

Stiff-Soft Hybrid Biomimetic Nano-Emulsion for Targeted Liver Delivery and Treatment of Early Nonalcoholic Fatty Liver Disease


Abstract
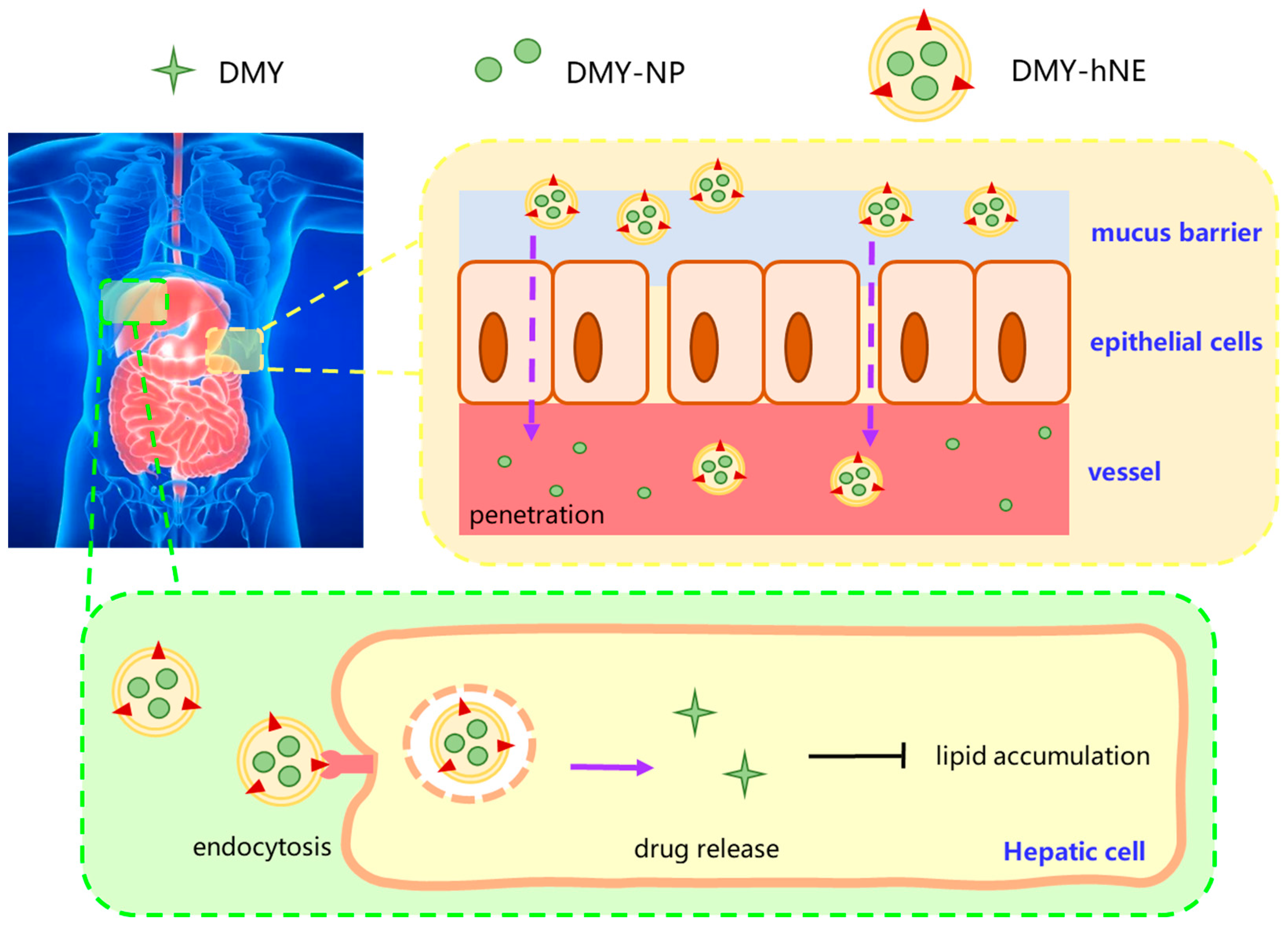
1. Introduction
2. Materials and Methods
2.1. Materials
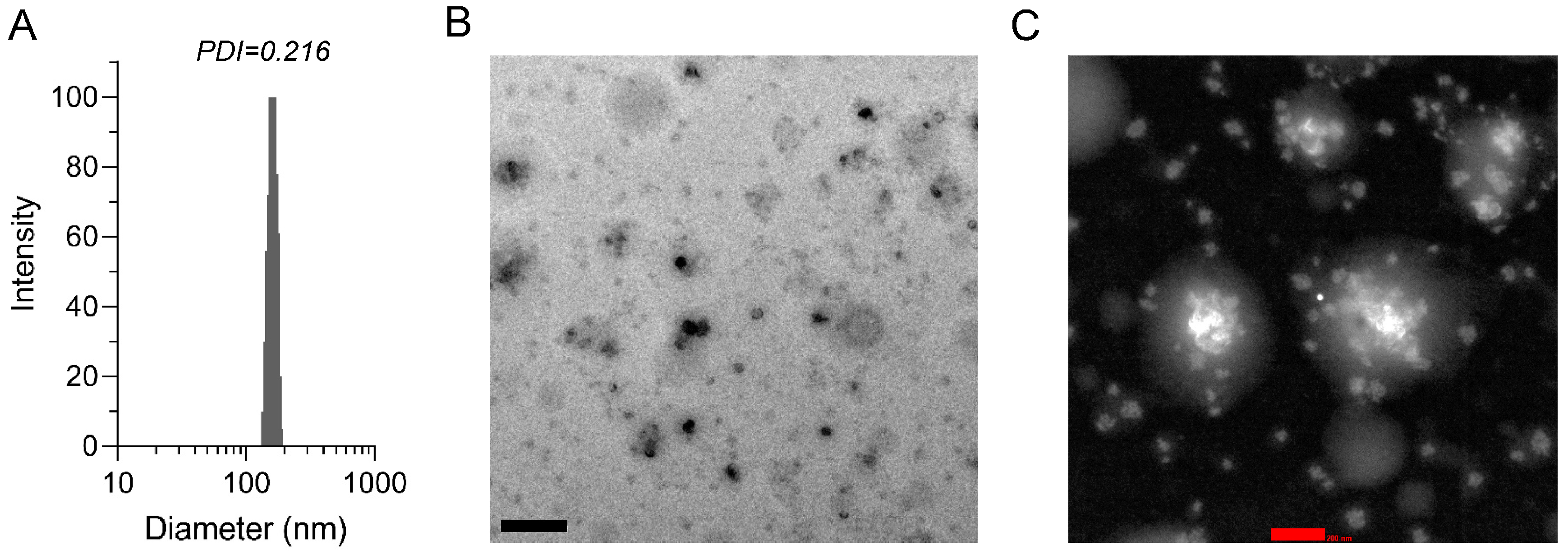
2.2. Preparation and Characterization of DMY-hNE
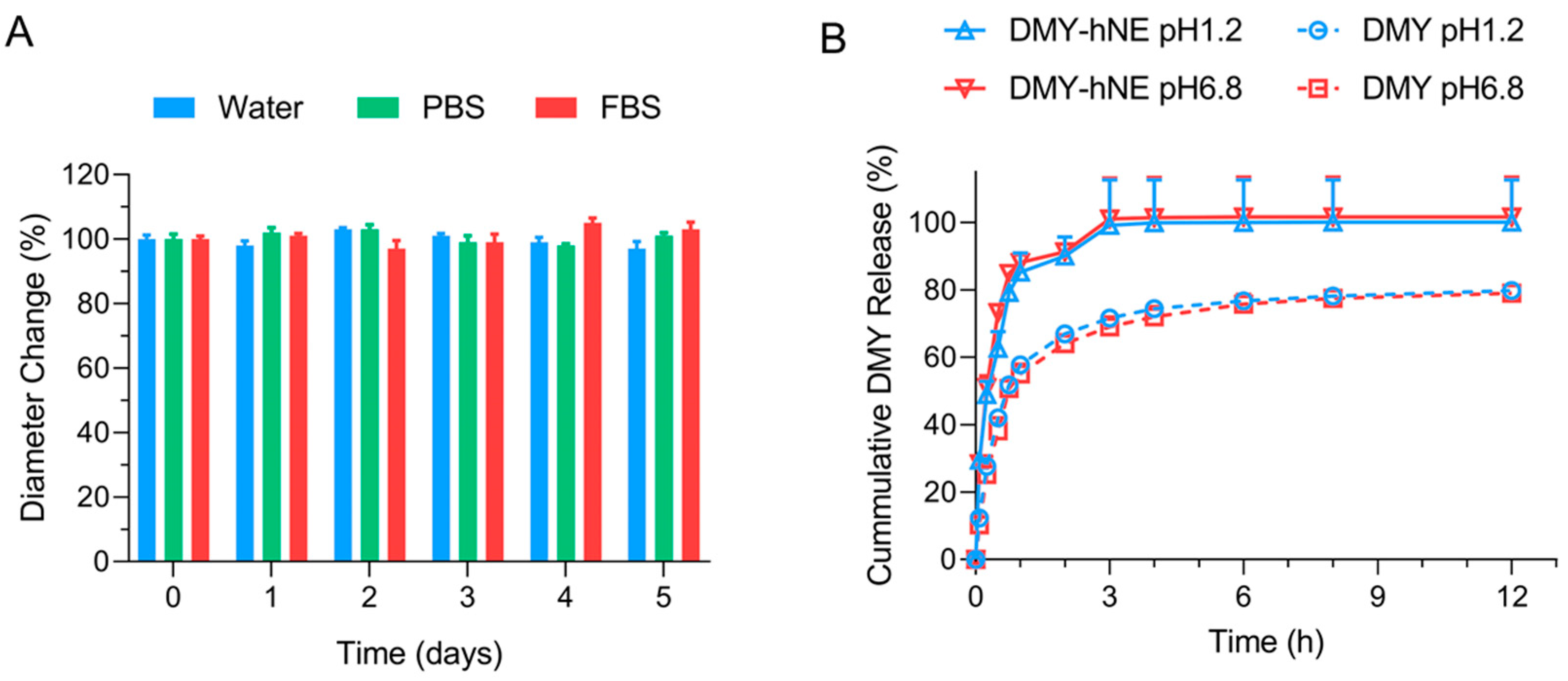
2.3. Stability
2.4. In Vitro Drug Release
2.5. In Vivo Drug Uptake
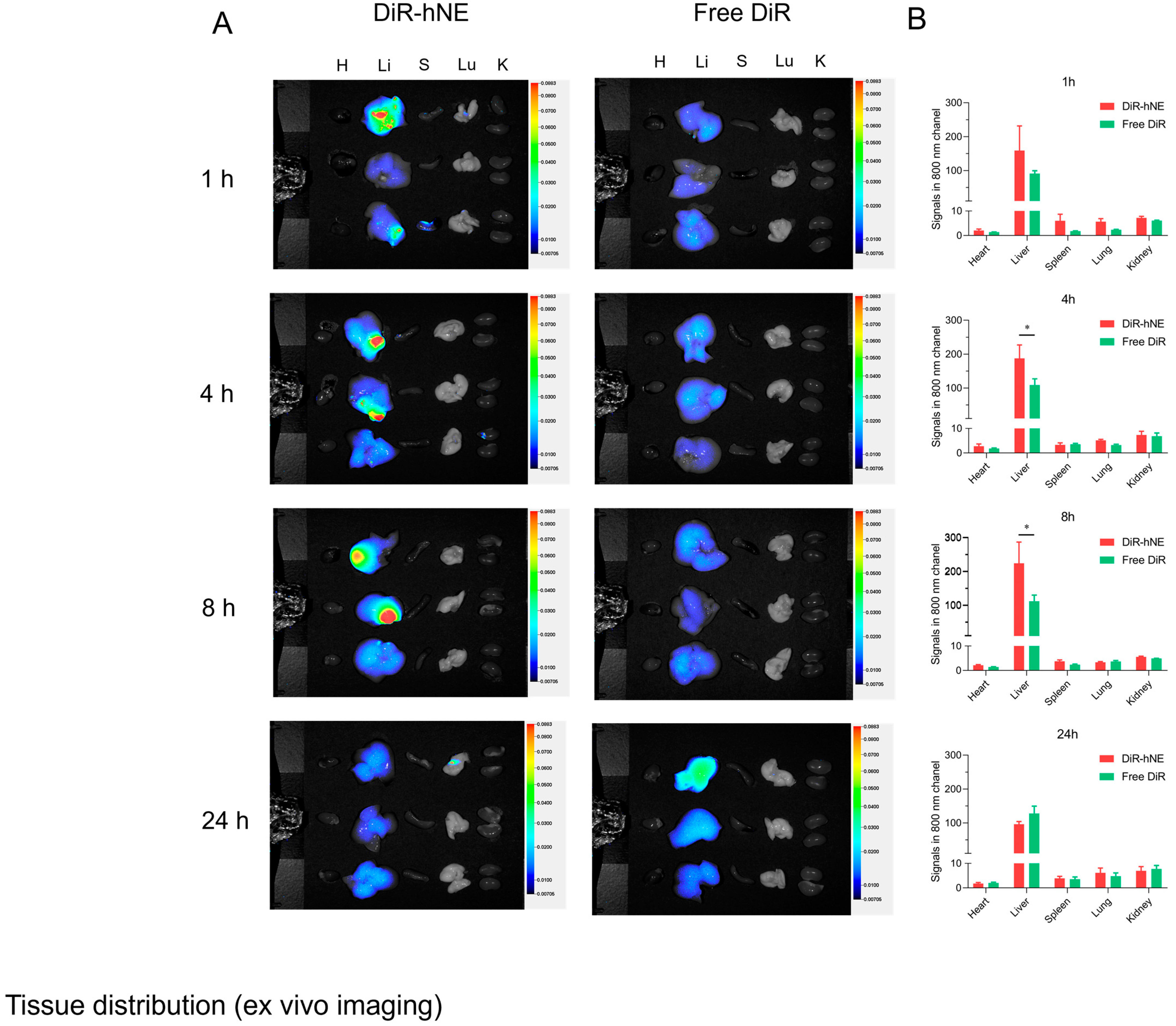
2.6. Ex Vivo Imaging
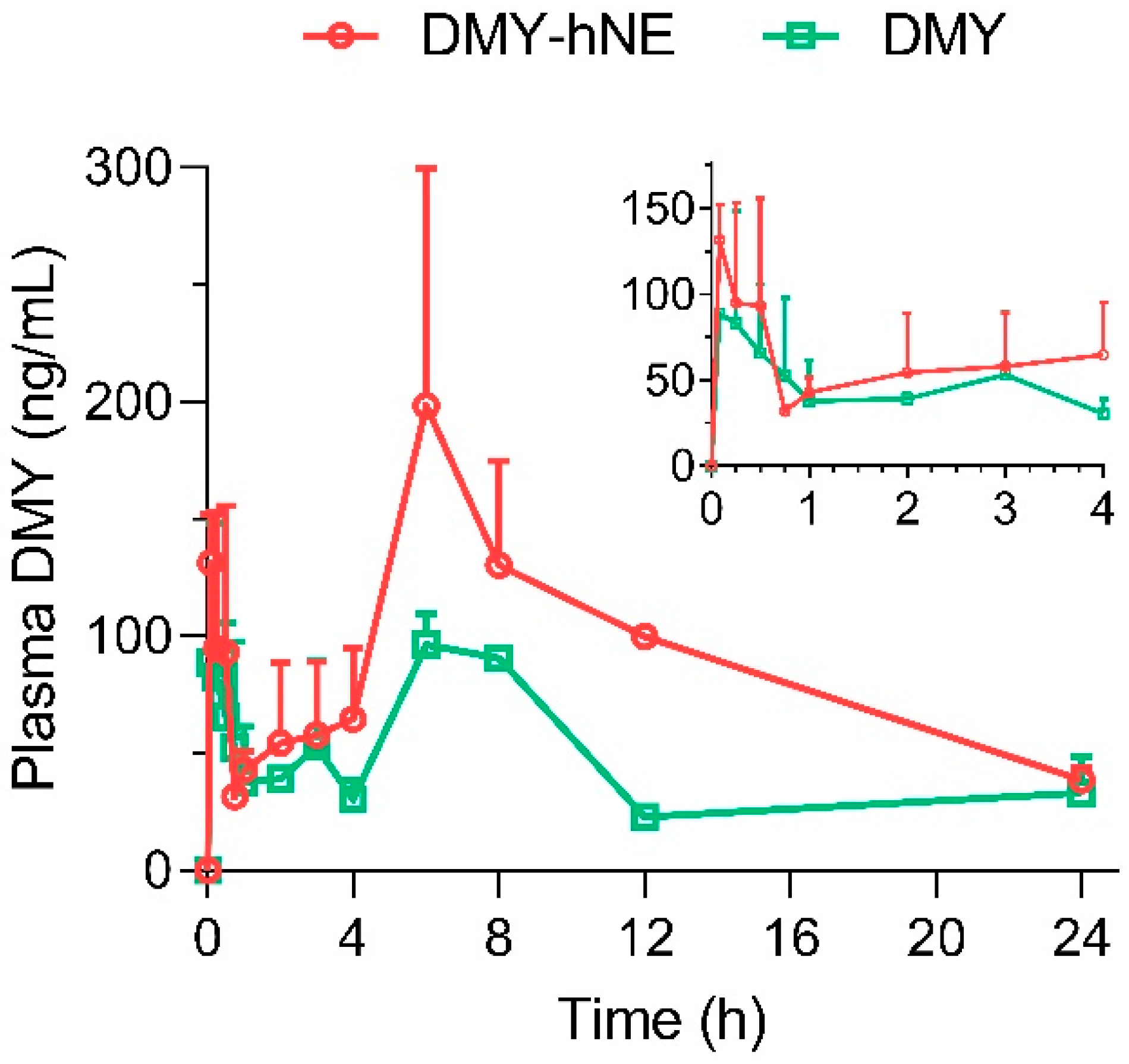
2.7. Pharmacokinetics
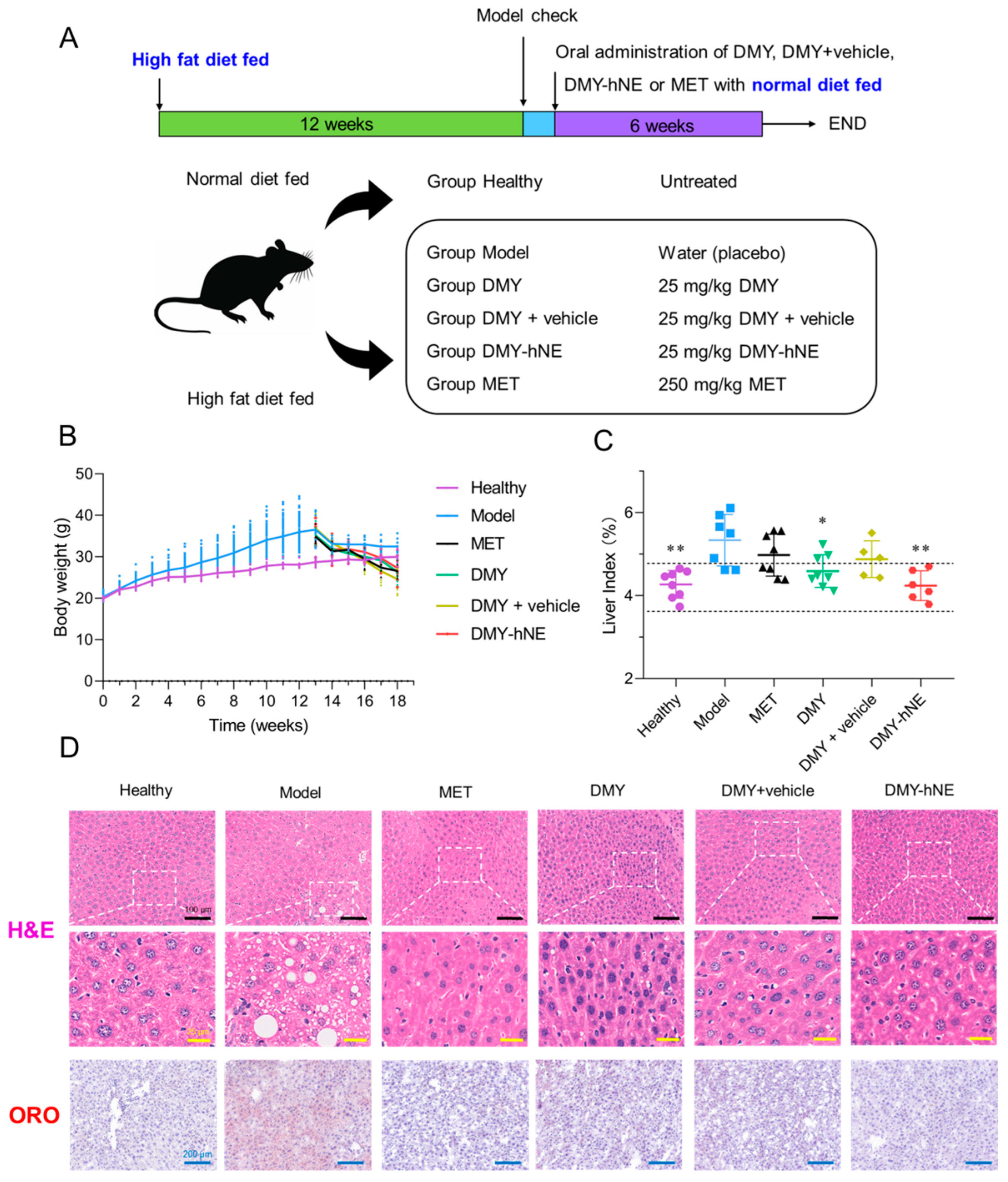
2.8. In Vivo Anti-NAFLD Efficacy
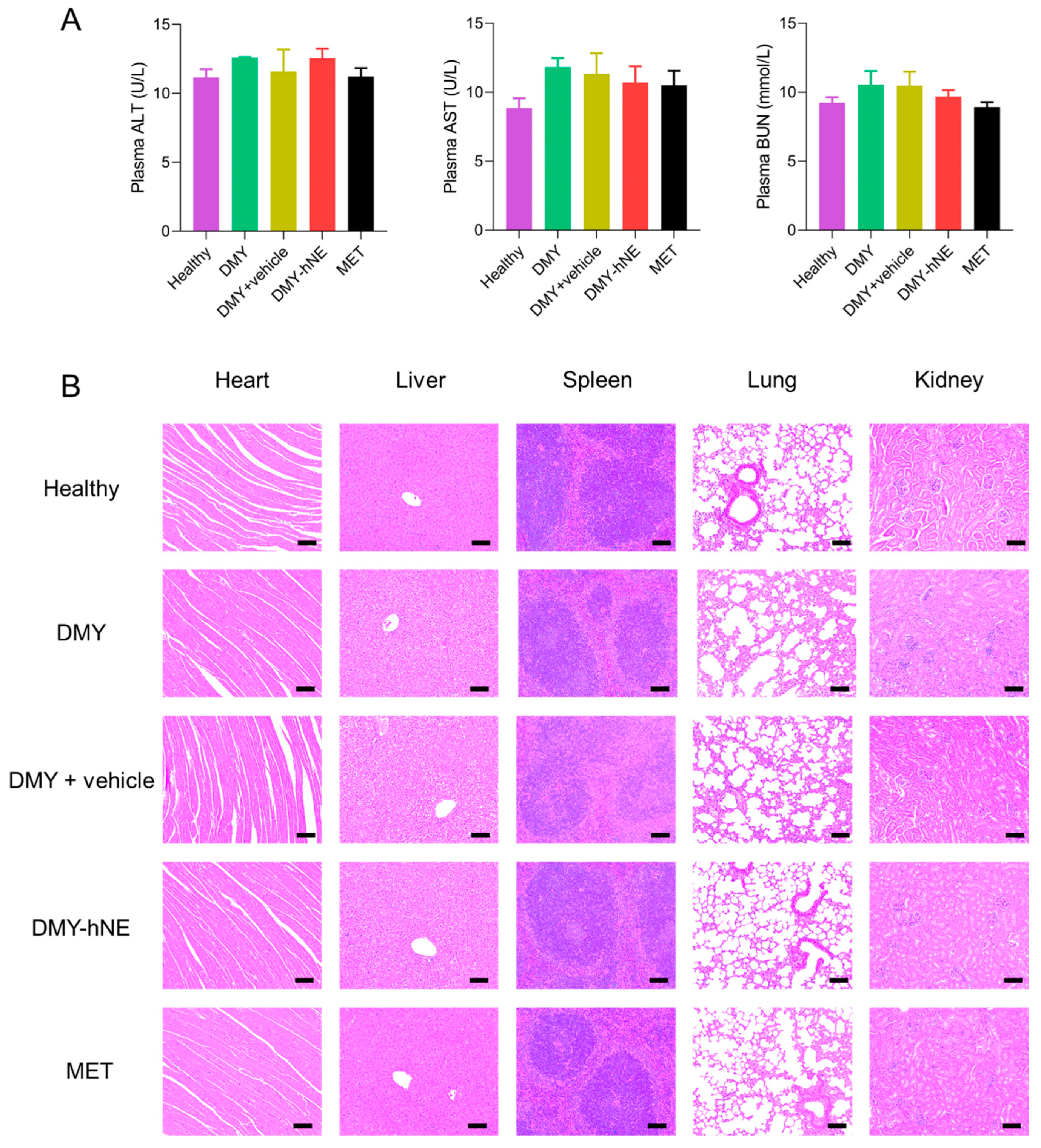
2.9. Safety Evaluation
2.10. Statistics
3. Results and Discussion
3.1. Preparation and Characterization of DMY-hNE
3.2. Stability and In Vitro DMY Release
3.3. Stomach and Small Intestine Uptake
3.4. In Vivo Liver Targeted Delivery
3.5. Pharmacokinetics
3.6. In Vivo Anti-NAFLD Efficacy of DMY-hNE
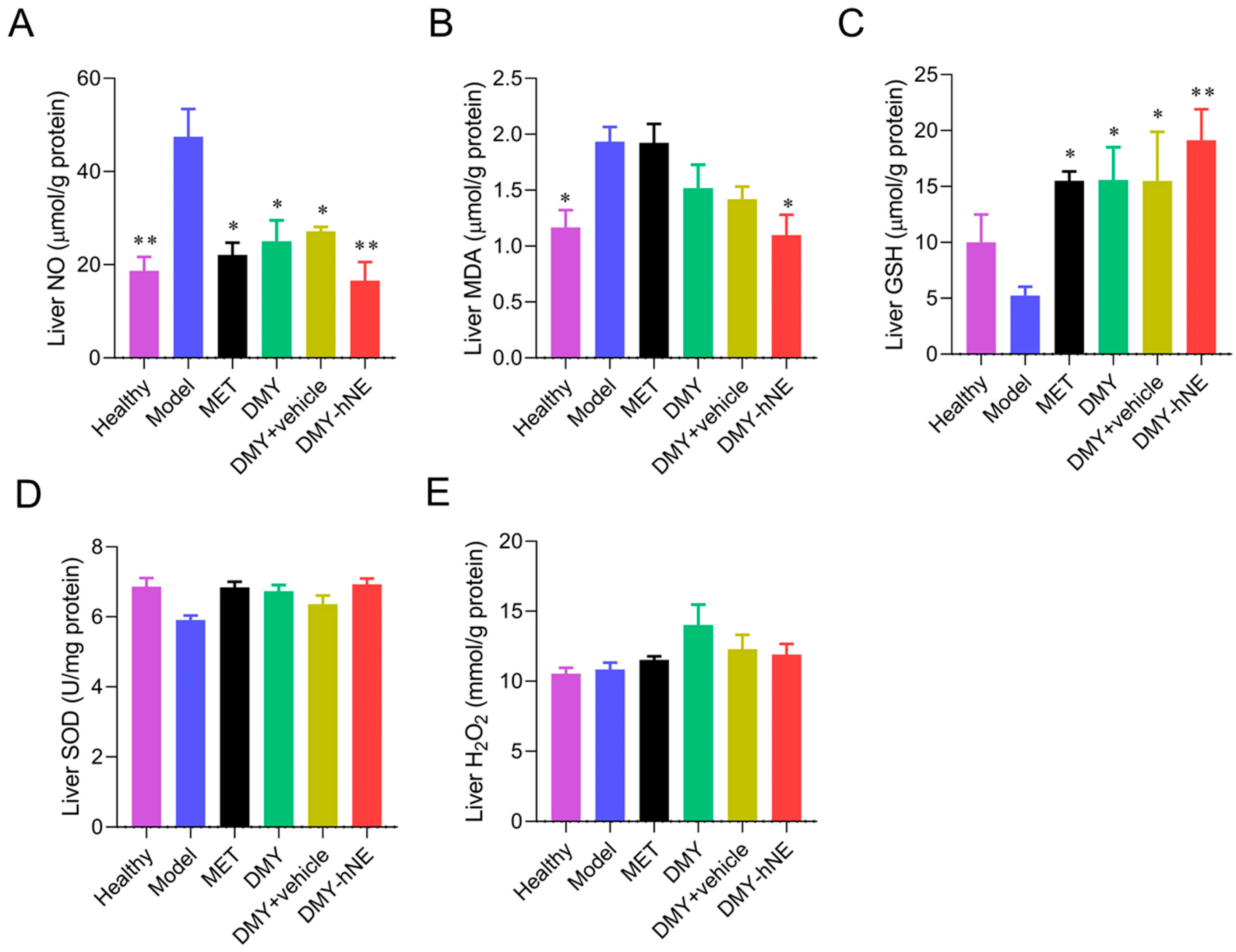
3.7. Biochemistry Analysis
3.8. Safety Evaluation
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Haigh, L.; Kirk, C.; El Gendy, K.; Gallacher, J.; Errington, L.; Mathers, J.C.; Anstee, Q.M. The Effectiveness and Acceptability of Mediterranean Diet and Calorie Restriction in Non-Alcoholic Fatty Liver Disease (NAFLD): A Systematic Review and Meta-Analysis. Clin. Nutr. 2022, 41, 1913–1931. [Google Scholar] [CrossRef] [PubMed]
- Henry, L.; Paik, J.; Younossi, Z.M. Review Article: The Epidemiologic Burden of Non-Alcoholic Fatty Liver Disease across the World. Aliment Pharmacol. Ther. 2022, 56, 942–956. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M. Non-Alcoholic Fatty Liver Disease—A Global Public Health Perspective. J. Hepatol. 2019, 70, 531–544. [Google Scholar] [CrossRef]
