
The Development of an Oral Solution Containing Nirmatrelvir and Ritonavir and Assessment of Its Pharmacokinetics and Stability

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Solubility Studies
2.3. Formulation Screening
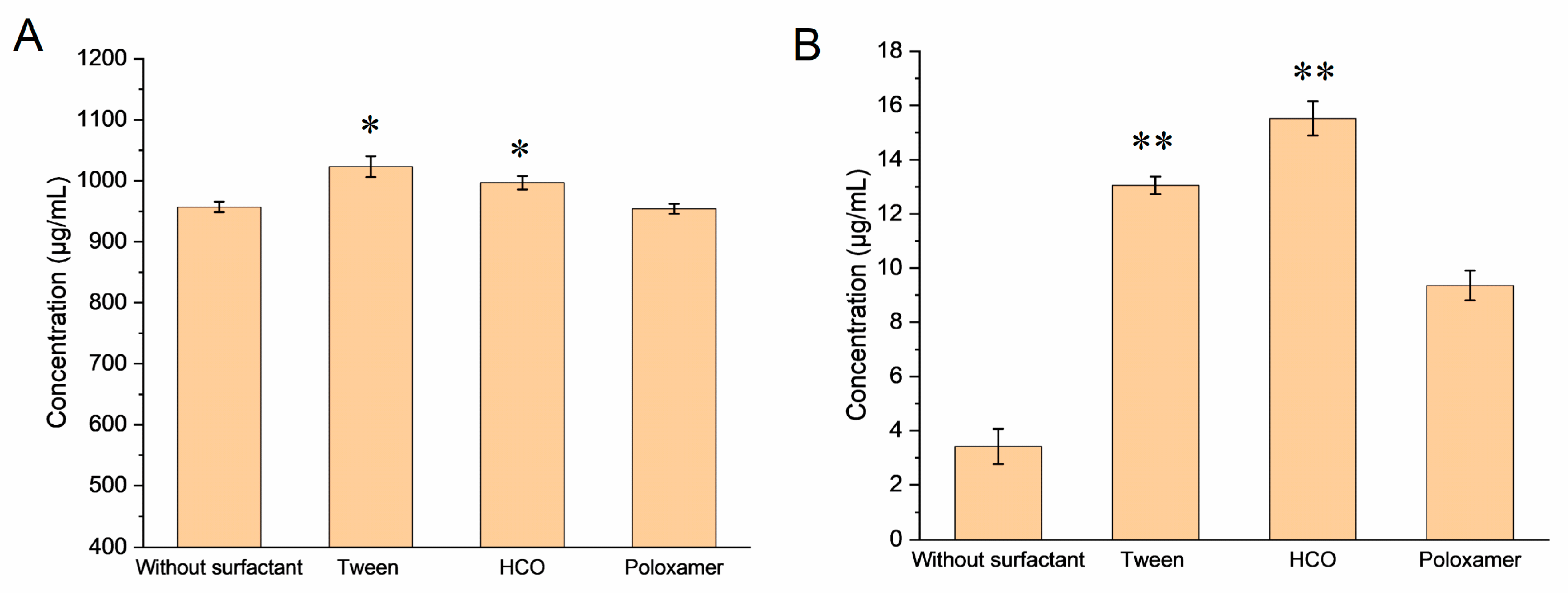
2.3.1. Solubilization Effect of Different Surfactants
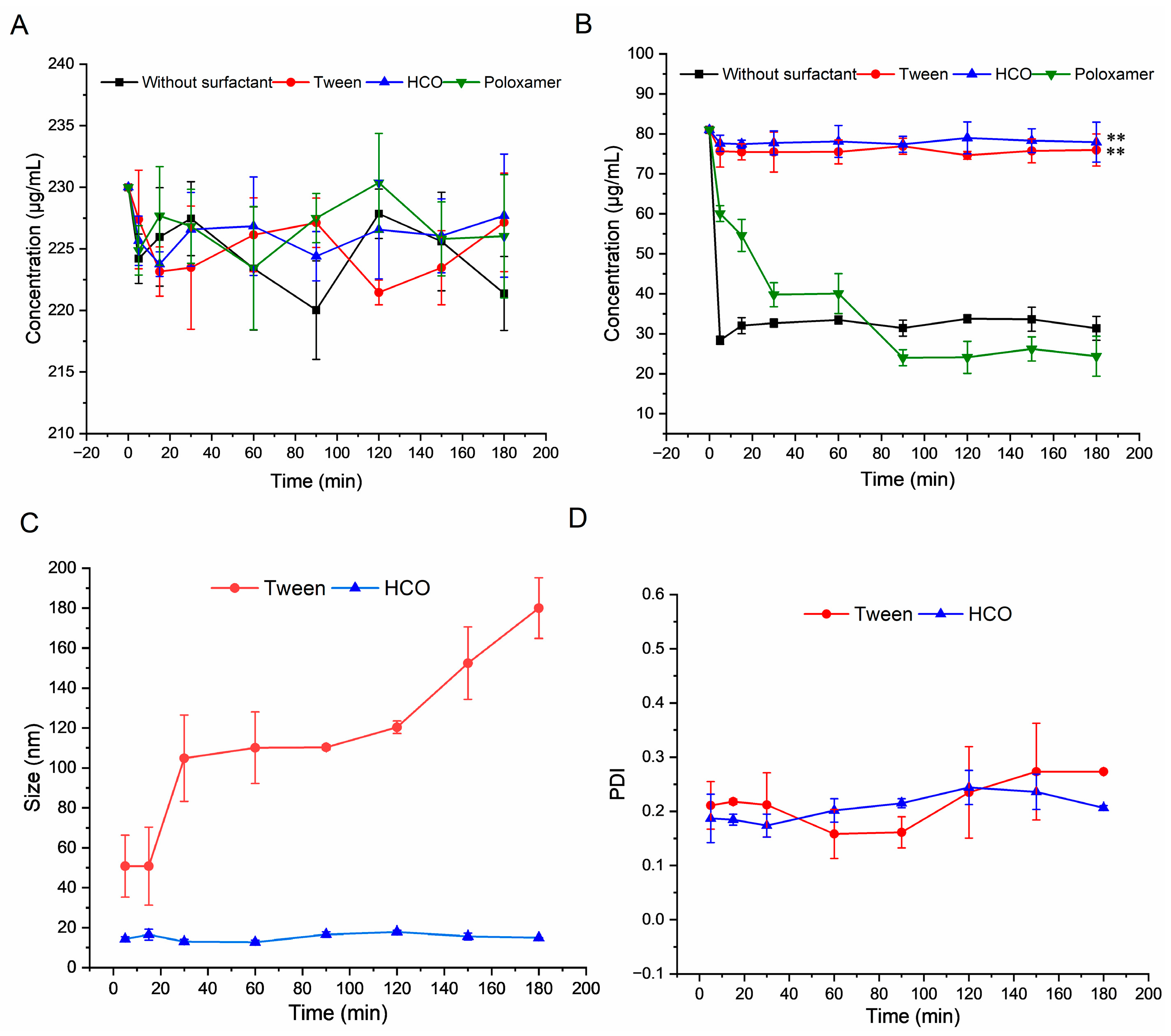
2.3.2. Recrystallization Inhibition Abilities of Different Surfactants
2.4. Formulations Preparation
2.5. Release under Non-Sink Conditions
2.6. In Vivo Pharmacokinetics in Rats
2.7. In Vivo Pharmacokinetics in a Beagle
2.8. Quantification of NRV and RTV in Plasma
2.9. Storage Stability
2.10. Statistical Analysis
3. Results and Discussion
3.1. Solubility of NRV and RTV
3.2. Screening of Surfactants
3.2.1. Solubilizing Effect of Different Surfactants
3.2.2. Inhibition of Recrystallization of NRV/RTV by Different Surfactants
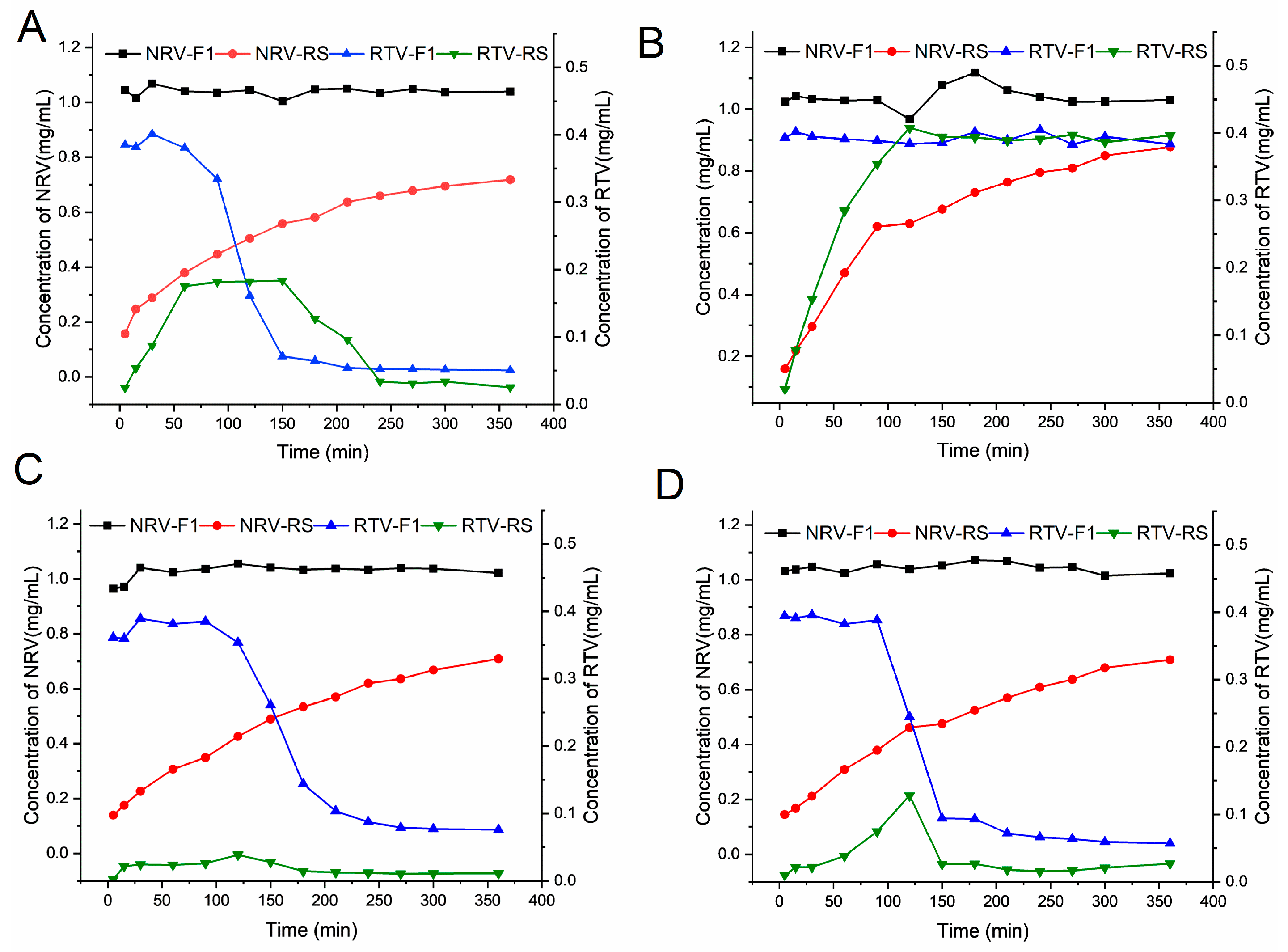
3.3. Release under Non-Sink Conditions
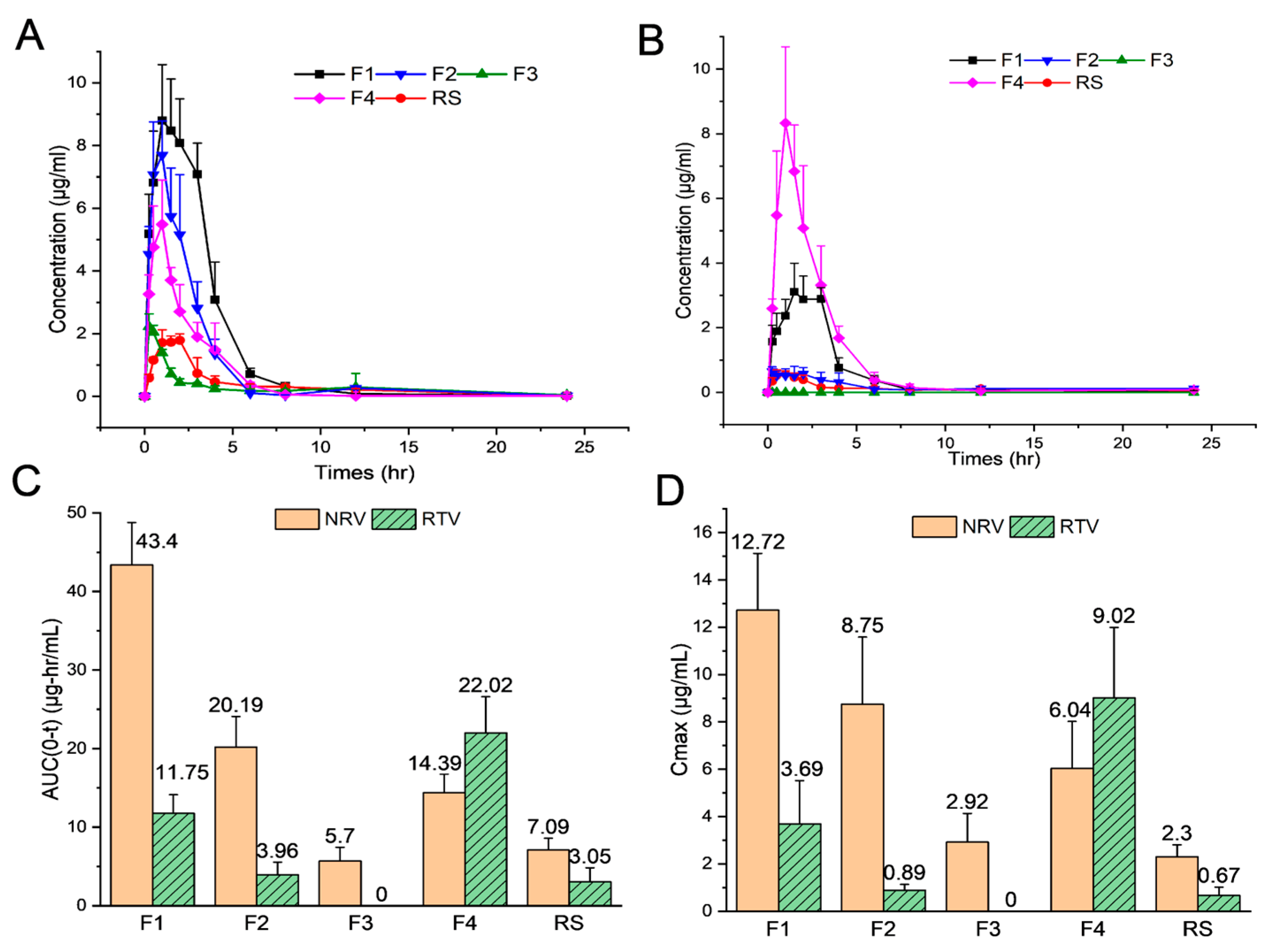
3.4. In Vivo Pharmacokinetics in Rats
