

Pharmaceutical Strategies to Improve Druggability of Potential Drug Candidates in Nonalcoholic Fatty Liver Disease Therapy


Abstract
1. Introduction
2. Therapeutic Drug Candidates for the Treatment of NAFLD
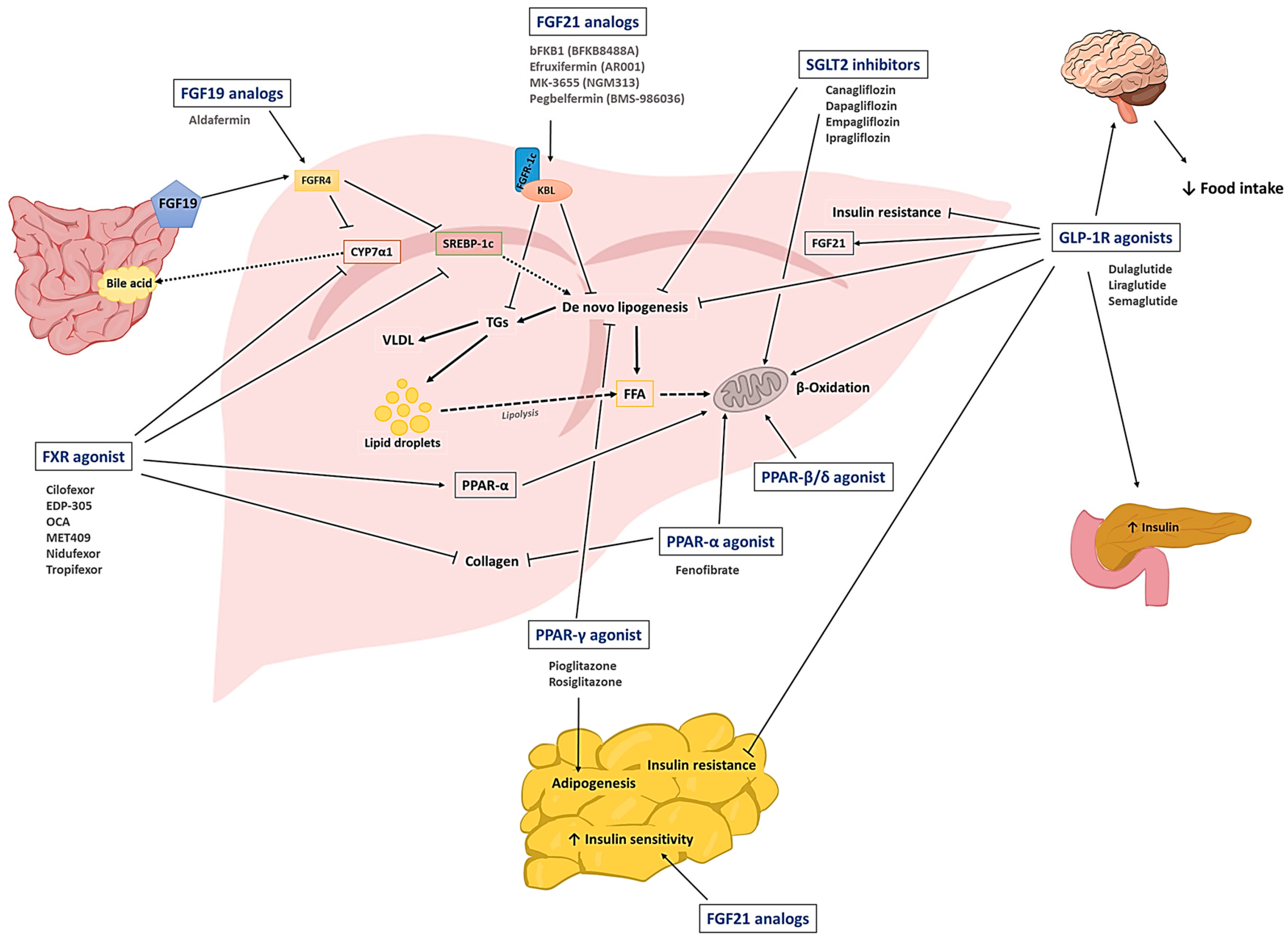
2.1. SGLT2 Inhibitors
2.2. GLP-1R Agonists
2.3. PPAR Agonists
2.4. Fibroblast Growth Factor (FGF) Analogs
2.5. FXR Agonists
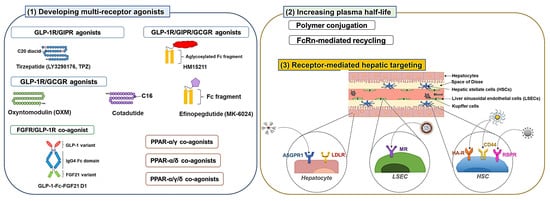
3. Strategies to Enhance the Efficacy of NAFLD Drug Candidates
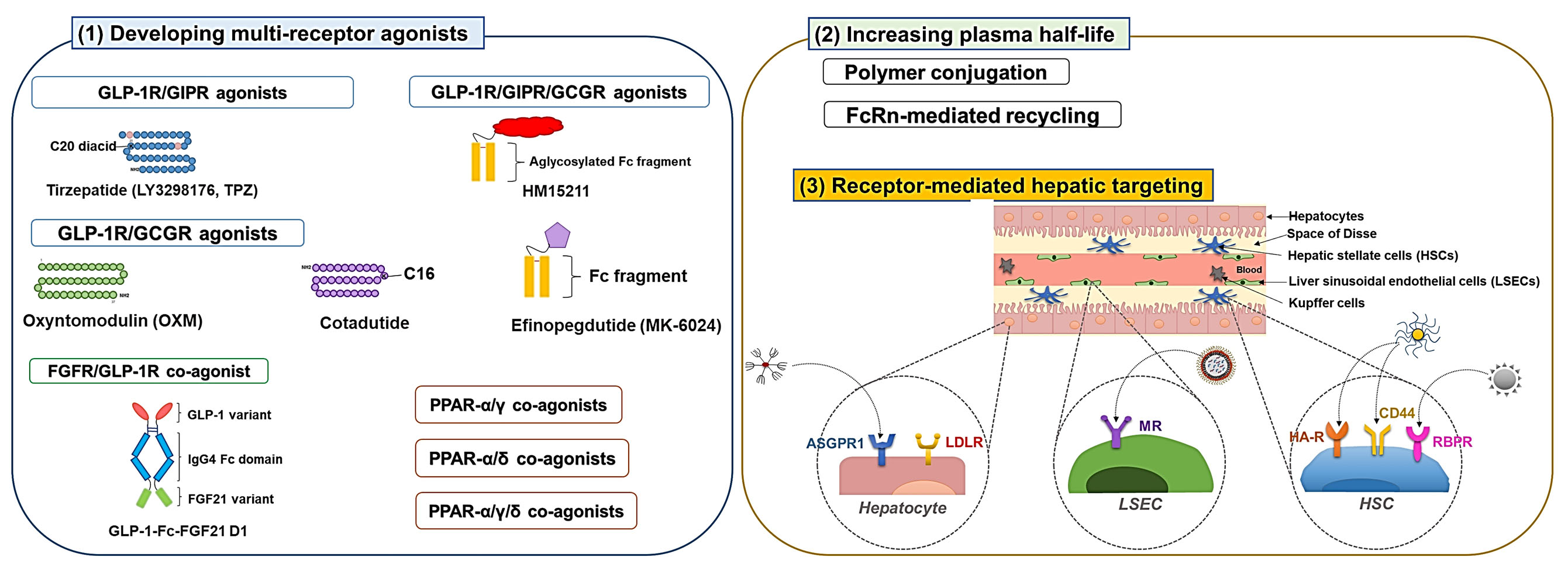
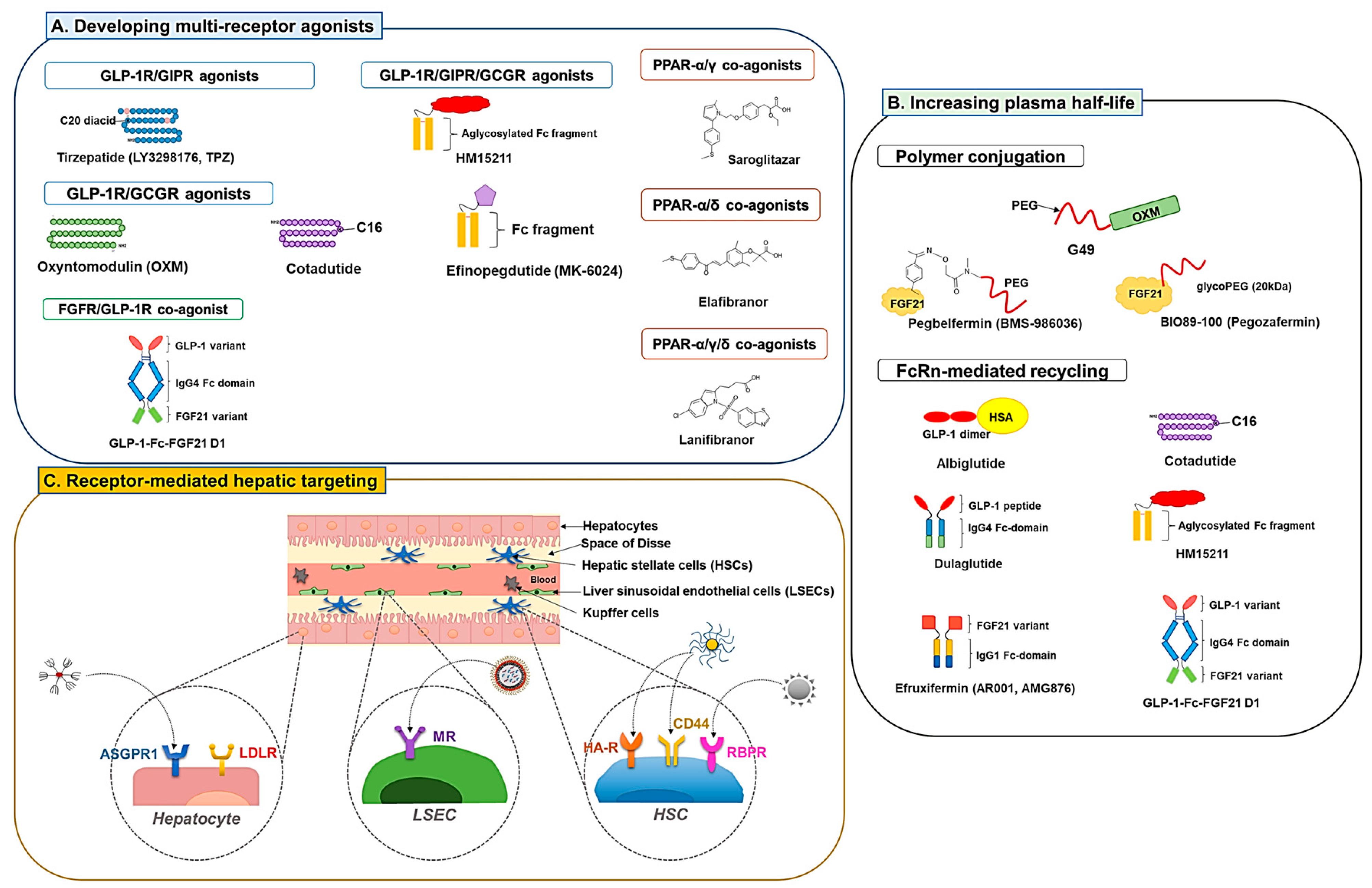
3.1. Developing Multiple Receptor Agonists