- Muzurovic, E.; Mikhailidis, D.P.; Mantzoros, C. Non-Alcoholic Fatty Liver Disease, Insulin Resistance, Metabolic Syndrome and Their Association with Vascular Risk. Metabolism 2021, 119, 154770. [Google Scholar] [CrossRef]
- Powell, E.E.; Wong, V.W.; Rinella, M. Non-Alcoholic Fatty Liver Disease. Lancet 2021, 397, 2212–2224. [Google Scholar] [CrossRef]
- Harrison, S.A.; Allen, A.M.; Dubourg, J.; Noureddin, M.; Alkhouri, N. Challenges and Opportunities in NASH Drug Development. Nat. Med. 2023, 29, 562–573. [Google Scholar] [CrossRef] [PubMed]
- Perry, R.J.; Samuel, V.T.; Petersen, K.F.; Shulman, G.I. The Role of Hepatic Lipids in Hepatic Insulin Resistance and Type 2 Diabetes. Nature 2014, 510, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The Diagnosis and Management of Nonalcoholic Fatty Liver Disease: Practice Guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef]
- Non-Alcoholic Fatty Liver Disease (NAFLD): Assessment and Management. Available online: https://www.nice.org.uk/guidance/ng49/resources/nonalcoholic-fatty-liver-disease-nafld-assessment-and-management-pdf-1837461227461 (accessed on 17 June 2024).
- Italian Association for the Study of the Liver (AISF). AISF Position Paper on Nonalcoholic Fatty Liver Disease (NAFLD): Updates and Future Directions. Dig. Liver Dis. 2017, 49, 471–483. [Google Scholar] [CrossRef]
- European Association for the Study of the Liver (EASL); European Association for the Study of Diabetes (EASD); European Association for the Study of Obesity (EASO). EASL–EASD–EASO Clinical Practice Guidelines for the Management of Non-Alcoholic Fatty Liver Disease. Diabetologia 2016, 59, 1121–1140. [Google Scholar] [CrossRef]
