
Combination of Betulinic Acid Fragments and Carbonic Anhydrase Inhibitors—A New Drug Targeting Approach


 , , , ,
, , , ,  and
and
Abstract
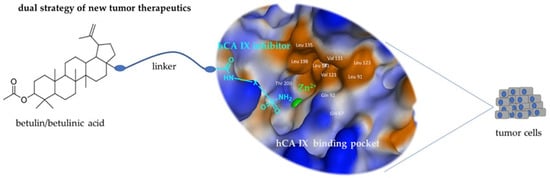
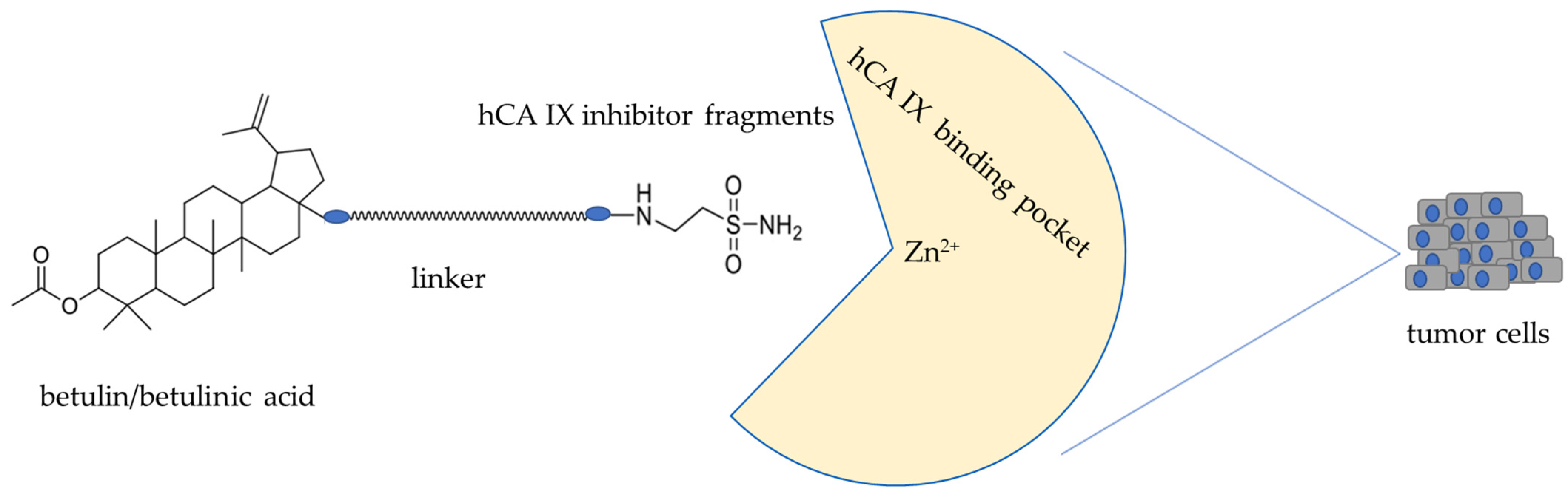
1. Introduction
2. Materials and Methods
2.1. Synthesis
2.1.1. General
2.1.2. General Procedure of Acetylation (GPA)
2.1.3. 3-O-Acetyl-betulin (1)
2.1.4. 3-O-Acetyl-betulinic Acid (2)
2.1.5. (3β)-28-{[2-(Aminosulfonyl)-ethyl]-amino}28-oxolup-20(29)-en-3-yl Acetate (3)
2.1.6. 4-{[(3beta)-3-(Acetyloxy)-lup-20(29)-en-28-yl]-oxy}-4-oxobutanoic Acid (4)
2.1.7. (3β)-3-(Acetyloxy)-lup-20(29)-en-28-yl 4-{[2-(aminosulfonyl)-ethyl]amino}-4-oxobutanoate (5)
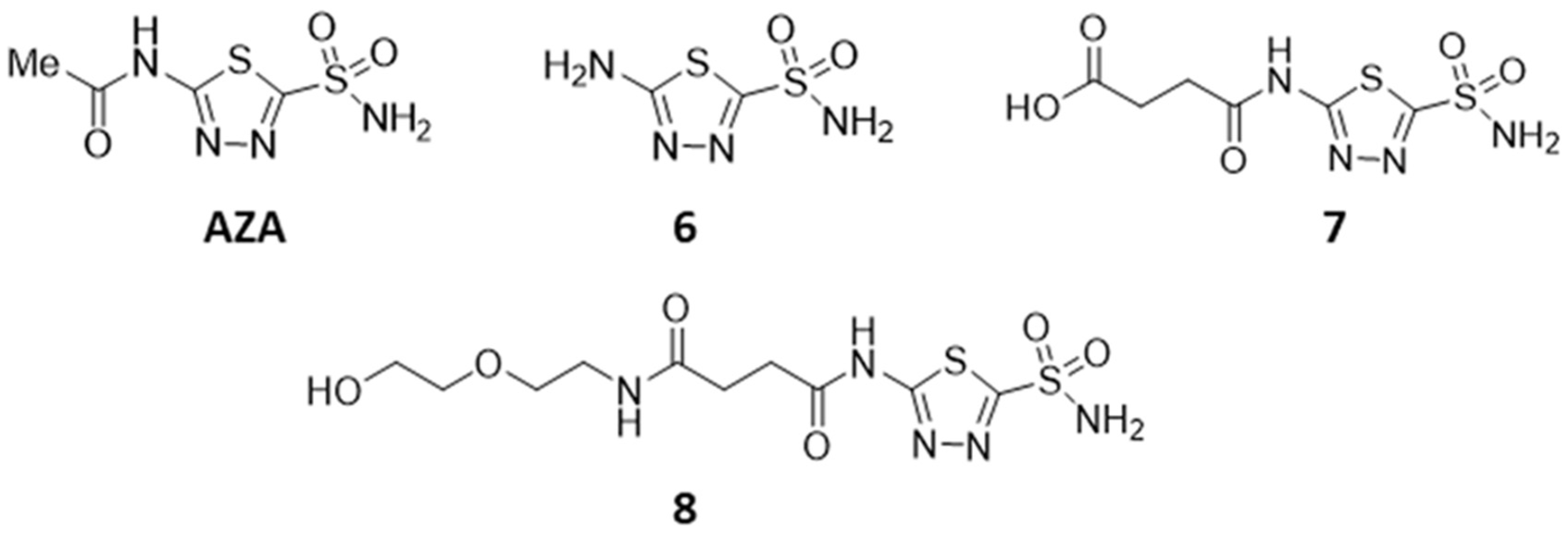
2.1.8. 5-Amino-1,3.4-thiadiazole-2-sulfonamide (6)
2.1.9. 4 {[5-(Amino sulfonyl)-1,3,4-thiadiazol-2-yl]-amino}-4-oxobutanoic Acid (7)
2.1.10. N-[5-(Aminosulfonyl)-1,3,4-thiadiazol-2-yl]-N’-[2-(2-hydroxyethoxy)-ethyl]-succinamide (8)
2.1.11. (3β)-3-(Acetyloxy)-lup-20(29)-en-28-yl-4-{[5-(aminosulfonyl)-1,3,4-thiadiazol-2-yl]-amino}-4-oxobutanoate (9)
2.1.12. (3β)-28-[2-(2-Hydroxyethoxy)-ethoxy]-lup-20(29)-en-3-yl Acetate (10)
2.1.13. 4-[2-2(2-{[(3β)-3-(Acetyloxy]-lup-20(29)-en-28-yl]-oxy}ethoxy)ethoxy]4-oxobutanoic Acid (11)
2.1.14. 2-(2-{[(3β)-3-Acetyloxy)-lup-20(29)-en-28-yl]oxy}ethoxy)ethyl 4-{[5-(amino-sulfonyl)-1,3,4-thiadiazol-2-yl}4-oxobutanoate (12)
2.2. Cell Lines and Treatment with Betulin and BA Derivatives
2.3. Cytotoxicity
2.4. Annexin V Assay
2.5. Cell Cycle
2.6. Human Carbonic Anhydrase IX (hCAIX) Inhibition
2.7. Cell-Based Measurement of hCA IX Inhibition
2.8. Computational Studies
3. Results and Discussion
3.1. Chemistry
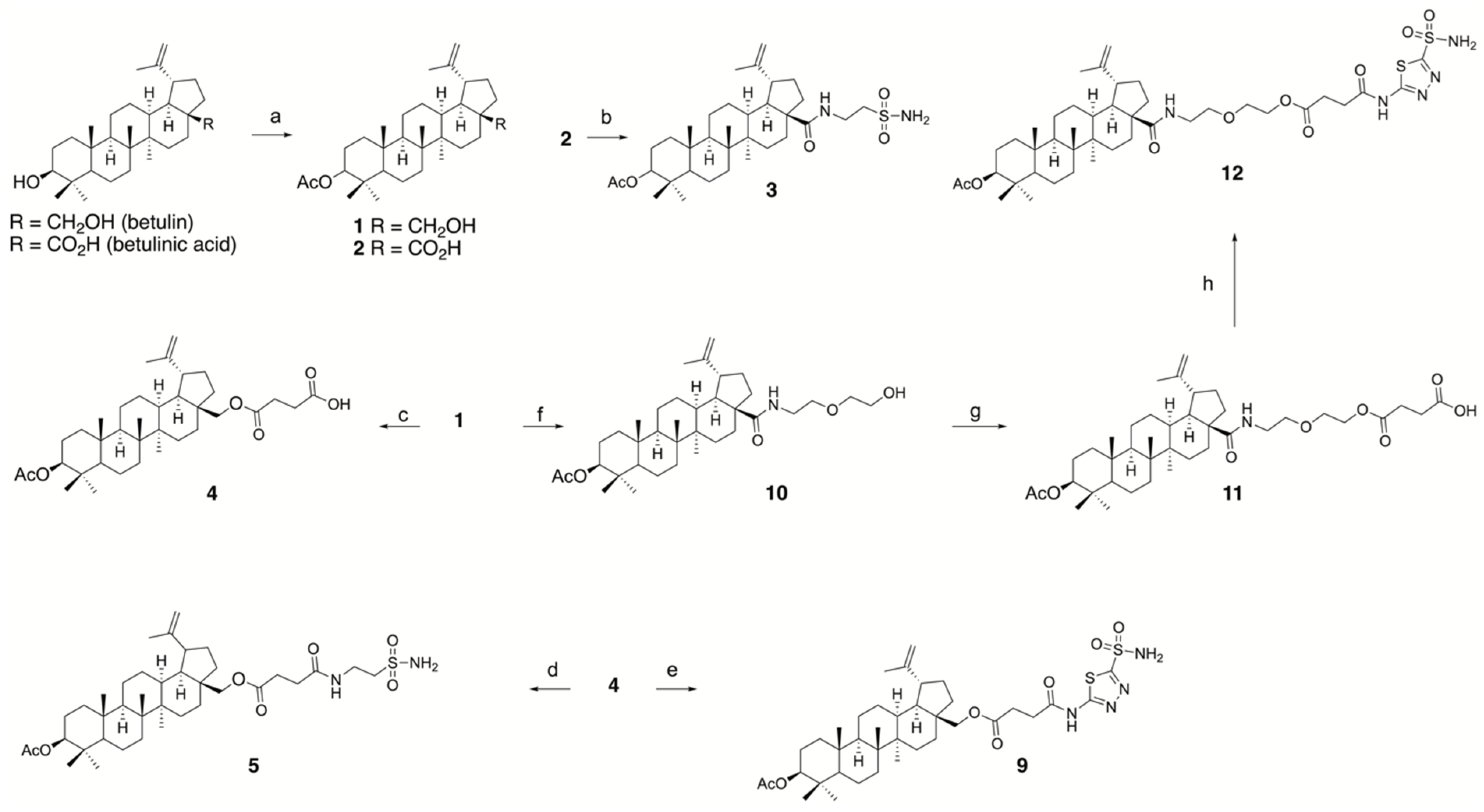
3.1.1. Synthesis of Conjugates
3.1.2. Synthesis of the Inhibitory Fragments of the Conjugate
3.2. Cytotoxic Effects of Conjugates and Fragments
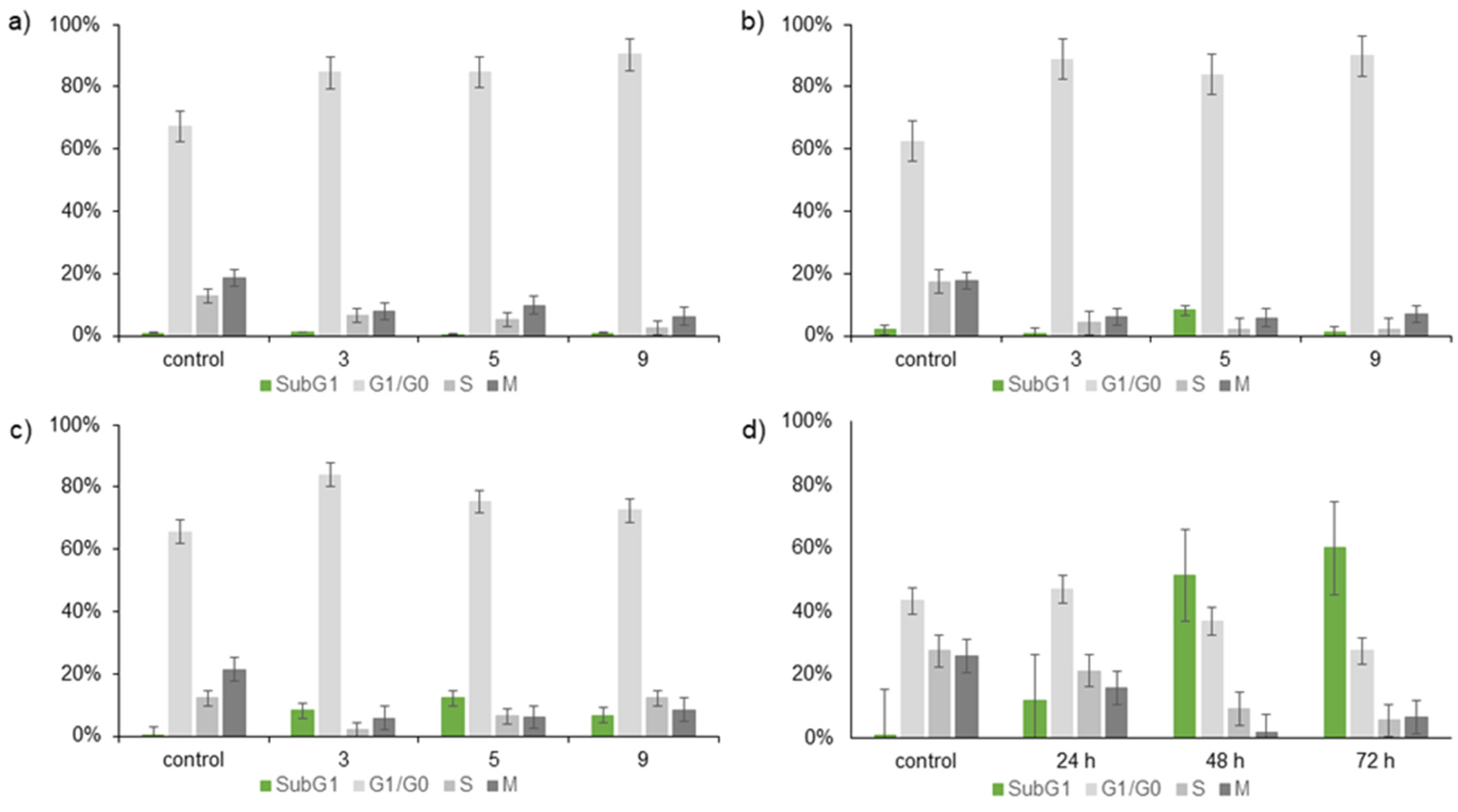
3.3. Annexin V Staining and Cell Cycle Analysis
