

Synthesis and Anticancer Activity of Novel Dual Inhibitors of Human Protein Kinases CK2 and PIM-1 †



Abstract
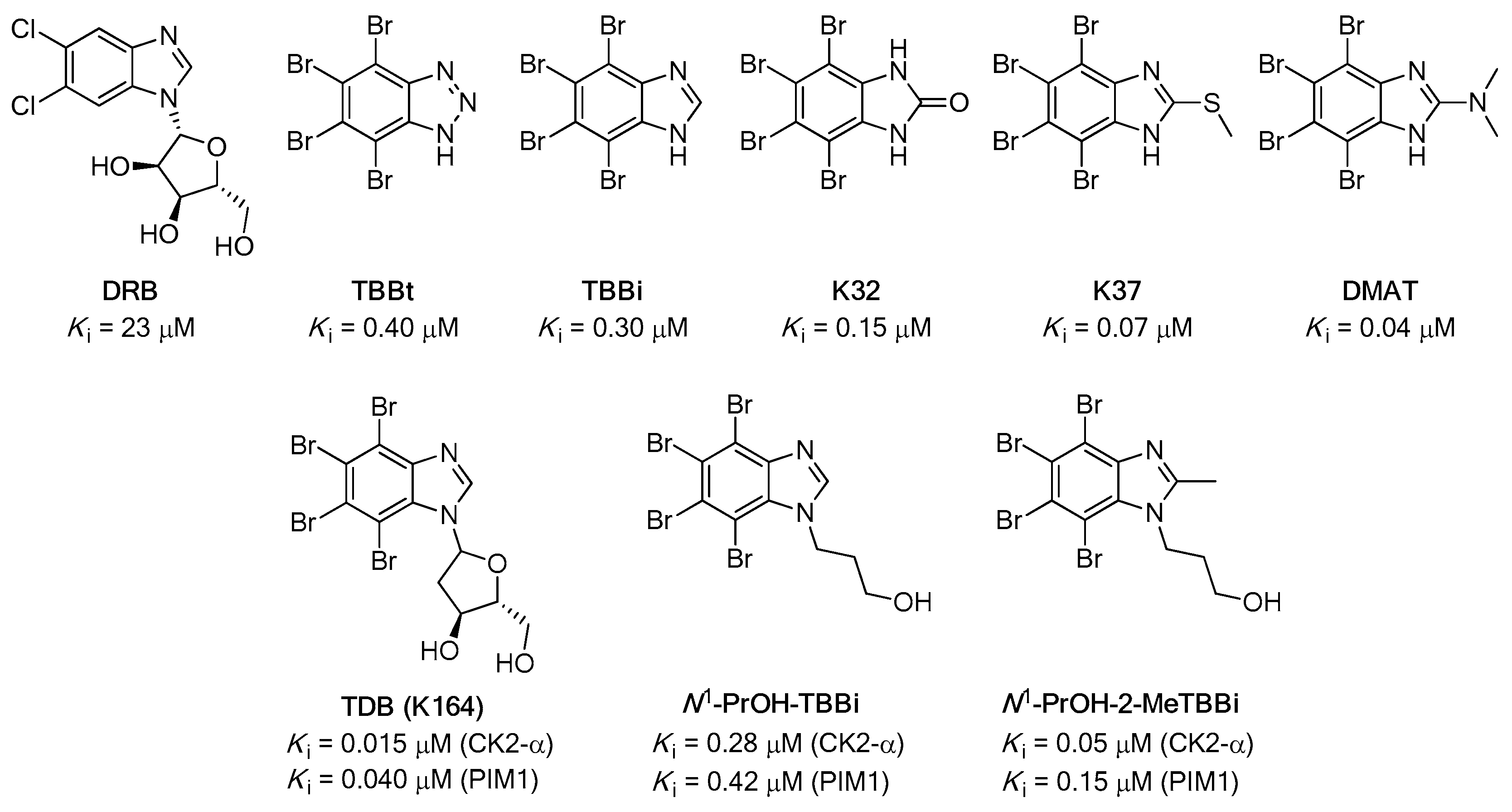
1. Introduction
2. Materials and Methods
2.1. Chemistry
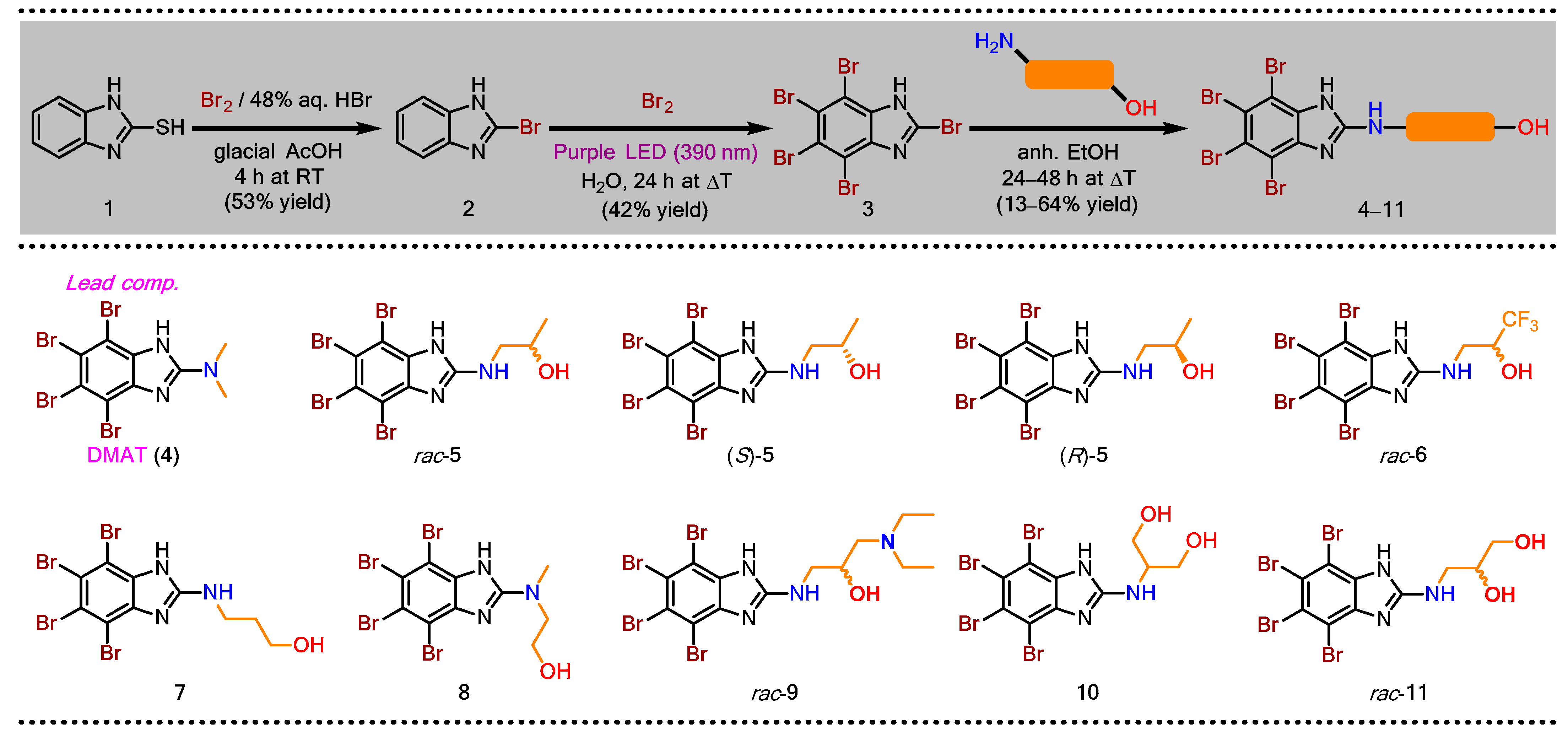
2.1.1. General Procedure for the Synthesis of 2-Bromo-1H-benzimidazole (2)
2.1.2. General Procedure for the Synthesis of 2,4,5,6,7-Pentabromo-1H-benzimidazole (3)
2.1.3. General Procedure for the Synthesis of Dual CK2/PIM-1 Inhibitors—TBBi Amino Alcohol Derivatives (4–11)
2.2. Biological Evaluation
2.2.1. Reagents and Antibodies
2.2.2. Cloning, Expression, and Purification of Human CK2α, holoCK2, and PIM-1
2.2.3. Inhibition of Recombinant CK2 and PIM-1
2.2.4. Cell Culture and Agent Treatment
2.2.5. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium Bromide (MTT)-Based Viability Assay
2.2.6. Detection of Apoptosis by Annexin V/propidium Iodide (PI) Labeling
2.2.7. Mitochondrial Membrane Potential (ΔΨm) Assay
2.2.8. Microscopic Examination
2.2.9. Fluorescence Intensity Assay
2.2.10. Detection of Cell Cycle Progression by Flow Cytometry
2.2.11. Western Blotting
2.2.12. Densitometry
2.2.13. Statistical Evaluation
2.3. Molecular Docking
2.3.1. Molecular Docking Preparation
2.3.2. Molecular Docking Procedure
3. Results
3.1. Chemical Synthesis
3.2. Biological Evaluation
3.2.1. Inhibition of Recombinant CK2 and PIM-1
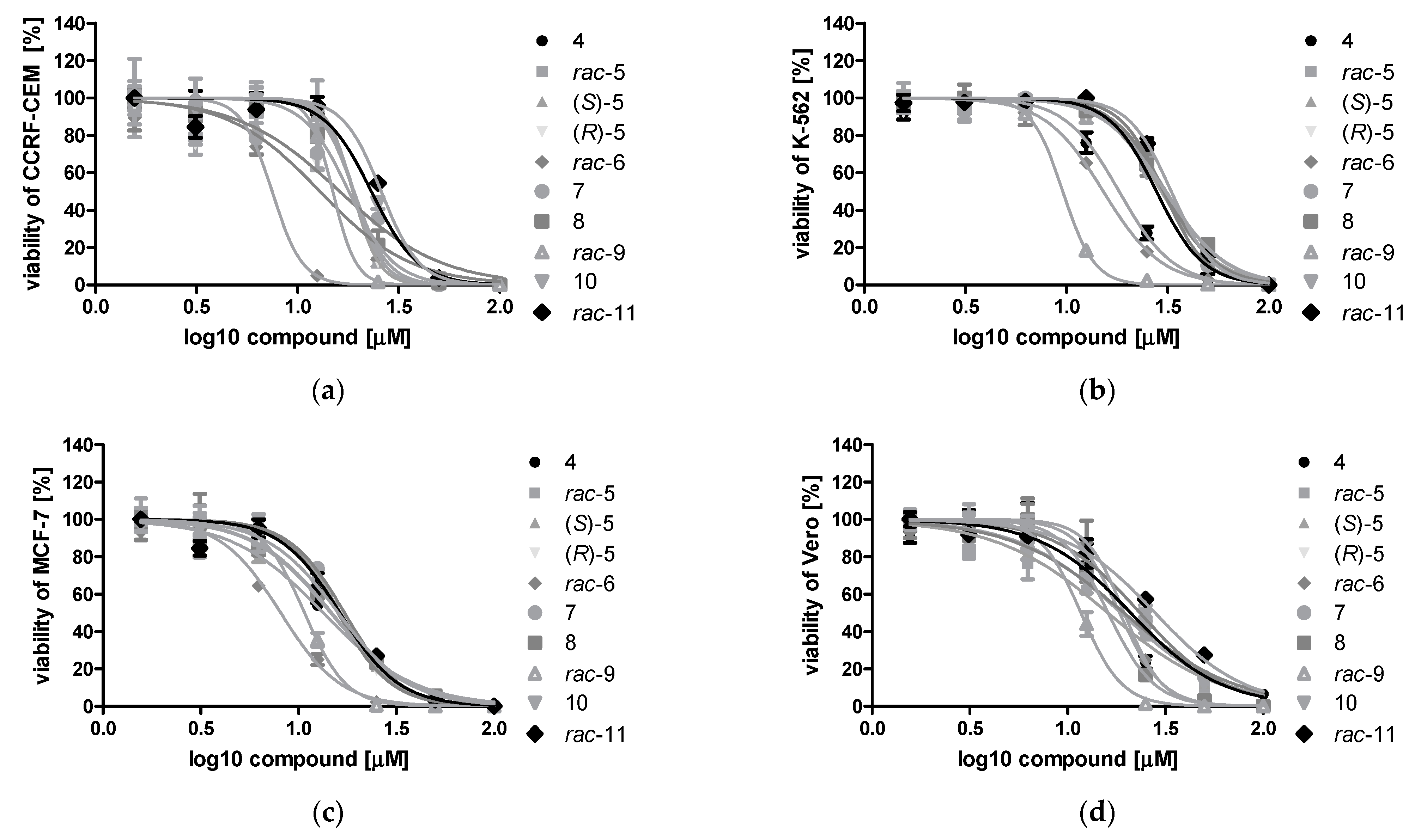
3.2.2. Cytotoxic Effect of DMAT Derivatives 4–11 toward Cancer Cell Lines: CCRF-CEM, K-562, MCF-7, and Non-Cancerous Vero Cells
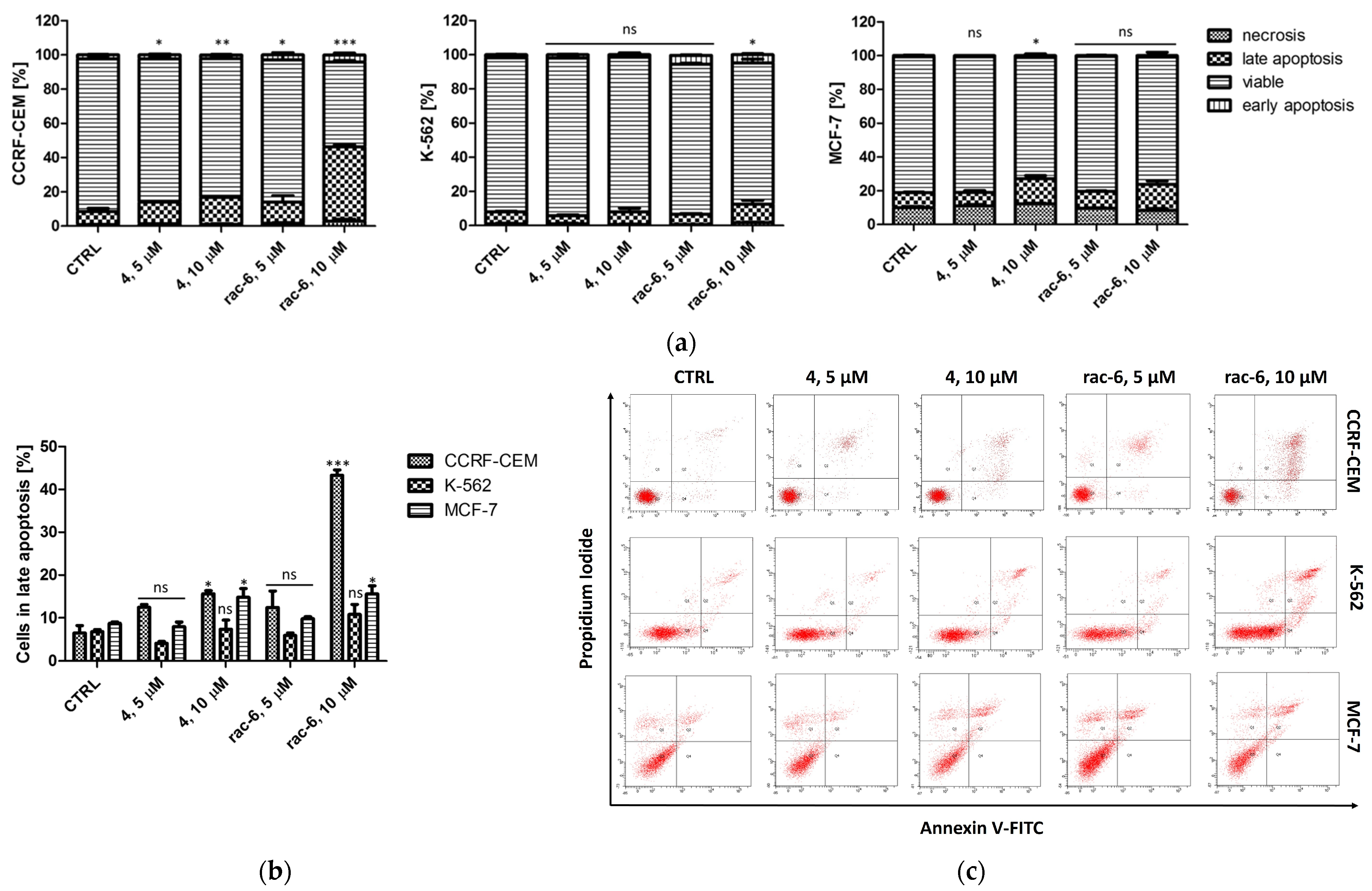
3.2.3. Induction of Apoptosis in CCRF-CEM, K-562, and MCF-7 Cells
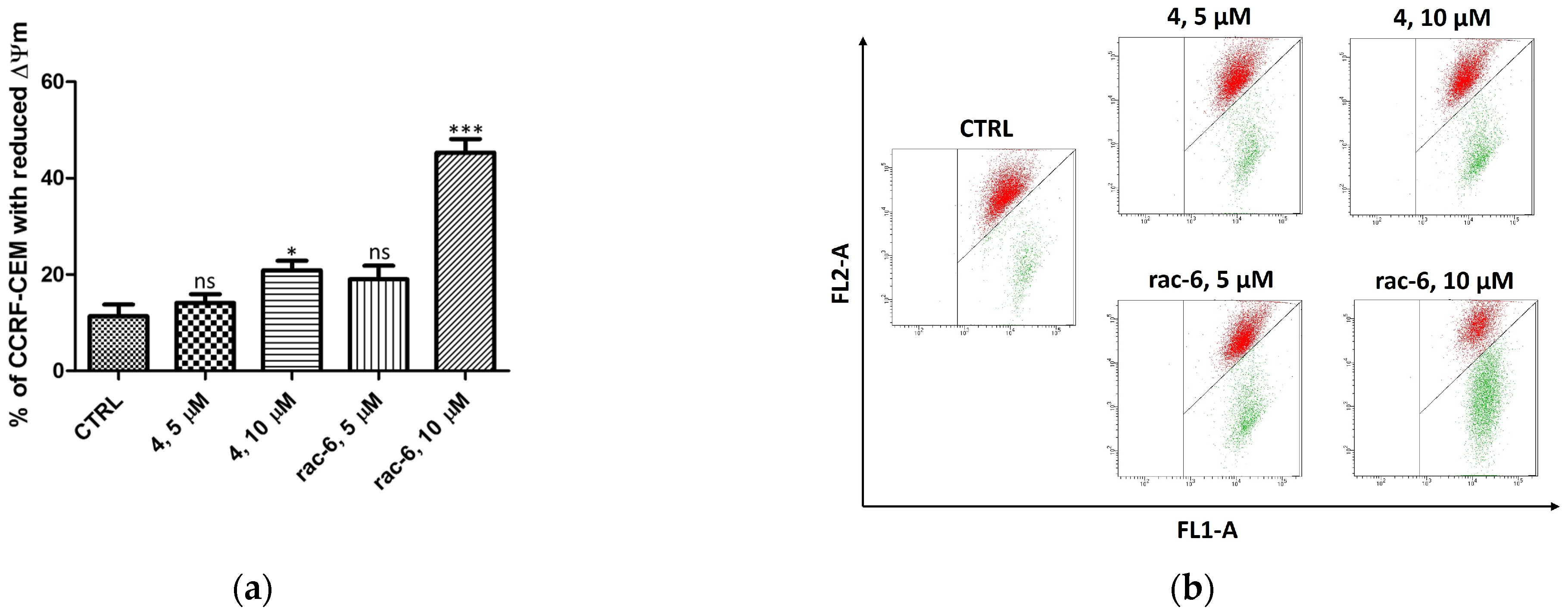
3.2.4. Mitochondrial Membrane Potential (ΔΨm) in CCRF-CEM
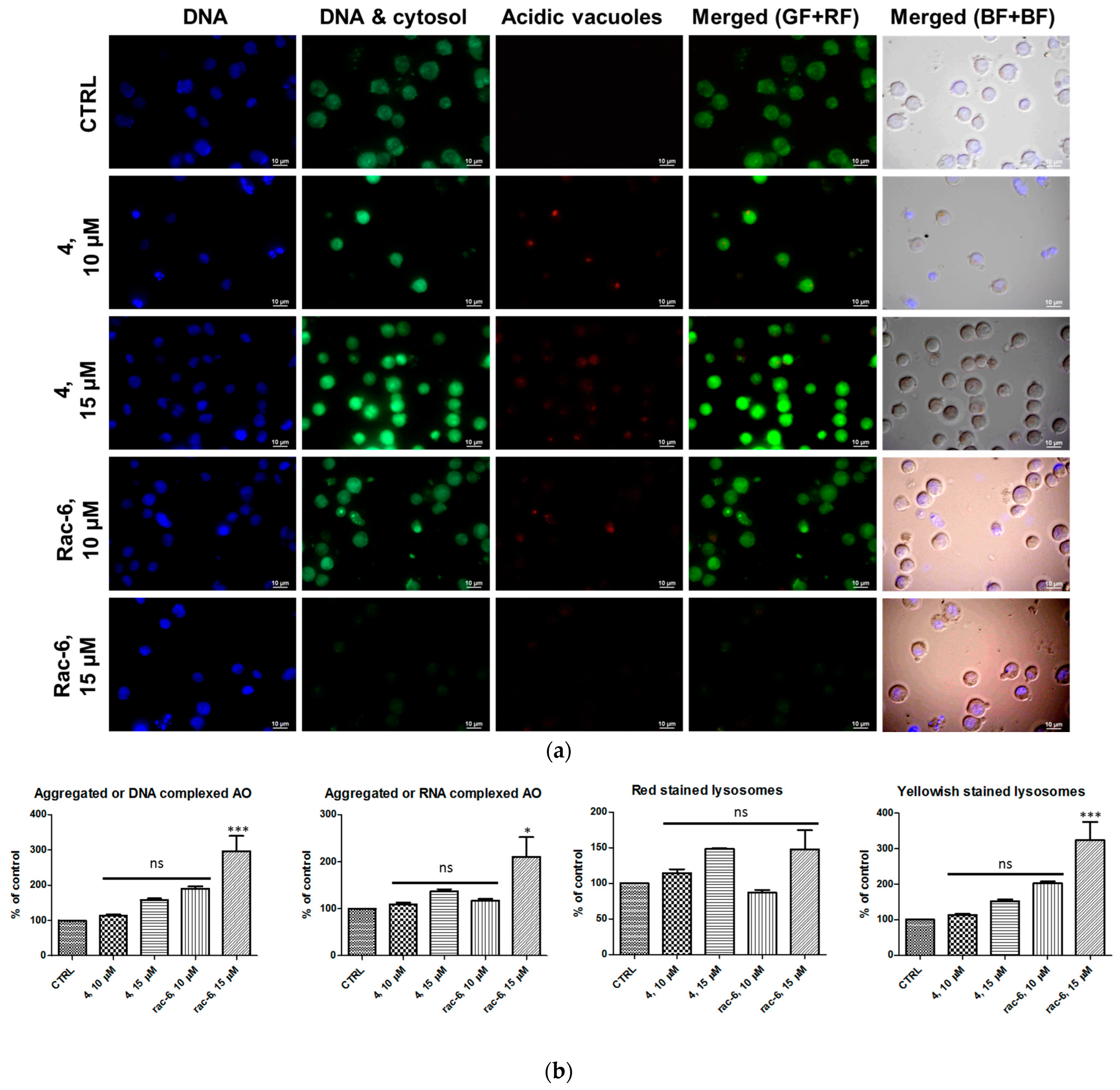
3.2.5. Detection of Autophagy in K-562 Cells
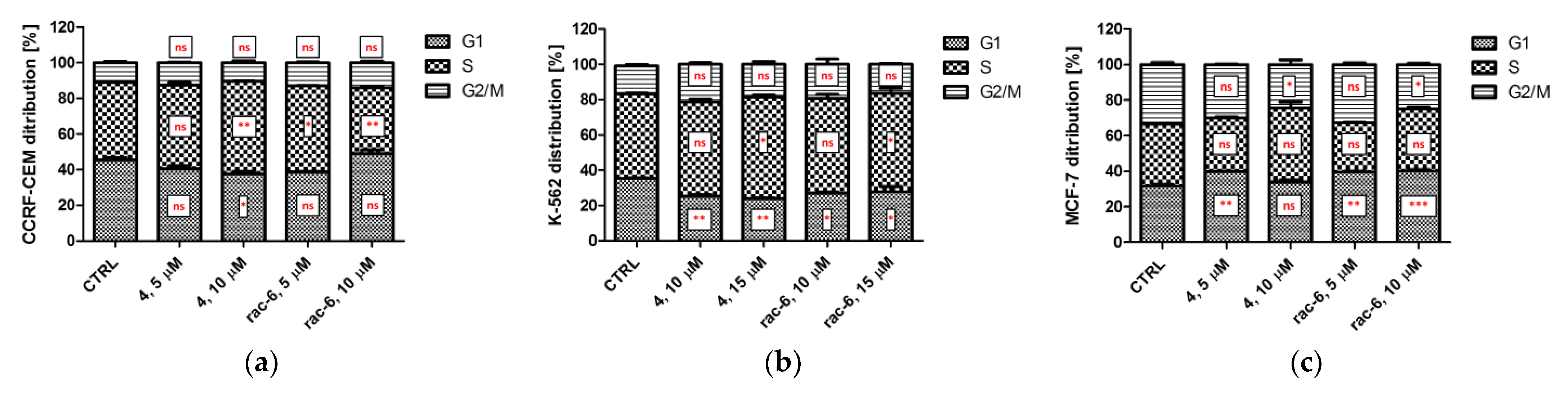
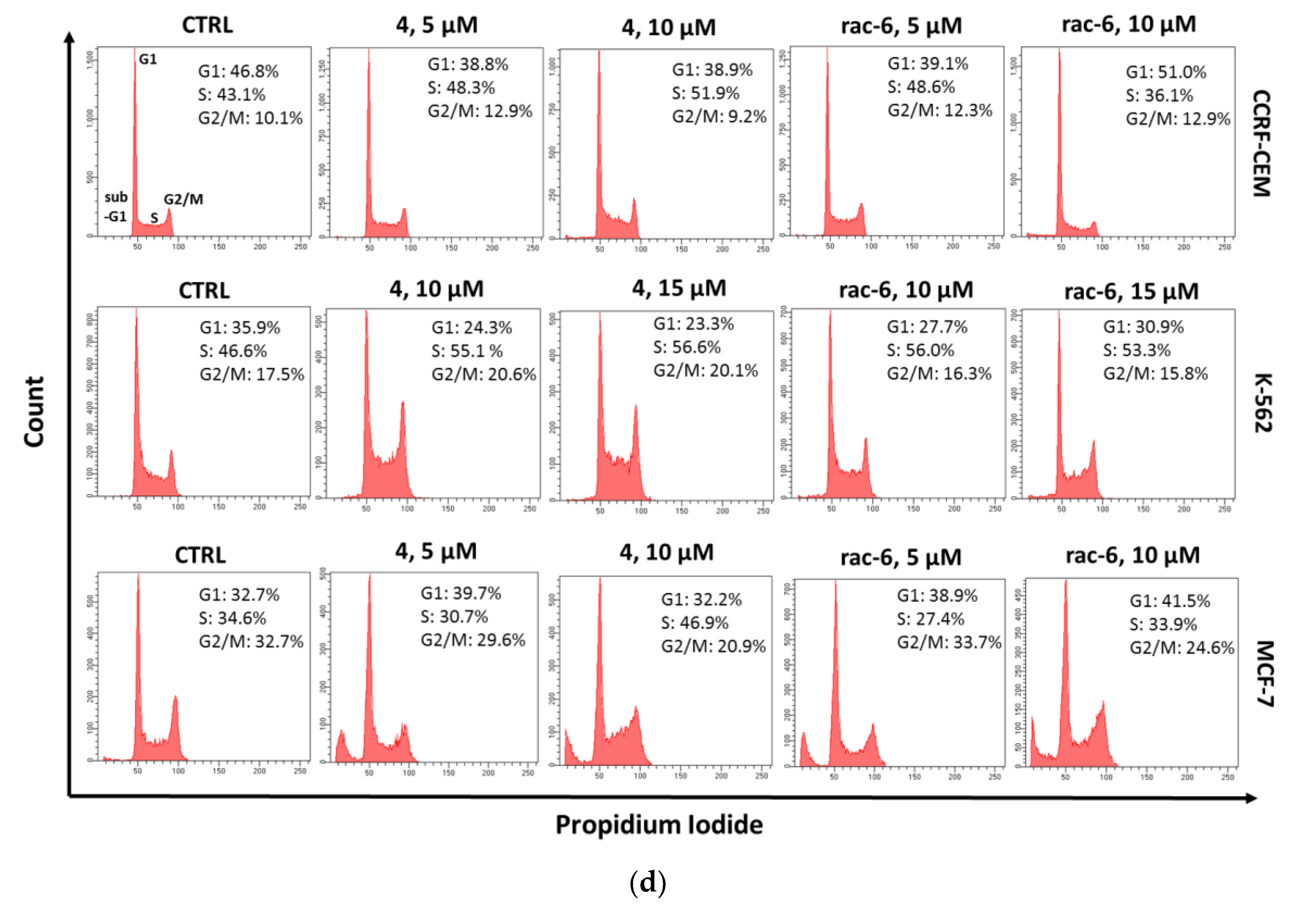
3.2.6. The Effect of 4 and rac-6 on Cell Cycle Progression in CCRF-CEM, K-562, and MCF-7 Cells
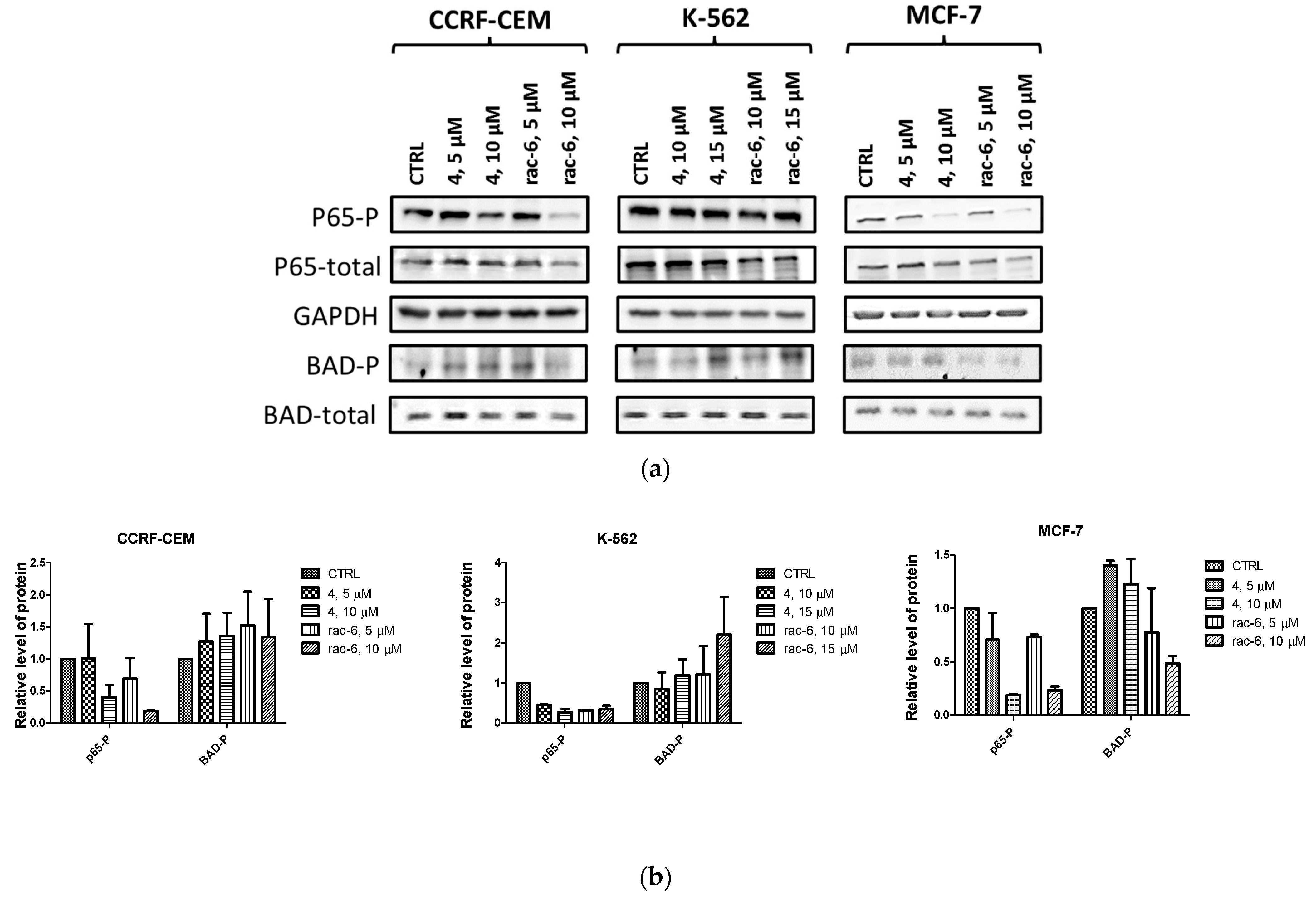
3.2.7. Intracellular Inhibition of Protein Kinase CK2 and PIM-1 in CCRF-CEM, K-562, and MCF-7 Cells
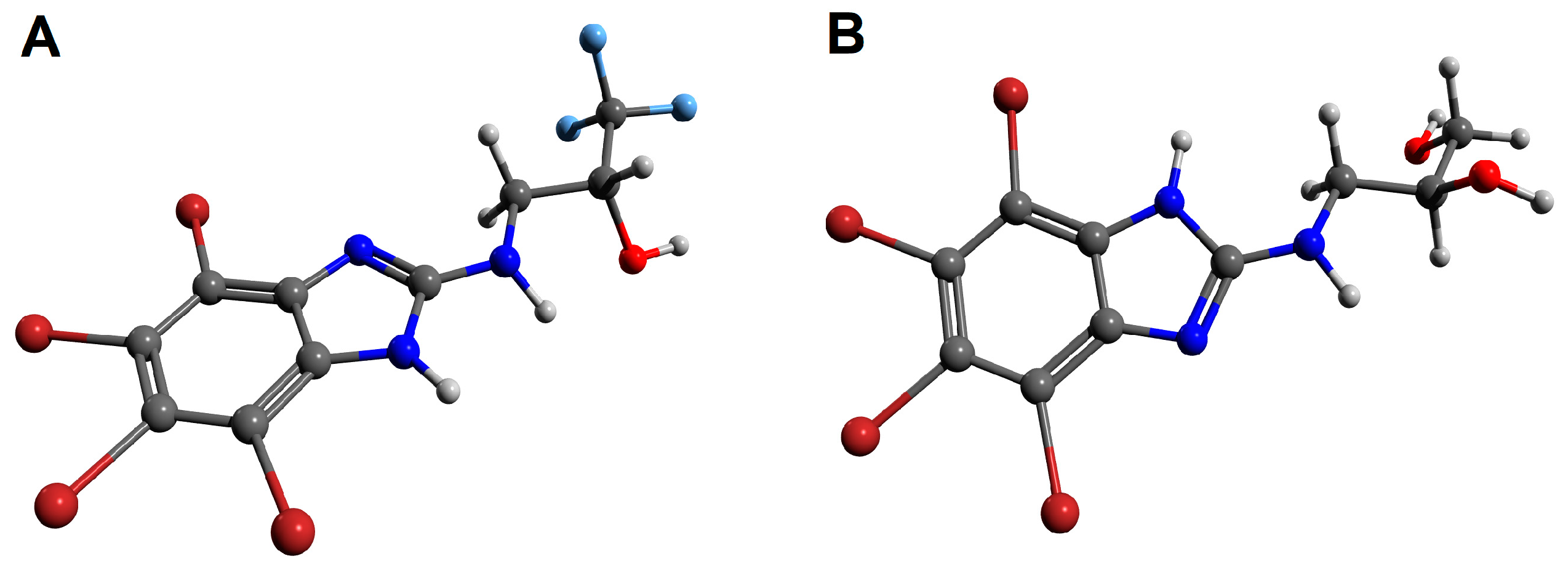
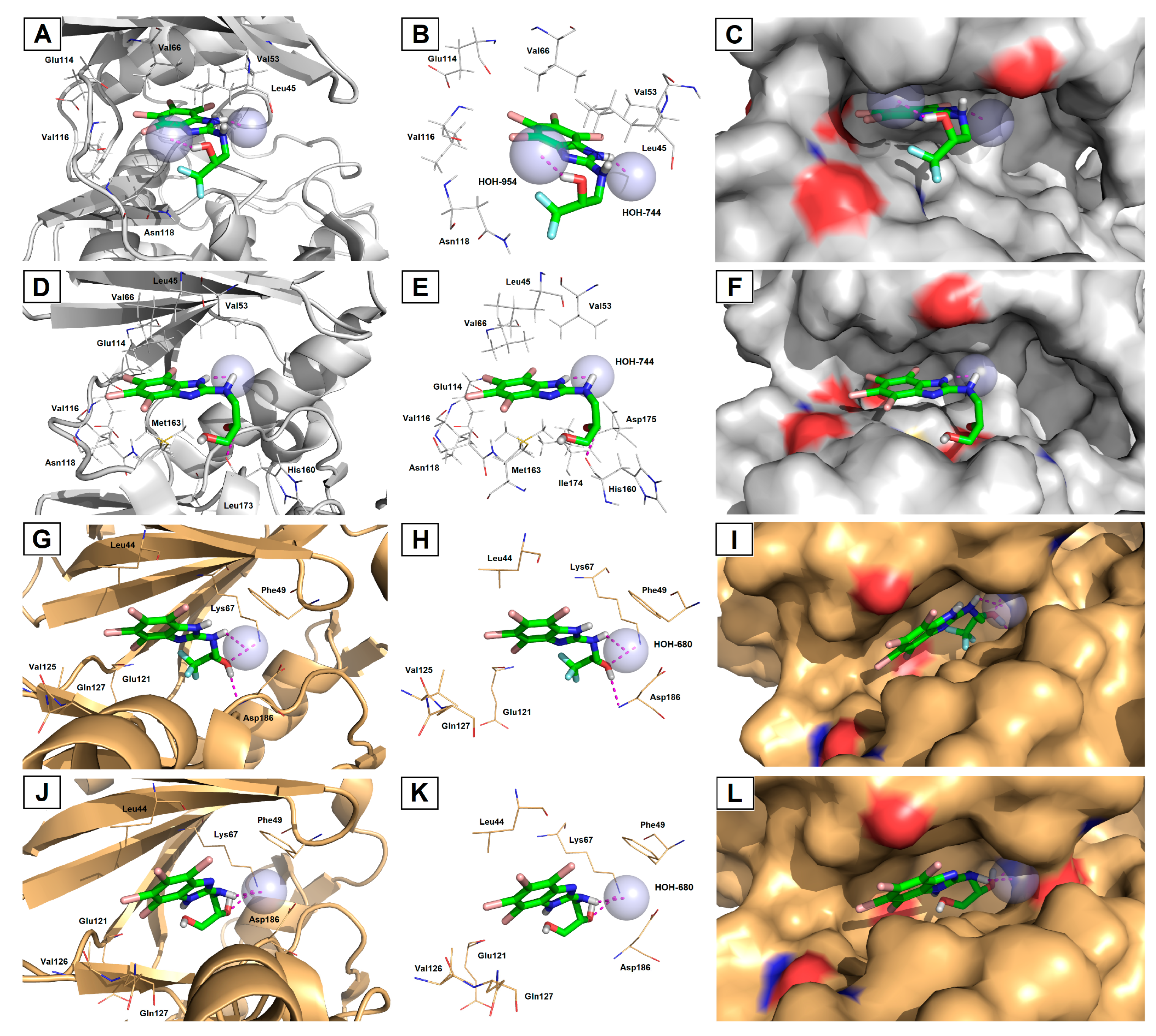
3.3. Molecular Docking
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Trembley, J.H.; Kren, B.T.; Afzal, M.; Scaria, G.A.; Klein, M.A.; Ahmed, K. Protein kinase CK2—Diverse roles in cancer cell biology and therapeutic promise. Mol. Cell. Biochem. 2023, 478, 899–926. [Google Scholar] [CrossRef] [PubMed]
