


Dendrimer-Derived Mimics of Host Defense Peptides Selectively Disrupt Cancer Cell Membranes for Melanoma Therapy

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture
2.3. CCK-8 Analysis
2.4. Live/Dead Staining Analysis
2.5. Flow Cytometry Analysis of Apoptosis
2.6. Scanning Electron Microscopy (SEM) Analysis
2.7. In Vivo Anticancer Analysis
2.8. Statistical Analysis
3. Results
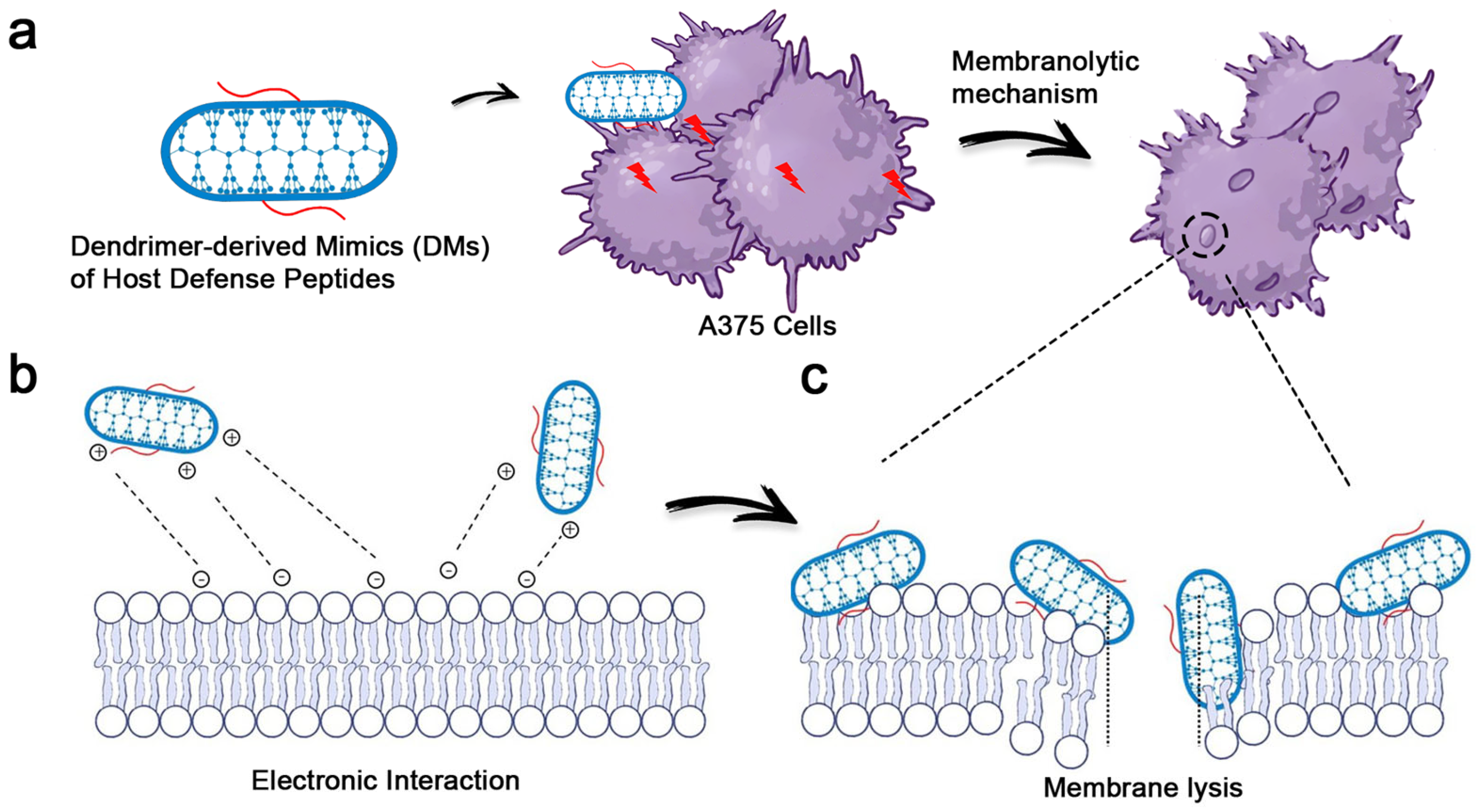
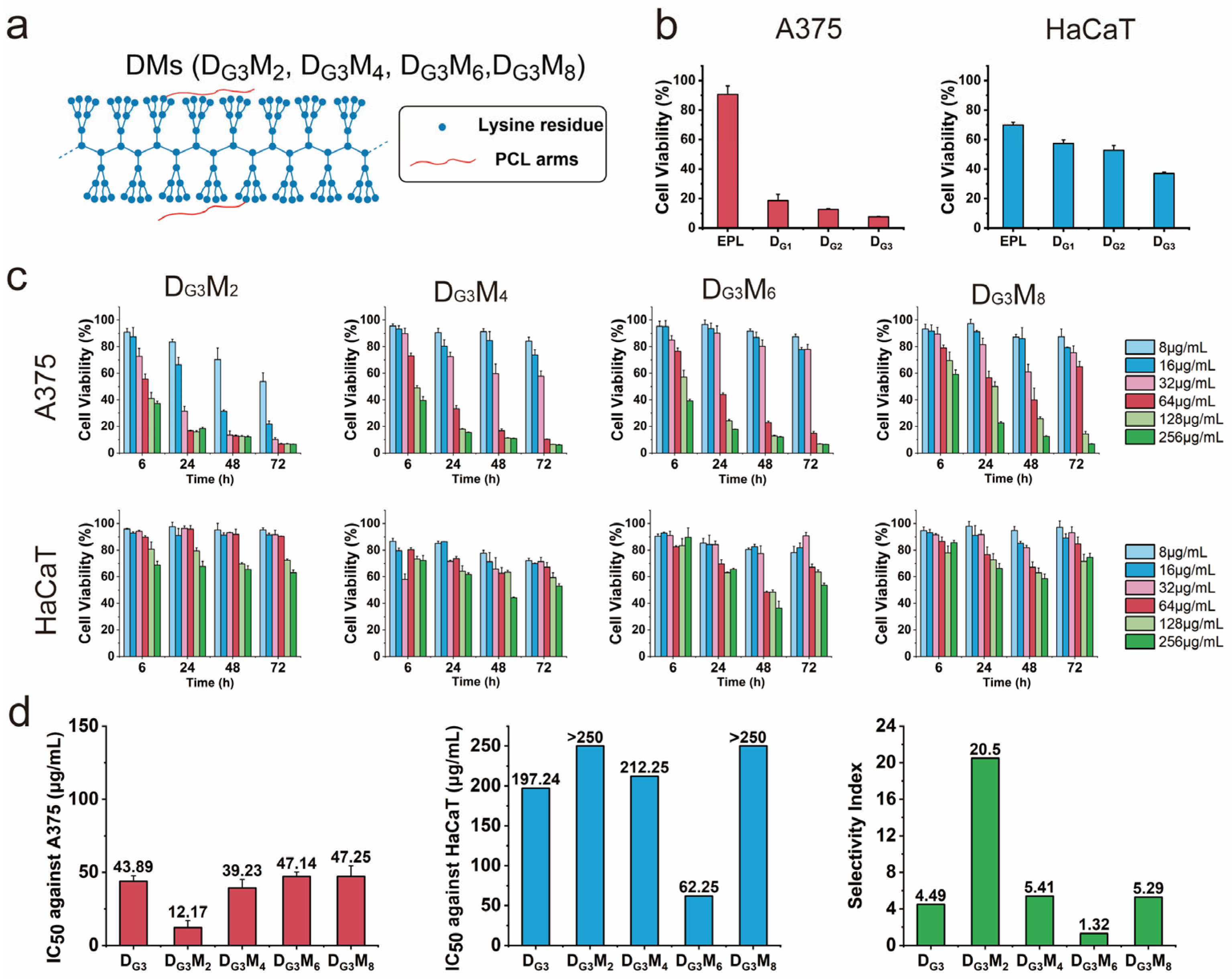
3.1. Potent Anticancer Activity and Remarkable Selectivity of DMs
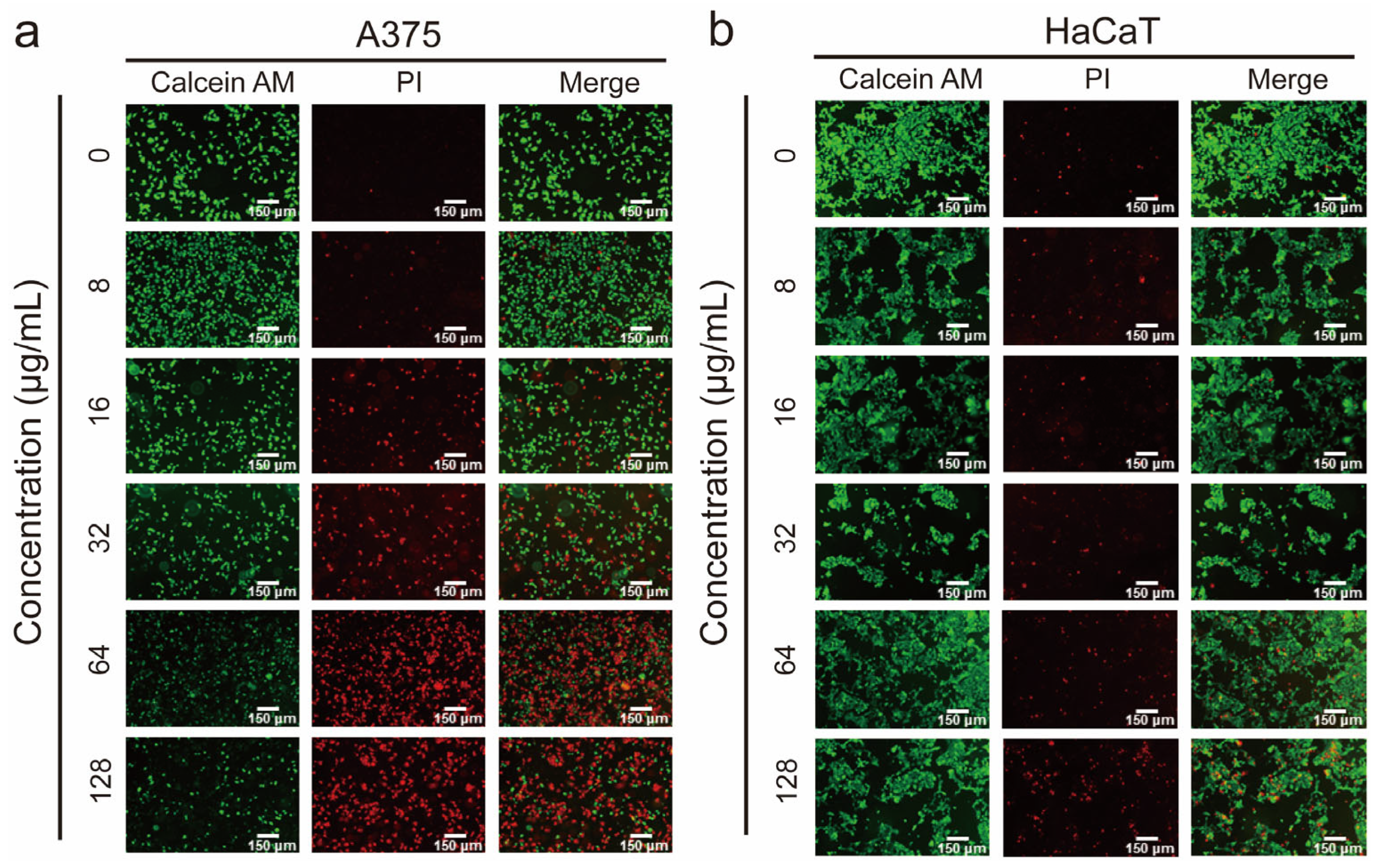
3.2. Demonstration of the Membranolytic Mechanism via the Live/Dead Staining Analysis
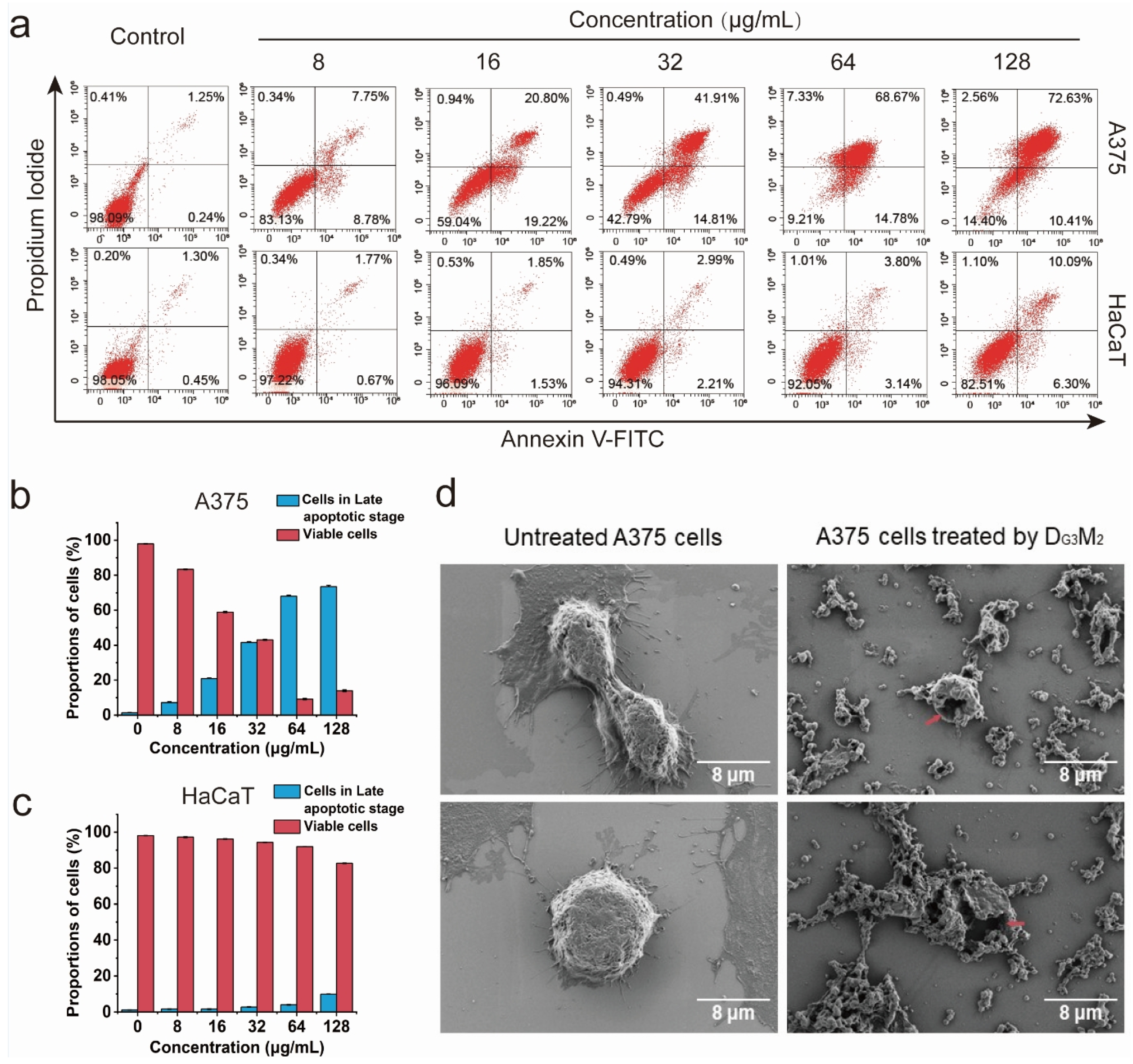
3.3. The Membranolytic Mechanism Indicated by Flow Cytometry Analysis
3.4. Clear Membrane Lysis Observed by SEM Analysis
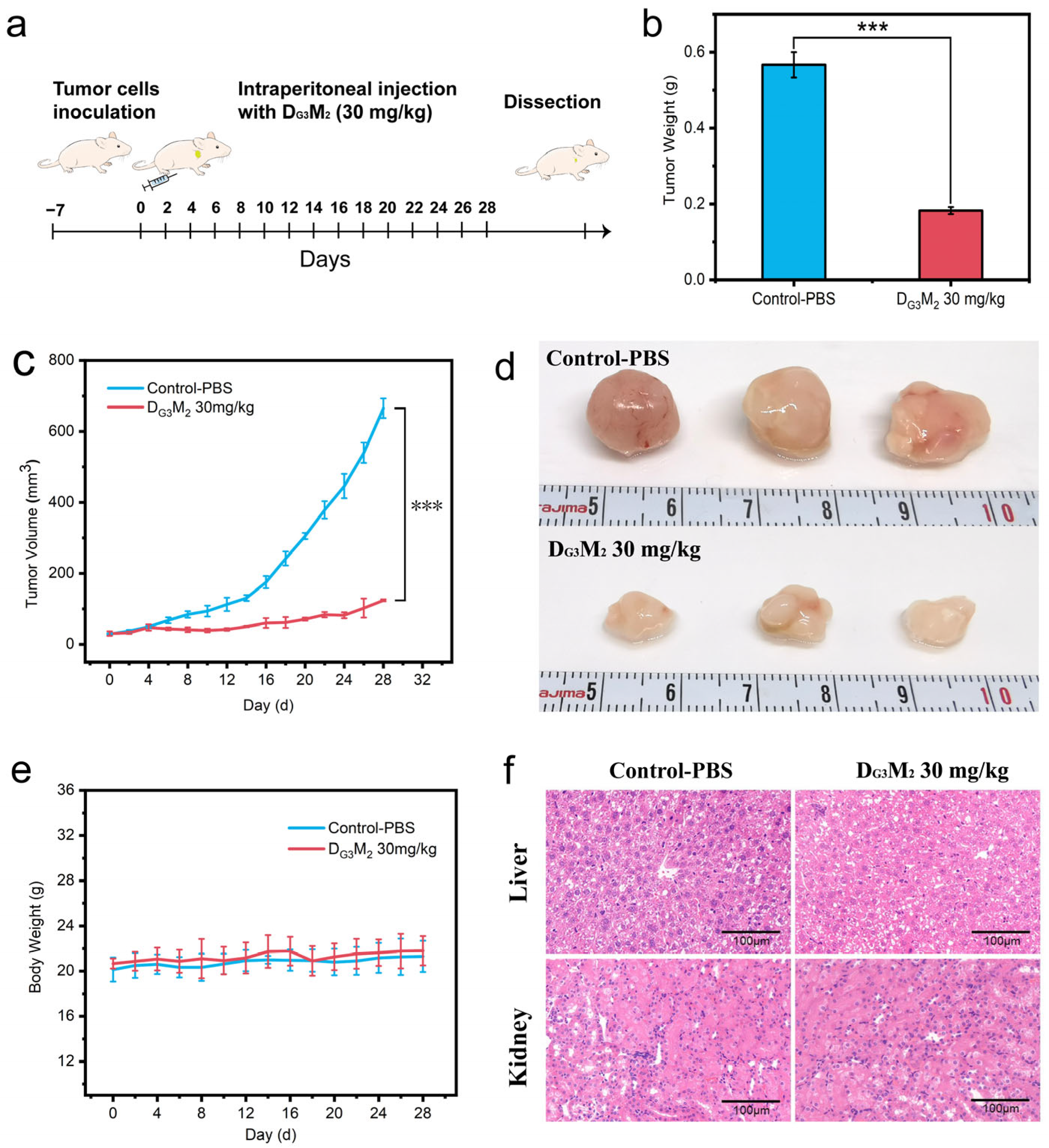
3.5. The Capability of DMs to Inhibit Tumor Growth in Vivo with Low Tissue Toxicity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Arnold, M.; Singh, D.; Laversanne, M.; Vignat, J.; Vaccarella, S.; Meheus, F.; Cust, A.E.; de Vries, E.; Whiteman, D.C.; Bray, F. Global Burden of Cutaneous Melanoma in 2020 and Projections to 2040. JAMA Dermatol. 