
Predicting the Release Mechanism of Amorphous Solid Dispersions: A Combination of Thermodynamic Modeling and In Silico Molecular Simulation

 , , , and
, , , and
Abstract
1. Introduction
2. Modeling, Materials, and Methods
2.1. Thermodynamic Modeling of Drug/Polymer/Water Phase Diagrams
2.1.1. Solid-Liquid Equilibrium
2.1.2. Liquid-Liquid Equilibrium
2.1.3. Glass-Transition
2.2. PC-SAFT
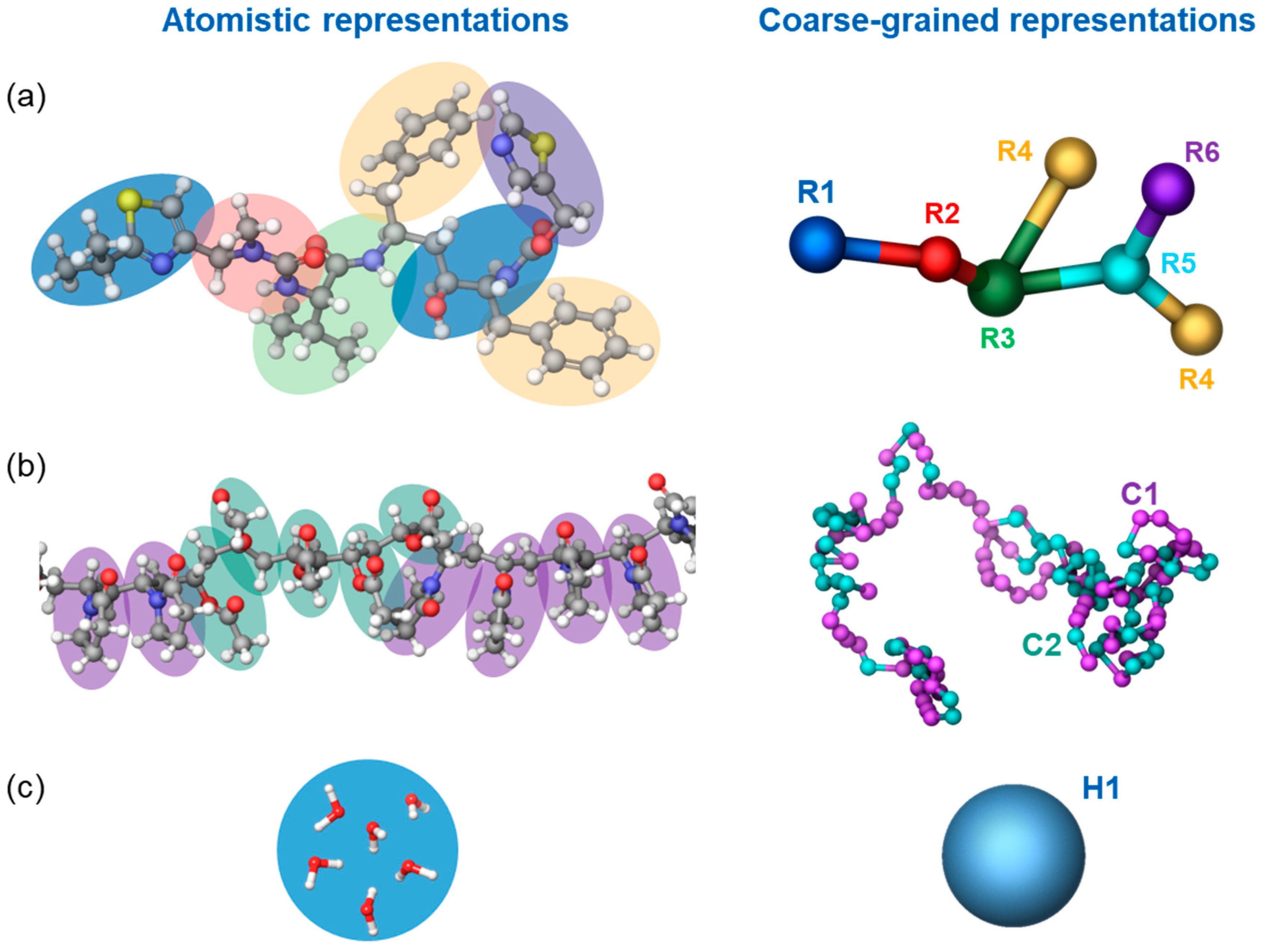
2.3. Molecular Simulation
2.3.1. Dissipative Particle Dynamics and Its Parameterization
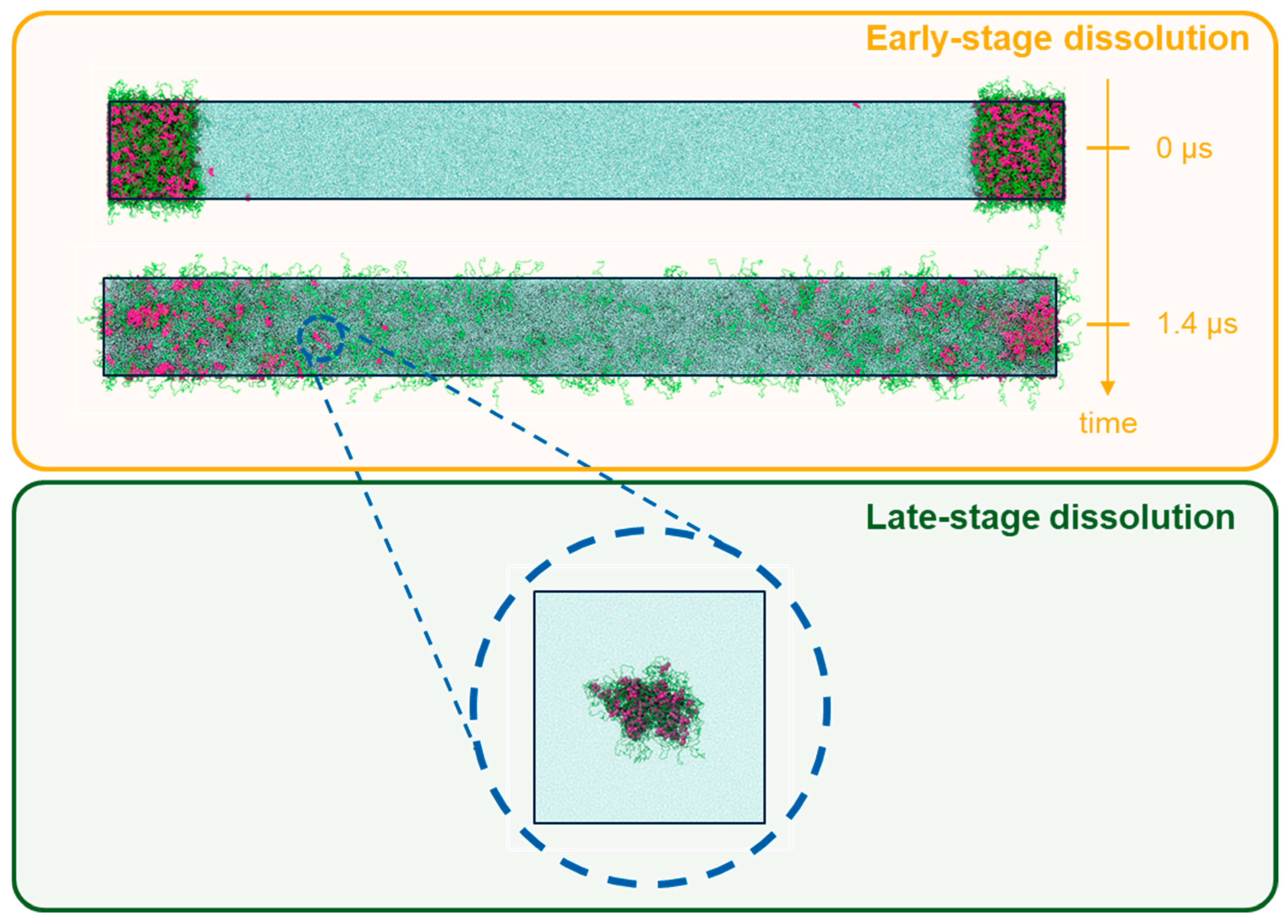
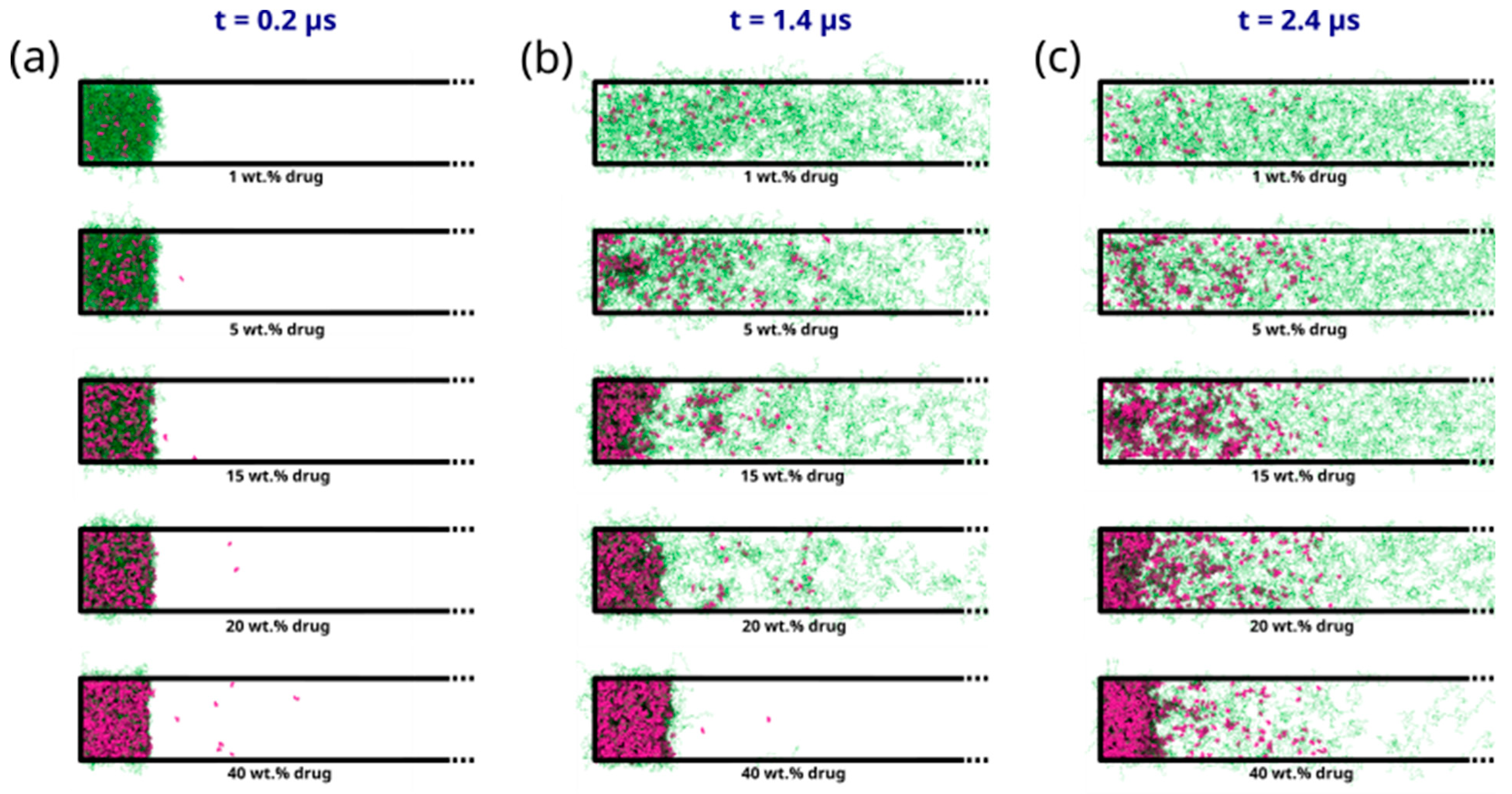
2.3.2. DPD Simulations of Early-Stage Dissolution
2.3.3. DPD Simulations of Late-Stage Dissolution
2.4. Materials and Experimental Methods
2.4.1. Preparation of ASD Discs
2.4.2. Microscopic Erosion Time Test (METT)
3. Results and Discussion
3.1. Thermodynamic Modeling of the ASD/Water Interface
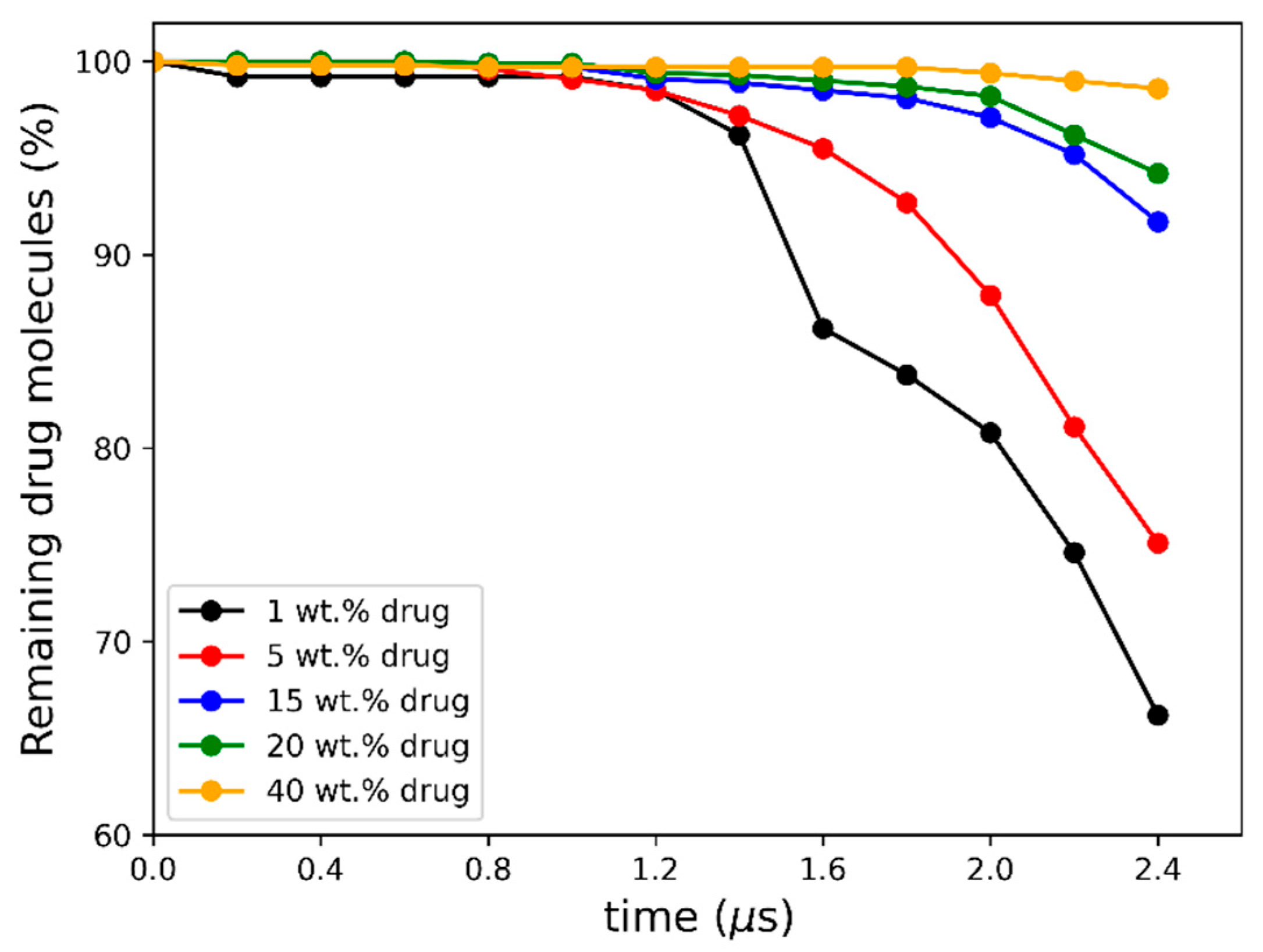
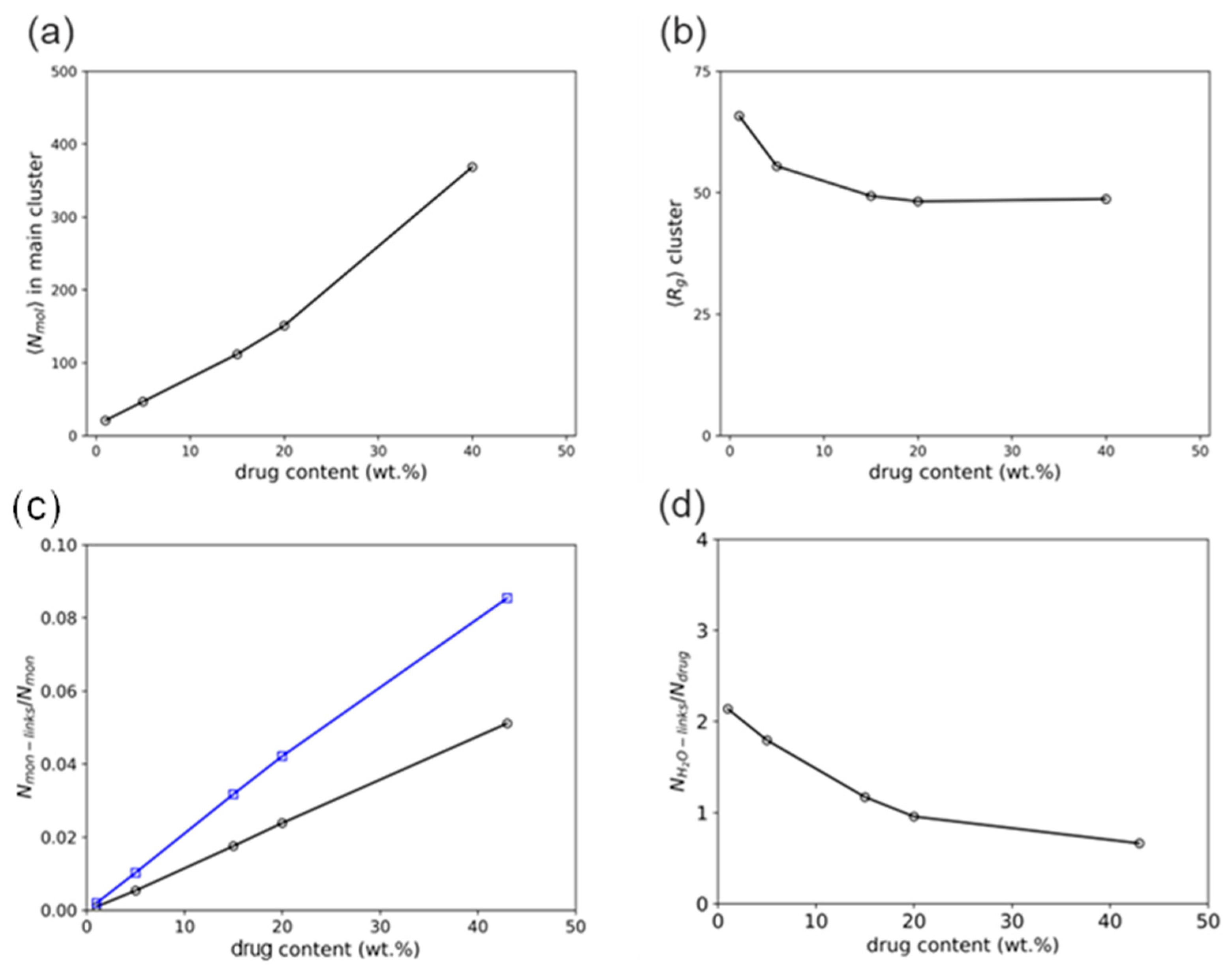
3.2. Molecular Simulations
4. Conclusions and Outlook
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| a | Helmholtz energy |
| AA | All-atom |
| Ai,Bi | Association sites A and B of molecule |
| bij | Equilibrium bond length |
| API | Active pharmaceutical ingredient |
| ASD | Amorphous solid dispersion |
| assoc | Association |
| Difference in solid and liquid heat capacity | |
| DPD | Dissipative particle dynamics |
| DL | Drug load |
| disp | Dispersion |
| eGT | Escape glass transition |
| FTIR | Fourier-transform infrared spectroscopy |
| h1+X | Enthalpy |
| hc | Hard-chain |
| Melting enthalpy | |
| K | Gordon-Taylor interaction parameter |
| kB | Boltzmann constant |
| kij | Binary interaction parameter |
| L | Liquid |
| LLPS | Liquid-liquid phase separation |
| LoR | Loss of Release |
| LJ | Lennard-Jones |
| m | Mass |
| MD | Molecular dynamics |
| METT | Microscopic Erosion Time Testing |
| mseg | Segment number |
| Mw | Molecular weight |
| Nassoc | Number of association sites |
| NPT | Thermal-isobaric (constant number of particles, pressure, and temperature) |
| NVT | Thermal-isochoric (constant number of particles, volume, and temperature) |
| PC-SAFT | Perturbed-Chain Statistical Associating Fluid Theory |
| PVPVA64 | Poly (vinylpyrrolidone-co-vinylacetate) |
| R | Universal gas constant |
