
N-(9-Acridinyl) Amino Acid Derivatives: Synthesis and In Vitro Evaluation of Anti-Toxoplasma gondii Activity

 ,
,  , , , and
, , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.1.1. Chemicals Used for Synthesis, Purification and Physiochemical Characterization
2.1.2. Cell Culture
2.1.3. Parasites
2.2. Methods
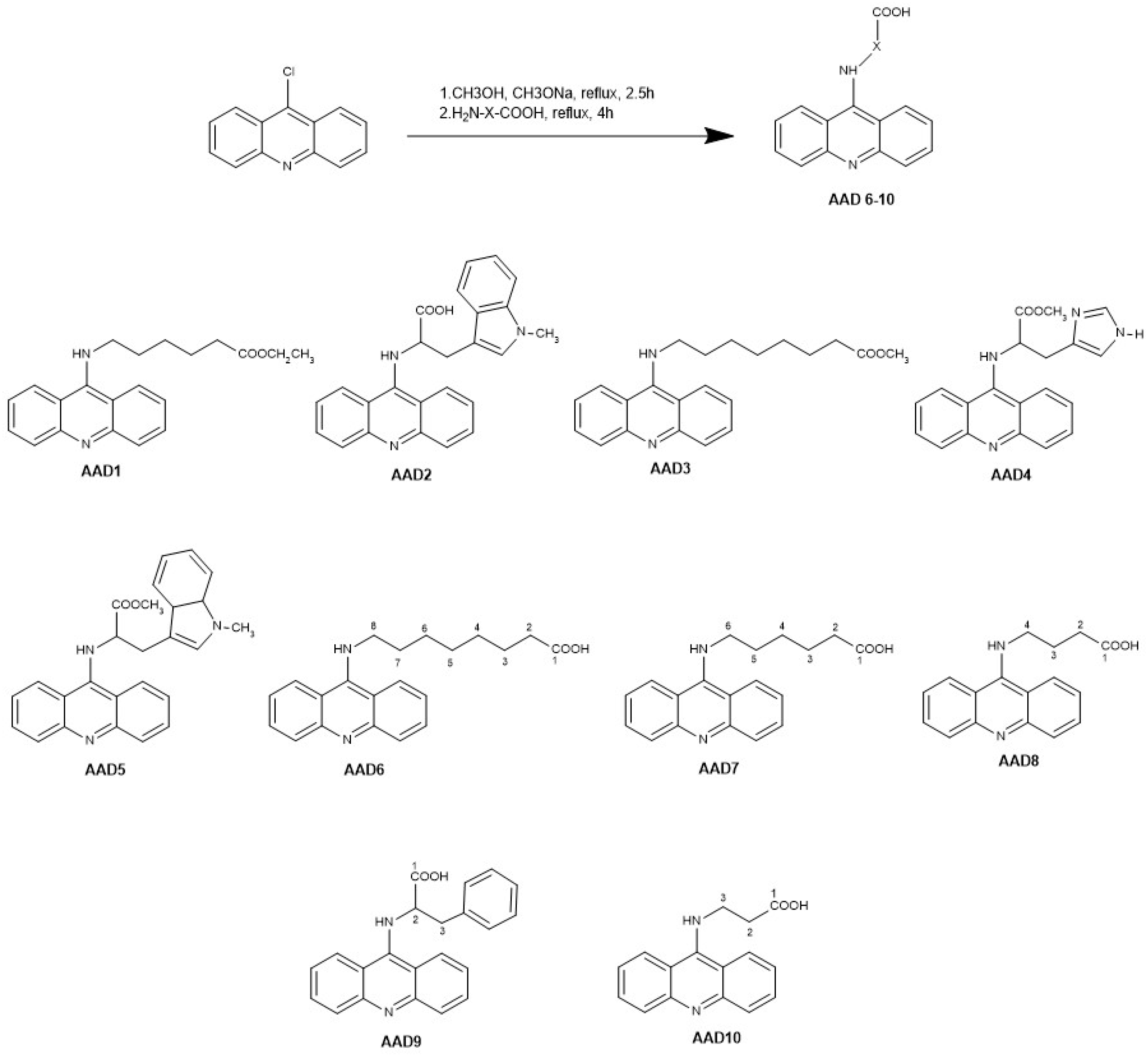
2.2.1. N-(9-Acrydinil) Amino Acid Derivatives Synthesis
2.2.2. Structural Characterization of Derivatives
2.2.3. Solubility and Solution Stability of the Synthesized Derivatives
2.2.4. Cytotoxicity Assay
2.2.5. Anti-T. gondii Activity Screening
2.2.6. Molecular Docking Analysis
3. Results
3.1. Cytotoxicity
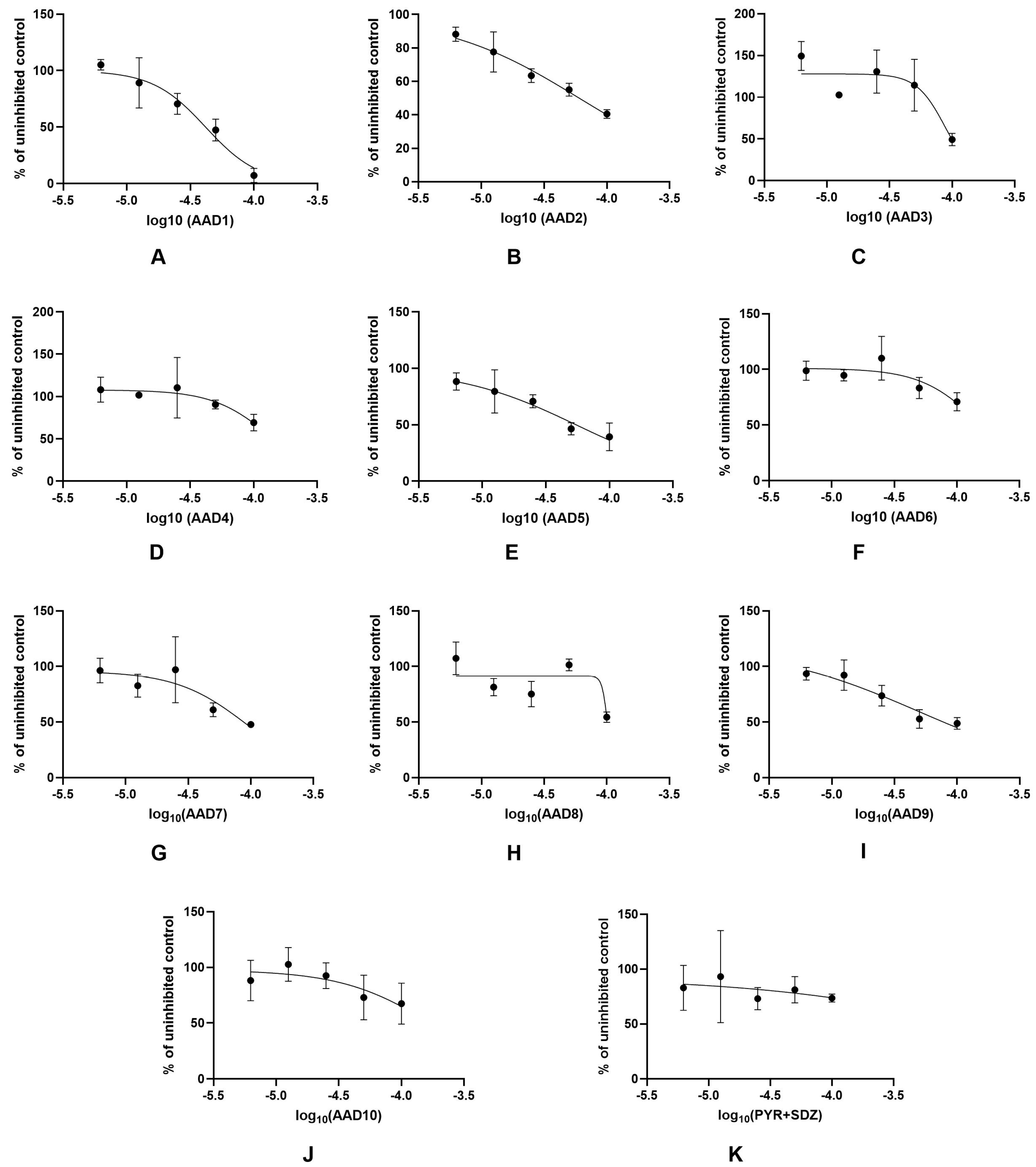
3.2. Anti-T. gondii Activity
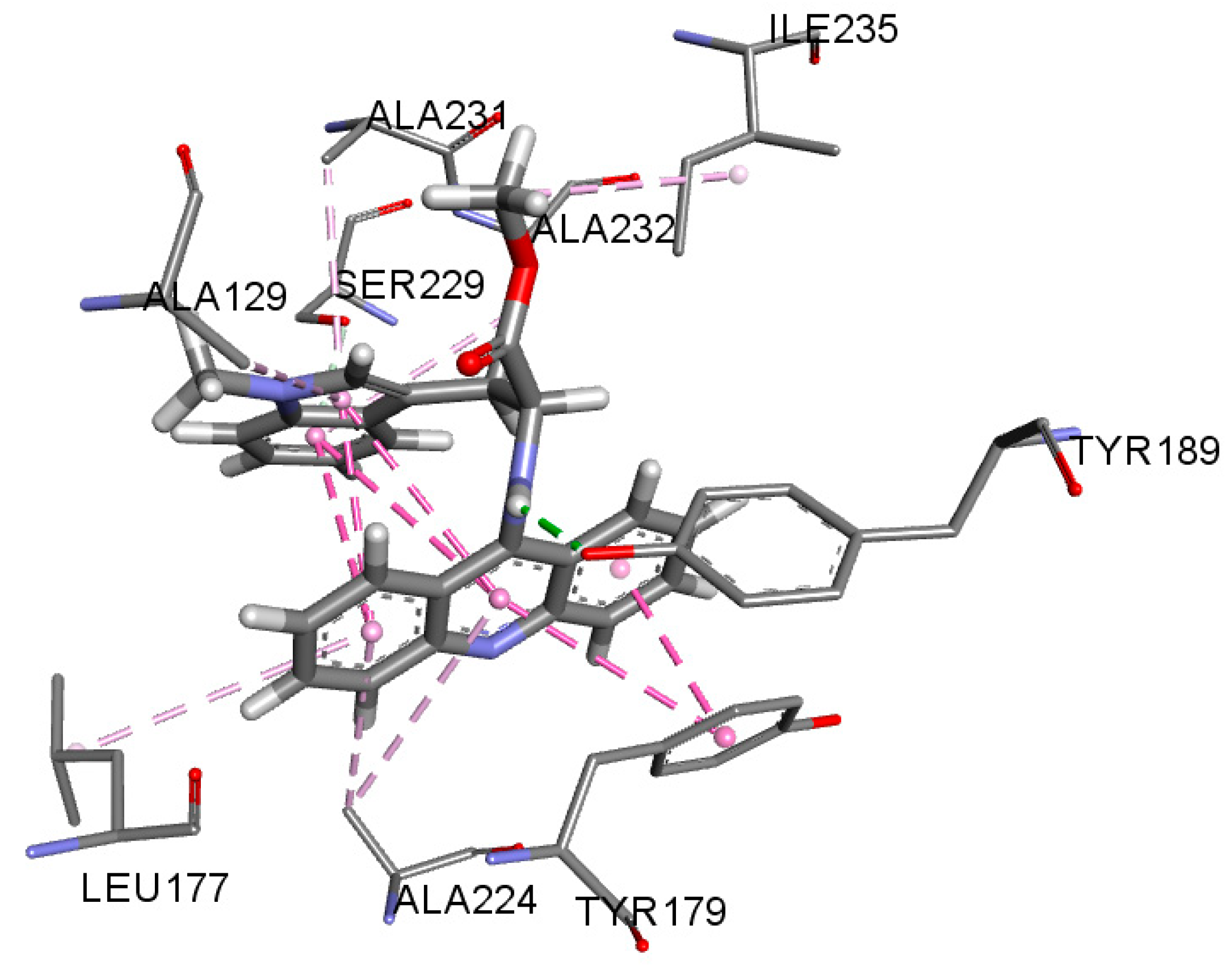
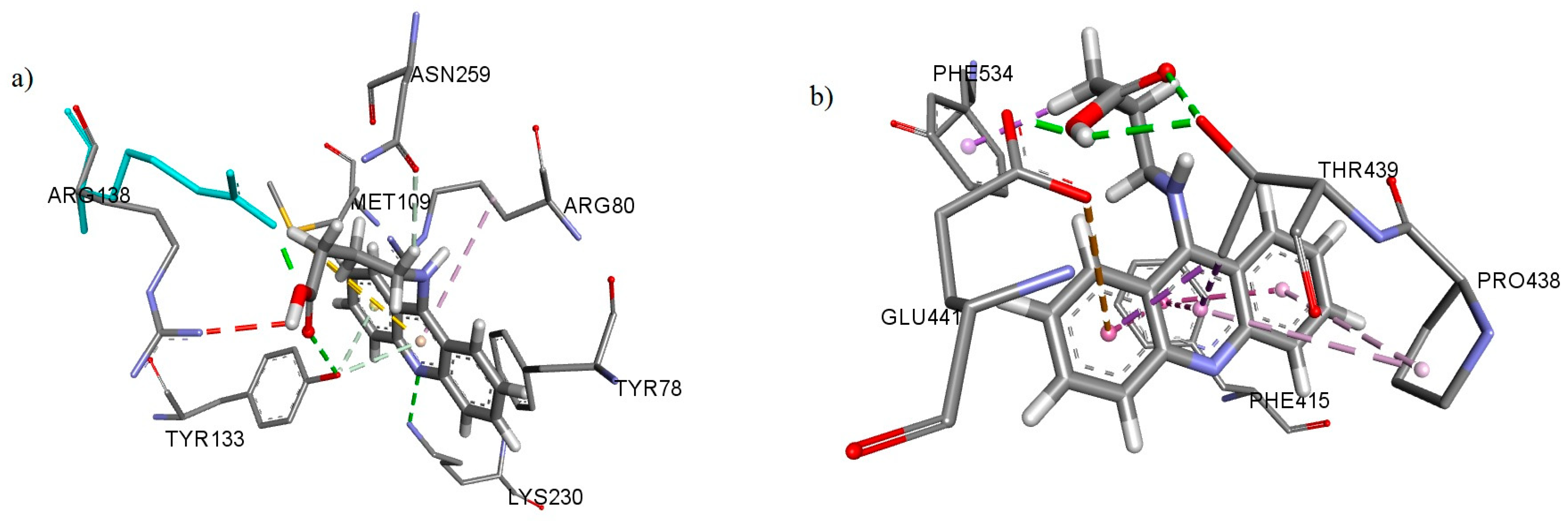
3.3. In Silico Analysis of Interactions with Potential Targets
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kumar, R.; Kaur, M.; Kumari, M. Acridine: A Versatile Heterocyclic Nucleus. Acta Pol. Pharm. 2012, 69, 3–9. [Google Scholar]
- Rupar, J.; Dobričić, V.; Grahovac, J.; Radulović, S.; Skok, Ž.; Ilaš, J.; Aleksić, M.; Brborić, J.; Čudina, O. Synthesis and Evaluation of Anticancer Activity of New 9-Acridinyl Amino Acid Derivatives. RSC Med. Chem. 2020, 11, 378–386. [Google Scholar] [CrossRef]
- Ehsanian, R.; Van Waes, C.; Feller, S.M. Beyond DNA Binding—A Review of the Potential Mechanisms Mediating Quinacrine’s Therapeutic Activities in Parasitic Infections, Inflammation, and Cancers. Cell Commun. Signal. 2011, 9, 13. [Google Scholar] [CrossRef] [PubMed]