- Walhekar, V.; Bagul, C.; Kumar, D.; Muthal, A.; Achaiah, G.; Kulkarni, R. Topical advances in PIM kinases and their inhibitors: Medicinal chemistry perspectives. Biochim. Biophys. Acta Rev. Cancer 2022, 1877, 188725. [Google Scholar] [CrossRef]
- Borgo, C.; Ruzzene, M. Role of protein kinases in antitumor drug resistance. J. Exp. Clin. Cancer Res. 2019, 38, 287. [Google Scholar] [CrossRef]
- Atkinson, E.L.; Iegre, J.; Brear, P.D.; Zhabina, E.Z.; Hyvönen, M.; Spring, D.R. Downfalls of Chemical Probes Acting at the Kinase ATP-Site: CK2 as a Case Study. Molecules 2021, 26, 1977. [Google Scholar] [CrossRef]
- Narlik-Grassow, M.; Blanco-Aparicio, C.; Carnero, A. The PIM family of serine/threonine kinases in cancer. Med. Res. Rev. 2014, 34, 136–159. [Google Scholar] [CrossRef] [PubMed]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef] [PubMed]
- Qian, K.C.; Wang, L.; Hickey, E.R.; Studts, J.; Barringer, K.; Peng, C.; Kronkaitis, A.; Li, J.; White, A.; Mische, S.; et al. Structural basis of constitutive activity and a unique nucleotide binding mode of human Pim-1 kinase. Biol. Chem. 2005, 280, 6130–6137. [Google Scholar] [CrossRef]
- Niefind, K.; Guerra, B.; Pinna, L.A.; Issinger, O.G.; Schomburg, D. Crystal structure of the catalytic subunit of protein kinase CK2 from Zea mays at 2.1 A resolution. EMBO J. 1998, 17, 2451–2462. [Google Scholar] [CrossRef]
- Pucko, E.B.; Ostrowski, R.P. Inhibiting CK2 among Promising Therapeutic Strategies for Gliomas and Several Other Neoplasms Emanuela, B. Pharmaceutics 2022, 14, 331. [Google Scholar] [CrossRef]
- Brault, L.; Gasser, C.; Bracher, F.; Huber, K.; Knapp, S.; Schwaller, J. PIM serine/threonine kinases in the pathogenesis and therapy of hematologic malignancies and solid cancers. Haematologica 2010, 95, 1004–1015. [Google Scholar] [CrossRef]
- Valdman, A.; Fang, X.; Pang, S.T.; Ekman, P.; Egevad, L. Pim-1 expression in prostatic intraepithelial neoplasia and human prostate cancer. Prostate 2004, 60, 367–371. [Google Scholar] [CrossRef] [PubMed]
- Tursynbay, Y.; Zhang, J.; Li, Z.; Tokay, T.; Zhumadilov, Z.; Wu, D.; Xie, Y. Pim-1 kinase as cancer drug target: An update (Review). Biomed. Rep. 2016, 4, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Yan, B.; Zemskova, M.; Holder, S.; Chin, V.; Kraft, A.; Koskinen, P.J.; Lilly, M.J. The PIM-2 kinase phosphorylates BAD on serine 112 and reverses BAD-induced cell death. Biol. Chem. 2003, 278, 45358–45367. [Google Scholar] [CrossRef] [PubMed]
- Channavajhala, P.; Seldin, D.C. Functional interaction of protein kinase CK2 and c-Myc in lymphomagenesis. Oncogene 2002, 21, 5280–5288. [Google Scholar] [CrossRef] [PubMed]