2022, 158, 495–503. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Cancer statistics for the year 2020: An overview. Int. J. Cancer 2021, 149, 778–789. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.F.; Li, Z.Y.; Yan, L.; Liu, Y.L.; Yang, H.J.; Li, H. Global, regional, and national cancer incidence and death for 29 cancer groups in 2019 and trends analysis of the global cancer burden, 1990–2019. J. Hematol. Oncol. 2021, 14, 197. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.L.; Hou, J.B.; Shi, S.M.; Du, J.; Liu, Y.D.; Huang, P.; Li, Q.; Liu, L.C.; Hu, H.R.; Ji, Y.C.; et al. CSN6 promotes melanoma proliferation and metastasis by controlling the UBR5-mediated ubiquitination and degradation of CDK9. Cell Death Dis. 2021, 12, 118. [Google Scholar] [CrossRef]
- Shirley, C.A.; Chhabra, G.; Amiri, D.; Chang, H.; Ahmad, N. Immune escape and metastasis mechanisms in melanoma: Breaking down the dichotomy. Front. Immunol. 2024, 15, 1336023. [Google Scholar] [CrossRef]
- Leonardi, G.C.; Falzone, L.; Salemi, R.; Zanghì, A.; Spandidos, D.A.; McCubrey, J.A.; Candido, S.; Libra, M. Cutaneous melanoma: From pathogenesis to therapy (Review). Int. J. Oncol. 2018, 52, 1071–1080. [Google Scholar] [CrossRef]
