

Towards DFO*12—Preliminary Results of a New Chelator for the Complexation of Actinium-225

 , , , , , ,
, , , , , , 
Abstract

1. Introduction
2. Materials and Methods
2.1. Materials
2.1.1. DFT Calculations
2.1.2. Monomer and Chelator Synthesis
2.1.3. 229Th/225Ac Generator
2.1.4. Radiochemistry
2.2. Methods
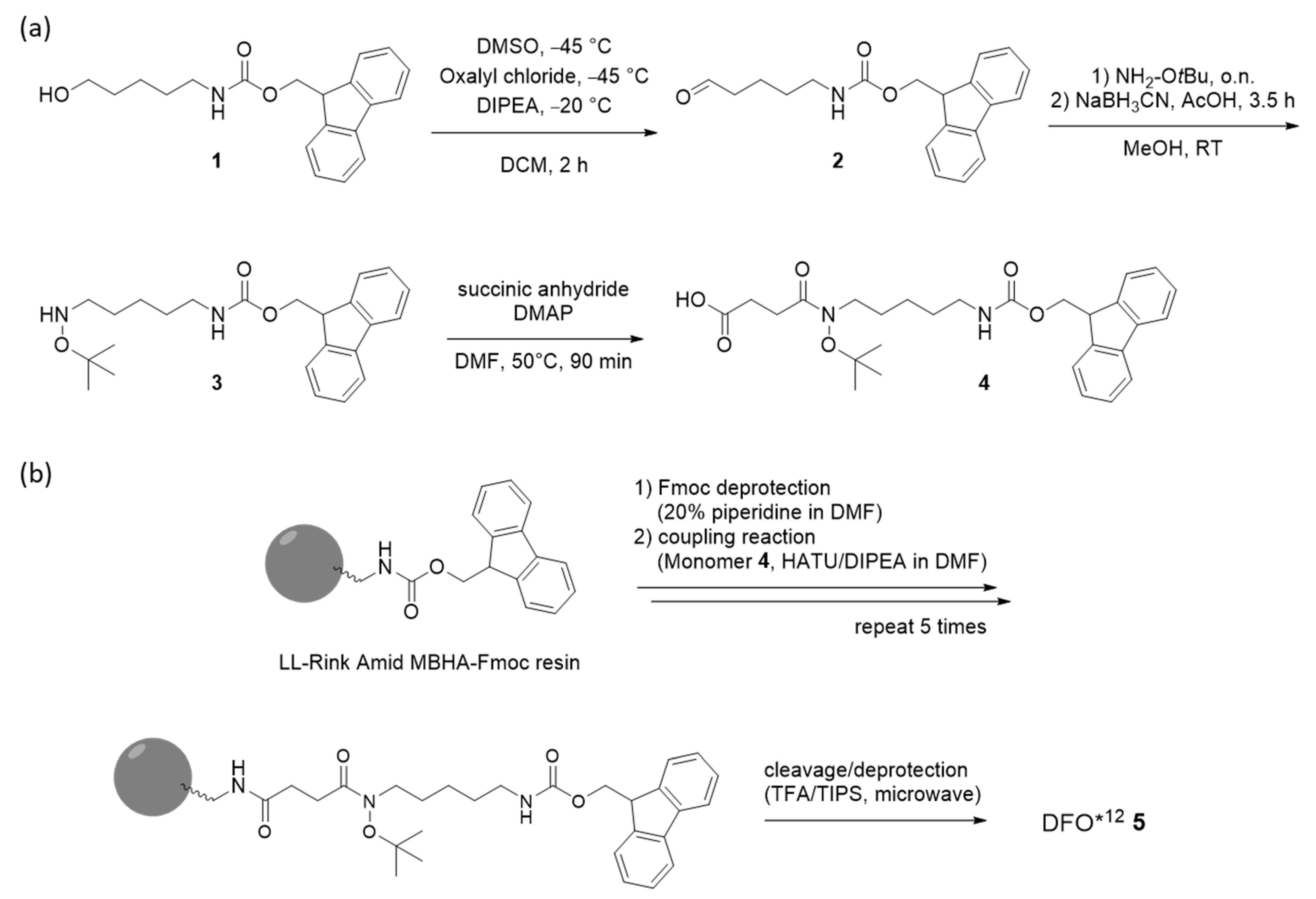
2.2.1. Monomer Synthesis
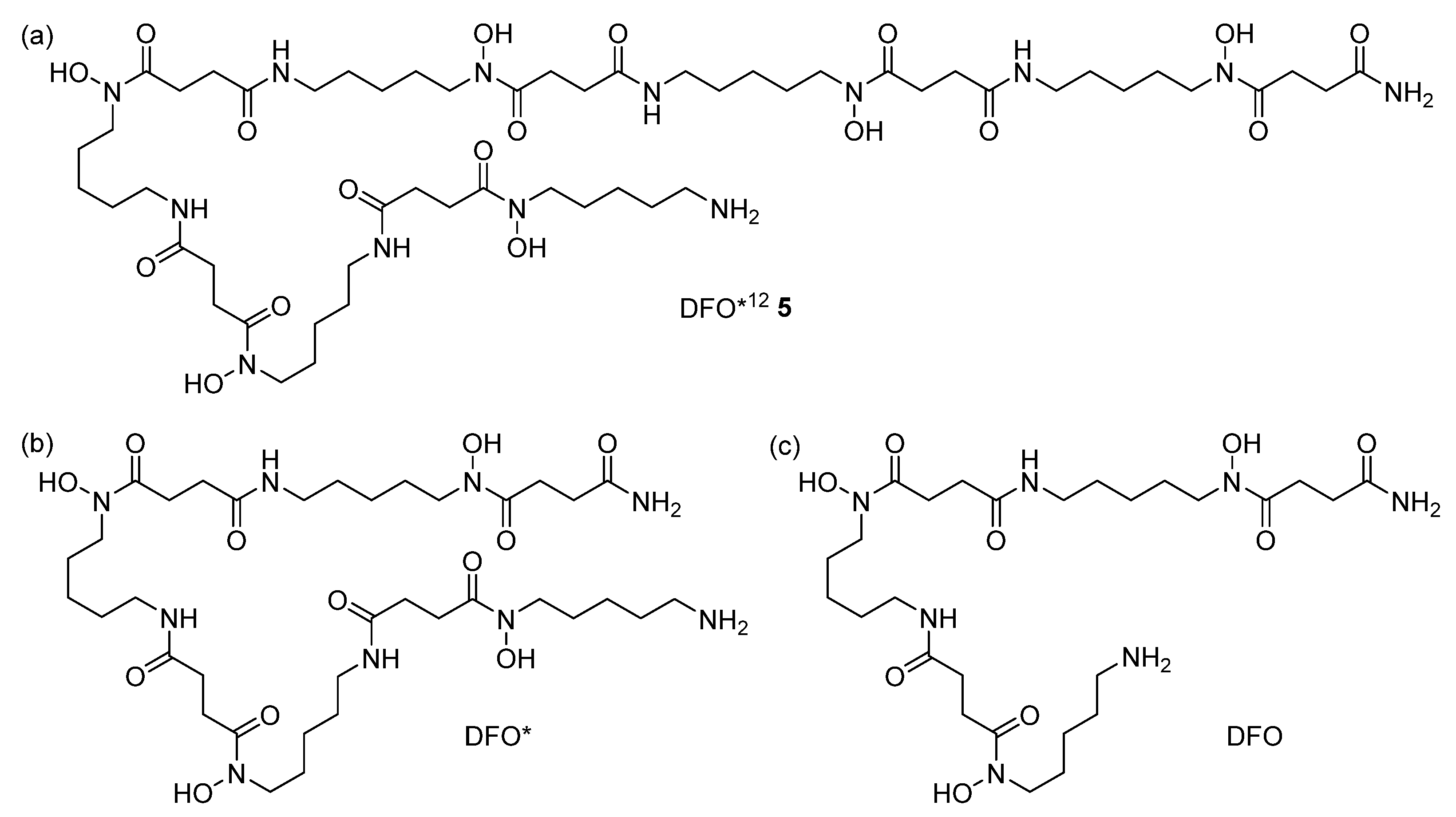
2.2.2. Chelator Synthesis
2.2.3. 229Th/225Ac Generator
2.2.4. Radiochemistry
3. Results and Discussion
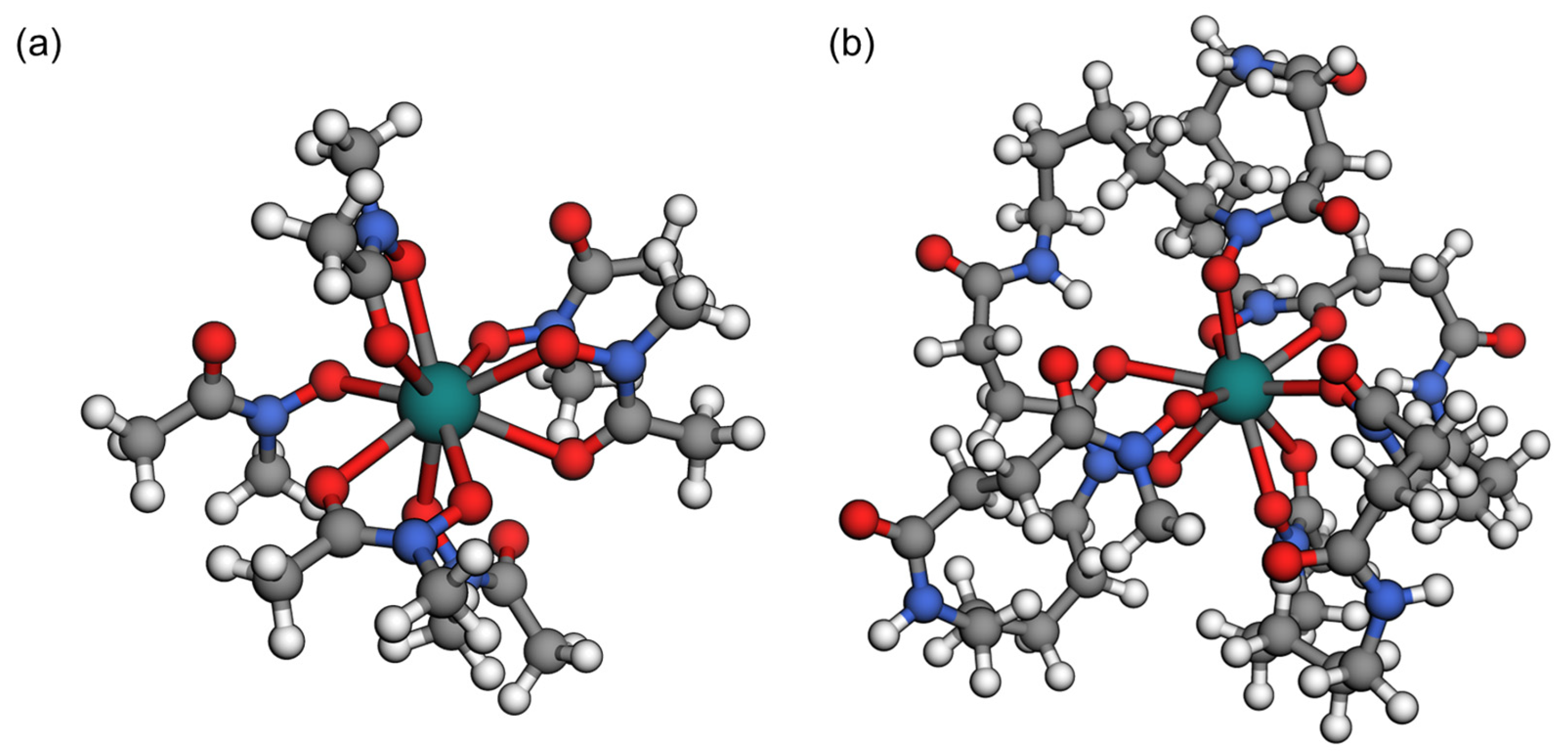
3.1. DFT Calculations
3.2. Monomer and Chelator Synthesis
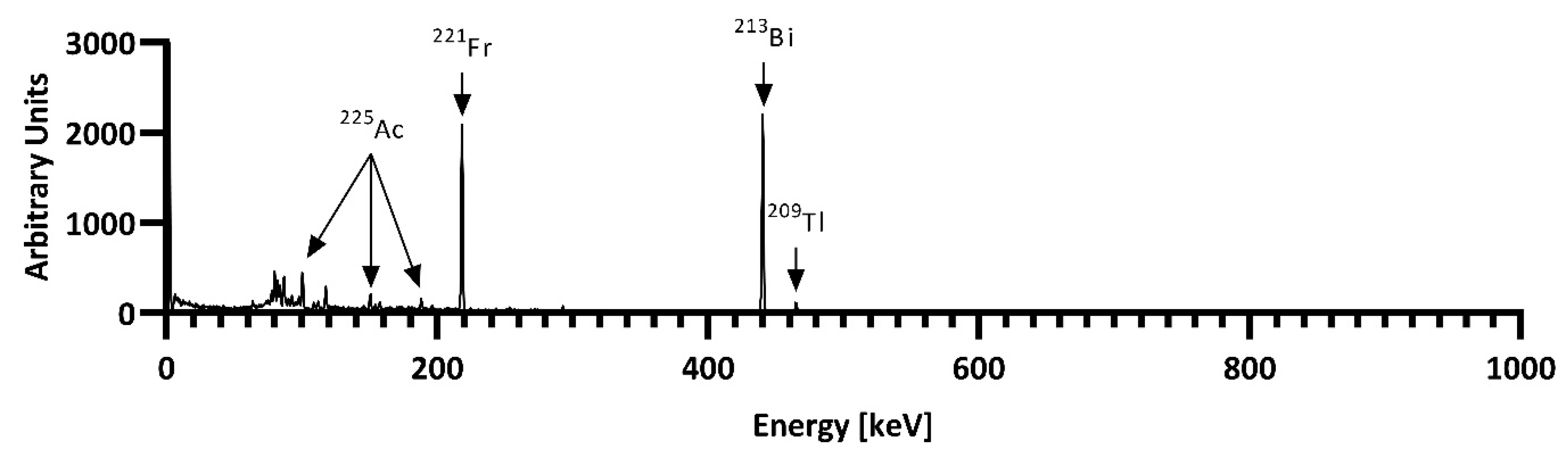
3.3. 229Th/225Ac Generator
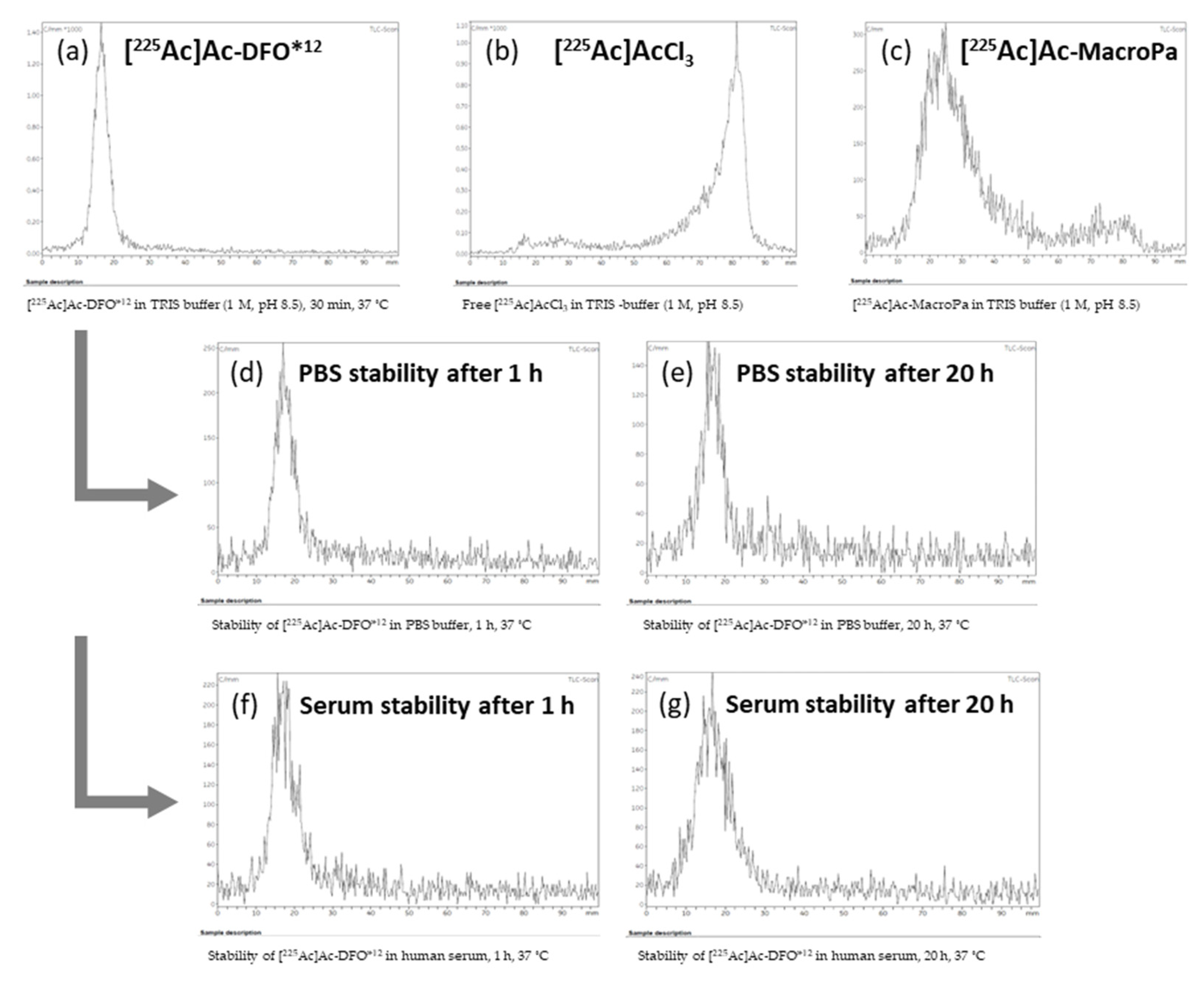
3.4. Radiochemistry
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Bn | Benzyl |
| DCM | Dichloromethane |
| DFO | Desferrioxamine B |
| DFT | Density functional theory |
| DIPEA | N,N-diisopropylethylamine |
| DMF | Dimethylformamide |
| DMSO | Dimethyl sulfoxide |
| DOTA | Dodecane tetraacetic acid |
| EDTA | Ethylenediaminetetraacetic acid |
| FA | Formic acid |
| Fmoc | Fluorenylmethoxycarbonyl |
| HATU | Hexafluorophosphate azabenzotriazole tetramethyl uronium |
| HRESI-MS | High-resolution electrospray mass spectrometry |
| MBHA | Methylbenzhydryl amine |
| NMR | Nuclear magnetic resonance spectroscopy |
| RCY | Radiochemical yield |
| Rf | Retention factor |
| RP-HPLC | Reversed-phase high-performance liquid chromatography |
| SI | Supporting information |
| SM | Starting material |
| TAT | Targeted alpha therapy |
| tBu | tert-Butyl |
| TLC | Thin-layer chromatography |
References
- Feuerecker, B.; Kratochwil, C.; Ahmadzadehfar, H.; Morgenstern, A.; Eiber, M.; Herrmann, K.; Pomykala, K.L. Clinical Translation of Targeted α-Therapy: An Evolution or a Revolution? J. Nucl. Med. 2023, 64, 685–692. [Google Scholar] [CrossRef] [PubMed]
- Jang, A.; Kendi, A.T.; Johnson, G.B.; Halfdanarson, T.R.; Sartor, O. Targeted Alpha-Particle Therapy: A Review of Current Trials. Int. J. Mol. Sci. 2023, 24, 11626. [Google Scholar] [CrossRef] [PubMed]
- Gape, P.M.D.; Schultz, M.K.; Stasiuk, G.J.; Terry, S.Y.A. Towards Effective Targeted Alpha Therapy for Neuroendocrine Tumours: A Review. Pharmaceuticals 2024, 17, 334. [Google Scholar] [CrossRef] [PubMed]
- Hurley, K.; Cao, M.; Huang, H.; Wang, Y. Targeted Alpha Therapy (TAT) with Single-Domain Antibodies (Nanobodies). Cancers 2023, 15, 3493. [Google Scholar] [CrossRef]
- Jalloul, W.; Ghizdovat, V.; Stolniceanu, C.R.; Ionescu, T.; Grierosu, I.C.; Pavaleanu, I.; Moscalu, M.; Stefanescu, C. Targeted Alpha Therapy: All We Need to Know about 225Ac’s Physical Characteristics and Production as a Potential Theranostic Radionuclide. Pharmaceuticals 2023, 16, 1679. [Google Scholar] [CrossRef]
- Mdanda, S.; Ngema, L.M.; Mdlophane, A.; Sathekge, M.M.; Zeevaart, J.R. Recent Innovations and Nano-Delivery of Actinium-225: A Narrative Review. Pharmaceutics 2023, 15, 1719. [Google Scholar] [CrossRef]
- Pouget, J.-P.; Constanzo, J. Revisiting the Radiobiology of Targeted Alpha Therapy. Front. Med. 2021, 8, 692436. [Google Scholar] [CrossRef]
- Parker, C.; Nilsson, S.; Heinrich, D.; Helle, S.I.; O’Sullivan, J.M.; Fosså, S.D.; Chodacki, A.; Wiechno, P.; Logue, J.; Seke, M.; et al. Alpha Emitter Radium-223 and Survival in Metastatic Prostate Cancer. N. Engl. J. Med. 2013, 369, 213–223. [Google Scholar] [CrossRef]
- RadioMedix and Orano Med Receive FDA Breakthrough Therapy Designation for AlphaMedixTM in Gastroenteropancreatic Neuroendocrine Tumors. Available online: https://www.oranomed.com/en/resources/news/2024/radiomedix-and-orano-med-receive-fda-breakthrough-therapy-designation-for-alphamedixtm-in-gastroenteropancreatic-neuroendocrine-tumors (accessed on 30 July 2024).
- Kratochwil, C.; Bruchertseifer, F.; Giesel, F.L.; Weis, M.; Verburg, F.A.; Mottaghy, F.; Kopka, K.; Apostolidis, C.; Haberkorn, U.; Morgenstern, A. 225Ac-PSMA-617 for PSMA-Targeted α-Radiation Therapy of Metastatic Castration-Resistant Prostate Cancer. J. Nucl. Med. 2016, 57, 1941–1944. [Google Scholar] [CrossRef]
- Kim, Y.-S.; Brechbiel, M.W. An Overview of Targeted Alpha Therapy. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2012, 33, 573–590. [Google Scholar] [CrossRef]
- Wacker, J.N.; Woods, J.J.; Rupert, P.B.; Peterson, A.; Allaire, M.; Lukens, W.W.; Gaiser, A.N.; Minasian, S.G.; Strong, R.K.; Abergel, R.J. Actinium Chelation and Crystallization in a Macromolecular Scaffold. Nat. Commun. 2024, 15, 5741. [Google Scholar] [CrossRef] [PubMed]
- Liubchenko, G.; Böning, G.; Zacherl, M.; Rumiantcev, M.; Unterrainer, L.M.; Gildehaus, F.J.; Brendel, M.; Resch, S.; Bartenstein, P.; Ziegler, S.I.; et al. Image-Based Dosimetry for [225Ac]Ac-PSMA-I&T Therapy and the Effect of Daughter-Specific Pharmacokinetics. Eur. J. Nucl. Med. Mol. Imaging 2024, 51, 2504–2514. [Google Scholar] [CrossRef] [PubMed]
- Stein, B.W.; Morgenstern, A.; Batista, E.R.; Birnbaum, E.R.; Bone, S.E.; Cary, S.K.; Ferrier, M.G.; John, K.D.; Pacheco, J.L.; Kozimor, S.A.; et al. Advancing Chelation Chemistry for Actinium and Other +3 F-Elements, Am, Cm, and La. J. Am. Chem. Soc. 2019, 141, 19404–19414. [Google Scholar] [CrossRef]
- Thiele, N.A.; Wilson, J.J. Actinium-225 for Targeted α Therapy: Coordination Chemistry and Current Chelation Approaches. Cancer Biother. Radiopharm. 2018, 33, 336–348. [Google Scholar] [CrossRef]
- Comba, P.; Jermilova, U.; Orvig, C.; Patrick, B.O.; Ramogida, C.F.; Rück, K.; Schneider, C.; Starke, M. Octadentate Picolinic Acid-Based Bispidine Ligand for Radiometal Ions. Chem. Eur. J. 2017, 23, 15945–15956. [Google Scholar] [CrossRef] [PubMed]
- Thiele, N.A.; Brown, V.; Kelly, J.M.; Amor-Coarasa, A.; Jermilova, U.; MacMillan, S.N.; Nikolopoulou, A.; Ponnala, S.; Ramogida, C.F.; Robertson, A.K.H.; et al. An Eighteen-Membered Macrocyclic Ligand for Actinium-225 Targeted Alpha Therapy. Angew. Chem. Int. Ed. 2017, 56, 14712–14717. [Google Scholar] [CrossRef]
- Kadassery, K.J.; King, A.P.; Fayn, S.; Baidoo, K.E.; MacMillan, S.N.; Escorcia, F.E.; Wilson, J.J. H2BZmacropa-NCS: A Bifunctional Chelator for Actinium-225 Targeted Alpha Therapy. Bioconjug. Chem. 2022, 33, 1222–1231. [Google Scholar] [CrossRef]
- Wharton, L.; Zhang, C.; Zeisler, J.; Rodríguez-Rodríguez, C.; Osooly, M.; Radchenko, V.; Yang, H.; Lin, K.-S.; Bénard, F.; Schaffer, P.; et al. H3TPAN-Triazole-Bn-NH2: Tripicolinate Clicked-Bifunctional Chelate for [225Ac]/[111In] Theranostics. Bioconjug. Chem. 2022, 33, 2381–2397. [Google Scholar] [CrossRef]
- Yang, H.; Wilson, J.J.; Orvig, C.; Li, Y.; Wilbur, D.S.; Ramogida, C.F.; Radchenko, V.; Schaffer, P. Harnessing α-Emitting Radionuclides for Therapy: Radiolabeling Method Review. J. Nucl. Med. 2022, 63, 5–13. [Google Scholar] [CrossRef]