- Klumpp, S.; Maurer, A.; Zhu, Y.; Aichele, D.; Pinna, L.A.; Krieglstein, J. Protein kinase CK2 phosphorylates BAD at threonine-117. J. Neurochem. Int. 2004, 45, 747–752. [Google Scholar] [CrossRef]
- Wang, J.; Kim, J.; Roh, M.; Franco, O.E.; Hayward, S.W.; Wills, M.L.; Abdulkadir, S.A. PIM-1 kinase synergizes with c-MYC to induce advanced prostate carcinoma. Oncogene 2010, 29, 2477–2487. [Google Scholar] [CrossRef]
- Laramas, M.; Pasquier, D.; Filhol, O.; Ringeisen, F.; Descotes, J.L.; Cochet, C. Nuclear localization of protein kinase CK2 catalytic subunit (CK2alpha) is associated with poor prognostic factors in human prostate cancer. Eur. J. Cancer 2007, 43, 928–934. [Google Scholar] [CrossRef]
- Cibull, T.L.; Jones, T.D.; Li, L.; Eble, J.N.; Baldridge, A.L.; Malott, S.R.; Luo, Y.; Cheng, L. Overexpression of Pim-1 during progression of prostatic adenocarcinoma. J. Clin. Pathol. 2006, 59, 285–288. [Google Scholar] [CrossRef]
- Trembley, J.H.; Kren, B.T.; Abedin, M.J.; Vogel, R.I.; Cannon, C.M.; Unger, G.M.; Ahmed, K. CK2 Molecular Targeting-Tumor Cell-Specific Delivery of RNAi in Various Models of Cancer. Pharmaceuticals 2017, 10, 25. [Google Scholar] [CrossRef]
- Ahmad, K.A.; Wang, G.; Slaton, J.; Unger, G.; Ahmed, K. Targeting CK2 for cancer therapy. Anticancer Drugs 2005, 16, 1037–1043. [Google Scholar] [CrossRef]
- Hu, X.F.; Li, J.; Vandervalk, S.; Wang, Z.; Magnuson, N.S.; Xing, P.X.J. PIM-1-specific mAb suppresses human and mouse tumor growth by decreasing PIM-1 levels, reducing Akt phosphorylation, and activating apoptosis. Clin. Invest. 2009, 119, 362–375. [Google Scholar] [CrossRef] [PubMed]
- Gowda, C.; Sachdev, M.; Muthusami, S.; Kapadia, M.; Petrovic-Dovat, L.; Hartman, M.; Ding, Y.; Song, C.; Payne, J.L.; Tan, B.H.; et al. Casein Kinase II (CK2) as a Therapeutic Target for Hematological Malignancies. Curr. Pharm. Des. 2017, 23, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Milani, M.; Venturini, S.; Bonardi, S.; Allevi, G.; Strina, C.; Cappelletti, M.R.; Corona, S.P.; Aguggini, S.; Bottini, A.; Berruti, A.; et al. Hypoxia-related biological markers as predictors of epirubicin-based treatment responsiveness and resistance in locally advanced breast cancer. Oncotarget 2017, 8, 78870–78881. [Google Scholar] [CrossRef] [PubMed]
- Garziera, M.; Scarabel, L.; Toffoli, G. Hypoxic Modulation of HLA-G Expression through the Metabolic Sensor HIF-1 in Human Cancer Cells. J. Immunol. Res. 2017, 2017, 4587520. [Google Scholar] [CrossRef]