- Alday, P.H.; Doggett, J.S. Drugs in Development for Toxoplasmosis: Advances, Challenges, and Current Status. Drug Des. Devel. Ther. 2017, 11, 273–293. [Google Scholar] [CrossRef]
- Bigna, J.J.; Tochie, J.N.; Tounouga, D.N.; Bekolo, A.O.; Ymele, N.S.; Youda, E.L.; Sime, P.S.; Nansseu, J.R. Global, Regional, and Country Seroprevalence of Toxoplasma gondii in Pregnant Women: A Systematic Review, Modelling, and Meta-Analysis. Sci. Rep. 2020, 10, 12102. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization & Food and Agriculture Organization of the United Nations, Multicriteria-Based Ranking for Risk Management of Food-Borne Parasites: Report of a Joint FAO/WHO Expert Meeting, 3–7 September 2012, FAO Headquarters, Rome, Italy. FAO, World Health Organization. Available online: https://apps.who.int/iris/handle/10665/112672 (accessed on 4 February 2025).
- Bouwknegt, M.; Devleesschauwer, B.; Graham, H.; Robertson, L.J.; van der Giessen, J.W. Euro-FBP Workshop Participants. Prioritisation of Food-Borne Parasites in Europe, 2016. Euro Surveill. 2018, 23, 17-00161. [Google Scholar] [CrossRef] [PubMed]
- Štajner, T.; Vujić, D.; Srbljanović, J.; Bauman, N.; Zečević, Ž.; Simić, M.; Djurković-Djaković, O. Risk of Reactivated Toxoplasmosis in Hematopoietic Stem Cell Transplant Recipients: A Prospective Cohort Study in a Setting Withholding Prophylaxis. Clin. Microbiol. Infect. 2022, 28, 733.e1–733.e5. [Google Scholar] [CrossRef]