- Curti, B.D.; Faries, M.B. Recent Advances in the Treatment of Melanoma. N. Engl. J. Med. 2021, 384, 2229–2240. [Google Scholar] [CrossRef]
- Rui, R.; Zhou, L.Q.; He, S.M. Cancer immunotherapies: Advances and bottlenecks. Front. Immunol. 2023, 14, 1212476. [Google Scholar] [CrossRef]
- Pham, J.P.; Joshua, A.M.; da Silva, I.P.; Dummer, R.; Goldinger, S.M. Chemotherapy in Cutaneous Melanoma: Is There Still a Role? Curr. Oncol. Rep. 2023, 25, 609–621. [Google Scholar] [CrossRef]
- Liu, Q.; Das, M.; Liu, Y.; Huang, L. Targeted drug delivery to melanoma. Adv. Drug Deliv. Rev. 2018, 127, 208–221. [Google Scholar] [CrossRef]
- Mundra, V.; Li, W.; Mahato, R.I. Nanoparticle-mediated drug delivery for treating melanoma. Nanomedicine 2015, 10, 2613–2633. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.T.; Zhu, Y.K.; Zhang, G.D.; Liu, Z.P. Renovation as innovation: Repurposing human antibacterial peptide LL-37 for cancer therapy. Front. Pharmacol. 2022, 13, 944147. [Google Scholar] [CrossRef] [PubMed]