- Chen, J.; Kobayashi, M.; Darmanin, S.; Qiao, Y.; Gully, C.; Zhao, R.; Yeung, S.C.; Lee, M.H. Pim-1 plays a pivotal role in hypoxia-induced chemoresistance. Oncogene 2009, 28, 2581–2592. [Google Scholar] [CrossRef]
- Mottet, D.; Ruys, S.P.; Demazy, C.; Raes, M.; Michiels, C. Role for casein kinase 2 in the regulation of HIF-1 activity. Int. J. Cancer. 2005, 117, 764–774. [Google Scholar] [CrossRef]
- Casillas, A.L.; Toth, R.K.; Sainz, A.G.; Singh, N.; Desai, A.A.; Kraft, A.S.; Warfel, N.A. Hypoxia-inducible PIM kinase expression promotes resistance to anti-angiogenic agents. Clin. Cancer Res. 2018, 24, 169–180. [Google Scholar] [CrossRef]
- Landesman-Bollag, E.; Song, D.H.; Romieu-Mourez, R.; Sussman, D.J.; Cardiff, R.D.; Sonenshein, G.E.; Seldin, D.C. Protein kinase CK2: Signaling and tumorigenesis in the mammary gland. Mol. Cell. Biochem 2001, 227, 153–165. [Google Scholar] [CrossRef]
- Hammerman, P.S.; Fox, C.J.; Cinalli, R.M.; Xu, A.; Wagner, J.D.; Lindsten, T.; Thompson, C.B. Lymphocyte transformation by Pim-2 is dependent on nuclear factor-kappaB activation. Cancer Res. 2004, 64, 8341–8348. [Google Scholar] [CrossRef]
- Zemskova, M.; Sahakian, E.; Bashkirova, S.; Lilly, M.J. The PIM-1 kinase is a critical component of a survival pathway activated by docetaxel and promotes survival of docetaxel-treated prostate cancer cells. Biol. Chem. 2008, 283, 20635–20644. [Google Scholar] [CrossRef]
- Sarno, S.; Papinutto, E.; Franchin, C.; Bain, J.; Elliott, M.; Meggio, F.; Kazimierczuk, Z.; Orzeszko, A.; Zanotti, G.; Battistutta, R.; et al. ATP site-directed inhibitors of protein kinase CK2: An update. Curr. Top. Med. Chem. 2011, 11, 1340–1351. [Google Scholar] [CrossRef] [PubMed]
- Gingipalli, L.; Block, M.H.; Bao, L.; Cooke, E.; Dakin, L.A.; Denz, C.R.; Ferguson, A.D.; Johannes, J.W.; Larsen, N.A.; Dowling, J.E.; et al. Discovery of 2,6-disubstituted pyrazine derivatives as inhibitors of CK2 and PIM kinases. Bioorg. Med. Chem. Lett. 2018, 28, 1336–1341. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, Y.; Wang, J.; Zhou, Z.; Cao, S.; Zhang, J. Strategies of Targeting CK2 in Drug Discovery: Challenges, Opportunities, and Emerging Prospects. J. Med. Chem. 2023, 66, 2257–2281. [Google Scholar] [CrossRef] [PubMed]
- Cozza, G.; Sarno, S.; Ruzzene, M.; Girardi, C.; Orzeszko, A.; Kazimierczuk, Z.; Zagotto, G.; Bonaiuto, E.; Di Paolo, M.L.; Pinna, L. Exploiting the repertoire of CK2 inhibitors to target DYRK and PIM kinases. Biochim. Biophys. Acta 2013, 1834, 1402–1409. [Google Scholar] [CrossRef] [PubMed]