- Leoni, S.; Tovoli, F.; Napoli, L.; Serio, I.; Ferri, S.; Bolondi, L. Current Guidelines for the Management of Non-Alcoholic Fatty Liver Disease: A Systematic Review with Comparative Analysis. World J. Gastroenterol. 2018, 24, 3361–3373. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.L.; Zhao, Y.F.; Zhang, M.Y.; Zhang, Y.L.; Ji, H.F.; Shen, L. Recent Advances in Research on Vine Tea, a Potential and Functional Herbal Tea with Dihydromyricetin and Myricetin as Major Bioactive Compounds. J. Pharm. Anal. 2021, 11, 555–563. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.R.; Wang, J.M.; Xiang, H.J.; Ding, P.L.; Wu, T.; Ji, G. Recent Update on Application of Dihydromyricetin in Metabolic Related Diseases. Biomed. Pharmacother. 2022, 148, 112771. [Google Scholar] [CrossRef]
- Zhang, J.Y.; Chen, Y.; Luo, H.Q.; Sun, L.L.; Xu, M.T.; Yu, J.; Zhou, Q.G.; Meng, G.L.; Yang, S.J. Recent Update on the Pharmacological Effects and Mechanisms of Dihydromyricetin. Front. Pharmacol. 2018, 9, 1204. [Google Scholar] [CrossRef]
- Chen, J.N.; Wang, X.T.; Xia, T.; Bi, Y.H.; Liu, B.; Fu, J.F.; Zhu, R.Z. Molecular Mechanisms and Therapeutic Implications of Dihydromyricetin in Liver Disease. Biomed. Pharmacother. 2021, 142, 111927. [Google Scholar] [CrossRef]
- Ruan, L.P.; Yu, B.Y.; Fu, G.M.; Zhu, D.N. Improving the Solubility of Ampelopsin by Solid Dispersions and Inclusion Complexes. J. Pharm. Biomed. Anal. 2005, 38, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Yin, X.L.; Wang, X.; Li, X.H. Determination of Dihydromyricetin in Rat Plasma by LC-MS/MS and Its Application to a Pharmacokinetic Study. Pharm. Biol. 2017, 55, 657–662. [Google Scholar] [CrossRef]