| res | Residual |
| Rg | Radius of gyration |
| RIT | Ritonavir |
| SLE | Solid liquid equilibrium |
| T | Temperature |
| Tg | Glass transition temperature |
| Melting temperature | |
| u/kB | Dispersion-energy parameter |
| VCM | Vacuum compression molding |
| w | Mass fraction |
| x | Mole fraction |
| γ | Activity coefficient |
| εAiBi/kB | Association-energy parameter |
| κAiBi | Association-volume parameter |
| ρ | Density |
| θij | Equilibrium angle |
| σ | Segment diameter |
References
- Leuner, C.; Dressmann, J. Improving drug solubility for oral delivery using solid dispersions. Eur. J. Pharm. Biopharm. 2000, 50, 47–60. [Google Scholar] [CrossRef]
- Vo, C.L.-N.; Park, C.; Lee, B.-J. Current trends and future perspectives of solid dispersions containing poorly water-soluble drugs. Eur. J. Pharm. Biopharm. 2013, 85, 799–813. [Google Scholar] [CrossRef]
- Zhang, J.; Han, R.; Chen, W.; Zhang, W.; Ji, Y.; Chen, L.; Pan, H.; Yang, X.; Pan, W.; Ouyang, D. Analysis of the Literature and Patents on Solid Dispersions from 1980 to 2015. Molecules 2018, 23, 1697. [Google Scholar] [CrossRef]
- van den Mooter, G. The use of amorphous solid dispersions: A formulation strategy to overcome poor solubility and dissolution rate. Drug Discov. Today 2012, 9, 79–85. [Google Scholar] [CrossRef]
- Punčochová, K.; Heng, J.Y.Y.; Beránek, J.; Stěpánek, F. Investigation of drug-polymer interaction in solid dispersions by vapour sorption methods. Int. J. Pharm. 2014, 469, 159–167. [Google Scholar] [CrossRef]
- Prudic, A.; Ji, Y.; Sadowski, G. Thermodynamic Phase Behavior of API/Polymer Solid Dispersions. Mol. Pharm. 2014, 11, 2294–2304. [Google Scholar] [CrossRef]
- Paus, R.; Ji, Y.; Braak, F.; Sadowski, G. Dissolution of Crystalline Pharmaceuticals: Experimental Investigation and Thermodynamic Modeling. Ind. Eng. Chem. Res. 2015, 54, 731–742. [Google Scholar] [CrossRef]
- Que, C.; Lou, X.; Zemlyanov, D.Y.; Mo, H.; Indulkar, A.S.; Gao, Y.; Zhang, G.G.; Taylor, L.S. Insights into the Dissolution Behavior of Ledipasvir–Copovidone Amorphous Solid Dispersions: Role of Drug Loading and Intermolecular Interactions. Mol. Pharm. 2019, 16, 5054–5067. [Google Scholar] [CrossRef]