- Cozza, G.; Girardi, C.; Ranchio, A.; Lolli, G.; Sarno, S.; Orzeszko, A.; Kazimierczuk, Z.; Battistutta, R.; Ruzzene, M.; Pinna, L.A. Cell-permeable dual inhibitors of protein kinases CK2 and PIM-1: Structural features and pharmacological potential. Cell. Mol. Life Sci. 2014, 71, 3173–3185. [Google Scholar] [CrossRef]
- Koronkiewicz, M.; Chilmonczyk, Z.; Kazimerczuk, Z.; Orzeszko, A. Deoxynucleosides with benzimidazoles as aglycone moiety are potent anticancer agents. Eur. J. Pharmacol. 2018, 820, 146–155. [Google Scholar] [CrossRef]
- Koronkiewicz, M.; Kazimierczuk, Z.; Orzeszko, A. Antitumor activity of the protein kinase inhibitor 1-(β-D-2′-deoxyribofuranosyl)-4,5,6,7-tetrabromo- 1H-benzimidazole in breast cancer cell lines. BMC Cancer 2022, 22, 1069. [Google Scholar] [CrossRef]
- Łukowska-Chojnacka, E.; Wińska, P.; Wielechowska, M.; Poprzeczko, M.; Bretner, M. Synthesis of novel polybrominated benzimidazole derivatives-potential CK2 inhibitors with anticancer and proapoptotic activity. Bioorg. Med. Chem. 2016, 24, 735–741. [Google Scholar] [CrossRef]
- Chojnacki, K.; Wińska, P.; Skierka, K.; Wielechowska, M.; Bretner, M. Synthesis, in vitro antiproliferative activity and kinase profile of new benzimidazole and benzotriazole derivatives. Bioorg. Chem. 2017, 72, 1–10. [Google Scholar] [CrossRef]
- Chojnacki, K.; Winska, P.; Karatsai, O.; Koronkiewicz, M.; Milner-Krawczyk, M.; Wielechowska, M.; Redowicz, M.J.; Bretner, M.; Borowiecki, P. Synthesis of Novel Acyl Derivatives of 3-(4,5,6,7-Tetrabromo-1H-benzimidazol-1-yl)propan-1-ols-Intracellular TBBi-Based CK2 Inhibitors with Proapoptotic Properties. Int. J. Mol. Sci. 2021, 22, 6261. [Google Scholar] [CrossRef]
- Ellingboe, J.W.; Spinelli, W.; Winkley, M.W.; Nguyen, T.T.; Parsons, R.W.; Moubarak, I.F.; Kitzen, J.M.; Von Engen, D.; Bagli, J.F. Class III antiarrhythmic activity of novel substituted 4-[(methylsulfonyl)amino]benzamides and sulfonamides. J. Med. Chem. 1992, 35, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Andrzejewska, M.; Pagano, M.A.; Meggio, F.; Brunati, A.M.; Kazimierczuk, Z. Polyhalogenobenzimidazoles: Synthesis and their inhibitory activity against casein kinases. Bioorg. Med. Chem. 2003, 11, 3997–4002. [Google Scholar] [CrossRef] [PubMed]
- Pagano, M.A.; Andrzejewska, M.; Ruzzene, M.; Sarno, S.; Cesaro, L.; Bain, J.; Elliott, M.; Meggio, F.; Kazimierczuk, Z.; Pinna, L.A. Optimization of protein kinase CK2 inhibitors derived from 4,5,6,7-tetrabromobenzimidazole. J. Med. Chem. 2004, 47, 6239–6247. [Google Scholar] [CrossRef] [PubMed]
- Borowiecki, P.; Wawro, A.; Wińska, P.; Wielechowska, M.; Bretner, M. Synthesis of novel chiral TBBt derivatives with hydroxyl moiety. Studies on inhibition of human protein kinase CK2α and cytotoxicity properties. Eur. J. Med. Chem. 2014, 84, 364–374. [Google Scholar] [CrossRef] [PubMed]