- Montoya, J.G.; Rosso, F. Diagnosis and Management of Toxoplasmosis. Clin. Perinatol. 2005, 32, 705–726. [Google Scholar] [CrossRef]
- Konstantinovic, N.; Guegan, H.; Štajner, T.; Belaz, S.; Robert-Gangneux, F. Treatment of Toxoplasmosis: Current Options and Future Perspectives. Food Waterborne Parasitol. 2019, 15, e00036. [Google Scholar] [CrossRef]
- Liu, S.; Cai, M.; Liu, Z.; Gao, W.; Li, J.; Li, Y.; Abudouxukuer, X.; Zhang, J. Comprehensive Insights into the Development of Antitoxoplasmosis Drugs: Current Advances, Obstacles, and Future Perspectives. J. Med. Chem. 2024, 67, 20740–20764. [Google Scholar] [CrossRef]
- Gallant, J. Get Rich Quick with Old Generic Drugs! The Pyrimethamine Pricing Scandal. Open Forum Infect. Dis. 2015, 2, ofv177. [Google Scholar] [CrossRef] [PubMed]
- Goswami, R.P.; Goswami, R.P.; Rahman, M.; Ray, Y.; Tripathi, S.K. Alternative Treatment Approach to Cerebral Toxoplasmosis in HIV/AIDS: Experience from a Resource-Poor Setting. Int. J. STD AIDS 2015, 26, 864–869. [Google Scholar] [CrossRef] [PubMed]
- Alday, P.H.; McConnell, E.V.; Boitz Zarella, J.M.; Dodean, R.A.; Kancharla, P.; Kelly, J.X.; Doggett, J.S. Acridones Are Highly Potent Inhibitors of Toxoplasma gondii Tachyzoites. ACS Infect. Dis. 2021, 7, 1877–1884. [Google Scholar] [CrossRef]
- Lyakhov, S.A.; Suveyzdis, Y.I.; Bykhovskaya, O.V.; Isko, N.M.; Andronati, S.A.; Litvinova, L.A. Biological Active Acridine Derivatives. Part 3: Acridinylamino Acids and Their Esters; Synthesis and Cytostatic Activity. Pharmazie 1997, 52, 560–561. [Google Scholar] [CrossRef]
