaded tetrahedral DNA nanostructure. Cell Prolif. 2019, 52, e12662. [Google Scholar] [CrossRef]
- Conway, B.R. Recent patents on ocular drug delivery systems. Recent Pat. Drug Deliv. Formul. 2008, 2, 1–8. [Google Scholar] [CrossRef]
- Stewart, E.A.; Samaranayake, G.J.; Browning, A.C.; Hopkinson, A.; Amoaku, W.M. Comparison of choroidal and retinal endothelial cells: Characteristics and response to VEGF isoforms and anti-VEGF treatments. Exp. Eye Res. 2011, 93, 761–766. [Google Scholar] [CrossRef]
- Campa, C. Effect of VEGF and anti-VEGF compounds on retinal pigment epithelium permeability: An in vitro study. Eur. J. Ophthalmol. 2013, 23, 690–696. [Google Scholar] [CrossRef]
- Lipp, M.; Bucher, F.; Parthasarathy, A.; Hos, D.; Onderka, J.; Cursiefen, C.; Bock, F. Blockade of the VEGF isoforms in inflammatory corneal hemangiogenesis and lymphangiogenesis. Graefes Arch. Clin. Exp. Ophthalmol. 2014, 252, 943–949. [Google Scholar] [CrossRef]
- Magnussen, A.L.; Rennel, E.S.; Hua, J.; Bevan, H.S.; Long, N.B.; Lehrling, C.; Gammons, M.; Floege, J.; Harper, S.J.; Agostini, H.T. VEGF-A165b is cytoprotective and antiangiogenic in the retina. Investig. Ophthalmol. Vis. Sci. 2010, 51, 4273–4281. [Google Scholar] [CrossRef]
- Huang, D.; Zhao, C.; Ju, R.; Kumar, A.; Tian, G.; Huang, L.; Zheng, L.; Li, X.; Liu, L.; Wang, S. VEGF-B inhibits hyperglycemia-and Macugen-induced retinal apoptosis. Sci. Rep. 2016, 6, 26059. [Google Scholar] [CrossRef]
- Eriksson, E.S.; Joshi, L.; Billeter, M.; Eriksson, L.A. De novo tertiary structure prediction using RNA123—Benchmarking and application to Macugen. J. Mol. Model. 2014, 20, 2389. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, S.; Amemiya, T.; Yabuki, Y.; Horimoto, K.; Fukui, K. ToGo-WF: Prediction of RNA tertiary structures and RNA–RNA/protein interactions using the KNIME workflow. J. Comput.-Aided Mol. Des. 2019, 33, 497–507. [Google Scholar] [CrossRef]
- Mullard, A. FDA approves second RNA aptamer. Nat. Rev. Drug Discov. 2023, 22, 774. [Google Scholar] [CrossRef] [PubMed]
- Al Shaer, D.; Al Musaimi, O.; Albericio, F.; de la Torre, B.G. 2023 FDA TIDES (Peptides and Oligonucleotides) Harvest. Pharmaceuticals 2024, 17, 243. [Google Scholar] [CrossRef] [PubMed]
- Danzig, C.J.; Khanani, A.M.; Loewenstein, A. C5 inhibitor avacincaptad pegol treatment for geographic atrophy: A comprehensive review. Immunotherapy 2024, 16, 779–790. [Google Scholar] [CrossRef]
- Raica, M.; Cimpean, A.M. Platelet-derived growth factor (PDGF)/PDGF receptors (PDGFR) axis as target for antitumor and antiangiogenic therapy. Pharmaceuticals 2010, 3, 572–599. [Google Scholar] [CrossRef]
- Jo, N.; Mailhos, C.; Ju, M.; Cheung, E.; Bradley, J.; Nishijima, K.; Robinson, G.S.; Adamis, A.P.; Shima, D.T. Inhibition of platelet-derived growth factor B signaling enhances the efficacy of anti-vascular endothelial growth factor therapy in multiple models of ocular neovascularization. Am. J. Pathol. 2006, 168, 2036–2053. [Google Scholar] [CrossRef]
- Nakamura, Y. Multiple therapeutic applications of RBM-007, an anti-FGF2 aptamer. Cells 2021, 10, 1617. [Google Scholar] [CrossRef]