- Chojnacki, K.; Wińska, P.; Wielechowska, M.; Łukowska-Chojnacka, E.; Tölzer, C.; Niefind, K.; Bretner, M. Biological properties and structural study of new aminoalkyl derivatives of benzimidazole and benzotriazole, dual inhibitors of CK2 and PIM1 kinases. Bioorg. Chem. 2018, 80, 266–275. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Dakin, L.A.; Block, M.H.; Chen, H.; Code, E.; Dowling, J.E.; Feng, X.; Ferguson, A.D.; Green, I.; Hird, A.W.; Howard, T.; et al. Discovery of novel benzylidene-1,3-thiazolidine-2,4-diones as potent and selective inhibitors of the PIM-1, PIM-2, and PIM-3 protein kinases. Bioorg. Med. Chem. Lett. 2012, 22, 4599–4604. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Yung-Chi, C.; Prusoff, W.H. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 per cent inhibition (IC50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar] [CrossRef] [PubMed]
- Thomé, M.P.; Filippi-Chiela, E.C.; Villodre, E.V.; Migliavaca, C.B.; Onzi, G.R.; Felipe, K.F.; Lenz, G. Ratiometric analysis of Acridine Orange staining in the study of acidic organelles and autophagy. J. Cell Sci. 2016, 129, 4622–4632. [Google Scholar] [CrossRef] [PubMed]
- Lozzio, C.B.; Lozzio, B.B. Human chronic myelogenous leukemia cell-line with positive Philadelphia-chromosome. Blood 1975, 45, 321–334. [Google Scholar] [CrossRef] [PubMed]
- Heriche, J.K.; Chambaz, E.M. Protein kinase CK2α is a target for the Abl and Bcr-Abl tyrosine kinases. Oncogene 1998, 17, 13–18. [Google Scholar] [CrossRef]
- Rai, Y.; Yadav, P.; Kumari, N.; Kalra, N.; Bhatt, A.N. Hexokinase II inhibition by 3-bromopyruvate sensitizes myeloid leukemic cells K-562 to anti-leukemic drug, daunorubicin. Biosci Rep. 2019, 39, BSR20190880. [Google Scholar] [CrossRef]
- Morotti, A.; Carrà, G.; Panuzzo, C.; Crivellaro, S.; Taulli, R.; Guerrasio, A.; Saglio, G. Protein Kinase CK2: A Targetable BCR-ABL Partner in Philadelphia Positive Leukemias. Adv. Hematol. 2015, 2015, 612567. [Google Scholar] [CrossRef]
- Hori, M.; Nogami, T.; Itabashi, M.; Yoshim, F.; Ono, H.; Koizumi, S. Expression of Bcl-2 in human breast cancer: Correlation between hormone receptor status, p53 protein accumulation and DNA strand breaks associated with apoptosis. Pathol. Int. 1997, 47, 757–762. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Denton, D.; Kumar, S. Autophagy-dependent cell death. Cell Death Differ. 2018, 26, 605–616. [Google Scholar] [CrossRef]