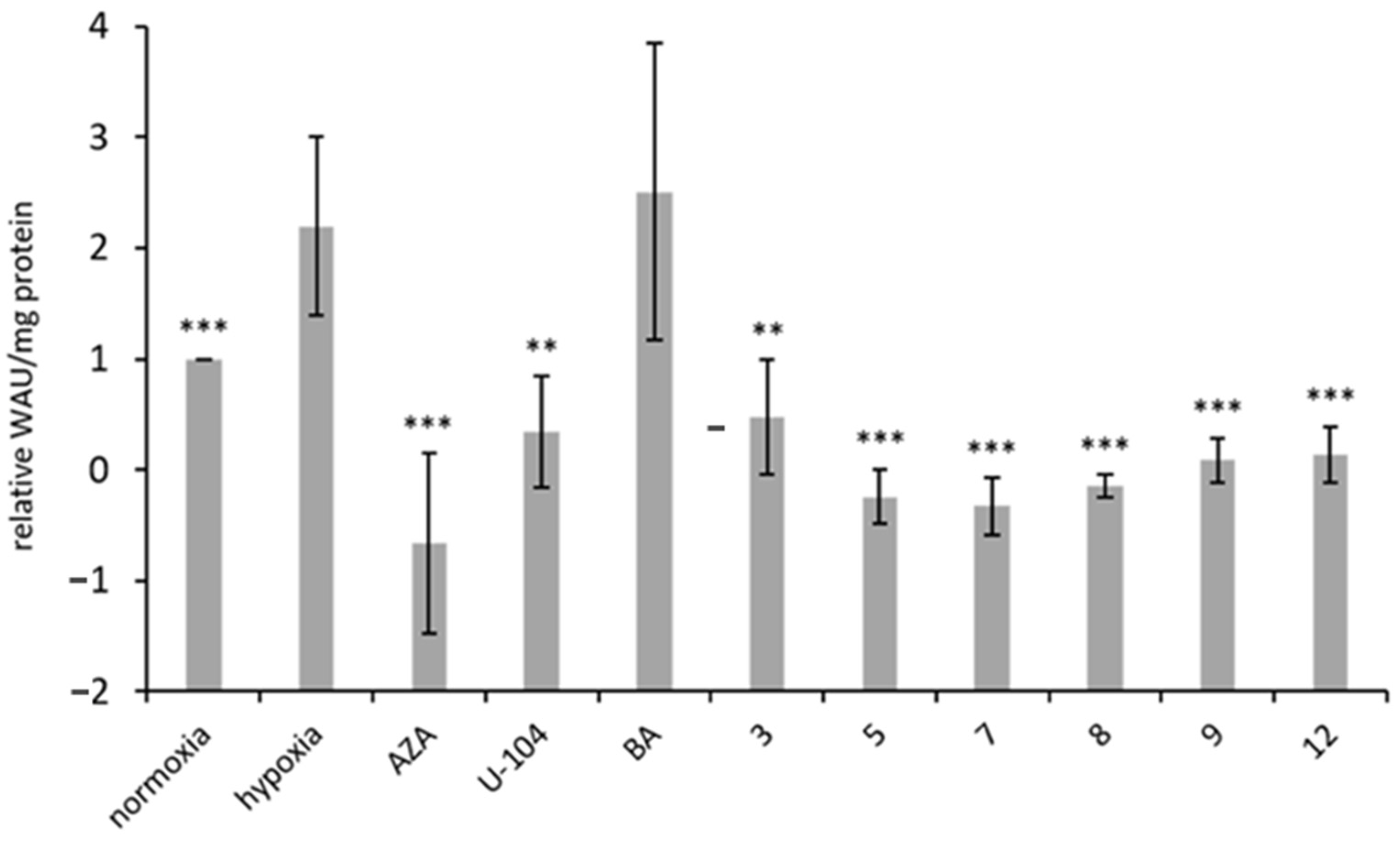
3.4. Inhibition of Carbonic Anhydrase IX
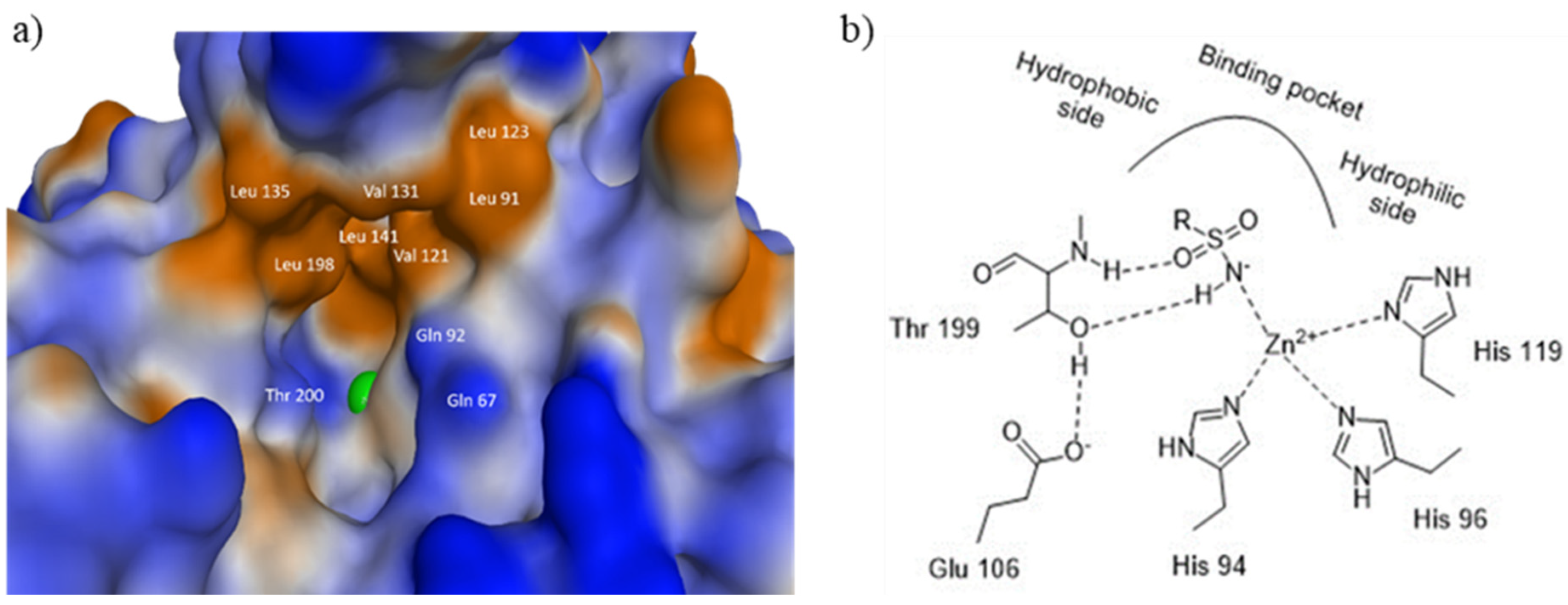
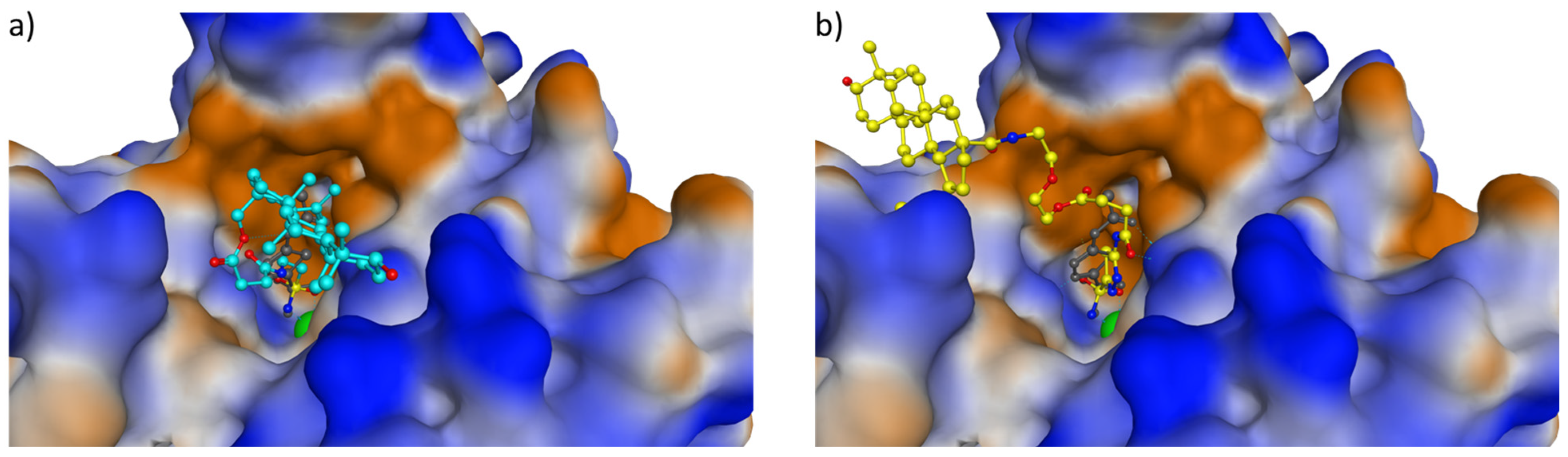
3.5. Computational Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gershenzon, J.; Dudareva, N. The function of terpene natural products in the natural world. Nat. Chem. Biol. 2007, 3, 408–414. [Google Scholar] [CrossRef]
- Pavlova, N.I.; Savinova, O.V.; Nikolaeva, S.N.; Boreko, E.I.; Flekhter, O.B. Antiviral activity of betulin, betulinic and betulonic acids against some enveloped and non-enveloped viruses. Fitoterapia 2003, 74, 489–492. [Google Scholar] [CrossRef]
- Baltina, L.A.; Flekhter, O.B.; Nigmatullina, L.R.; Boreko, E.I.; Pavlova, N.I.; Nikolaeva, S.N.; Savinova, O.V.; Tolstikov, G.A. Lupane triterpenes and derivatives with antiviral activity. Bioorg. Med. Chem. Lett. 2003, 13, 3549–3552. [Google Scholar] [CrossRef]
- Zdzisińska, B.; Rzeski, W.; Paduch, R.; Szuster-Ciesielska, A.; Kaczor, J.; Wejksza, K.; Kandefer-Szerszeń, M. Differential effect of betulin and betulinic acid on cytokine production in human whole blood cell cultures. Pol. J. Pharmacol. 2003, 55, 235–238. [Google Scholar]
- Takada, Y.; Aggarwal, B.B. Betulinic acid suppresses carcinogen-induced NF-kappa B activation through inhibition of I kappa B alpha kinase and p65 phosphorylation: Abrogation of cyclooxygenase-2 and matrix metalloprotease-9. J. Immunol. 2003, 171, 3278–3286. [Google Scholar] [CrossRef]
- Kashiwada, Y.; Hashimoto, F.; Cosentino, L.M.; Chen, C.H.; Garrett, P.E.; Lee, K.H. Betulinic acid and dihydrobetulinic acid derivatives as potent anti-HIV agents. J. Med. Chem. 1996, 39, 1016–1017. [Google Scholar] [CrossRef]
- Ren, W.; Qin, L.; Xu, Y.; Cheng, N. Inhibition of betulinic acid to growth and angiogenesis of human colorectal cancer cell in nude mice. Chin.-Ger. J. Clin. Oncol. 2010, 9, 153–157. [Google Scholar] [CrossRef]