- Ahuja, N.; Katare, O.P.; Singh, B. Studies on dissolution enhancement and mathematical modeling of drug release of a poorly water-soluble drug using water-soluble carriers. Eur. J. Pharm. Biopharm. 2007, 65, 26–38. [Google Scholar] [CrossRef]
- Faiz Afzal, M.A.; Lehmkemper, K.; Sobich, E.; Hughes, T.F.; Giesen, D.J.; Zhang, T.; Krauter, C.M.; Winget, P.; Degenhardt, M.; Kyeremateng, S.O.; et al. Molecular-Level Examination of Amorphous Solid Dispersion Dissolution. Mol. Pharm. 2021, 18, 3999–4014. [Google Scholar] [CrossRef]
- Ji, Y.; Paus, R.; Prudic, A.; Luebbert, C.; Sadowski, G. A Novel Approach for Analyzing the Dissolution Mechanism of Solid Dispersions. Pharm. Res. 2015, 32, 2559–2578. [Google Scholar] [CrossRef]
- Lehmkemper, K.; Kyeremateng, S.O.; Heinzerling, O.; Degenhardt, M.; Sadowski, G. Impact of Polymer Type and Relative Humidity on the Long-Term Physical Stability of Amorphous Solid Dispersions. Mol. Pharm. 2017, 14, 4374–4386. [Google Scholar] [CrossRef]
- Hancock, B.C.; Shamblin, S.L.; Zografi, G. Molecular Mobility of Amorphous Pharmaceutical Solids Below Their Glass Transition Temperatures. Pharm. Res. 1995, 12, 799–806. [Google Scholar] [CrossRef]
- Yang, R.; Zhang, G.G.Z.; Zemlyanov, D.Y.; Purohit, H.S.; Taylor, L.S. Release Mechanisms of Amorphous Solid Dispersions: Role of Drug-Polymer Phase Separation and Morphology. J. Pharm. Sci. 2023, 112, 304–317. [Google Scholar] [CrossRef]
- Bochmann, E.S.; Steidel, A.; Rosenblatt, K.M.; Gessner, D.; Liepold, B. Assessment of the amorphous solid dispersion erosion behavior following a novel small-scale predictive approach. Eur. J. Pharm. Sci. 2021, 158, 105682. [Google Scholar] [CrossRef]
- Pudlas, M.; Kyeremateng, S.O.; Williams, L.A.M.; Kimber, J.A.; van Lishaut, H.; Kazarian, S.G.; Woehrle, G.H. Analyzing the impact of different excipients on drug release behavior in hot-melt extrusion formulations using FTIR spectroscopic imaging. Eur. J. Pharm. Sci. 2015, 67, 21–31. [Google Scholar]
- Punčochová, K.; Ewing, A.V.; Gajdošová, M.; Pekárek, T.; Beránek, J.; Kazarian, S.G.; Štěpánek, F. The Combined Use of Imaging Approaches to Assess Drug Release from Multicomponent Solid Dispersions. Pharm. Res. 2017, 34, 990–1001. [Google Scholar] [CrossRef]
- Punčochová, K.; Ewing, A.V.; Gajdošová, M.; Sarvašová, N.; Kazarian, S.G.; Beránek, J.; Štěpánek, F. Identifying the mechanisms of drug release from amorphous solid dispersions using MRI and ATR-FTIR spectroscopic imaging. Int. J. Pharm. 2015, 483, 256–267. [Google Scholar] [CrossRef]