- Bellodi, C.; Lidonnici, M.; Hamilton, A.; Helgason, V.; Soliera, A.R.; Ronchetti, M.; Galavotti, S.; Young, K.W.; Selmi, T.; Yacobi, R.; et al. Targeting autophagy. J. Clin. Investig. 2009, 119, 1109–1123. [Google Scholar] [CrossRef]
- Lin, Q.; Meloni, D.; Pan, Y.; Xia, M.; Rodgers, J.; Shepard, S.; Li, M.; Galya, L.; Metcalf, B.; Yue, T.-Y.; et al. Enantioselective synthesis of Janus kinase inhibitor INCB018424 via an organocatalytic aza-Michael reaction. Org. Lett. 2009, 11, 1999–2002. [Google Scholar] [CrossRef] [PubMed]
- Izeradjene, K.; Douglas, L.; Delaney, A.; Houghton, J.A. Casein kinase ii (CK2) enhances death-inducing signaling complex (disc) activity in trail-induced apoptosis in human colon carcinoma cell lines. Oncogene 2005, 24, 2050–2058. [Google Scholar] [CrossRef] [PubMed]
- Trembley, J.H.; Chen, Z.; Unger, G.; Slaton, J.; Kren, B.T.; Van Waes, C.; Ahmed, K. Emergence of protein kinase ck2 as a key target in cancer therapy. BioFactors 2010, 36, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; He, M.; Li, L.; Liang, Z.; Zou, Z.; Tao, A. Cell-in-cell death is not restricted by caspase-3 deficiency in mcf-7 cells. J. Breast Cancer 2016, 19, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Dowbenko, D.; Lasky, L.A. AKT/PKB phosphorylation of p21Cip/WAF1 enhances protein stability of p21Cip/WAF1 and promotes cell survival. J. Biol. Chem. 2002, 277, 11352–11361. [Google Scholar] [CrossRef] [PubMed]
- Wińska, P.; Skierka, K.; Łukowska-Chojnacka, E.; Koronkiewicz, M.; Cieśla, J.; Bretner, M. Effect of Simultaneous Inhibition of Protein Kinase CK2 and Thymidylate Synthase in Leukemia and Breast Cancer Cells. Anticancer Res. 2018, 38, 4617–4627. [Google Scholar] [CrossRef]
- Wińska, P.; Widło, Ł.; Skierka, K.; Krzyśko, A.; Koronkiewicz, M.; Cieśla, J.M.; Cieśla, J.; Bretner, M. Simultaneous Inhibition of Protein Kinase CK2 and Dihydrofolate Reductase Results in Synergistic Effect on Acute Lymphoblastic Leukemia Cells. Anticancer Res. 2019, 39, 3531–3542. [Google Scholar] [CrossRef]
- Wińska, P.; Karatsai, O.; Staniszewska, M.; Koronkiewicz, M.; Chojnacki, K.; Rędowicz, M.J. Synergistic Interactions of 5- Fluorouracil with Inhibitors of Protein Kinase CK2 Correlate with p38 MAPK Activation and FAK Inhibition in the Triple-Negative Breast Cancer Cell Line. Int. J. Mol. Sci. 2020, 21, 6234. [Google Scholar] [CrossRef]
- Daina, A.; Blatter, M.-C.; Baillie Gerritsen, V.; Palagi, P.M.; Marek, D.; Xenarios, I.; Schwede, T.; Michielin, O.; Zoete, V. Drug design workshop: A web-based educational tool to introduce computer-aided drug design to the general public. J. Chem. Educ. 2017, 94, 335–344. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cpd. | Ki (µM) | ||