- Dehelean, C.A.; Feflea, S.; Ganta, S.; Amiji, M. Anti-angiogenic effects of betulinic acid administered in nanoemulsion formulation using chorioallantoic membrane assay. J. Biomed. Nanotechnol. 2011, 7, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Pisha, E.; Chai, H.; Lee, I.S.; Chagwedera, T.E.; Farnsworth, N.R.; Cordell, G.A.; Beecher, C.W.; Fong, H.H.; Kinghorn, A.D.; Brown, D.M. Discovery of betulinic acid as a selective inhibitor of human melanoma that functions by induction of apoptosis. Nat. Med. 1995, 1, 1046–1051. [Google Scholar] [CrossRef]
- Zuco, V.; Supino, R.; Righetti, S.C.; Cleris, L.; Marchesi, E.; Gambacorti-Passerini, C.; Formelli, F. Selective cytotoxicity of betulinic acid on tumor cell lines, but not on normal cells. Cancer Lett. 2002, 175, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Gülçin, I.; Beydemir, S.; Büyükokuroğlu, M.E. In vitro and in vivo effects of dantrolene on carbonic anhydrase enzyme activities. Biol. Pharm. Bull. 2004, 27, 613–616. [Google Scholar] [CrossRef] [PubMed]
- Hisar, O.; Sukru, S.; Gulcin, I.; Hisar, S.A.; Yanik, T.; Omer, O.I. The effects of melatonin hormone on carbonic anhydrase enzyme activity in rainbow trout (Oncorhynchus mykiss) erythrocytes in vitro and in vivo. Turk. J. Vet. Anim. Sci. 2005, 29, 841–845. [Google Scholar]
- Frost, S.C.; McKenna, R. Carbonic Anhydrase: Mechanism, Regulation, Links to Disease, and Industrial Applications; Springer: Dordrecht, The Netherlands, 2014; ISBN 978-94-007-7358-5. [Google Scholar]
- Fisher, S.Z.; Maupin, C.M.; Budayova-Spano, M.; Govindasamy, L.; Tu, C.; Agbandje-McKenna, M.; Silverman, D.N.; Voth, G.A.; McKenna, R. Atomic crystal and molecular dynamics simulation structures of human carbonic anhydrase II: Insights into the proton transfer mechanism. Biochemistry 2007, 46, 2930–2937. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. How many carbonic anhydrase inhibition mechanisms exist? J. Enzyme Inhib. Med. Chem. 2016, 31, 345–360. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Structure and function of carbonic anhydrases. Biochem. J. 2016, 473, 2023–2032. [Google Scholar] [CrossRef] [PubMed]