- Bahuguna, A.; Khan, I.; Bajpai, V.K.; Kang, S.C. MTT Assay to Evaluate the Cytotoxic Potential of a Drug. Bangladesh J. Pharmacol. 2017, 12, 115–118. [Google Scholar] [CrossRef]
- Bessa, G.d.L.; Vitor, R.W.d.A.; Lobo, L.M.S.; Rêgo, W.M.F.; de Souza, G.C.A.; Lopes, R.E.N.; Costa, J.G.L.; Martins-Duarte, E.S. In Vitro and In Vivo Susceptibility to Sulfadiazine and Pyrimethamine of Toxoplasma gondii Strains Isolated from Brazilian Free Wild Birds. Sci. Rep. 2023, 13, 7359. [Google Scholar] [CrossRef]
- Zaki, L.; Ghaffarifar, F.; Sharifi, Z.; Horton, J.; Sadraei, J. Effect of Imiquimod on Tachyzoites of Toxoplasma gondii and Infected Macrophages in Vitro and in BALB/c Mice. Front. Cell Infect. Microbiol. 2020, 10, 387. [Google Scholar] [CrossRef]
- MAKE Receptor, version 3.2.0.2. OpenEye Scientific Software: Santa Fe, NM, USA. Available online: https://www.eyesopen.com/ (accessed on 22 January 2025).
- FRED, version 3.2.0.2. OpenEye Scientific Software: Santa Fe, NM, USA. Available online: https://www.eyesopen.com/ (accessed on 22 January 2025).
- McGann, M. FRED Pose Prediction and Virtual Screening Accuracy. J. Chem. Inf. Model. 2011, 51, 578–596. [Google Scholar] [CrossRef]
- McGann, M. FRED and HYBRID Docking Performance on Standardized Datasets. J. Comput.-Aided Mol. Des. 2012, 26, 897–906. [Google Scholar] [CrossRef]
- Fonte, M.; Tassi, N.; Gomes, P.; Teixeira, C. Acridine-Based Antimalarials: From the Very First Synthetic Antimalarial to Recent Developments. Molecules 2021, 26, 600. [Google Scholar] [CrossRef]