- Tres, F.; Treacher, K.; Booth, J.; Hughes, L.P.; Wren, S.A.C.; Aylott, J.W.; Burley, J.C. Real time Raman imaging to understand dissolution performance of amorphous solid dispersions. J. Control. Release 2014, 188, 53–60. [Google Scholar] [CrossRef]
- Novakovic, D.; Isomäki, A.; Pleunis, B.; Fraser-Miller, S.J.; Peltonen, L.; Laaksonen, T.; Strachan, C.J. Understanding Dissolution and Crystallization with Imaging: A Surface Point of View. Mol. Pharm. 2018, 15, 5361–5373. [Google Scholar] [CrossRef]
- Shi, C.; Li, L.; Zhang, G.G.Z.; Borchardt, T.B. Direct Visualization of Drug-Polymer Phase Separation in Ritonavir-Copovidone Amorphous Solid Dispersions Using in situ Synchrotron X-ray Fluorescence Imaging of Thin Films. Mol. Pharm. 2019, 16, 4751–4754. [Google Scholar] [CrossRef] [PubMed]
- Krummnow, A.; Danzer, A.; Voges, K.; Dohrn, S.; Kyeremateng, S.O.; Degenhardt, M.; Sadowski, G. Explaining the Release Mechanism of Ritonavir/PVPVA Amorphous Solid Dispersions. Pharmaceutics 2022, 14, 1904. [Google Scholar] [CrossRef] [PubMed]
- Krummnow, A.; Danzer, A.; Voges, K.; Kyeremateng, S.O.; Degenhardt, M.; Sadowski, G. Kinetics of Water-Induced Amorphous Phase Separation in Amorphous Solid Dispersions via Raman Mapping. Pharmaceutics 2023, 15, 1395. [Google Scholar] [CrossRef] [PubMed]
- Dohrn, S.; Kyeremateng, S.O.; Bochmann, E.; Sobich, E.; Wahl, A.; Liepold, B.; Sadowski, G.; Degenhardt, M. Thermodynamic Modeling of the Amorphous Solid Dispersion-Water Interfacial Layer and Its Impact on the Release Mechanism. Pharmaceutics 2023, 15, 1539. [Google Scholar] [CrossRef] [PubMed]
- Luebbert, C.; Stoyanov, E. Tailored ASD destabilization—Balancing shelf life stability and dissolution performance with hydroxypropyl cellulose. Int. J. Pharm. X 2023, 5, 100187. [Google Scholar] [CrossRef]
- Kwei, T.K. The effect of hydrogen bonding on the glass transition temperatures of polymer mixtures. J. Polym. Sci. 1983, 22, 307–313. [Google Scholar] [CrossRef]
- Indulkar, A.S.; Lou, X.; Zhang, G.G.; Taylor, L.S. Insights into the Dissolution Mechanism of Ritonavir–Copovidone Amorphous Solid Dispersions: Importance of Congruent Release for Enhanced Performance. Mol. Pharm. 2019, 16, 1327–1339. [Google Scholar] [CrossRef]