- Riedl, S.; Zweytick, D.; Lohner, K. Membrane-active host defense peptides—Challenges and perspectives for the development of novel anticancer drugs. Chem. Phys. Lipids 2011, 164, 766–781. [Google Scholar] [CrossRef]
- Kardani, K.; Bolhassani, A. Antimicrobial/anticancer peptides: Bioactive molecules and therapeutic agents. Immunotherapy 2021, 13, 669–684. [Google Scholar] [CrossRef]
- Szlasa, W.; Zendran, I.; Zalesinska, A.; Tarek, M.; Kulbacka, J. Lipid composition of the cancer cell membrane. J. Bioenerg. Biomembr. 2020, 52, 321–342. [Google Scholar] [CrossRef]
- Tornesello, A.L.; Borrelli, A.; Buonaguro, L.; Buonaguro, F.M.; Tornesello, M.L. Antimicrobial Peptides as Anticancer Agents: Functional Properties and Biological Activities. Molecules 2020, 25, 2850. [Google Scholar] [CrossRef]
- Quemé-Peña, M.; Ricci, M.; Juhász, T.; Horváti, K.; Bosze, S.; Biri-Kovács, B.; Szeder, B.; Zsila, F.; Beke-Somfai, T. Old Polyanionic Drug Suramin Suppresses Detrimental Cytotoxicity of the Host Defense Peptide LL-37. ACS Pharmacol. Transl. Sci. 2021, 4, 155–167. [Google Scholar] [CrossRef]
- Mookherjee, N.; Anderson, M.A.; Haagsman, H.P.; Davidson, D.J. Antimicrobial host defence peptides: Functions and clinical potential. Nat. Rev. Drug Discov. 2020, 19, 311–332. [Google Scholar] [CrossRef]