|---|---|---|---|
| CK2α | CK2α2β2 | PIM-1 | |
| 4 | 0.223 | 0.056 | 0.052 |
| rac-5 | 0.296 | 0.127 | 0.064 |
| (S)-5 | 0.271 | 0.167 | 0.099 |
| (R)-5 | 0.394 | 0.104 | 0.089 |
| rac-6 | 0.294 | 0.112 | 0.067 |
| 7 | 0.156 | 0.114 | 0.082 |
| 8 | 0.256 | 0.072 | 0.071 |
| rac-9 | 1.671 | 0.605 | 0.267 |
| 10 | 0.139 | 0.124 | 0.093 |
| rac-11 | 0.151 | 0.089 | 0.073 |
| Cpd. | Cell Line | SI * | logP ** | |||||
|---|---|---|---|---|---|---|---|---|
| CCRF-CEM | K-562 | MCF-7 | Vero | CCRF-CEM | K-562 | MCF-7 | ||
| 4 | 18.94 ± 0.88 | 22.54 ± 6.09 | 14.68 ± 0.27 | 18.72 ± 0.33 | 0.99 | 0.83 | 1.27 | 5.39 |
| rac-5 | 21.70 ± 0.86 | 38.70 ± 8.08 | 16.86 ± 0.30 | 22.58 ± 0.13 | 1.04 | 0.58 | 1.34 | 4.41 |
| (S)-5 | 23.21± 4.01 | 39.28 ± 9.05 | 16.55 ± 0.30 | 25.20 ± 0.54 | 1.11 | 0.64 | 1.52 | 4.41 |
| (R)-5 | 22.31 ± 0.10 | 39.59 ± 8.11 | 16.09 ± 0.19 | 28.30 ± 0.43 | 1.27 | 0.71 | 1.76 | 4.41 |
| rac-6 | 11.83 ± 0.40 | 17.74 ± 3.54 | 9.66 ± 0.31 | 20.38 ± 0.41 | 1.72 | 1.15 | 2.11 | 5.05 |
| 7 | 25.64 ± 0.54 | 33.70 ± 4.92 | 18.21 ± 0.30 | 29.46 ± 0.12 | 1.15 | 0.87 | 1.62 | 4.19 |
| 8 | 17.37 ± 0.27 | 40.02 ± 7.45 | 15.11 ± 0.39 | 16.07 ± 0.27 | 0.92 | 0.40 | 1.06 | 4.88 |
| rac-9 | 13.59 ± 0.84 | 11.61 ± 2.85 | 10.80 ± 0.23 | 12.01 ± 0.21 | 0.88 | 1.03 | 1.11 | 4.74 |
| 10 | 24.47 ± 0.21 | 38.06 ± 5.56 | 16.65 ± 0.23 | 25.19 ± 0.10 | 1.03 | 0.66 | 1.51 | 3.55 |
| rac-11 | 33.80 ± 0.26 | 41.49 ± 8.45 | 17.20 ± 0.39 | 41.53 ± 0.10 | 1.23 | 1.00 | 2.41 | 3.55 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wińska, P.; Wielechowska, M.; Koronkiewicz, M.; Borowiecki, P. Synthesis and Anticancer Activity of Novel Dual Inhibitors of Human Protein Kinases CK2 and PIM-1. Pharmaceutics 2023, 15, 1991. https://doi.org/10.3390/pharmaceutics15071991
Wińska P, Wielechowska M, Koronkiewicz M, Borowiecki P. Synthesis and Anticancer Activity of Novel Dual Inhibitors of Human Protein Kinases CK2 and PIM-1. Pharmaceutics. 2023; 15(7):1991. https://doi.org/10.3390/pharmaceutics15071991
Chicago/Turabian StyleWińska, Patrycja, Monika Wielechowska, Mirosława Koronkiewicz, and Paweł Borowiecki. 2023. "Synthesis and Anticancer Activity of Novel Dual Inhibitors of Human Protein Kinases CK2 and PIM-1" Pharmaceutics 15, no. 7: 1991. https://doi.org/10.3390/pharmaceutics15071991
APA StyleWińska, P., Wielechowska, M., Koronkiewicz, M., & Borowiecki, P. (2023). Synthesis and Anticancer Activity of Novel Dual Inhibitors of Human Protein Kinases CK2 and PIM-1. Pharmaceutics, 15(7), 1991. https://doi.org/10.3390/pharmaceutics15071991