- Pereira, D.S.; Maturi, R.K.; Akita, K.; Mahesh, V.; Bhisitkul, R.B.; Nishihata, T.; Sakota, E.; Ali, Y.; Nakamura, E.; Bezwada, P. Clinical proof of concept for anti-FGF2 therapy in exudative age-related macular degeneration (nAMD): Phase 2 trials in treatment-naïve and anti-VEGF pretreated patients. Eye 2024, 38, 1140–1148. [Google Scholar] [CrossRef]
- Pereira, D.S.; Akita, K.; Bhisitkul, R.B.; Nishihata, T.; Ali, Y.; Nakamura, E.; Nakamura, Y. Safety and tolerability of intravitreal umedaptanib pegol (anti-FGF2) for neovascular age-related macular degeneration (nAMD): A phase 1, open-label study. Eye 2024, 38, 1149–1154. [Google Scholar] [CrossRef]
- Leaderer, D.; Cashman, S.M.; Kumar-Singh, R. Topical application of a G-Quartet aptamer targeting nucleolin attenuates choroidal neovascularization in a model of age-related macular degeneration. Exp. Eye Res. 2015, 140, 171–178. [Google Scholar] [CrossRef]
- Barrera-Ocampo, A. Monoclonal antibodies and aptamers: The future therapeutics for Alzheimer’s disease. Acta Pharm. Sin. B 2024, 14, 2795–2814. [Google Scholar] [CrossRef]
- Bates, P.J.; Laber, D.A.; Miller, D.M.; Thomas, S.D.; Trent, J.O. Discovery and development of the G-rich oligonucleotide AS1411 as a novel treatment for cancer. Exp. Mol. Pathol. 2009, 86, 151–164. [Google Scholar] [CrossRef]
- Zhang, Y.; Lai, B.S.; Juhas, M. Recent advances in aptamer discovery and applications. Molecules 2019, 24, 941. [Google Scholar] [CrossRef]
- Kong, A.H.-Y.; Wu, A.J.; Ho, O.K.-Y.; Leung, M.M.-K.; Huang, A.S.; Yu, Y.; Zhang, G.; Lyu, A.; Li, M.; Cheung, K.-H. Exploring the potential of aptamers in targeting neuroinflammation and neurodegenerative disorders: Opportunities and challenges. Int. J. Mol. Sci. 2023, 24, 11780. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Duan, Q.; Ran, C.; Fu, T.; Liu, Y.; Tan, W. Recent progress of aptamer–drug conjugates in cancer therapy. Acta Pharm. Sin. B 2023, 13, 1358–1370. [Google Scholar] [CrossRef]
- Mahmoudi, A.; Alavizadeh, S.H.; Hosseini, S.A.; Meidany, P.; Doagooyan, M.; Abolhasani, Y.; Saadat, Z.; Amani, F.; Kesharwani, P.; Gheybi, F. Harnessing aptamers against COVID-19: A therapeutic strategy. Drug Discov. Today 2023, 28, 103663. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Liu, S.; Wei, X.; Wan, S.; Huang, M.; Song, T.; Lu, Y.; Weng, X.; Lin, Z.; Chen, H. Aptamer blocking strategy inhibits SARS-CoV-2 virus infection. Angew. Chem. Int. Ed. 2021, 60, 10266–10272. [Google Scholar] [CrossRef]
- Park, J.-P.; Shin, H.J.; Park, S.-G.; Oh, H.-K.; Choi, C.-H.; Park, H.-J.; Kook, M.-S.; Ohk, S.-H. Screening and development of DNA aptamers specific to several oral pathogens. J. Microbiol. Biotechnol. 2015, 25, 393–398. [Google Scholar] [CrossRef]
- Kazemi, M.S.; Shoari, A.; Salehibakhsh, N.; Aliabadi, H.A.M.; Abolhosseini, M.; Arab, S.S.; Ahmadieh, H.; Kanavi, M.R.; Behdani, M. Anti-angiogenic biomolecules in neovascular age-related macular degeneration; therapeutics and drug delivery systems. Int. J. Pharm. 2024, 659, 124258. [Google Scholar] [CrossRef]
- Shigdar, S.; Macdonald, J.; O’Connor, M.; Wang, T.; Xiang, D.; Al Shamaileh, H.; Qiao, L.; Wei, M.; Zhou, S.-F.; Zhu, Y. Aptamers as theranostic agents: Modifications, serum stability and functionalisation. Sensors 2013, 13, 13624–13637. [Google Scholar] [CrossRef] [PubMed]