- Lindskog, S. Structure and mechanism of carbonic anhydrase. Pharmacol. Ther. 1997, 74, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Vanchanagiri, K.; Emmerich, D.; Bruschke, M.; Bache, M.; Seifert, F.; Csuk, R.; Vordermark, D.; Paschke, R. Synthesis and biological investigation of new carbonic anhydrase IX (CAIX) inhibitors. Chem. Biol. Interact. 2018, 284, 12–23. [Google Scholar] [CrossRef]
- Petrenko, M.; Güttler, A.; Pflüger, E.; Serbian, I.; Kahnt, M.; Eiselt, Y.; Keßler, J.; Funtan, A.; Paschke, R.; Csuk, R.; et al. MSBA-S—A pentacyclic sulfamate as a new option for radiotherapy of human breast cancer cells. Eur. J. Med. Chem. 2021, 224, 113721. [Google Scholar] [CrossRef]
- Winum, J.-Y.; Pastorekova, S.; Jakubickova, L.; Montero, J.-L.; Scozzafava, A.; Pastorek, J.; Vullo, D.; Innocenti, A.; Supuran, C.T. Carbonic anhydrase inhibitors: Synthesis and inhibition of cytosolic/tumor-associated carbonic anhydrase isozymes I, II, and IX with bis-sulfamates. Bioorg. Med. Chem. Lett. 2005, 15, 579–584. [Google Scholar] [CrossRef]
- Güttler, A.; Eiselt, Y.; Funtan, A.; Thiel, A.; Petrenko, M.; Keßler, J.; Thondorf, I.; Paschke, R.; Vordermark, D.; Bache, M. Betulin Sulfonamides as Carbonic Anhydrase Inhibitors and Anticancer Agents in Breast Cancer Cells. Int. J. Mol. Sci. 2021, 22, 8808. [Google Scholar] [CrossRef]
- Patel, T.K.; Adhikari, N.; Amin, S.A.; Biswas, S.; Jha, T.; Ghosh, B. Small molecule drug conjugates (SMDCs): An emerging strategy for anticancer drug design and discovery. New J. Chem. 2021, 45, 5291–5321. [Google Scholar] [CrossRef]