- Fan, L.; Tong, Q.; Dong, W.W.; Yang, G.J.; Hou, X.L.; Xiong, W.; Shi, C.Y.; Fang, J.G.; Wang, W.Q. Tissue Distribution, Excretion, and Metabolic Profile of Dihydromyricetin, a Flavonoid from Vine Tea (Ampelopsis grossedentata) after Oral Administration in Rats. J. Agric. Food Chem. 2017, 65, 4597–4604. [Google Scholar] [CrossRef]
- Luo, F.; Zeng, D.D.; Chen, R.X.; Zafar, A.; Weng, L.; Wang, W.X.; Tian, Y.B.; Hasan, M.; Shu, X.G. PEGylated Dihydromyricetin-Loaded Nanoliposomes Coated with Tea Saponin Inhibit Bacterial Oxidative Respiration and Energy Metabolism. Food Funct. 2021, 12, 9007–9017. [Google Scholar] [CrossRef]
- Ye, J.; Bao, S.; Zhao, S.Y.; Zhu, Y.J.; Ren, Q.; Li, R.; Xu, X.H.; Zhang, Q. Self-Assembled Micelles Improve the Oral Bioavailability of Dihydromyricetin and Anti-Acute Alcoholism Activity. AAPS PharmSciTech 2021, 22, 111. [Google Scholar] [CrossRef]
- Yan, Q.H.; Li, M.Q.; Dong, L.Y.; Luo, J.; Zhong, X.H.; Shi, F.; Ye, G.; Zhao, L.; Fu, H.; Shu, G.; et al. Preparation, Characterization and Protective Effect of Chitosan—Tripolyphosphate Encapsulated Dihydromyricetin Nanoparticles on Acute Kidney Injury Caused by Cisplatin. Int. J. Biol. Macromol. 2023, 245, 125569. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.C.; Su, H.; Zheng, G.D.; Wang, W.J.; Yuan, E.; Zhang, Q.F. Fabrication and Characterization of Dihydromyricetin Encapsulated Zein-Caseinate Nanoparticles and Its Bioavailability in Rat. Food Chem. 2020, 330, 127245. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.Y.; Shi, C.Y.; Zhou, X.Y.; Lin, T.; Gong, Y.S.; Yin, M.X.; Fan, L.; Wang, W.Q.; Fang, J.G. Preparation of a Nanoscale Dihydromyricetin-Phospholipid Complex to Improve the Bioavailability: In Vitro and in Vivo Evaluations. Eur. J. Pharm. Sci. 2019, 138, 104994. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.R.; Zhang, H.; Shi, H.Y.; Zhang, D.; Zhang, Z.; Liu, H. Strategic Developments in the Drug Delivery of Natural Product Dihydromyricetin: Applications, Prospects, and Challenges. Drug Deliv. 2022, 29, 3052–3070. [Google Scholar] [CrossRef] [PubMed]
- Alqahtani, M.S.; Kazi, M.; Alsenaidy, M.A.; Ahmad, M.Z. Advances in Oral Drug Delivery. Front. Pharmacol. 2021, 12, 618411. [Google Scholar] [CrossRef]
- Ensign, L.M.; Cone, R.; Hanes, J. Oral Drug Delivery with Polymeric Nanoparticles: The Gastrointestinal Mucus Barriers. Adv. Drug Deliv. Rev. 2012, 64, 557–570. [Google Scholar] [CrossRef]
- Miyazaki, K.; Sasaki, A.; Mizuuchi, H. Advances in the Evaluation of Gastrointestinal Absorption Considering the Mucus Layer. Pharmaceutics 2023, 15, 2714. [Google Scholar] [CrossRef]
- Yu, M.R.; Xu, L.; Tian, F.L.; Su, Q.; Zheng, N.; Yang, Y.W.; Wang, J.L.; Wang, A.H.; Zhu, C.L.; Guo, S.Y.; et al. Rapid Transport of Deformation-Tuned Nanoparticles across Biological Hydrogels and Cellular Barriers. Nat. Commun. 2018, 9, 2607. [Google Scholar] [CrossRef]
- Yu, Y.L.; Xing, L.Y.; Li, L.; Wu, J.W.; He, J.H.; Huang, Y. Coordination of Rigidity Modulation and Targeting Ligand Modification on Orally-Delivered Nanoparticles for the Treatment of Liver Fibrosis. J. Control. Release 2022, 341, 215–226. [Google Scholar] [CrossRef]