- Ergene, C.; Yasuhara, K.; Palermo, E.F. Biomimetic antimicrobial polymers: Recent advances in molecular design. Polym. Chem. 2018, 9, 2407–2427. [Google Scholar] [CrossRef]
- Kalelkar, P.P.; Riddick, M.; García, A.J. Biomaterial-based antimicrobial therapies for the treatment of bacterial infections. Nat. Rev. Mater. 2022, 7, 39–54. [Google Scholar] [CrossRef] [PubMed]
- Dias, A.P.; Santos, S.D.; da Silva, J.V.; Parise, R.; Ferreira, E.I.; El Seoud, O.; Giarolla, J. Dendrimers in the context of nanomedicine. Int. J. Pharm. 2020, 573, 118814. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, A.S. Dendrimers for Drug Delivery. Molecules 2018, 23, 938. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, B.X.; Qiu, L.; Qiao, X.; Yang, H. Dendrimer-based drug delivery systems: History, challenges, and latest developments. J. Biol. Eng. 2022, 16, 18. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.S.; Yang, D.J.; Zhu, J.M.; Huang, S.T.; Chen, S.J.; Zeng, J.; Xu, J.; He, J.; Zhou, C.C. Mimics of Host Defense Peptides Derived from Dendronized Polylysines for Antibacterial and Anticancer Therapy. ACS Macro Lett. 2024, 13, 1156–1163. [Google Scholar] [CrossRef] [PubMed]