- Krall, N.; Pretto, F.; Decurtins, W.; Bernardes, G.J.L.; Supuran, C.T.; Neri, D. A small-molecule drug conjugate for the treatment of carbonic anhydrase IX expressing tumors. Angew. Chem. Int. Ed. Engl. 2014, 53, 4231–4235. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, S.; Ma, P.; Jiang, Y.; Cheng, K.; Yu, Y.; Jiang, N.; Miao, H.; Tang, Q.; Liu, F.; et al. Drug conjugate-based anticancer therapy—Current status and perspectives. Cancer Lett. 2023, 552, 215969. [Google Scholar] [CrossRef]
- Wiemann, J.; Heller, L.; Perl, V.; Kluge, R.; Ströhl, D.; Csuk, R. Betulinic acid derived hydroxamates and betulin derived carbamates are interesting scaffolds for the synthesis of novel cytotoxic compounds. Eur. J. Med. Chem. 2015, 106, 194–210. [Google Scholar] [CrossRef]
- Loesche, A.; Kahnt, M.; Serbian, I.; Brandt, W.; Csuk, R. Triterpene-Based Carboxamides Act as Good Inhibitors of Butyrylcholinesterase. Molecules 2019, 24, 948. [Google Scholar] [CrossRef]
- Roblin, R.O.; Clapp, J.W. The Preparation of Heterocyclic Sulfonamides 1. J. Am. Chem. Soc. 1950, 72, 4890–4892. [Google Scholar] [CrossRef]
- Liebscher, G.; Vanchangiri, K.; Mueller, T.; Feige, K.; Cavalleri, J.-M.V.; Paschke, R. In vitro anticancer activity of Betulinic acid and derivatives thereof on equine melanoma cell lines from grey horses and in vivo safety assessment of the compound NVX-207 in two horses. Chem. Biol. Interact. 2016, 246, 20–29. [Google Scholar] [CrossRef]
- Kraft, O.; Hartmann, A.-K.; Brandt, S.; Hoenke, S.; Heise, N.V.; Csuk, R.; Mueller, T. Asiatic acid as a leading structure for derivatives combining sub-nanomolar cytotoxicity, high selectivity, and the ability to overcome drug resistance in human preclinical tumor models. Eur. J. Med. Chem. 2023, 250, 115189. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef] [PubMed]
- Bache, M.; Münch, C.; Güttler, A.; Wichmann, H.; Theuerkorn, K.; Emmerich, D.; Paschke, R.; Vordermark, D. Betulinyl Sulfamates as Anticancer Agents and Radiosensitizers in Human Breast Cancer Cells. Int. J. Mol. Sci. 2015, 16, 26249–26262. [Google Scholar] [CrossRef]
- Costa, E.C.; Moreira, A.F.; de Melo-Diogo, D.; Gaspar, V.M.; Carvalho, M.P.; Correia, I.J. 3D tumor spheroids: An overview on the tools and techniques used for their analysis. Biotechnol. Adv. 2016, 34, 1427–1441. [Google Scholar] [CrossRef]
- Nunes, A.S.; Barros, A.S.; Costa, E.C.; Moreira, A.F.; Correia, I.J. 3D tumor spheroids as in vitro models to mimic in vivo human solid tumors resistance to therapeutic drugs. Biotechnol. Bioeng. 2019, 116, 206–226. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.J.; Gieling, R.G. Preclinical Evaluation of Ureidosulfamate Carbonic Anhydrase IX/XII Inhibitors in the Treatment of Cancers. Int. J. Mol. Sci. 2019, 20, 6080. [Google Scholar] [CrossRef]
- Bortner, C.D.; Oldenburg, N.B.; Cidlowski, J.A. The role of DNA fragmentation in apoptosis. Trends Cell Biol. 1995, 5, 21–26. [Google Scholar] [CrossRef]
- Cocca, B.A.; Cline, A.M.; Radic, M.Z. Blebs and apoptotic bodies are B cell autoantigens. J. Immunol. 2002, 169, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Demchenko, A.P. The change of cellular membranes on apoptosis: Fluorescence detection. Exp. Oncol. 2012, 34, 263–268. [Google Scholar]
- Mourdjeva, M.; Kyurkchiev, D.; Mandinova, A.; Altankova, I.; Kehayov, I.; Kyurkchiev, S. Dynamics of membrane translocation of phosphatidylserine during apoptosis detected by a monoclonal antibody. Apoptosis 2005, 10, 209–217. [Google Scholar] [CrossRef]
- Vermes, I.; Haanen, C.; Steffens-Nakken, H.; Reutelingsperger, C. A novel assay for apoptosis. Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V. J. Immunol. Methods 1995, 184, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Kommera, H.; Kaluderović, G.N.; Kalbitz, J.; Dräger, B.; Paschke, R. Small structural changes of pentacyclic lupane type triterpenoid derivatives lead to significant differences in their anticancer properties. Eur. J. Med. Chem. 2010, 45, 3346–3353. [Google Scholar] [CrossRef]
- Yeruva, L.; Elegbede, J.A.; Carper, S.W. Methyl jasmonate decreases membrane fluidity and induces apoptosis through tumor necrosis factor receptor 1 in breast cancer cells. Anticancer. Drugs 2008, 19, 766–776. [Google Scholar] [CrossRef] [PubMed]
- Gooch, J.L.; Yee, D. Strain-specific differences in formation of apoptotic DNA ladders in MCF-7 breast cancer cells. Cancer Lett. 1999, 144, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Jänicke, R.U.; Sprengart, M.L.; Wati, M.R.; Porter, A.G. Caspase-3 is required for DNA fragmentation and morphological changes associated with apoptosis. J. Biol. Chem. 1998, 273, 9357–9360. [Google Scholar] [CrossRef]
- Verpoorte, J.A.; Mehta, S.; Edsall, J.T. Esterase Activities of Human Carbonic Anhydrases B and C. J. Biol. Chem. 1967, 242, 4221–4229. [Google Scholar] [CrossRef]
- Pocker, Y.; Stone, J.T. The catalytic versatility of erythrocyte carbonic anhydrase. 3. Kinetic studies of the enzyme-catalyzed hydrolysis of p-nitrophenyl acetate. Biochemistry 1967, 6, 668–678. [Google Scholar] [CrossRef]
- Supuran, C.T. Carbonic anhydrases: Novel therapeutic applications for inhibitors and activators. Nat. Rev. Drug Discov. 2008, 7, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An overview of the Amber biomolecular simulation package. WIREs Comput. Mol. Sci. 2013, 3, 198–210. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | A375 | MCF-7 | A2780 |
|---|---|---|---|
| AZA | >100 | >100 | >100 |
| BA | 13.1 ± 0.9 | 13.0 ± 1.0 | 12.5 ± 1.8 |
| 3 | 6.4 ± 0.7 | 7.5 ± 0.7 | 10.4 ± 0.8 |
| 4 | 12.1 ± 1.5 | 9.8 ± 0.6 | 12.3 ± 3.9 |
| 5 | 9.4 ± 0.5 | 8.6 ± 0.7 | 11.4 ± 1.4 |
| 6 | >100 | >100 | >100 |
| 7 | >100 | >100 | >100 |
| 8 | >100 | >100 | >100 |
| 9 | 7.2 ± 0.6 | 6.2 ± 0.2 | 8.7 ± 2.0 |
| 10 | 4.3 ± 0.2 | 5.9 ± 0.2 | 9.5 ± 0.9 |
| 11 | 10.5 ± 0.6 | 7.3 ± 0.4 | 12.3 ± 1.5 |