- Valdés, A.F. Acridine and Acridinones: Old and New Structures With Antimalarial Activity. Open Med. Chem. J. 2011, 5, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, C.; Vale, N.; Pérez, B.; Gomes, A.; Gomes, J.R.; Gomes, P. “Recycling” Classical Drugs for Malaria. Chem. Rev. 2014, 114, 11164–11220. [Google Scholar] [CrossRef] [PubMed]
- Rupar, J.; Dobričić, V.; Brborić, J.; Čudina, O.; Aleksić, M.M. Square Wave Voltammetric Study of Interaction between 9-Acridinyl Amino Acid Derivatives and DNA. Bioelectrochemistry 2023, 149, 108323. [Google Scholar] [CrossRef] [PubMed]
- Boissavy, T.; Rotili, D.; Mouveaux, T.; Roger, E.; Aliouat, E.M.; Pierrot, C.; Valente, S.; Mai, A.; Gissot, M. Hydroxamate-Based Compounds Are Potent Inhibitors of Toxoplasma gondii HDAC Biological Activity. Antimicrob. Agents Chemother. 2023, 67, e0066123. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Q.; Li, H.; Cong, H.; Qu, Y. In Vitro and In Vivo Anti-Toxoplasma Activities of HDAC Inhibitor Panobinostat on Experimental Acute Ocular Toxoplasmosis. Front. Cell. Infect. Microbiol. 2022, 12, 1002817. [Google Scholar] [CrossRef]
- Cui, Z.; Li, X.; Li, L.; Zhang, B.; Gao, C.; Chen, Y.; Tan, C.; Liu, H.; Xie, W.; Yang, T.; et al. Design, Synthesis, and Evaluation of Acridine Derivatives as Multi-Target Src and MEK Kinase Inhibitors for Anti-Tumor Treatment. Bioorg. Med. Chem. 2016, 24, 261–269. [Google Scholar] [CrossRef]
- Doggett, J.S.; Ojo, K.K.; Fan, E.; Maly, D.J.; Van Voorhis, W.C. Bumped Kinase Inhibitor 1294 Treats Established Toxoplasma gondii Infection. Antimicrob. Agents Chemother. 2014, 58, 3547–3549. [Google Scholar] [CrossRef]
- Alday, P.H.; Nilsen, A.; Doggett, J.S. Structure–Activity Relationships of Toxoplasma gondii Cytochrome bc1 Inhibitors. Expert Opin. Drug Discov. 2022, 17, 997–1011. [Google Scholar] [CrossRef]
- Chavalitshewinkoon, P.; Wilairat, P.; Gamage, S.; Denny, W.; Figgitt, D.; Ralph, R. Structure–Activity Relationships and Modes of Action of 9-Anilinoacridines against Chloroquine-Resistant Plasmodium falciparum in Vitro. Antimicrob. Agents Chemother. 1993, 37, 403–406. [Google Scholar] [CrossRef]
- Swale, C.; Bellini, V.; Bowler, M.W.; Flore, N.; Brenier-Pinchart, M.-P.; Cannella, D.; Belmudes, L.; Mas, C.; Couté, Y.; Laurent, F.; et al. Altiratinib Blocks Toxoplasma gondii and Plasmodium falciparum Development by Selectively Targeting a Spliceosome Kinase. Sci. Transl. Med. 2022, 14, eabn3231. [Google Scholar] [CrossRef]
- Ramakrishnan, S.; Docampo, M.D.; MacRae, J.I.; Ralton, J.E.; Rupasinghe, T.; McConville, M.J.; Striepen, B. The Intracellular Parasite Toxoplasma gondii Depends on the Synthesis of Long-Chain and Very Long-Chain Unsaturated Fatty Acids Not Supplied by the Host Cell. Mol. Microbiol. 2015, 97, 64–76. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Rodríguez, C.; Conter, C.; Oyenarte, I.; Favretto, F.; Quintana, I.; Martinez-Chantar, M.L.; Astegno, A.; Martínez-Cruz, L.A. Structural Basis of the Inhibition of Cystathionine γ-Lyase from Toxoplasma gondii by Propargylglycine and Cysteine. Protein Sci. 2023, 32, e4619. [Google Scholar] [CrossRef] [PubMed]