- Burton, K.A.; Ashack, K.A.; Khachemoune, A. Cutaneous Squamous Cell Carcinoma: A Review of High-Risk and Metastatic Disease. Am. J. Clin. Dermatol. 2016, 17, 491–508. [Google Scholar] [CrossRef]
- Seidel, J.A.; Otsuka, A.; Kabashima, K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, efficacy, and Limitations. Front. Oncol. 2018, 8, 86. [Google Scholar] [CrossRef]
- Janaszewska, A.; Lazniewska, J.; Trzepinski, P.; Marcinkowska, M.; Klajnert-Maculewicz, B. Cytotoxicity of Dendrimers. Biomolecules 2019, 9, 330. [Google Scholar] [CrossRef]
- Chis, A.A.; Dobrea, C.; Morgovan, C.; Arseniu, A.M.; Rus, L.L.; Butuca, A.; Juncan, A.M.; Totan, M.; Vonica-Tincu, A.L.; Cormos, G.; et al. Applications and Limitations of Dendrimers in Biomedicine. Molecules 2020, 25, 3982. [Google Scholar] [CrossRef]
- Tian, Y.H.; Li, S.P.; Song, J.; Ji, T.J.; Zhu, M.T.; Anderson, G.J.; Wei, J.Y.; Nie, G.J. A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials 2014, 35, 2383–2390. [Google Scholar] [CrossRef]