- Bege, M.; Borbás, A. Rise and fall of fomivirsen, the first approved gene silencing medicine—A historical review. Acta Pharm. Hung. 2022, 92, 38–44. [Google Scholar] [CrossRef]
- Tadayoni, R.; Sararols, L.; Weissgerber, G.; Verma, R.; Clemens, A.; Holz, F.G. Brolucizumab: A newly developed anti-VEGF molecule for the treatment of neovascular age-related macular degeneration. Ophthalmologica 2021, 244, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Rossi, J. Aptamers as targeted therapeutics: Current potential and challenges. Nat. Rev. Drug Discov. 2017, 16, 181–202. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.X.; Kwon, Y.J. Aptamers: The “evolution” of SELEX. Methods 2016, 106, 21–28. [Google Scholar] [CrossRef]
- Cesarini, V.; Appleton, S.L.; de Franciscis, V.; Catalucci, D. The recent blooming of therapeutic aptamers. Mol. Asp. Med. 2025, 102, 101350. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, Z.; Zhang, W.; Li, Y.; Feng, Y.; Lv, S.; Diao, H.; Luo, Z.; Yan, P.; He, M. AptaDiff: De novo design and optimization of aptamers based on diffusion models. Brief. Bioinform. 2024, 25, bbae517. [Google Scholar] [CrossRef]
- Buglak, A.A.; Samokhvalov, A.V.; Zherdev, A.V.; Dzantiev, B.B. Methods and applications of in silico aptamer design and modeling. Int. J. Mol. Sci. 2020, 21, 8420. [Google Scholar] [CrossRef]
- Stuber, A.; Nakatsuka, N. Aptamer Renaissance for Neurochemical Biosensing. ACS Nano 2024, 18, 2552–2563. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Properties | Aptamers | Antibodies |
|---|---|---|
| chemical structure | oligonucleotide (RNA or DNA) | protein |
| size | 6–30 kDa | 150–180 kDa |
| target | wide range of molecules a | wide range of molecules b |
| specificity | high | high |
| affinity | high | high |
| chemical stability | higher | lower |
| thermal stability | stable up to 80 °C c | Low d |
| stability in biological systems | Low | higher |
| batch-to-batch variation | lower | higher |
| shelf life | long | shorter |
| immunogenicity | low, even in excess of therapeutic doses e | higher, especially in high doses f |
| synthesis | chemical (SPOS or enzymatic) and, to a lesser extent, biological | biological |
| development and synthesis cost | cheaper | more extensive |
| usage of modified building blocks | possible | not possible |
| after synthesis modifications | easier | more difficult |
| Phase | Type | Outcome | Dosage | Duration | N | Results | Ref |
|---|---|---|---|---|---|---|---|
| Ia | multicenter, open-label, dose-escalation, and safety study | 3 lines or greater increase in vision on ETDRS chart | single dose varying from 0.25 to 3.0 mg | 3 months | 15 | 4/15 patients (26,7%) a | [56] |
| II | multicenter, open-label repeat dose study | 3 lines or greater increase in vision on ETDRS chart | 3 mg 3 times at 28-day intervals | 3 months | 10 + 11 b | 25% and 60% a,b | [59] |
| III | prospective, randomized, double-blind, multicenter, dose-ranging, controlled | losing < 15 letters | 0.3, 1, or 3 mg, or sham | 54 weeks | 1186 (294, 300, 296, 296) | 70%, 71%, 65%, 55% | [61] |
| Type | Outcome | Duration | N | Results | Ref |
|---|---|---|---|---|---|
| Multicenter, randomized, double-masked, sham-controlled study | mean change in VA loss > 15 letters progressing to legal blindness | 48 weeks a | 111 vs. 116 | maintained 7% vs. 14% 34%→35% vs. 24%→38% | [68] |
| Exploratory analysis of the VISION study results | loss < 15 letters | 54 weeks | 34 and 30 b | 76% vs. 50% and 80% vs. 57% compared to sham | [75] |
| Respective review of data | gain > 3 lines stabilized VA loss > 3 lines | 6–14 months | 90 | 20%, 70%, 10% | [76] |
| Interventional case report | CNV progression | 6 weeks | 2 | CNV progressed in both cases | [77] |
| Prospective, nonrandomized, observational case series | CFT leakage VA | 12 weeks | 41 | 340 ± 24 μm→299 ± 14 μm 100%→81% 20/116→20/120 | [79] |
| Longitudinal observation | mean logMAR VA change | 30.5 ± 8 weeks | 14 | 0.67 ± 0.3→0.74 ± 0.16 | [80] |
| Retrospective data analysis | mean VA change | 6 month | 50 | 0.37 ± 0.24→0.4 ± 0.26 | [81] |
| Noncontrolled study | loss < 3 lines | 24 weeks | 27 | 89% | [82] |
| Retrospective, interventional, comparative study | mean logMAR VA change foveal thickness | 12 months | 43 | 1.25 ± 0.43→0.83 ± 0.44 452.3 ± 44.83 μm→274.3 ± 13.33 | [83] |
| Retrospective review | mean VA change gain > 3 lines loss < 3 lines | 21 weeks | 73 | −0.68 lines 16% 70% | [84] |
| Retrospective review | median VA change | 24 months | 5 | 20/200→20/60 | [85] |
| Retrospective review | loss < 15 letters | 54 weeks | 62 | 90% | [86] |
| Noncontrolled study | mean VA change gain > 3 lines loss > 6 lines | 5.2 ± 1.5 months | 41 | −0.03 lines 17.1% 7.31% | [87] |
| Noncontrolled study | loss < 15 letters gain > 0 letters gain > 15 letters | 52 weeks | 56 | 79% 43% 9% | [89] |
| Retrospective, interventional case series | mean VA improvement | 6 months | 22 | 2.2 lines or 0.7 lines c | [91] |
| Prospective, open-label, non-comparative, observational case series | mean VA change CNV area change | 6 months | 7 | −5 letters 1.4 mm2→2.7 mm2 | [92] |
| Randomized comparative | mean VA change | 13.93 ± 5.87 months | 3 × 15 d | 0.3, 1.0, 2.2 d | [93] |
| Randomized comparative | stable VA | 6 months | 13: 18: 17 e | 61.5%, 55.5%, 76.5% e | [94] |
| Phase IV, prospective, uncontrolled study | mean VA improvement | 54 weeks | 568 | 15.9 letters | [95] |
| Prospective, uncontrolled study | mean logMAR VA | 54 weeks | 75 | 0.26 ± 0.24→0.29 ± 0.28 f | [96] |
| Prospective, uncontrolled study | mean logMAR VA CFT | 3 years | 16 | 0.24 ± 0.25→0.25 ± 0.28 f 232 ± 54 μm→210 ± 59 μm f | [97] |
| Retrospective review | loss < 15 letters | 10–16 months | 80 | 97% | [98] |
| Type | Outcome | Duration | N | Results | Ref |
|---|---|---|---|---|---|
| Phase II, randomized, double-masked, dose-ranging controlled trial | median VA gain > 10 letters mean CRT change | 36 weeks | 172 | 20/50 vs. 20/63 a 34% vs. 10% a −68 μm vs. +4 μm a | [100] |
| Retrospective analysis of a randomized clinical trial. | changes in retinal neovascularization | 36 weeks | 16 | 62% showed regression in the pegaptanib group and 0 in the sham group | [101] |