- Deac, A.; Luebbert, C.; Qi, Q.; Courtney, R.M.; Indulkar, A.S.; Gao, Y.; Zhang, G.G.Z.; Sadowski, G.; Taylor, L.S. Dissolution Mechanisms of Amorphous Solid Dispersions: Application of Ternary Phase Diagrams to Explain Release Behavior. Mol. Pharm. 2024, 21, 1900–1918. [Google Scholar] [CrossRef]
- Yani, Y.; Kanaujia, P.; Chow, P.S.; Tan, R.B.H. Effect of API-Polymer Miscibility and Interaction on the Stabilization of Amorphous Solid Dispersion: A Molecular Simulation Study. Ind. Eng. Chem. Res. 2017, 56, 12698–12707. [Google Scholar] [CrossRef]
- Gupta, K.M.; Chin, X.; Kanaujia, P. Molecular Interactions between APIs and Enteric Polymeric Excipients in Solid Dispersion: Insights from Molecular Simulations and Experiments. Pharmaceutics 2023, 15, 1164. [Google Scholar] [CrossRef]
- Walden, D.M.; Bundey, Y.; Jagarapu, A.; Antontsev, V.; Chakravarty, K.; Varshney, J. Molecular Simulation and Statistical Learning Methods toward Predicting Drug-Polymer Amorphous Solid Dispersion Miscibility, Stability, and Formulation Design. Molecules 2021, 26, 182. [Google Scholar] [CrossRef] [PubMed]
- Kapourani, A.; Vardaka, E.; Katopodis, K.; Kachrimanis, K.; Barmpalexis, P. Rivaroxaban polymeric amorphous solid dispersions: Moisture-induced thermodynamic phase behavior and intermolecular interactions. Eur. J. Pharm. Biopharm. 2019, 145, 98–112. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.; Ouyang, D. Investigating the molecular dissolution process of binary solid dispersions by molecular dynamics simulations. Asian J. Pharm. Sci. 2018, 13, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Jarray, A.; Gerbaud, V.; Hemati, M. Structure of aqueous colloidal formulations used in coating and agglomeration processes: Mesoscale model and experiments. Powder Technol. 2016, 291, 244–261. [Google Scholar] [CrossRef]
- Jarray, A.; Gerbaud, V.; Hemati, M. Polymer-plasticizer compatibility during coating formulation: A multi-scale investigation. Prog. Org. Coat. 2016, 101, 195–206. [Google Scholar] [CrossRef]
- Dohrn, S.; Luebbert, C.; Lehmkemper, K.; Kyeremateng, S.O.; Degenhardt, M.; Sadowski, G. Phase behavior of pharmaceutically relevant polymer/solvent mixtures. Int. J. Pharm. 2020, 577, 119065. [Google Scholar] [CrossRef]
- Dohrn, S.; Luebbert, C.; Lehmkemper, K.; Kyeremateng, S.O.; Degenhardt, M.; Sadowski, G. Solvent influence on the phase behavior and glass transition of Amorphous Solid Dispersions. Eur. J. Pharm. Biopharm. 2021, 158, 132–142. [Google Scholar] [CrossRef]
- Gross, J.; Sadowski, G. Application of the Perturbed-Chain SAFT Equation of State to Associating Systems. Ind. Eng. Chem. Res. 2002, 41, 5510–5515. [Google Scholar] [CrossRef]
- Simha, R.; Boyer, R.F. On a General Relation Involving the Glass Temperature and Coefficients of Expansion of Polymers. J. Chem. Phys. 1962, 37, 1003–1007. [Google Scholar] [CrossRef]
- Six, K.; Verreck, G.; Peeters, J.; Brewster, M.; van den Mooter, G. Increased physical stability and improved dissolution properties of itraconazole, a class II drug, by solid dispersions that combine fast- and slow-dissolving polymers. J. Pharm. Sci. 2004, 93, 124–131. [Google Scholar] [CrossRef]
- Prudic, A.; Kleetz, T.; Korf, M.; Ji, Y.; Sadowski, G. Influence of Copolymer Composition on the Phase Behavior of Solid Dispersions. Mol. Pharm. 2014, 11, 4189–4198. [Google Scholar] [CrossRef] [PubMed]
- Hancock, B.C.; Zografi, G. The relationship between the glass transition temperature and the water content of amorphous pharmaceutical solids. Pharm. Res. 1994, 11, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Hallbrucker, A.; Mayer, E.; Johari, G.P. The heat capacity and glass transition of hyperquenched glassy water. Philos. Mag. B 1989, 60, 179–187. [Google Scholar] [CrossRef]
- Lehmkemper, K.; Kyeremateng, S.O.; Heinzerling, O.; Degenhardt, M.; Sadowski, G. Long-Term Physical Stability of PVP- and PVPVA-Amorphous Solid Dispersions. Mol. Pharm. 2017, 14, 157–171. [Google Scholar] [CrossRef] [PubMed]
- Cameretti, L.F.; Sadowski, G. Modeling of aqueous amino acid and polypeptide solutions with PC-SAFT. Chem. Eng. Process. 2008, 47, 1018–1025. [Google Scholar] [CrossRef]
- Berthelot, D. Sur le mélange des gaz. Comptes Rendus Hebd. Séances L’académie Sci. 1898, 126, 1703–1855. [Google Scholar]
- Lorentz, H.A. Ueber die Anwendung des Satzes vom Virial in der kinetischen Theorie der Gase. Ann. Phys. 1881, 248, 127–136. [Google Scholar] [CrossRef]
- Wolbach, J.P.; Sandler, S.I. Using Molecular Orbital Calculations to Describe the Phase Behavior of Cross-associating Mixtures. Ind. Eng. Chem. Res. 1998, 37, 2917–2928. [Google Scholar] [CrossRef]
- Schrödinger Materials Science Suite, Version 23-3; Schrödinger Materials Maestro-Materials Science: New York, NY, USA, 2019.
- Lu, C.; Wu, C.; Ghoreishi, D.; Chen, W.; Wang, L.; Damm, W.; Ross, G.A.; Dahlgren, M.K.; Russell, E.; Bargen, C.D.; et al. OPLS4: Improving Force Field Accuracy on Challenging Regimes of Chemical Space. J. Chem. Theory Comput. 2021, 17, 4291–4300. [Google Scholar] [CrossRef]
- Predescu, C.; Lerer, A.K.; Lippert, R.A.; Towles, B.; Grossman, J.P.; Dirks, R.M.; Shaw, D.E. The u-series: A separable decomposition for electrostatics computation with improved accuracy. J. Chem. Phys. 2020, 152, 084113. [Google Scholar] [CrossRef]
- Nosé, S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 1984, 81, 511–519. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A Gen. Phys. 1985, 31, 1695–1697. [Google Scholar] [CrossRef]
- Schrödinger: Maestro-Desmond Interoperability Tools, v. 2023-1; Schrodinger: New York, NY, USA, 2023.
- Bowers, K.J.; Chow, E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Kllepeis, J.L.; Kolossváry, I. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In Proceedings of the ACM/IEEE Conference on Supercomputing (SC06), Tampa, Florida, 11–17 November 2006. [Google Scholar]
- Groot, R.D.; Warren, P.B. Dissipative particle dynamics: Bridging the gap between atomistic and mesoscopic simulation. J. Chem. Phys. 1997, 107, 4423–4435. [Google Scholar] [CrossRef]
- Yang, R.; Zhang, G.G.Z.; Zemlyanov, D.Y.; Purohit, H.S.; Taylor, L.S. Drug Release from Surfactant-Containing Amorphous Solid Dispersions: Mechanism and Role of Surfactant in Release Enhancement. Pharm. Res. 2023, 40, 2817–2845. [Google Scholar] [CrossRef] [PubMed]
- Deac, A.; Qi, Q.; Indulkar, A.S.; Purohit, H.S.; Gao, Y.; Zhang, G.G.Z.; Taylor, L.S. Dissolution Mechanisms of Amorphous Solid Dispersions: Role of Drug Load and Molecular Interactions. Mol. Pharm. 2023, 20, 722–737. [Google Scholar] [CrossRef]
- Indulkar, A.S.; Lou, X.; Zhang, G.G.Z.; Taylor, L.S. Role of Surfactants on Release Performance of Amorphous Solid Dispersions of Ritonavir and Copovidone. Pharm. Res. 2022, 39, 381–397. [Google Scholar] [CrossRef]
 ) and drug-rich (
) and drug-rich ( ) phases, respectively, after LLPS at eGT.