- Schweizer, F. Cationic amphiphilic peptides with cancer-selective toxicity. Eur. J. Pharmacol. 2009, 625, 190–194. [Google Scholar] [CrossRef]
- Gajski, G.; Garaj-Vrhovac, V. Melittin: A lytic peptide with anticancer properties. Environ. Toxicol. Pharmacol. 2013, 36, 697–705. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.Y.; He, J.; Zhou, C.C. Strategies from nature: Polycaprolactone-based mimetic antimicrobial peptide block copolymers with low cytotoxicity and excellent antibacterial efficiency. Polym. Chem. 2019, 10, 945–953. [Google Scholar] [CrossRef]
- Shao, N.; Yuan, L.; Ma, P.C.; Zhou, M.; Xiao, X.M.; Cong, Z.H.; Wu, Y.M.; Xiao, G.H.; Fei, J.; Liu, R.H. Heterochiral β-Peptide Polymers Combating Multidrug-Resistant Cancers Effectively without Inducing Drug Resistance. J. Am. Chem. Soc. 2022, 144, 7283–7294. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Kizhakkedathu, J.N.; Straus, S.K. Antimicrobial Peptides: Diversity, Mechanism of Action and Strategies to Improve the Activity and Biocompatibility In Vivo. Biomolecules 2018, 8, 4. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qian, Y.; Yang, D.; Lin, X.; Shen, C.; Zhang, J.; Xu, J.; Zhao, Y.; Zhu, L.; Kong, H.; Zhang, M.; et al. Dendrimer-Derived Mimics of Host Defense Peptides Selectively Disrupt Cancer Cell Membranes for Melanoma Therapy. Pharmaceutics 2025, 17, 361. https://doi.org/10.3390/pharmaceutics17030361
Qian Y, Yang D, Lin X, Shen C, Zhang J, Xu J, Zhao Y, Zhu L, Kong H, Zhang M, et al. Dendrimer-Derived Mimics of Host Defense Peptides Selectively Disrupt Cancer Cell Membranes for Melanoma Therapy. Pharmaceutics. 2025; 17(3):361. https://doi.org/10.3390/pharmaceutics17030361
Chicago/Turabian StyleQian, Yusheng, Danjing Yang, Xiangyu Lin, Chenyun Shen, Jieping Zhang, Jin Xu, Yan Zhao, Ling Zhu, Haoran Kong, Mingyu Zhang, and et al. 2025. "Dendrimer-Derived Mimics of Host Defense Peptides Selectively Disrupt Cancer Cell Membranes for Melanoma Therapy" Pharmaceutics 17, no. 3: 361. https://doi.org/10.3390/pharmaceutics17030361
APA StyleQian, Y., Yang, D., Lin, X., Shen, C., Zhang, J., Xu, J., Zhao, Y., Zhu, L., Kong, H., Zhang, M., Zhu, Y., Zhou, C., & He, J. (2025). Dendrimer-Derived Mimics of Host Defense Peptides Selectively Disrupt Cancer Cell Membranes for Melanoma Therapy. Pharmaceutics, 17(3), 361. https://doi.org/10.3390/pharmaceutics17030361