- Mao, Y.L.; Feng, S.; Li, S.; Zhao, Q.F.; Di, D.H.; Liu, Y.F.; Wang, S.L. Chylomicron-Pretended Nano-Bio Self-Assembling Vehicle to Promote Lymphatic Transport and GALTs Target of Oral Drugs. Biomaterials 2019, 188, 173–186. [Google Scholar] [CrossRef]
- Zheng, B.; Pan, F.; Shi, M.F.; He, C.P.; He, B.B.; Wang, R.R.; Ren, G.L.; Yang, S.; Zhang, S.Q. 2-Monoacylglycerol Mimetic Liposomes to Promote Intestinal Lymphatic Transport for Improving Oral Bioavailability of Dihydroartemisinin. Int. J. Nanomed. 2024, 19, 5273–5295. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.G.; Xiong, W.; Perumalla, S.R.; Fang, J.G.; Sun, C.C. Solid-state characterization of optically pure (+) Dihydromyricetin extracted from Ampelopsis grossedentata leaves. Int. J. Pharm. 2016, 511, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.F.; Zhu, M.L.; Zhou, S.; Feng, W.Y.; Chen, H.Q. Rapamycin-Loaded mPEG-PLGA Nanoparticles Ameliorate Hepatic Steatosis and Liver Injury in Non-Alcoholic Fatty Liver Disease. Front. Chem. 2020, 8, 407. [Google Scholar] [CrossRef] [PubMed]
- Vilar-Gomez, E.; Calzadilla-Bertot, L.; Wong, V.W.S.; Castellanos, M.; Fuente, R.A.L.; Eslam, M.; Wong, G.L.H.; George, J.; Romero-Gomez, M.; Adams, L.A. Type 2 Diabetes and Metformin Use Associate with Outcomes of Patients with Nonalcoholic Steatohepatitis–Related, Child–Pugh A Cirrhosis. Clin. Gastroenterol. Hepatol. 2021, 19, 136–145.e6. [Google Scholar] [CrossRef]
- Lyu, Q.Y.; Chen, L.; Lin, S.Y.; Cao, H.; Teng, H. A Designed Self-Microemulsion Delivery System for Dihydromyricetin and Its Dietary Intervention Effect on High-Fat-Diet Fed Mice. Food Chem. 2022, 390, 132954. [Google Scholar] [CrossRef]
- Liu, L.; Sun, S.; Li, X.H. A Network Pharmacology-Based Approach to Explore the Effect of Dihydromyricetin on Non-Alcoholic Fatty Liver Rats via Regulating PPARG and CASP3. Mol. Cell. Probes 2023, 71, 101926. [Google Scholar] [CrossRef]
- Mi, H.Y.; Salick, M.R.; Jing, X.; Jacques, B.R.; Crone, W.C.; Peng, X.F.; Turng, L.S. Characterization of Thermoplastic Polyurethane/Polylactic Acid (TPU/PLA) Tissue Engineering Scaffolds Fabricated by Microcellular Injection Molding. Mater. Sci. Eng. C Mater. Biol. Appl. 2013, 33, 4767–4776. [Google Scholar] [CrossRef]
- Li, X.Y.; Montague, E.C.; Pollinzi, A.; Lofts, A.; Hoare, T. Design of Smart Size-, Surface-, and Shape-Switching Nanoparticles to Improve Therapeutic Efficacy. Small 2022, 18, e2104631. [Google Scholar] [CrossRef]
- Shushunova, N.A.; Mayorova, O.A.; Prikhozhdenko, E.S.; Goryacheva, O.A.; Kulikov, O.A.; Plastun, V.O.; Gusliakova, O.A.; Muslimov, A.R.; Inozemtseva, O.A.; Pyataev, N.A.; et al. Targeted Therapy for Glomerulonephritis Using Arterial Delivery of Encapsulated Etanercept. Int. J. Mol. Sci. 2023, 24, 2784. [Google Scholar] [CrossRef]
- Porter, C.J.H.; Trevaskis, N.L.; Charman, W.N. Lipids and Lipid-Based Formulations: Optimizing the Oral Delivery of Lipophilic Drugs. Nat. Rev. Drug Discov. 2007, 6, 231–248. [Google Scholar] [CrossRef]
- Eng, J.M.; Estall, J.L. Diet-Induced Models of Non-Alcoholic Fatty Liver Disease: Food for Thought on Sugar, Fat, and Cholesterol. Cells 2021, 10, 1805. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.X.; Zhang, W.X.; Yang, L.; Zhao, W.J.; Liu, Z.J.; Wang, E.; Wang, J. Phytochemical Gallic Acid Alleviates Nonalcoholic Fatty Liver Disease via AMPK-ACC-PPARa Axis through Dual Regulation of Lipid Metabolism and Mitochondrial Function. Phytomedicine 2023, 109, 154589. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Unit | DMY-hNE | DMY |
|---|---|---|---|
| AUC [A] | μg L−1 h | 2121.80 ± 74.20 [**] | 1065.70 ± 90.34 |
| MRT0-t [B] | h | 10.08 ± 0.56 [ns] | 10.74 ± 0.23 |
| t1/2 [C] | h | 8.71 ± 3.47 [ns] | 11.71 ± 4.55 |
| CL [D] | L h−1 kg−1 | 7.66 ± 0.63 [ns] | 12.65 ± 4.28 |
| V [E] | L kg−1 | 94.75 ± 30.51 [*] | 199.76 ± 10.85 |
| Cmax [F] | μg L−1 | 215.66 ± 76.47 [ns] | 117.34 ± 16.76 |
| Tmax 1 [G] | h | 0.25 | 0.25 |
| Tmax 2 | h | 6 | 6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Yin, M.; Tian, M.; Fang, J.; Xu, H. Stiff-Soft Hybrid Biomimetic Nano-Emulsion for Targeted Liver Delivery and Treatment of Early Nonalcoholic Fatty Liver Disease. Pharmaceutics 2024, 16, 1303. https://doi.org/10.3390/pharmaceutics16101303
Li J, Yin M, Tian M, Fang J, Xu H. Stiff-Soft Hybrid Biomimetic Nano-Emulsion for Targeted Liver Delivery and Treatment of Early Nonalcoholic Fatty Liver Disease. Pharmaceutics. 2024; 16(10):1303. https://doi.org/10.3390/pharmaceutics16101303
Chicago/Turabian StyleLi, Juan, Mingxing Yin, Maoxian Tian, Jianguo Fang, and Hanlin Xu. 2024. "Stiff-Soft Hybrid Biomimetic Nano-Emulsion for Targeted Liver Delivery and Treatment of Early Nonalcoholic Fatty Liver Disease" Pharmaceutics 16, no. 10: 1303. https://doi.org/10.3390/pharmaceutics16101303
APA StyleLi, J., Yin, M., Tian, M., Fang, J., & Xu, H. (2024). Stiff-Soft Hybrid Biomimetic Nano-Emulsion for Targeted Liver Delivery and Treatment of Early Nonalcoholic Fatty Liver Disease. Pharmaceutics, 16(10), 1303. https://doi.org/10.3390/pharmaceutics16101303