3.1.1. GLP-1R Co-Agonists
3.1.2. PPAR Dual and Pan Agonists
3.1.3. FGFR Dual Agonists
3.2. Development of Long-Acting Derivatives
3.2.1. Polymer Conjugation
3.2.2. Exploiting Neonatal Fc Receptor (FcRn)-Mediated Recycling
3.3. Receptor-Mediated Hepatic Targeting
4. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Clark, J.M.; Brancati, F.L.; Diehl, A.M. Nonalcoholic fatty liver disease. Gastroenterology 2002, 122, 1649–1657. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Afolabi, B.I.; Ibitoye, B.O.; Ikem, R.T.; Omisore, A.D.; Idowu, B.M.; Soyoye, D.O. The relationship between glycaemic control and non-alcoholic fatty liver disease in Nigerian type 2 diabetic patients. J. Natl. Med. Assoc. 2018, 110, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Wang, J.; Zeng, J.; Cao, X.; He, Y. Association of NAFLD with diabetes and the impact of BMI changes: A 5-year cohort study based on 18,507 elderly. J. Clin. Endocrinol. Metab. 2017, 102, 1309–1316. [Google Scholar] [CrossRef]
- Younossi, Z.M. Non-alcoholic fatty liver disease–a global public health perspective. J. Hepatol. 2019, 70, 531–544. [Google Scholar] [CrossRef]
- