
Transcriptional and Post-Transcriptional Anticholestatic Mechanisms of Obeticholic Acid in Lipopolysaccharide-Induced Cholestasis

 ,
,  , and
, and
Abstract
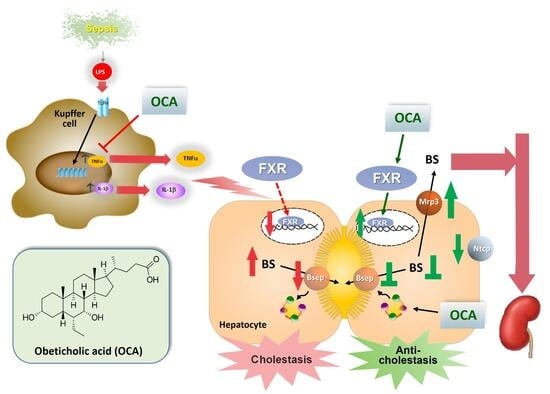
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Animals and Treatments
- LPS group: Animals were administered with OCA vehicle (saline), at the daily dose of 4.5 mL/kg of b.w, i.p., for 6 days, and with 3 single, i.p., injections of LPS (dissolved in saline), according to the following schedule: 2 LPS doses of 2.5 mg/kg/day of b.w. at 8:00 a.m. of the last 2 days, and an additional LPS dose of 5 mg/kg of b.w. at 8:00 p.m. of the last day [29].
- OCA + LPS group: Animals were injected with OCA, i.p., at the dose of 20 mg/kg of b.w., dissolved in saline solution (pH = 7.4) for 6 days; this was the minimum period of time required for OCA to reproducibly improve expression of its model target gene, Bsep. Animals were also administered 3 doses of LPS, as described above.
- Control group: Animals were injected only with OCA and LPS vehicle (saline solution).
- OCA group: Animals were injected only with OCA for 6 days, as described above.
2.3. Surgical Procedures
2.4. Bile/Plasma Samples Collection and Analytical Assessments
2.5. Mrp2 and Bsep Transport Activity Studies
2.6. Real-Time PCR Analysis of Canalicular and Basolateral Hepatocellular Transporters
2.7. Transporter Protein Expression in Liver
2.8. Immunofluorescence Detection of Bsep and Mrp2
2.9. Statistical Analysis
3. Results
3.1. Serum Parameters of Inflammatory Cholestasis
3.2. Biliary Parameters of Cholestasis
3.3. Bsep and Mrp2 Transport Function
3.4. Canalicular and Basolateral Hepatocellular Transporter Gene Expression
3.5. Canalicular and Basolateral Transporter Protein Expression
3.6. Bsep and Mrp2 Localization by Immunofluorescence Followed by Confocal Microscopy
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ALP | Alkaline phosphatase |
| BF | Bile flow |
| BS | Bile salt |
| Bsep | Bile salt export pump |
| BSP | Bromosulfophthalein |
| FXR | Farsenoid X receptor |
| GSH | Glutathione |
| LPS | Lipopolysaccharides |
| Mrp2 | Multidrug resistance-associated protein 2 |
| Mrp3 | Multidrug resistance-associated protein 3 |
| Ntcp | Na+/taurocholate cotransporting polypeptide |
| Oatp2, | Organic anion-transporting polypeptide |
| RXRα, | 9-cis-retinoic acid receptor-α |
| TC | Taurocholate |
References
- Roma, M.G.; Crocenzi, F.A.; Sanchez Pozzi, E.A. Hepatocellular transport in acquired cholestasis: New insights into functional, regulatory and therapeutic aspects. Clin. Sci. 2008, 114, 567–588. [Google Scholar] [CrossRef]
- Trauner, M.; Fickert, P.; Stauber, R.E. Inflammation-induced cholestasis. J. Gastroenterol. Hepatol. 1999, 14, 946–959. [Google Scholar] [CrossRef] [PubMed]
- Bhogal, H.K.; Sanyal, A.J. The molecular pathogenesis of cholestasis in sepsis. Front. Biosci. (Elite Ed.) 2013, 5, 87–96. [Google Scholar] [CrossRef]
- Rodriguez-Garay, E.A. Cholestasis: Human disease and experimental animal models. Ann. Hepatol. 2003, 2, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Cherrington, N.J.; Slitt, A.L.; Li, N.; Klaassen, C.D. Lipopolysaccharide-mediated regulation of hepatic transporter mRNA levels in rats. Drug Metab. Dispos. 2004, 32, 734–741. [Google Scholar] [CrossRef] [PubMed]
- Donner, M.G.; Warskulat, U.; Saha, N.; Haussinger, D. Enhanced expression of basolateral multidrug resistance protein isoforms Mrp3 and Mrp5 in rat liver by LPS. Biol. Chem. 2004, 385, 331–339. [Google Scholar] [CrossRef]
- Kosters, A.; Karpen, S.J. Bile acid transporters in health and disease. Xenobiotica 2008, 38, 1043–1071. [Google Scholar] [CrossRef]
- Arrese, M.; Karpen, S.J. Nuclear receptors, inflammation, and liver disease: Insights for cholestatic and fatty liver diseases. Clin. Pharmacol. Ther. 2010, 87, 473–478. [Google Scholar] [CrossRef]
- Geier, A.; Wagner, M.; Dietrich, C.G.; Trauner, M. Principles of hepatic organic anion transporter regulation during cholestasis, inflammation and liver regeneration. Biochim. Biophys. Acta 2007, 1773, 283–308. [Google Scholar] [CrossRef]
- Kosters, A.; Karpen, S.J. The role of inflammation in cholestasis: Clinical and basic aspects. Semin. Liver Dis. 2010, 30, 186–194. [Google Scholar] [CrossRef]
- Liu, N.; Meng, Z.; Lou, G.; Zhou, W.; Wang, X.; Zhang, Y.; Zhang, L.; Liu, X.; Yen, Y.; Lai, L.; et al. Hepatocarcinogenesis in FXR-/-mice mimics human HCC progression that operates through HNF1alpha regulation of FXR expression. Mol. Endocrinol. 2012, 26, 775–785. [Google Scholar] [CrossRef]
- Wang, Y.D.; Chen, W.D.; Wang, M.; Yu, D.; Forman, B.M.; Huang, W. Farnesoid X receptor antagonizes nuclear factor kappaB in hepatic inflammatory response. Hepatology 2008, 48, 1632–1643. [Google Scholar] [CrossRef]
- Roma, M.G.; Barosso, I.R.; Miszczuk, G.S.; Crocenzi, F.A.; Sánchez Pozzi, E.J. Dynamic localization of hepatocellular transporters: Role in biliary excretion and impairment in cholestasis. Curr. Med. Chem. 2019, 26, 1113–1154. [Google Scholar] [CrossRef]
- Elferink, M.G.; Olinga, P.; Draaisma, A.L.; Merema, M.T.; Faber, K.N.; Slooff, M.J.; Meijer, D.K.; Groothuis, G.M.M. LPS-induced downregulation of MRP2 and BSEP in human liver is due to a posttranscriptional process. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 287, G1008–G1016. [Google Scholar] [CrossRef]
- Yang, Q.L.; Yang, F.; Gong, J.T.; Tang, X.W.; Wang, G.Y.; Wang, Z.T.; Yang, L. Sweroside ameliorates alpha-naphthylisothiocyanate-induced cholestatic liver injury in mice by regulating bile acids and suppressing pro-inflammatory responses. Acta Pharmacol. Sin. 2016, 37, 1218–1228. [Google Scholar] [CrossRef] [PubMed]
- Fiorucci, S.; Clerici, C.; Antonelli, E.; Orlandi, S.; Goodwin, B.; Sadeghpour, B.M.; Sabatino, G.; Russo, G.; Castellani, D.; Willson, T.M.; et al. Protective effects of 6-ethyl chenodeoxycholic acid, a farnesoid X receptor ligand, in estrogen-induced cholestasis. J. Pharmacol. Exp. Ther. 2005, 313, 604–612. [Google Scholar] [CrossRef]
- Pellicciari, R.; Fiorucci, S.; Camaioni, E.; Clerici, C.; Costantino, G.; Maloney, P.R.; Morelli, A.; Parks, D.J.; Willson, T.M. 6alpha-ethyl-chenodeoxycholic acid (6-ECDCA), a potent and selective FXR agonist endowed with anticholestatic activity. J. Med. Chem. 2002, 45, 3569–3572. [Google Scholar] [CrossRef]
- Pellicciari, R.; Costantino, G.; Camaioni, E.; Sadeghpour, B.M.; Entrena, A.; Willson, T.M.; Fiorucci, S.; Clerici, C.; Gioiello, A. Bile acid derivatives as ligands of the farnesoid X receptor. Synthesis, evaluation, and structure-activity relationship of a series of body and side chain modified analogues of chenodeoxycholic acid. J. Med. Chem. 2004, 47, 4559–4569. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Wang, L.; Woo, E.; He, X.; Yang, G.; Bowlus, C.; Leung, P.S.C.; Gershwin, M.E. Clinical management of primary biliary cholangitis-strategies and evolving trends. Clin. Rev. Allergy Immunol. 2020, 59, 175–194. [Google Scholar] [CrossRef]
- Kjaergaard, K.; Frisch, K.; Sorensen, M.; Munk, O.L.; Hofmann, A.F.; Horsager, J.; Schacht, A.C.; Erickson, M.; Shapiro, D.; Keiding, S. Obeticholic acid improves hepatic bile acid excretion in patients with primary biliary cholangitis. J. Hepatol. 2021, 74, 58–65. [Google Scholar] [CrossRef] [PubMed]
- van Golen, R.F.; Olthof, P.B.; Lionarons, D.A.; Reiniers, M.J.; Alles, L.K.; Uz, Z.; de Haan, L.; Ergin, B.; de Waart, D.R.; Maas, A.; et al. FXR agonist obeticholic acid induces liver growth but exacerbates biliary injury in rats with obstructive cholestasis. Sci. Rep. 2018, 8, 16529. [Google Scholar] [CrossRef]
- Renga, B.; Mencarelli, A.; Migliorati, M.; Cipriani, S.; D’Amore, C.; Distrutti, E.; Fiorucci, S. SHP-dependent and -independent induction of peroxisome proliferator-activated receptor-gamma by the bile acid sensor farnesoid X receptor counter-regulates the pro-inflammatory phenotype of liver myofibroblasts. Inflamm. Res. 2011, 60, 577–587. [Google Scholar] [CrossRef]
- Gao, X.; Fu, T.; Wang, C.; Ning, C.; Kong, Y.; Liu, Z.; Sun, H.; Ma, X.; Liu, K.; Meng, Q. Computational discovery and experimental verification of farnesoid X receptor agonist auraptene to protect against cholestatic liver injury. Biochem. Pharmacol. 2017, 146, 127–138. [Google Scholar] [CrossRef]
- Koelfat, K.V.K.; Visschers, R.G.J.; Hodin, C.; de Waart, D.R.; van Gemert, W.G.; Cleutjens, J.P.M.; Gijbels, M.J.; Shiri-Sverdlov, R.; Mookerjee, R.P.; Lenaerts, K.; et al. FXR agonism protects against liver injury in a rat model of intestinal failure-associated liver disease. J. Clin. Transl. Res. 2018, 3, 318–327. [Google Scholar] [CrossRef]
- Xiong, X.; Ren, Y.; Cui, Y.; Li, R.; Wang, C.; Zhang, Y. Obeticholic acid protects mice against lipopolysaccharide-induced liver injury and inflammation. Biomed. Pharmacother. 2017, 96, 1292–1298. [Google Scholar] [CrossRef]
- Koch, A.; Horn, A.; Duckers, H.; Yagmur, E.; Sanson, E.; Bruensing, J.; Buendgens, L.; Voigt, S.; Trautwein, C.; Tacke, F. Increased liver stiffness denotes hepatic dysfunction and mortality risk in critically ill non-cirrhotic patients at a medical ICU. Crit. Care 2011, 15, R266. [Google Scholar] [CrossRef]
- Recknagel, P.; Gonnert, F.A.; Westermann, M.; Lambeck, S.; Lupp, A.; Rudiger, A.; Dyson, A.; Carré, J.E.; Kortgen, A.; Krafft, C.; et al. Liver dysfunction and phosphatidylinositol-3-kinase signalling in early sepsis: Experimental studies in rodent models of peritonitis. PLoS Med. 2012, 9, e1001338. [Google Scholar] [CrossRef] [PubMed]
- Hao, H.; Cao, L.; Jiang, C.; Che, Y.; Zhang, S.; Takahashi, S.; Wang, G.; Gonzalez, F.J. Farnesoid X receptor regulation of the NLRP3 inflammasome underlies cholestasis-associated sepsis. Cell. Metab. 2017, 25, 856–867.e5. [Google Scholar] [CrossRef]
- Razori, M.V.; Martin, P.L.; Maidagan, P.M.; Barosso, I.R.; Ciriaci, N.; Andermatten, R.B.; Sanchez Pozzi, E.J.; Basiglio, C.L.; Ruiz, M.L.; Roma, M.G. Spironolactone ameliorates lipopolysaccharide-induced cholestasis in rats by improving Mrp2 function: Role of transcriptional and post-transcriptional mechanisms. Life Sci. 2020, 259, 118352. [Google Scholar] [CrossRef] [PubMed]
- Tietze, F. Enzymic method for quantitative determination of nanogram amounts of total and oxidized glutathione: Applications to mammalian blood and other tissues. Anal. Biochem. 1969, 27, 502–522. [Google Scholar] [CrossRef] [PubMed]
- Griffith, O.W. Determination of glutathione and glutathione disulfide using glutathione reductase and 2-vinylpyridine. Anal. Biochem. 1980, 106, 207–212. [Google Scholar] [CrossRef]
- Gerloff, T.; Stieger, B.; Hagenbuch, B.; Madon, J.; Landmann, L.; Roth, J.; Hofmann, A.F.; Meier, P.J. The sister of P-glycoprotein represents the canalicular bile salt export pump of mammalian liver. J. Biol. Chem. 1998, 273, 10046–10050. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Konig, J.; Keppler, D. Vectorial transport by double-transfected cells expressing the human uptake transporter SLC21A8 and the apical export pump ABCC2. Mol. Pharmacol. 2001, 60, 934–943. [Google Scholar] [CrossRef]
- Yokota, M.; Iga, T.; Sugiyama, Y.; Suyama, A.; Awazu, S.; Hanano, M. Comparative hepatic transport of sulfobromophthalein and its glutathione conjugate in rats. J. Pharmacobiodyn. 1981, 4, 287–293. [Google Scholar] [CrossRef]
- Razori, M.V.; Maidagan, P.M.; Ciriaci, N.; Andermatten, R.B.; Barosso, I.R.; Martin, P.L.; Basiglio, C.L.; Sanchez Pozzi, E.J.; Ruiz, M.L.; Roma, M.G. Anticholestatic mechanisms of ursodeoxycholic acid in lipopolysaccharide-induced cholestasis. Biochem. Pharmacol. 2019, 168, 48–56. [Google Scholar] [CrossRef]
- Ruiz, M.L.; Villanueva, S.S.; Luquita, M.G.; Ikushiro, S.; Mottino, A.D.; Catania, V.A. Beneficial effect of spironolactone administration on ethynylestradiol-induced cholestasis in the rat: Involvement of up-regulation of multidrug resistance-associated protein 2. Drug Metab. Dispos. 2007, 35, 2060–2066. [Google Scholar] [CrossRef]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef]
- Kubitz, R.; Helmer, A.; Haussinger, D. Biliary transport systems: Short-term regulation. Methods Enzymol. 2005, 400, 542–557. [Google Scholar] [CrossRef] [PubMed]
- Crocenzi, F.A.; Mottino, A.D.; Cao, J.; Veggi, L.M.; Sánchez Pozzi, E.J.; Vore, M.; Coleman, R.; Roma, M.G. Estradiol-17beta-D-glucuronide induces endocytic internalization of Bsep in rats. Am. J. Physiol. Gastrointest. Liver Physiol. 2003, 285, G449–G459. [Google Scholar] [CrossRef] [PubMed]
- Charan, J.; Kantharia, N.D. How to calculate sample size in animal studies? J. Pharmacol. Pharmacother. 2013, 4, 303–306. [Google Scholar] [CrossRef]
- Moss, D.W. Physicochemical and pathophysiological factors in the release of membrane-bound alkaline phosphatase from cells. Clin. Chim. Acta 1997, 257, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Hirohashi, T.; Suzuki, H.; Takikawa, H.; Sugiyama, Y. ATP-dependent transport of bile salts by rat multidrug resistance-associated protein 3 (Mrp3). J. Biol. Chem. 2000, 275, 2905–2910. [Google Scholar] [CrossRef]
- Denk, G.U.; Soroka, C.J.; Takeyama, Y.; Chen, W.S.; Schuetz, J.D.; Boyer, J.L. Multidrug resistance-associated protein 4 is up-regulated in liver but down-regulated in kidney in obstructive cholestasis in the rat. J. Hepatol. 2004, 40, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Whitehead, M.W.; Hainsworth, I.; Kingham, J.G. The causes of obvious jaundice in South West Wales: Perceptions versus reality. Gut 2001, 48, 409–413. [Google Scholar] [CrossRef]
- Peng, M.; Deng, F.; Qi, D.; Hu, Z.; Zhang, L. The Hyperbilirubinemia and Potential Predictors Influence on Long-Term Outcomes in Sepsis: A Population-Based Propensity Score-Matched Study. Front. Med. 2021, 8, 713917. [Google Scholar] [CrossRef]
- Geier, A.; Fickert, P.; Trauner, M. Mechanisms of disease: Mechanisms and clinical implications of cholestasis in sepsis. Nat. Clin. Pract. Gastroenterol. Hepatol. 2006, 3, 574–585. [Google Scholar] [CrossRef]
- Hatoff, D.E.; Hardison, W.G. Bile acids modify alkaline phosphatase induction and bile secretion pressure after bile duct obstruction in the rat. Gastroenterology 1981, 80, 666–672. [Google Scholar] [CrossRef]
- Cai, S.Y.; Boyer, J.L. The role of bile acids in cholestatic liver injury. Ann. Transl. Med. 2021, 9, 737. [Google Scholar] [CrossRef]
- Akita, H.; Suzuki, H.; Hirohashi, T.; Takikawa, H.; Sugiyama, Y. Transport activity of human MRP3 expressed in Sf9 cells: Comparative studies with rat MRP3. Pharm. Res. 2002, 19, 34–41. [Google Scholar] [CrossRef]
- Guo, H.L.; Hassan, H.M.; Zhang, Y.; Dong, S.Z.; Ding, P.P.; Wang, T.; Sun, L.X.; Zhang, L.-Y.; Jiang, Z.Z. Pyrazinamide induced rat cholestatic liver injury through inhibition of FXR regulatory effect on bile acid synthesis and transport. Toxicol. Sci. 2016, 152, 417–428. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ananthanarayanan, M.; Balasubramanian, N.; Makishima, M.; Mangelsdorf, D.J.; Suchy, F.J. Human bile salt export pump promoter is transactivated by the farnesoid X receptor/bile acid receptor. J. Biol. Chem. 2001, 276, 28857–28865. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Lo, J.L.; Huang, L.; Zhao, A.; Metzger, E.; Adams, A.; Meink, P.T.; Wright, S.D.; Cui, J. Lithocholic acid decreases expression of bile salt export pump through farnesoid X receptor antagonist activity. J. Biol. Chem. 2002, 277, 31441–31447. [Google Scholar] [CrossRef]
- de Haan, L.R.; Verheij, J.; van Golen, R.F.; Horneffer-van der Sluis, V.; Lewis, M.R.; Beuers, U.H.W.; van Gulik, T.M.; Olde Damink, S.W.M.; Schaap, F.G.; Heger, M.; et al. Unaltered liver regeneration in post-cholestatic rats treated with the FXR agonist obeticholic acid. Biomolecules 2021, 11, 260. [Google Scholar] [CrossRef]
- Ijssennagger, N.; Janssen, A.W.F.; Milona, A.; Ramos Pittol, J.M.; Hollman, D.A.A.; Mokry, M.; Betzel, B.; Berends, F.J.; Janssen, I.M.; van Mil, S.W.C.; et al. Gene expression profiling in human precision cut liver slices in response to the FXR agonist obeticholic acid. J. Hepatol. 2016, 64, 1158–1166. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, G.; Cheung, A.K.; Piquette-Miller, M. Inflammatory cytokines, but not bile acids, regulate expression of murine hepatic anion transporters in endotoxemia. J. Pharmacol. Exp. Ther. 2002, 303, 273–281. [Google Scholar] [CrossRef]
- Beigneux, A.P.; Moser, A.H.; Shigenaga, J.K.; Grunfeld, C.; Feingold, K.R. The acute phase response is associated with retinoid X receptor repression in rodent liver. J. Biol. Chem. 2000, 275, 16390–16399. [Google Scholar] [CrossRef] [PubMed]
- Kong, B.; Zhu, Y.; Li, G.; Williams, J.A.; Buckley, K.; Tawfik, O.; Luyendyk, J.P.; Guo, G.L. Mice with hepatocyte-specific FXR deficiency are resistant to spontaneous but susceptible to cholic acid-induced hepatocarcinogenesis. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 310, G295–G302. [Google Scholar] [CrossRef]
- Balasubramaniyan, N.; Luo, Y.; Sun, A.Q.; Suchy, F.J. SUMOylation of the farnesoid X receptor (FXR) regulates the expression of FXR target genes. J. Biol. Chem. 2013, 288, 13850–13862. [Google Scholar] [CrossRef]
- Vavassori, P.; Mencarelli, A.; Renga, B.; Distrutti, E.; Fiorucci, S. The bile acid receptor FXR is a modulator of intestinal innate immunity. J. Immunol. 2009, 183, 6251–6261. [Google Scholar] [CrossRef]
- Geier, A.; Dietrich, C.G.; Voigt, S.; Ananthanarayanan, M.; Lammert, F.; Schmitz, A.; Trauner, M.; Wasmuth, H.E.; Boraschi, D.; Natarajan, B.; et al. Cytokine-dependent regulation of hepatic organic anion transporter gene transactivators in mouse liver. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 289, G831–G841. [Google Scholar] [CrossRef]
- Kim, M.S.; Shigenaga, J.; Moser, A.; Feingold, K.; Grunfeld, C. Repression of farnesoid X receptor during the acute phase response. J. Biol. Chem. 2003, 278, 8988–8995. [Google Scholar] [CrossRef]
- Wang, Y.D.; Chen, W.D.; Li, C.; Guo, C.; Li, Y.; Qi, H.; Shen, H.; Kong, J.; Long, X.; Yuan, F.; et al. Farnesoid X receptor antagonizes JNK signaling pathway in liver carcinogenesis by activating SOD3. Mol. Endocrinol. 2015, 29, 322–331. [Google Scholar] [CrossRef]
- Zhang, Y.; Jackson, J.P.; St Claire, R.L., 3rd; Freeman, K.; Brouwer, K.R.; Edwards, J.E. Obeticholic acid, a selective farnesoid X receptor agonist, regulates bile acid homeostasis in sandwich-cultured human hepatocytes. Pharmacol. Res. Perspect. 2017, 5, e00329. [Google Scholar] [CrossRef]
- Hayashi, H.; Sugiyama, Y. 4-phenylbutyrate enhances the cell surface expression and the transport capacity of wild-type and mutated bile salt export pumps. Hepatology 2007, 45, 1506–1516. [Google Scholar] [CrossRef] [PubMed]
- Aida, K.; Hayashi, H.; Inamura, K.; Mizuno, T.; Sugiyama, Y. Differential roles of ubiquitination in the degradation mechanism of cell surface-resident bile salt export pump and multidrug resistance-associated protein 2. Mol. Pharmacol. 2014, 85, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Saadane, A.; Delautier, D.; Leboucher, J.; Kharbajou, M.; Feldmann, G.; Lardeux, B.; Bleiberg-Daniel, F. Stimulation of liver RNA and protein breakdown in endotoxemic rats: Role of glucocorticoids. Shock 1999, 11, 429–435. [Google Scholar]
- Borst, P.; de Wolf, C.; van de Wetering, K. Multidrug resistance-associated proteins 3, 4, and 5. Pflug. Arch. 2007, 453, 661–673. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Hill, C.E. Nifedipine modulation of biliary GSH and GSSG/conjugate efflux in normal and regenerating rat liver. Am. J. Physiol. Gastrointest. Liver. Physiol. 2001, 281, G85–G94. [Google Scholar] [CrossRef] [PubMed]
- Lauterburg, B.H.; Smith, C.V.; Hughes, H.; Mitchell, J.R. Biliary excretion of glutathione and glutathione disulfide in the rat. Regulation and response to oxidative stress. J. Clin. Investig. 1984, 73, 124–133. [Google Scholar] [CrossRef]
- Sakaguchi, S.; Furusawa, S. Oxidative stress and septic shock: Metabolic aspects of oxygen-derived free radicals generated in the liver during endotoxemia. FEMS Immunol. Med. Microbiol. 2006, 47, 167–177. [Google Scholar] [CrossRef]
- Nomoto, M.; Miyata, M.; Yin, S.; Kurata, Y.; Shimada, M.; Yoshinari, K.; Gonzalez, F.J.; Suzuki, K.; Shibasaki, S.; Kurosawa, T.; et al. Bile acid-induced elevated oxidative stress in the absence of farnesoid X receptor. Biol. Pharm. Bull. 2009, 32, 172–178. [Google Scholar] [CrossRef]
- Moseley, R.H.; Wang, W.; Takeda, H.; Lown, K.; Shick, L.; Ananthanarayanan, M.; Suchy, F.J. Effect of endotoxin on bile acid transport in rat liver: A potential model for sepsis-associated cholestasis. Am. J. Physiol. 1996, 271, G137–G146. [Google Scholar] [CrossRef]
- Green, R.M.; Beier, D.; Gollan, J.L. Regulation of hepatocyte bile salt transporters by endotoxin and inflammatory cytokines in rodents. Gastroenterology 1996, 111, 193–198. [Google Scholar] [CrossRef]
- Wagner, M.; Zollner, G.; Trauner, M. Nuclear receptor regulation of the adaptive response of bile acid transporters in cholestasis. Semin. Liver Dis. 2010, 30, 160–177. [Google Scholar] [CrossRef]
- Denson, L.A.; Sturm, E.; Echevarria, W.; Zimmerman, T.L.; Makishima, M.; Mangelsdorf, D.J.; Karpen, S.J. The orphan nuclear receptor, shp, mediates bile acid-induced inhibition of the rat bile acid transporter, ntcp. Gastroenterology 2001, 121, 140–147. [Google Scholar] [CrossRef]
- Zinchuk, V.; Zinchuk, O.; Okada, T. Experimental LPS-induced cholestasis alters subcellular distribution and affects colocalization of Mrp2 and Bsep proteins: A quantitative colocalization study. Microsc. Res. Tech. 2005, 67, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Vos, T.A.; Hooiveld, G.J.; Koning, H.; Childs, S.; Meijer, D.K.; Moshage, H.; Jansen, P.L.; Müller, M. Up-regulation of the multidrug resistance genes, Mrp1 and Mdr1b, and down-regulation of the organic anion transporter, Mrp2, and the bile salt transporter, Spgp, in endotoxemic rat liver. Hepatology 1998, 28, 1637–1644. [Google Scholar] [CrossRef] [PubMed]
- Miszczuk, G.S.; Barosso, I.R.; Larocca, M.C.; Marrone, J.; Marinelli, R.A.; Boaglio, A.C.; Sanchez Pozzi, E.J.; Roma, M.G.; Crocenzi, F.A. Mechanisms of canalicular transporter endocytosis in the cholestatic rat liver. Biochim. Biophys-Acta Mol. Basis Dis. 2018, 1864, 1072–1085. [Google Scholar] [CrossRef] [PubMed]
- Crocenzi, F.A.; Mottino, A.D.; Sanchez Pozzi, E.J.; Pellegrino, J.M.; Rodriguez Garay, E.A.; Milkiewicz, P.; Vore, M.; Coleman, R.; Roma, M.G. Impaired localisation and transport function of canalicular Bsep in taurolithocholate induced cholestasis in the rat. Gut 2003, 52, 1170–1177. [Google Scholar] [CrossRef][Green Version]
- Kurz, A.K.; Graf, D.; Schmitt, M.; Vom Dahl, S.; Haussinger, D. Tauroursodesoxycholate-induced choleresis involves p38(MAPK) activation and translocation of the bile salt export pump in rats. Gastroenterology 2001, 121, 407–419. [Google Scholar] [CrossRef]
- Schliess, F.; Kurz, A.K.; vom Dahl, S.; Haussinger, D. Mitogen-activated protein kinases mediate the stimulation of bile acid secretion by tauroursodeoxycholate in rat liver. Gastroenterology 1997, 113, 1306–1314. [Google Scholar] [CrossRef]
- Graf, D.; Kurz, A.K.; Reinehr, R.; Fischer, R.; Kircheis, G.; Haussinger, D. Prevention of bile acid-induced apoptosis by betaine in rat liver. Hepatology 2002, 36, 829–839. [Google Scholar] [CrossRef]
- Kubitz, R.; Wettstein, M.; Warskulat, U.; Haussinger, D. Regulation of the multidrug resistance protein 2 in the rat liver by lipopolysaccharide and dexamethasone. Gastroenterology 1999, 116, 401–410. [Google Scholar] [CrossRef]
- Nolan, J.P. Intestinal endotoxins as mediators of hepatic injury--an idea whose time has come again. Hepatology 1989, 10, 887–891. [Google Scholar] [CrossRef] [PubMed]
- Nolan, J.P. Endotoxin, reticuloendothelial function, and liver injury. Hepatology 1981, 1, 458–465. [Google Scholar] [CrossRef]
- Kessoku, T.; Kobayashi, T.; Imajo, K.; Tanaka, K.; Yamamoto, A.; Takahashi, K.; Kasai, Y.; Ozaki, A.; Iwaki, M.; Nogami, A.; et al. Endotoxins and non-alcoholic fatty liver disease. Front. Endocrinol. 2021, 12, 770986. [Google Scholar] [CrossRef]
- Latham, P.S.; Menkes, E.; Phillips, M.J.; Jeejeebhoy, K.N. Hyperalimentation-associated jaundice: An example of a serum factor inducing cholestasis in rats. Am. J. Clin. Nutr. 1985, 41, 61–65. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gen Target | Primer | Primer Sequence (5′ ⟶ 3′) |
|---|---|---|
| Bsep | Forward | CCGAAGGCTCAGGGTATTGG |
| Reverse | ATCAGGTGACATGGTGGCAG | |
| Mrp2 | Forward | ACCTTCCACGTAGTGATCCT |
| Reverse | ACCTGCTAAGATGGACGGTC | |
| Mrp3 | Forward | GTGCTGAAGAATTTGACTCTG |
| Reverse | GACCAGGACCCGGTTGTAGTC | |
| Mrp4 | Forward | CTGCGGTCACAGTCCTCTTT |
| Reverse | GTGCAGAGTCTGGGAAGCAT | |
| Ntcp | Forward | TCCTTCCCCTAATGGCCTGA |
| Reverse | CGTCGACGTTCGTTCCTTTT | |
| Oatp2 | Forward | TCCTCTGCATGTGCAAAACTTC |
| Reverse | TGGCACACTCTGAAGAGTCTAAT | |
| 18s rRNA | Forward | GTAACCCGTTGAACCCCATT |
| Reverse | CCATCCAATCGGTAGTAGCG |
| Control | OCA | LPS | OCA + LPS | |
|---|---|---|---|---|
| ALP (U/L) | 170 ± 8 | 146 ± 7 * | 368 ± 52 * | 183 ± 18 # |
| IL-1β (pg/mL) | 26 ± 24 | 36 ± 22 | 860 ± 153 * | 266 ± 56 *# |
| TNF-α (pg/mL) | 21 ± 3 | 41 ± 8 | 52 ± 8 * | 13 ± 3 # |
| Biliary Parameters | Control | OCA | LPS | OCA + LPS |
|---|---|---|---|---|
| Bile flow (μL/min per g liver) | 3.3 ± 0.2 | 3.2 ± 0.1 | 2.3 ± 0.1 * | 2.5 ± 0.1 * |
| Total BS output (nmol/min per g liver) | 45 ± 6 | 13 ± 2 * | 25 ± 3 * | 7 ± 1 *# |
| Total GSH output (nmol/min per g liver) | 1534 ± 171 | 1682 ± 230 | 578 ± 47 * | 787 ± 50 *# |
| mRNA Levels (% of Mean Control Values) | Control | OCA | LPS | OCA + LPS |
|---|---|---|---|---|
| Bsep | 100 ± 7 | 112 ± 13 | 40 ± 9 * | 79 ± 19 # |
| Mrp2 | 100 ± 14 | 132 ± 9 | 47 ± 4 * | 71 ± 5 # |
| Mrp3 | 100 ± 10 | 215 ± 54 * | 179 ± 60 * | 289 ± 29 *# |
| Mrp4 | 100 ± 12 | 101 ± 4 | 100 ± 11 | 105 ± 12 |
| Oatp2 | 100 ± 11 | 151 ± 17 * | 5 ± 1 * | 4 ± 1 * |
| Ntcp | 100 ± 3 | 158 ± 20 * | 45 ± 2 * | 68 ± 7 *# |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Razori, M.V.; Hillotte, G.L.; Martín, P.L.; Barosso, I.R.; Basiglio, C.L.; Ruiz, M.L.; Roma, M.G. Transcriptional and Post-Transcriptional Anticholestatic Mechanisms of Obeticholic Acid in Lipopolysaccharide-Induced Cholestasis. Pharmaceutics 2025, 17, 1393. https://doi.org/10.3390/pharmaceutics17111393
Razori MV, Hillotte GL, Martín PL, Barosso IR, Basiglio CL, Ruiz ML, Roma MG. Transcriptional and Post-Transcriptional Anticholestatic Mechanisms of Obeticholic Acid in Lipopolysaccharide-Induced Cholestasis. Pharmaceutics. 2025; 17(11):1393. https://doi.org/10.3390/pharmaceutics17111393
Chicago/Turabian StyleRazori, María Valeria, Geraldine L. Hillotte, Pamela L. Martín, Ismael R. Barosso, Cecilia L. Basiglio, María Laura Ruiz, and Marcelo G. Roma. 2025. "Transcriptional and Post-Transcriptional Anticholestatic Mechanisms of Obeticholic Acid in Lipopolysaccharide-Induced Cholestasis" Pharmaceutics 17, no. 11: 1393. https://doi.org/10.3390/pharmaceutics17111393
APA StyleRazori, M. V., Hillotte, G. L., Martín, P. L., Barosso, I. R., Basiglio, C. L., Ruiz, M. L., & Roma, M. G. (2025). Transcriptional and Post-Transcriptional Anticholestatic Mechanisms of Obeticholic Acid in Lipopolysaccharide-Induced Cholestasis. Pharmaceutics, 17(11), 1393. https://doi.org/10.3390/pharmaceutics17111393