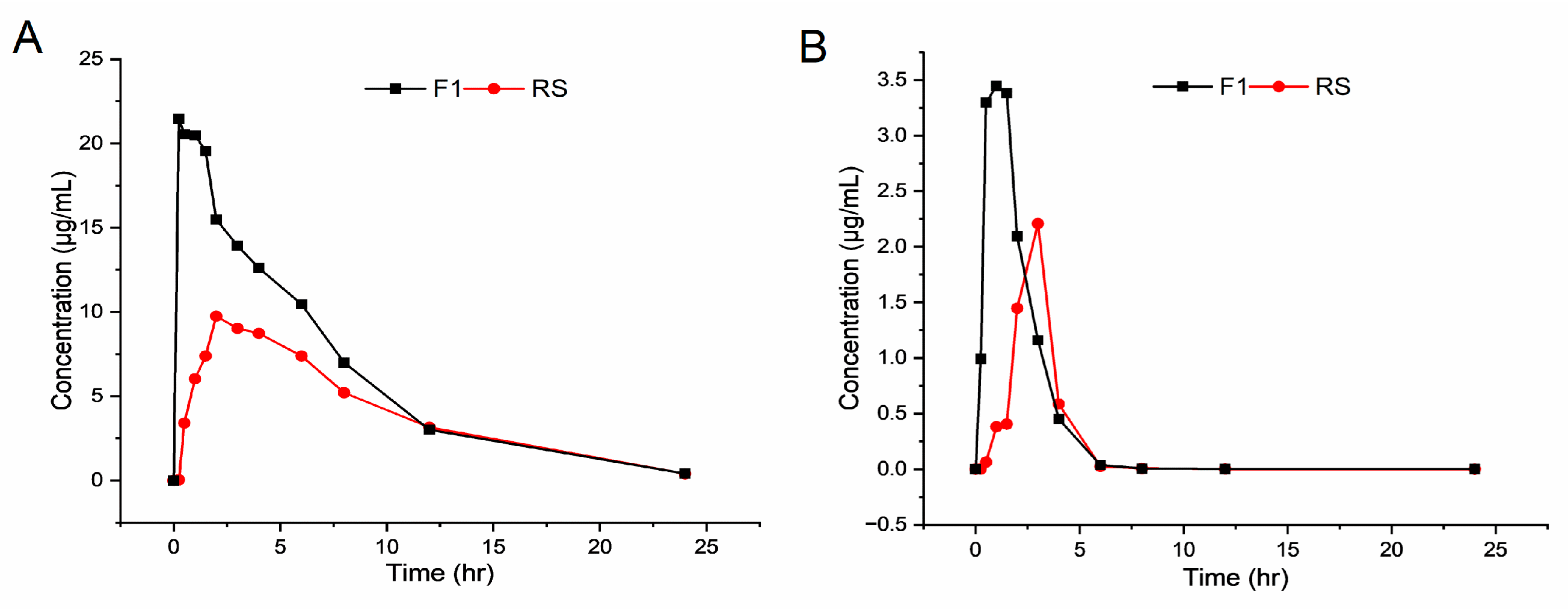
3.5. In Vivo Pharmacokinetics in a Beagle
3.6. Stability Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. WHO Coronavirus (COVID-19) Dashboard. Available online: https://covid19.who.int/ (accessed on 5 October 2022).
- Chen, N.; Zhou, M.; Dong, X.; Qu, J.; Gong, F.; Han, Y.; Qiu, Y.; Wang, J.; Liu, Y.; Wei, Y.; et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet 2020, 395, 507–513. [Google Scholar] [CrossRef]
- Khamis, F.; Memish, Z.; Bahrani, M.A.; Dowaiki, S.A.; Pandak, N.; Bolushi, Z.A.; Salmi, I.A.; Al-Zakwani, I. Prevalence and predictors of in-hospital mortality of patients hospitalized with COVID-19 infection. J. Infect. Public Health 2021, 14, 759–765. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. Paxlovid® (Nirmatrelvir Co-Packaged with Ritonavir) for the Treatment of Mild-to-Moderate Coronavirus Disease 2019 (COVID-19) in Certain Adults and Pediatric Patients. Available online: https://www.fda.gov/media/155049/download (accessed on 8 October 2023).
- Walsh, J.; Ranmal, S.R.; Ernest, T.B.; Liu, F. Patient acceptability, safety and access: A balancing act for selecting age-appropriate oral dosage forms for paediatric and geriatric populations. Int. J. Pharm. 2018, 536, 547–562. [Google Scholar] [CrossRef]
- Hammond, J.; Leister-Tebbe, H.; Gardner, A.; Abreu, P.; Bao, W.; Wisemandle, W.; Baniecki, M.; Hendrick, V.M.; Damle, B.; Simón-Campos, A.; et al. Oral Nirmatrelvir for High-Risk, Nonhospitalized Adults with COVID-19. N. Engl. J. Med. 2022, 386, 1397–1408. [Google Scholar] [CrossRef]
- Burdet, C.; Ader, F. Real-world effectiveness of oral antivirals for COVID-19. Lancet 2022, 400, 1175–1176. [Google Scholar] [CrossRef]
- Japan Pharmaceuticals and Medical Devices Agency, The interview form of Paxlovid®. Available online: https://www.pmda.go.jp/PmdaSearch/iyakuDetail/GeneralList/62501B5 (accessed on 9 October 2023).
- EMA European Medicines Agency. Overview of Paxlovid®. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/paxlovid (accessed on 9 October 2023).
- Owen, D.R.; Allerton, C.M.N.; Anderson, A.S.; Aschenbrenner, L.; Avery, M.; Berritt, S.; Boras, B.; Cardin, R.D.; Carlo, A.; Coffman, K.J.; et al. An oral SARS-CoV-2 Mpro inhibitor clinical candidate for the treatment of COVID-19. Science 2021, 374, 1586–1593. [Google Scholar] [CrossRef]
- Hilgenfeld, R. From SARS to MERS: Crystallographic studies on coronaviral proteases enable antiviral drug design. FEBS J. 2014, 281, 4085–4096. [Google Scholar] [CrossRef]
- Sevrioukova, I.F.; Poulos, T.L. Structure and mechanism of the complex between cytochrome P4503A4 and ritonavir. Proc. Natl. Acad. Sci. USA 2010, 107, 18422–18427. [Google Scholar] [CrossRef]
- Awad, A.; Madla, C.M.; Gavins, F.K.H.; Allahham, N.; Trenfield, S.J.; Basit, A.W. Chapter 20—Liquid dosage forms. In The Science and Practice of Pharmacy, 23rd ed.; Adejare, A.B.T.-R., Ed.; Elsevier: Cambridge, MA, USA, 2021; pp. 359–379. [Google Scholar]
- Chu, J.N.; Traverso, G. Foundations of gastrointestinal-based drug delivery and future developments. Nat. Rev. Gastroenterol. Hepatol. 2022, 19, 219–238. [Google Scholar] [CrossRef]
- Takagi, T.; Ramachandran, C.; Bermejo, M.; Yamashita, S.; Yu, L.X.; Amidon, G.L. A provisional biopharmaceutical classification of the top 200 oral drug products in the United States, Great Britain, Spain, and Japan. Mol. Pharm. 2006, 3, 631–643. [Google Scholar] [CrossRef]
- Salunke, S.; O’Brien, F.; Cheng Thiam Tan, D.; Harris, D.; Math, M.-C.; Ariën, T.; Klein, S.; Timpe, C. Oral drug delivery strategies for development of poorly water soluble drugs in paediatric patient population. Adv. Drug Del. Rev. 2022, 190, 114507. [Google Scholar] [CrossRef]
- Trivedi, J.S.; Yue, Z. Solubilization using cosolvent approach. In Water-Insoluble Drug Formulation, 3rd ed.; Liu, R., Ed.; CRC Press: Boca Raton, FL, USA, 2018; pp. 177–209. [Google Scholar]
- Salunke, S.; O’Brien, F.; Cheng Thiam Tan, D.; Harris, D.; Math, M.-C.; Ariën, T.; Klein, S.; Timpe, C. Liquisolid technique and its applications in pharmaceutics. Asian J. Pharm. Sci. 2017, 12, 115–123. [Google Scholar]
- Price, D.J.; Ditzinger, F.; Koehl, N.J.; Jankovic, S.; Tsakiridou, G.; Nair, A.; Holm, R.; Kuentz, M.; Dressman, J.B.; Saal, C. Approaches to increase mechanistic understanding and aid in the selection of precipitation inhibitors for supersaturating formulations—A PEARRL review. J. Pharm. Pharmacol. 2018, 71, 483–509. [Google Scholar] [CrossRef]
- Zhu, S.; Yu, R.; Qian, G.; Deng, L. A supersaturating drug delivery system to enhance the oral bioavailability of nilotinib. J. Drug Deliv. Sci. Technol. 2022, 68, 103038. [Google Scholar] [CrossRef]
- Xi, Z.; Fei, Y.; Wang, Y.; Lin, Q.; Ke, Q.; Feng, G.; Xu, L. Solubility improvement of curcumin by crystallization inhibition from polymeric surfactants in amorphous solid dispersions. J. Drug Deliv. Sci. Technol. 2023, 83, 104351. [Google Scholar] [CrossRef]
- Strickley, R.G. Solubilizing excipients in oral and injectable formulations. Pharm. Res. 2004, 21, 201–230. [Google Scholar] [CrossRef]
- Chen, J.; Ormes, J.D.; Higgins, J.D.; Taylor, L.S. Impact of Surfactants on the Crystallization of Aqueous Suspensions of Celecoxib Amorphous Solid Dispersion Spray Dried Particles. Mol. Pharm. 2015, 12, 533–541. [Google Scholar] [CrossRef]
- Zhang, W.; Hate, S.S.; Russell, D.J.; Hou, H.H.; Nagapudi, K. Impact of Surfactant and Surfactant-Polymer Interaction on Desupersaturation of Clotrimazole. J. Pharm. Sci. 2019, 108, 3262–3271. [Google Scholar] [CrossRef]
- Alqahtani, M.S.; Kazi, M.; Alsenaidy, M.A.; Ahmad, M.Z. Advances in Oral Drug Delivery. Front. Pharmacol. 2021, 12, 618411. [Google Scholar] [CrossRef]
- Riebesehl, B.U. Chapter 29—Drug Delivery with Organic Solvents or Colloidal Dispersed Systems. In The Practice of Medicinal Chemistry, 4th ed.; Wermuth, C.G., Ed.; Academic Press: San Diego, CA, USA, 2015; pp. 699–722. [Google Scholar]
- U.S. Food and Drug Administration. The Label of NORVIR®: Drugs @ FDA. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/022417Orig1s025,020659Orig1s073,209512Orig1s008lbl.pdf (accessed on 4 September 2023).
- Baka, E.; Comer, J.E.A.; Takács-Novák, K. Study of equilibrium solubility measurement by saturation shake-flask method using hydrochlorothiazide as model compound. J. Pharm. Biomed. Anal. 2008, 46, 335–341. [Google Scholar] [CrossRef]
- Bevernage, J.; Brouwers, J.; Brewster, M.E.; Augustijns, P. Evaluation of gastrointestinal drug supersaturation and precipitation: Strategies and issues. Int. J. Pharm. 2013, 453, 25–35. [Google Scholar] [CrossRef]
- Schver, G.C.R.; Lee, M.P.I. On the usefulness of sink index in characterizing the degree of nonsinkness in dissolution studies. Int. J. Pharm. 2021, 605, 120845. [Google Scholar] [CrossRef]
- Sun, D.D.; Wen, H.; Taylor, L.S. Non-Sink Dissolution Conditions for Predicting Product Quality and In Vivo Performance of Supersaturating Drug Delivery Systems. J. Pharm. Sci. 2016, 105, 2477–2488. [Google Scholar] [CrossRef]
- Guan, Q.; Ma, Q.; Zhao, Y.; Jiang, X.; Zhang, H.; Liu, M.; Wang, Z.; Han, J. Cellulose derivatives as effective recrystallization inhibitor for ternary ritonavir solid dispersions: In vitro-in vivo evaluation. Carbohydr. Polym. 2021, 273, 118562. [Google Scholar] [CrossRef]
- Guyon, J.; Novion, M.; Fulda, V.; Ducint, D.; Molimard, M.; Couzi, L.; Kaminski, H.; Salvo, F.; Bouchet, S. A UPLC-MS/MS Method for Plasma Biological Monitoring of Nirmatrelvir and Ritonavir in the Context of SARS-CoV-2 Infection and Application to a Case. J. Am. Soc. Mass Spectrom. 2022, 33, 1975–1981. [Google Scholar] [CrossRef]
- Cirri, M.; Maestrelli, F.; Mennini, N.; Di Cesare Mannelli, L.; Micheli, L.; Ghelardini, C.; Mura, P. Development of a stable oral pediatric solution of hydrochlorothiazide by the combined use of cyclodextrins and hydrophilic polymers. Int. J. Pharm. 2020, 587, 119692. [Google Scholar] [CrossRef]
- Binson, G.; Beuzit, K.; Migeot, V.; Marco, L.; Troussier, B.; Venisse, N.; Dupuis, A. Preparation and Physicochemical Stability of Liquid Oral Dosage Forms Free of Potentially Harmful Excipient Designed for Pediatric Patients. Pharmaceutics 2019, 11, 190. [Google Scholar] [CrossRef]
- Fuss, D.; Gondé, H.; Lamoureux, F.; Pereira, T.; Colnot, M.; Buchbinder, N.; Coquard, A.; Varin, R.; Hervouët, C. Stability study of a compounded oral solution of nicardipine for the treatment of hypertension in children. Eur. J. Pharm. Sci. 2021, 160, 105738. [Google Scholar] [CrossRef]
- Boscolo, O.; Perra, F.; Salvo, L.; Buontempo, F.; Lucangioli, S. Formulation and Stability Study of Omeprazole Oral Liquid Suspension for Pediatric Patients. Hosp. Pharm. 2020, 55, 314–322. [Google Scholar] [CrossRef]
- Wu, H.; Wang, Z.; Zhao, Y.; Gao, Y.; Wang, L.; Zhang, H.; Bu, R.; Ding, Z.; Han, J. Effect of Different Seed Crystals on the Supersaturation State of Ritonavir Tablets Prepared by Hot-Melt Extrusion. Eur. J. Pharm. Sci. 2023, 185, 106440. [Google Scholar] [CrossRef]
- Attebäck, M.; Hedin, B.; Mattsson, S. Formulation Optimization of Extemporaneous Oral Liquids Containing Naloxone and Propranolol for Pediatric Use. Sci. Pharm. 2022, 90, 15. [Google Scholar] [CrossRef]
- Correa-Soto, C.E.; Gao, Y.; Indulkar, A.S.; Zhang, G.G.Z.; Taylor, L.S. Role of surfactants in improving release from higher drug loading amorphous solid dispersions. Int. J. Pharm. 2022, 625, 122120. [Google Scholar] [CrossRef]