| 12 | >100 | >100 | 35.1 ± 3.4 |
| Compound | MDA-MB-231 | Hs578T |
|---|---|---|
| U-104 | 33.5 ± 7.6 | 83.9 ± 12.4 |
| 5 | 10.3 ± 2.7 | 30.1 ± 0.7 |
| 7 | >100 | >100 |
| 8 | >100 | >100 |
| 9 | 8.1 ± 1.4 | 28.4 ± 0.3 |
| 12 | >100 | >100 |
| Compound | Ki Values [µM] of hCA IX |
|---|---|
| AZA | 0.094 ± 0.027 |
| U-104 | 0.128 ± 0.045 |
| BA | n.i. |
| 3 | 1.04 ± 0.39 |
| 5 | 1.1 ± 0.31 |
| 6 | 0.185 ± 0.04 |
| 7 | 0.129 ± 0.05 |
| 8 | 0.146 ± 0.05 |
| 9 | 1.25 ± 0.25 |
| 12 | r.a. (10 µM): 60% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bache, M.; Heise, N.V.; Thiel, A.; Funtan, A.; Seifert, F.; Petrenko, M.; Güttler, A.; Brandt, S.; Mueller, T.; Vordermark, D.; et al. Combination of Betulinic Acid Fragments and Carbonic Anhydrase Inhibitors—A New Drug Targeting Approach. Pharmaceutics 2024, 16, 401. https://doi.org/10.3390/pharmaceutics16030401
Bache M, Heise NV, Thiel A, Funtan A, Seifert F, Petrenko M, Güttler A, Brandt S, Mueller T, Vordermark D, et al. Combination of Betulinic Acid Fragments and Carbonic Anhydrase Inhibitors—A New Drug Targeting Approach. Pharmaceutics. 2024; 16(3):401. https://doi.org/10.3390/pharmaceutics16030401
Chicago/Turabian StyleBache, Matthias, Niels V. Heise, Andreas Thiel, Anne Funtan, Franziska Seifert, Marina Petrenko, Antje Güttler, Sarah Brandt, Thomas Mueller, Dirk Vordermark, and et al. 2024. "Combination of Betulinic Acid Fragments and Carbonic Anhydrase Inhibitors—A New Drug Targeting Approach" Pharmaceutics 16, no. 3: 401. https://doi.org/10.3390/pharmaceutics16030401
APA StyleBache, M., Heise, N. V., Thiel, A., Funtan, A., Seifert, F., Petrenko, M., Güttler, A., Brandt, S., Mueller, T., Vordermark, D., Thondorf, I., Csuk, R., & Paschke, R. (2024). Combination of Betulinic Acid Fragments and Carbonic Anhydrase Inhibitors—A New Drug Targeting Approach. Pharmaceutics, 16(3), 401. https://doi.org/10.3390/pharmaceutics16030401