- Manickam, Y.; Malhotra, N.; Mishra, S.; Babbar, P.; Dusane, A.; Laleu, B.; Bellini, V.; Hakimi, M.A.; Bougdour, A.; Sharma, A. Double Drugging of Prolyl-tRNA Synthetase Provides a New Paradigm for Anti-Infective Drug Development. PLoS Pathog. 2022, 18, e1010363. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound (AAD) | CC50 VALUE (µM) |
|---|---|
| AAD1 | 41.72 |
| AAD2 | 60.22 |
| AAD3 | 88.37 |
| AAD4 | 133.70 |
| AAD5 | 54.66 |
| AAD6 | 154.10 |
| AAD7 | 92.95 |
| AAD8 | 102.00 |
| AAD9 | 50.19 |
| AAD10 | 176.80 |
| PYR+SDZ | 2056.00 |
| Sample | Viability Reduction of T. gondii Tachyzoites (%) |
|---|---|
| AAD1 | 24.7 |
| AAD2 | 25.8 |
| AAD3 | 15.2 |
| AAD4 | 4.5 |
| AAD5 | 0.0 |
| AAD6 | 22.0 |
| AAD7 | 20.5 |
| AAD8 | 33.3 |
| AAD9 | 21.9 |
| AAD10 | 21.9 |
| PYR+SDZ | 35.6 |
| 7Q4A | 2O2S | 5E5I | 8BIS | 4WG3 | 4KYA | 5XIG | |
|---|---|---|---|---|---|---|---|
| Co-crystallized ligand | −13.95 | −10.77 | −10.16 | −5.75 | −13.60 | −15.21 | −10.54 |
| AAD1 | −14.34 | −11.92 | −7.91 | −3.27 | −12.19 | −10.64 | −9.37 |
| AAD2 | −13.61 | −12.68 | −9.62 | −1.88 | −10.86 | −11.88 | −10.57 |
| AAD3 | −14.13 | −10.20 | −7.86 | −1.83 | −12.76 | −11.15 | −8.69 |
| AAD4 | −10.58 | −12.04 | −10.77 | −6.24 | −11.69 | −12.26 | −9.25 |
| AAD5 | −14.07 | −14.74 | −8.86 | −1.61 | −11.50 | −11.76 | −9.36 |
| AAD6 | −12.91 | −11.52 | −8.75 | −3.10 | −11.59 | −12.00 | −9.09 |