) and drug-rich () phases, respectively, after LLPS at eGT.
) phases, respectively, after LLPS at eGT.
) and drug-rich () phases, respectively, after LLPS at eGT.


 ) and drug-rich (
) and drug-rich ( ) phases after LLPS at the interface. A schematic connection between the hydration pathway and the MD dissolution simulation of 20 wt% DL ASD is shown on the right.
) and drug-rich () phases after LLPS at the interface. A schematic connection between the hydration pathway and the MD dissolution simulation of 20 wt% DL ASD is shown on the right.
) phases after LLPS at the interface. A schematic connection between the hydration pathway and the MD dissolution simulation of 20 wt% DL ASD is shown on the right.
) and drug-rich () phases after LLPS at the interface. A schematic connection between the hydration pathway and the MD dissolution simulation of 20 wt% DL ASD is shown on the right.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Component i | |||
|---|---|---|---|
| ritonavir [37] | 398.15 | 63.15 | 224 |
| ritonavir | 1151 [37] | 323.5 [37] |
| PVPVA64 | 1190 [40] | 384.15 [41] |
| water | 1000 [42] | 138.00 [43] |
| Mixture | /K |
|---|---|
| ritonavir/PVPVA64 | −18.83 [22] |
| ritonavir/water | −304.87 [22] |
| PVPVA64/water | 34.82 [22] |
| ritonavir [37] | 721 | 0.0220 | 3.900 | 305.787 | 1041.0 | 0.02 | 4/4 |
| PVPVA64 [44] | 65000 | 0.0372 | 2.947 | 205.271 | 0 | 0.02 | 653/653 |
| water [45] | 18.015 | 0.0669 | * | 353.950 | 2425.7 | 0.0451 | 1/1 |
| Mixture | ||
|---|---|---|
| ritonavir/PVPVA64 [37] | 0 | 0.019 |
| ritonavir/water [37] | 0.00006 | −0.059 |
| PVPVA64/water [44] | 0 | −0.156 |
) and drug-rich () phases, respectively, after LLPS.
) and drug-rich () phases, respectively, after LLPS.| Ritonavir DL in the Dry ASD | Phase | Phase Mass Fraction at eGT | Phase Composition (Mass Fraction) at eGT | Phase Tg [°C] | |||
|---|---|---|---|---|---|---|---|
| Water | Ritonavir | PVPVA64 | |||||
| 5 wt% |  | Polymer-rich phase | 0.98 | 0.11 | 0.05 | 0.84 | 37.41 |
 | drug-rich phase | 0.02 | 0.06 | 0.24 | 0.71 | 50.38 | |
| 15 wt% |  | Polymer-rich phase | 0.58 | 0.12 | 0.03 | 0.85 | 32.40 |
 | drug-rich phase | 0.43 | 0.05 | 0.29 | 0.66 | 49.77 | |
| 20 wt% |  | Polymer-rich phase | 0.46 | 0.13 | 0.03 | 0.84 | 28.71 |
 | drug-rich phase | 0.54 | 0.05 | 0.31 | 0.64 | 48.68 | |
| 40 wt% |  | Polymer-rich phase | 0.13 | 0.16 | 0.01 | 0.83 | 18.51 |
 | drug-rich phase | 0.87 | 0.04 | 0.44 | 0.52 | 45.99 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Walter, S.; Mileo, P.G.M.; Afzal, M.A.F.; Kyeremateng, S.O.; Degenhardt, M.; Browning, A.R.; Shelley, J.C. Predicting the Release Mechanism of Amorphous Solid Dispersions: A Combination of Thermodynamic Modeling and In Silico Molecular Simulation. Pharmaceutics 2024, 16, 1292. https://doi.org/10.3390/pharmaceutics16101292
Walter S, Mileo PGM, Afzal MAF, Kyeremateng SO, Degenhardt M, Browning AR, Shelley JC. Predicting the Release Mechanism of Amorphous Solid Dispersions: A Combination of Thermodynamic Modeling and In Silico Molecular Simulation. Pharmaceutics. 2024; 16(10):1292. https://doi.org/10.3390/pharmaceutics16101292
Chicago/Turabian StyleWalter, Stefanie, Paulo G. M. Mileo, Mohammad Atif Faiz Afzal, Samuel O. Kyeremateng, Matthias Degenhardt, Andrea R. Browning, and John C. Shelley. 2024. "Predicting the Release Mechanism of Amorphous Solid Dispersions: A Combination of Thermodynamic Modeling and In Silico Molecular Simulation" Pharmaceutics 16, no. 10: 1292. https://doi.org/10.3390/pharmaceutics16101292
APA StyleWalter, S., Mileo, P. G. M., Afzal, M. A. F., Kyeremateng, S. O., Degenhardt, M., Browning, A. R., & Shelley, J. C. (2024). Predicting the Release Mechanism of Amorphous Solid Dispersions: A Combination of Thermodynamic Modeling and In Silico Molecular Simulation. Pharmaceutics, 16(10), 1292. https://doi.org/10.3390/pharmaceutics16101292