- Timur, B.; Yilmaz Usta, D.; Teksin, Z.S. Investigation of the effect of colloidal structures formed during lipolysis of lipid-based formulation on exemestane permeability using the in vitro lipolysis-permeation model. J. Drug Deliv. Sci. Technol. 2022, 77, 103797. [Google Scholar] [CrossRef]
- Szymczyk, K.; Szaniawska, M.; Krawczyk, J. Temperature Effect on the Adsorption and Volumetric Properties of Aqueous Solutions of Kolliphor®ELP. Molecules 2020, 25, 743. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, Z.; Luo, L.; Li, K.; Zi, T.; Ren, J.; Pan, L.; Wang, Z.; Wang, Z.; Liu, M.; et al. Impact of Poloxamer on Crystal Nucleation and Growth of Amorphous Clotrimazole. Pharmaceutics 2023, 15, 2164. [Google Scholar] [CrossRef]
- Taylor, L.S.; Zhang, G.G.Z. Physical chemistry of supersaturated solutions and implications for oral absorption. Adv. Drug Deliv. Rev. 2016, 101, 122–142. [Google Scholar] [CrossRef]
- Xu, S.; Dai, W.-G. Drug precipitation inhibitors in supersaturable formulations. Int. J. Pharm. 2013, 453, 36–43. [Google Scholar] [CrossRef]
- Anby, M.U.; Williams, H.D.; McIntosh, M.; Benameur, H.; Edwards, G.A.; Pouton, C.W.; Porter, C.J.H. Lipid Digestion as a Trigger for Supersaturation: Evaluation of the Impact of Supersaturation Stabilization on the in Vitro and in Vivo Performance of Self-Emulsifying Drug Delivery Systems. Mol. Pharm. 2012, 9, 2063–2079. [Google Scholar] [CrossRef]
- Brouwers, J.; Brewster, M.E.; Augustijns, P. Supersaturating Drug Delivery Systems: The Answer to Solubility-Limited Oral Bioavailability? J. Pharm. Sci. 2009, 98, 2549–2572. [Google Scholar] [CrossRef]
- Siepmann, J.; Siepmann, F. Mathematical modeling of drug dissolution. Int. J. Pharm. 2013, 453, 12–24. [Google Scholar] [CrossRef]
- Wu, H.; Wang, Z.; Zhao, Y.; Gao, Y.; Zhang, H.; Wang, L.; Wang, Z.; Han, J. Effect of Span 20 Feeding Zone in the Twin Screw Extruder on the Properties of Amorphous Solid Dispersion of Ritonavir. Pharmaceutics 2023, 15, 441. [Google Scholar] [CrossRef]
- Ilevbare, G.A.; Liu, H.; Pereira, J.; Edgar, K.J.; Taylor, L.S. Influence of Additives on the Properties of Nanodroplets Formed in Highly Supersaturated Aqueous Solutions of Ritonavir. Mol. Pharm. 2013, 10, 3392–3403. [Google Scholar] [CrossRef]
- Ilevbare, G.A.; Taylor, L.S. Liquid-Liquid Phase Separation in Highly Supersaturated Aqueous Solutions of Poorly Water-Soluble Drugs: Implications for Solubility Enhancing Formulations. Cryst. Growth Des. 2013, 13, 1497–1509. [Google Scholar] [CrossRef]
- Lentz, K.A.; Plum, J.; Steffansen, B.; Arvidsson, P.O.; Omkvist, D.H.O.; Pedersen, A.J.; Sennbro, C.J.; Pedersen, G.P.; Jacobsen, J. Predicting in vivo performance of fenofibrate amorphous solid dispersions using in vitro non-sink dissolution and dissolution permeation setup. Int. J. Pharm. 2021, 610, 121174. [Google Scholar] [CrossRef]
- Girardin, F.; Manuel, O.; Marzolini, C.; Buclin, T. Evaluating the risk of drug-drug interactions with pharmacokinetic boosters: The case of ritonavir-enhanced nirmatrelvir to prevent severe COVID-19. Clin. Microbiol. Infect. 2022, 28, 1044–1046. [Google Scholar] [CrossRef]
- Secretan, P.H.; Annereau, M.; Kini-Matondo, W.; Prost, B.; Prudhomme, J.; Bournane, L.; Paul, M.; Yagoubi, N.; Sadou-Yayé, H.; Do, B. Unequal Behaviour between Hydrolysable Functions of Nirmatrelvir under Stress Conditions: Structural and Theoretical Approaches in Support of Preformulation Studies. Pharmaceutics 2022, 14, 1720. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Components | Quantities | Functional Category |
|---|---|---|
| NRV | 300 mg | API |
| RTV | 100 mg | API |
| Ethanol | 3.0 mL | Co-solvent |
| Propylene glycol | 1.9 mL | Co-solvent |
| Surfactant | 0.7 mL | Solubilizer |
| Water | 1.40 mL | Solvent |
| Components | F1 a | F2 a | F3 a | F4 a | RS b |
|---|---|---|---|---|---|
| Dose of NRV (mg) | 300 | 300 | 300 | 150 | 300 |
| Dose of RTV (mg) | 100 | 50 | 0 | 100 | 100 |
| Blank solvent (mL) | 7.0 | 7.0 | 7.0 | 7.0 | – |
| Concentration of NRV (Mean ± SD, µg/mL) | Concentration of RTV (Mean ± SD, µg/mL) | |
|---|---|---|
| pH 1.2 | 997.7 ± 6.5 | 381.9 ± 5.8 |
| pH 2.0 | 983.2 ± 5.4 | 17.5 ± 0.7 |
| pH 3.8 | 974.7 ± 4.8 | 3.6 ± 0.3 |
| pH 4.5 | 973.5 ± 5.3 | 3.6 ± 0.4 |
| pH 6.8 | 957.8 ± 7.5 | 3.3 ± 0.5 |
| pH 7.4 | 998.8 ± 6.2 | 3.4 ± 0.6 |
| Parameters | NRV | RTV | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| F1 | F2 | F3 | F4 | RS | F1 | F2 | F3 | F4 | RS | |
| Cmax (µg/mL) | 12.72 ± 2.41 | 8.75 ± 3.83 | 2.92 ± 1.22 | 6.04 ± 1.98 | 2.30 ± 0.51 | 3.69 ± 1.83 | 0.89 ± 0.25 | 0 | 9.02 ± 2.97 | 0.67 ± 0.35 |
| Tmax (h) | 1.50 ± 0.39 | 0.95 ± 0.67 | 0.63 ± 0.43 | 0.75 ± 0.35 | 2.38 ± 0.43 | 2.00 ± 0.97 | 1.50 ± 0.50 | 0 | 1.60 ± 1.39 | 2.30 ± 1.21 |
| t1/2 (h) | 4.21 ± 1.14 | 3.57 ± 0.75 | 2.38 ± 1.19 | 2.42 ± 0.38 | 4.52 ± 1.21 | 9.26 ± 1.83 | 11.95 ± 4.35 | 0 | 4.92 ± 3.09 | 8.84 ± 2.17 |
| AUC(0–t) (µg·h/mL) | 43.40 ± 5.39 | 20.19 ± 3.91 | 5.70 ± 1.74 | 14.39 ± 2.37 | 7.09 ± 1.51 | 11.75 ± 2.38 | 3.96 ± 1.59 | 0 | 22.02 ± 4.59 | 3.05 ± 1.78 |
| Parameters | NRV | RTV | ||
|---|---|---|---|---|
| F1 | RS | F1 | RS | |
| Cmax (µg/mL) | 25.76 | 9.75 | 3.45 | 2.21 |
| Tmax (h) | 0.25 | 2.00 | 1.00 | 3.00 |
| t1/2 (h) | 3.64 | 4.27 | 2.05 | 3.77 |
| AUC(0–t) (µg·h/mL) | 174.55 | 95.36 | 8.44 | 4.68 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.; Ding, Z.; Wang, Z.; Zhao, Y.; Wu, H.; Wei, Q.; Gao, L.; Han, J. The Development of an Oral Solution Containing Nirmatrelvir and Ritonavir and Assessment of Its Pharmacokinetics and Stability. Pharmaceutics 2024, 16, 109. https://doi.org/10.3390/pharmaceutics16010109
Wang L, Ding Z, Wang Z, Zhao Y, Wu H, Wei Q, Gao L, Han J. The Development of an Oral Solution Containing Nirmatrelvir and Ritonavir and Assessment of Its Pharmacokinetics and Stability. Pharmaceutics. 2024; 16(1):109. https://doi.org/10.3390/pharmaceutics16010109
Chicago/Turabian StyleWang, Lili, Zhuang Ding, Zhengping Wang, Yanna Zhao, Hengqian Wu, Qipeng Wei, Lingfeng Gao, and Jun Han. 2024. "The Development of an Oral Solution Containing Nirmatrelvir and Ritonavir and Assessment of Its Pharmacokinetics and Stability" Pharmaceutics 16, no. 1: 109. https://doi.org/10.3390/pharmaceutics16010109
APA StyleWang, L., Ding, Z., Wang, Z., Zhao, Y., Wu, H., Wei, Q., Gao, L., & Han, J. (2024). The Development of an Oral Solution Containing Nirmatrelvir and Ritonavir and Assessment of Its Pharmacokinetics and Stability. Pharmaceutics, 16(1), 109. https://doi.org/10.3390/pharmaceutics16010109