- Hassan, M.; Bokhari, T.H.; Lodhi, N.A.; Khosa, M.K.; Usman, M. A Review of Recent Advancements in Actinium-225 Labeled Compounds and Biomolecules for Therapeutic Purposes. Chem. Biol. Drug Des. 2023, 102, 1276–1292. [Google Scholar] [CrossRef]
- Feiner, I.V.J.; Brandt, M.; Cowell, J.; Demuth, T.; Vugts, D.; Gasser, G.; Mindt, T.L. The Race for Hydroxamate-Based Zirconium-89 Chelators. Cancers 2021, 13, 4466. [Google Scholar] [CrossRef]
- Ferrier, M.G.; Batista, E.R.; Berg, J.M.; Birnbaum, E.R.; Cross, J.N.; Engle, J.W.; La Pierre, H.S.; Kozimor, S.A.; Lezama Pacheco, J.S.; Stein, B.W.; et al. Spectroscopic and Computational Investigation of Actinium Coordination Chemistry. Nat. Commun. 2016, 7, 12312. [Google Scholar] [CrossRef] [PubMed]
- Tomeček, J.; Li, C.; Schreckenbach, G. Actinium Coordination Chemistry: A Density Functional Theory Study with Monodentate and Bidentate Ligands. J. Comput. Chem. 2023, 44, 334–345. [Google Scholar] [CrossRef] [PubMed]
- Neese, F. Software Update: The ORCA Program System—Version 5.0. WIREs Comput. Mol. Sci. 2022, 12, e1606. [Google Scholar] [CrossRef]
- Becke, A.D. Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Perdew, J.P. Density-Functional Approximation for the Correlation Energy of the Inhomogeneous Electron Gas. Phys. Rev. B 1986, 33, 8822–8824. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Aebersold, L.E.; Wilson, A.K. Considering Density Functional Approaches for Actinide Species: The An66 Molecule Set. J. Phys. Chem. A 2021, 125, 7029–7037. [Google Scholar] [CrossRef]
- van Lenthe, E.; Baerends, E.J.; Snijders, J.G. Relativistic Total Energy Using Regular Approximations. J. Chem. Phys. 1994, 101, 9783–9792. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M. Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Pantazis, D.A.; Neese, F. All-Electron Scalar Relativistic Basis Sets for the Actinides. J. Chem. Theory Comput. 2011, 7, 677–684. [Google Scholar] [CrossRef]
- Weigend, F. Accurate Coulomb-Fitting Basis Sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef]
- Patra, M.; Bauman, A.; Mari, C.; Fischer, C.A.; Blacque, O.; Häussinger, D.; Gasser, G.; Mindt, T.L. An Octadentate Bifunctional Chelating Agent for the Development of Stable Zirconium-89 Based Molecular Imaging Probes. Chem. Commun. 2014, 50, 11523–11525. [Google Scholar] [CrossRef]
- Briand, M.; Aulsebrook, M.L.; Mindt, T.L.; Gasser, G. A Solid Phase-Assisted Approach for the Facile Synthesis of a Highly Water-Soluble Zirconium-89 Chelator for Radiopharmaceutical Development. Dalton Trans. 2017, 46, 16387–16389. [Google Scholar] [CrossRef] [PubMed]
- Brandt, M.; Cowell, J.; Aulsebrook, M.L.; Gasser, G.; Mindt, T.L. Radiolabelling of the Octadentate Chelators DFO* and oxoDFO* with Zirconium-89 and Gallium-68. JBIC J. Biol. Inorg. Chem. 2020, 25, 789–796. [Google Scholar] [CrossRef]
- Guarrochena, X.; Kronberger, J.; Tieber, M.; Ciesielski, P.; Mindt, T.L.; Feiner, I.V.J. Straightforward Synthesis of DFO*—An Octadentate Chelator for Zirconium-89. ChemMedChem 2024, 19, e202300495. [Google Scholar] [CrossRef]
- Omura, K.; Swern, D. Oxidation of Alcohols by “Activated” Dimethyl Sulfoxide. a Preparative, Steric and Mechanistic Study. Tetrahedron 1978, 34, 1651–1660. [Google Scholar] [CrossRef]
- Abdel-Magid, A.F.; Carson, K.G.; Harris, B.D.; Maryanoff, C.A.; Shah, R.D. Reductive Amination of Aldehydes and Ketones with Sodium Triacetoxyborohydride. Studies on Direct and Indirect Reductive Amination Procedures1. J. Org. Chem. 1996, 61, 3849–3862. [Google Scholar] [CrossRef]
- Zielinska, B.; Apostolidis, C.; Bruchertseifer, F.; Morgenstern, A. An Improved Method for the Production of Ac-225/Bi-213 from Th-229 for Targeted Alpha Therapy. Solvent Extr. Ion Exch. 2007, 25, 339–349. [Google Scholar] [CrossRef]
- Eichrom. 225Ac/225Ra Generator, AN-1614-11; University Lane: Lisle, IL, USA, 2018; Available online: https://www.eichrom.com/wp-content/uploads/2018/03/AN-1614_Ac-225-Generator_V11.pdf (accessed on 18 February 2025).
- Kelly, J.M.; Amor-Coarasa, A.; Sweeney, E.; Wilson, J.J.; Causey, P.W.; Babich, J.W. A Suitable Time Point for Quantifying the Radiochemical Purity of 225Ac-Labeled Radiopharmaceuticals. EJNMMI Radiopharm. Chem. 2021, 6, 38. [Google Scholar] [CrossRef] [PubMed]
- International Atomic Energy Agency. Production and Quality Control of Actinium-225 Radiopharmaceuticals; Iaea Tecdoc Series; International Atomic Energy Agency: Vienna, Austria, 2024; ISBN 978-92-0-121324-2. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Buffer | pH | Temperature (°C) | Time (h) | RCY |
|---|---|---|---|---|
| NaOAc (1.5 M) | 4.5 | 37 | 0.5 | quant. |
| NH4OAc (1 M) | 7.3 | 60 | 24.0 | - |
| HEPES (0.5 M) | 7.4 | 60 | 24.0 | - |
| TRIS (1 M) | 7.4 | 60 | 3.5 | 12% |
| TRIS (1 M) | 8.5 | 37 | 0.5 | quant. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feiner, I.V.J.; Svatunek, D.; Pressler, M.; Demuth, T.; Guarrochena, X.; Sterba, J.H.; Dorudi, S.; Pichler, C.; Denk, C.; Mindt, T.L. Towards DFO*12—Preliminary Results of a New Chelator for the Complexation of Actinium-225. Pharmaceutics 2025, 17, 320. https://doi.org/10.3390/pharmaceutics17030320
Feiner IVJ, Svatunek D, Pressler M, Demuth T, Guarrochena X, Sterba JH, Dorudi S, Pichler C, Denk C, Mindt TL. Towards DFO*12—Preliminary Results of a New Chelator for the Complexation of Actinium-225. Pharmaceutics. 2025; 17(3):320. https://doi.org/10.3390/pharmaceutics17030320
Chicago/Turabian StyleFeiner, Irene V. J., Dennis Svatunek, Martin Pressler, Tori Demuth, Xabier Guarrochena, Johannes H. Sterba, Susanne Dorudi, Clemens Pichler, Christoph Denk, and Thomas L. Mindt. 2025. "Towards DFO*12—Preliminary Results of a New Chelator for the Complexation of Actinium-225" Pharmaceutics 17, no. 3: 320. https://doi.org/10.3390/pharmaceutics17030320
APA StyleFeiner, I. V. J., Svatunek, D., Pressler, M., Demuth, T., Guarrochena, X., Sterba, J. H., Dorudi, S., Pichler, C., Denk, C., & Mindt, T. L. (2025). Towards DFO*12—Preliminary Results of a New Chelator for the Complexation of Actinium-225. Pharmaceutics, 17(3), 320. https://doi.org/10.3390/pharmaceutics17030320