| Retrospective analysis | mean VA change (logMAR) | varying | 13 12 15 | −0.16 ± 0.15 (pegaptanib) −0.06 ± 0.14 (combined treatment) +0.03 ± 0.13 (photocoagulation) | [103] |
| Retrospective analysis | mean CRT change | varying | 13 12 15 | −146.77 ± 93.39 (pegaptanib) −71.67 ± 105 (combined treatment) +19.2 ± 54.2 (photocoagulation) | [103] |
| Prospective, interventional, single case report | CRT VA | 42 months | 1 | 511 μm→376 μm stable | [104] |
| Case report | VA | 15 months | 1 | 30/200→180/200 | [105] |
| Retrospective analysis | mean VA mean CRT | 6–12 months | 63 | 0.66 ± 0.37→0.53 ± 0.37 533.1 ± 123.1→373.7 ± 135.8 | [106] |
| Prospective, randomized, controlled, exploratory study | NV regression VA change | 30 weeks | 20 | complete regression in all eyes vs. in 20% of eyes +5.8 letters vs. −6.0 letters | [107] |
| Phase II/III, sham-controlled trial | gained letters | 2 year | 207 | 6.1 vs. 1.3 | [109] |
| Randomized controlled trial | gain > 10 letters | 54 weeks | 260 | 36.8% vs. 19.7% | [110] |
| Prospective, interventional study | FAZ size change | 36 weeks | 30 | no significant change | [111] |
| Longitudinal, interventional, non-randomized study | mean reduction in CFT VA | 48 weeks | 30 | 551.5 ± 129.8→237.4 ± 41.1 18.2 ± 8.5 →25.5 ± 8.4 letters | [112] |
| Prospective case series | VA change | 18 weeks | 8 | no significant change | [113] |
| Treatment | Outcome | Duration | N | Results | Ref |
|---|---|---|---|---|---|
| Pegaptanib (0.3 mg) vs. sham | loss < 15 letters | 54 weeks | 1186 | 70% vs. 55% | [61] |
| Ranibizumab (0.3 mg) Ranibizumab (0.5 mg) PDT | loss < 15 letters | 12 months | 423 | 94.3% 96.4% 64.3% | [208] |
| Ranibizumab (0.3 mg) ranibizumab (0.5 mg) Verteporfin | loss < 15 letters | 24 months | 716 | 90% 92% 52.9% | [209] |
| Ranibizumab + PDT vs. sham + PDT | loss < 15 letters | 12 months | 162 | 90.5% vs. 67.9% | [210] |
| Ranibizumab + PDT vs. sham + PDT | loss < 15 letters | 24 months | 162 | 88% vs. 75% | [211] |
| Bevacizumab (no control) | mean VA | 8 weeks | 81 | 20/200→20/125 | [214] |
| Bevacizumab vs. ST | loss < 15 letters | 54 weeks | 131 | 91% vs. 67% | [216] |
| Bevacizumab vs. pegaptanib | retinal edema reduction | 54 weeks | 122 | −0.82 mm3 vs. −0.31 mm3 | [217] |
| Bevacizumab vs. pegaptanib | total retinal volume reduction | 3 months | 53 | −0.88 ± 1.4 mm3 vs. −0.07 ± 0.5 mm3 | [218] |
| Bevacizumab vs. pegaptanib | gained letters FT change | 6 months | 15 | +7.2 vs. +9.1 −52.9 μm vs. −88.2 μm | [219] |
| Pegaptanib vs. ranibizumab | mean VA change | 12 months | 81 | −0.095→−0.18 vs. −0.077→−0.11 | [221] |
| Treatment/Delivery System | Disease | Advantages | Route of Administration | Ref |
|---|---|---|---|---|
| Conventional | ||||
| AMD | - | Intravitreal injection | |
| Less vitreal efflux | [261] | ||
| Less vitreal efflux | [262] | ||
| Advanced | ||||
| AMD | Sustained release of pegaptanib Co-encapsulation with trehalose maintains aptamer integrity after the release | Intravitreal injection | [58,266] |
| Cancer | Overcoming poor pharmacokinetic lifetimes and enhancing therapeutic potential | Intraperitoneal or Intravenous injection | [264] |
| Enhancing therapeutic potential by improving pegaptanib’s binding affinity and serum stability | - | [265] | |
| Aptamer | Disease | Development Stage and Trial ID | Route of Administration |
|---|---|---|---|
| Pegaptanib | Neovascular AMD | FDA-approved | Intravitreal |
| Avacincaptad pegol | GA secondary to AMD | FDA-approved | Intravitreal |
| Autosomal recessive Stargardt disease | Phase IIb [NCT03364153] | ||
| Umedaptanib pegol (APT-F2P, RBM-007) | Neovascular AMD | Phase II [NCT04200248, NCT04640272] | Intravitreal |
| Achondroplasia | Phase II [jRCT2031220338] | Subcutaneous | |
| AS1411 | Acute Myeloid Leukemia | Phase II [NCT00512083] | Intravenous |
| Advanced solid tumors | Phase I [NCT00881244] | ||
| BC-007 | Dilated cardiomyopathy | Phase II [NCT04192214] | Intravenous |
| Long COVID-19 | Phase II [NCT05911009] | ||
| Rondaptivon pegol (BT200) | Von Willebrand Diseases, Hemophilia A | Phase II [NCT04677803] | Subcutaneous or intravenous |
| Von-Willebrand disease Type 2B | Phase II [2024-518294-34-01] | ||
| AON-D21 | Community-acquired pneumonia | Phase II [NCT05962606] | Intravenous |
| Olaptesed pegol (NOX-A12) | Glioblastoma | Phase I/I [NCT04121455] | Intravenous |
| Colorectal and pancreatic cancer | Phase I/II [NCT03168139] | ||
| Chronic Lymphocytic Leukemia | Phase II [NCT01486797] | ||
| Multiple Myeloma | Phase II [NCT01521533] | ||
| NOX-E36 | Type 2 Diabetes Mellitus | Phase I/II [NCT01085292] | Subcutaneous |
| Type 2 Diabetes Mellitus and Albuminuria | Phase II [NCT01547897] | ||
| Lexaptepid Pegol (NOX-H94) | Anemia, end-stage renal disease | Phase I/II [NCT02079896] | Subcutaneous or intravenous |
| Anemia of Chronic Diseases | Phase II [NCT01691040] | ||
| ApTOLL | Acute ischemic stroke | Phase Ib/IIa [NCT04734548] | Intravenous |
| BB-031 | Acute ischemic stroke | Phase II [NCT06226805] | Intravenous |
| AM003 | Solid tumors | Phase I [NCT06258330] | Intratumor route |
| Apta-1 | Sepsis, Inflammation | Phase I [ISRCTN15455814] | Intravenous |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bege, M.; Ghanem Kattoub, R.; Borbás, A. The 20th Anniversary of Pegaptanib (MacugenTM), the First Approved Aptamer Medicine: History, Recent Advances and Future Prospects of Aptamers in Therapy. Pharmaceutics 2025, 17, 394. https://doi.org/10.3390/pharmaceutics17030394
Bege M, Ghanem Kattoub R, Borbás A. The 20th Anniversary of Pegaptanib (MacugenTM), the First Approved Aptamer Medicine: History, Recent Advances and Future Prospects of Aptamers in Therapy. Pharmaceutics. 2025; 17(3):394. https://doi.org/10.3390/pharmaceutics17030394
Chicago/Turabian StyleBege, Miklós, Rasha Ghanem Kattoub, and Anikó Borbás. 2025. "The 20th Anniversary of Pegaptanib (MacugenTM), the First Approved Aptamer Medicine: History, Recent Advances and Future Prospects of Aptamers in Therapy" Pharmaceutics 17, no. 3: 394. https://doi.org/10.3390/pharmaceutics17030394
APA StyleBege, M., Ghanem Kattoub, R., & Borbás, A. (2025). The 20th Anniversary of Pegaptanib (MacugenTM), the First Approved Aptamer Medicine: History, Recent Advances and Future Prospects of Aptamers in Therapy. Pharmaceutics, 17(3), 394. https://doi.org/10.3390/pharmaceutics17030394