| AAD7 | −12.67 | −12.67 | −8.77 | −6.21 | −11.99 | −10.67 | −10.08 |
| AAD8 | −12.18 | −11.80 | −8.14 | −7.03 | −11.11 | −10.40 | −10.05 |
| AAD9 | −11.94 | −13.28 | −7.54 | −3.87 | −11.15 | −11.76 | −8.72 |
| AAD10 | −10.26 | −11.48 | −7.56 | −5.38 | −11.26 | −10.12 | −9.84 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zlatković, Đ.; Dobričić, V.; Srbljanović, J.; Lijeskić, O.; Bauman, N.; Ćirković, V.; Štajner, T. N-(9-Acridinyl) Amino Acid Derivatives: Synthesis and In Vitro Evaluation of Anti-Toxoplasma gondii Activity. Pharmaceutics 2025, 17, 374. https://doi.org/10.3390/pharmaceutics17030374
Zlatković Đ, Dobričić V, Srbljanović J, Lijeskić O, Bauman N, Ćirković V, Štajner T. N-(9-Acridinyl) Amino Acid Derivatives: Synthesis and In Vitro Evaluation of Anti-Toxoplasma gondii Activity. Pharmaceutics. 2025; 17(3):374. https://doi.org/10.3390/pharmaceutics17030374
Chicago/Turabian StyleZlatković, Đorđe, Vladimir Dobričić, Jelena Srbljanović, Olivera Lijeskić, Neda Bauman, Vladimir Ćirković, and Tijana Štajner. 2025. "N-(9-Acridinyl) Amino Acid Derivatives: Synthesis and In Vitro Evaluation of Anti-Toxoplasma gondii Activity" Pharmaceutics 17, no. 3: 374. https://doi.org/10.3390/pharmaceutics17030374
APA StyleZlatković, Đ., Dobričić, V., Srbljanović, J., Lijeskić, O., Bauman, N., Ćirković, V., & Štajner, T. (2025). N-(9-Acridinyl) Amino Acid Derivatives: Synthesis and In Vitro Evaluation of Anti-Toxoplasma gondii Activity. Pharmaceutics, 17(3), 374. https://doi.org/10.3390/pharmaceutics17030374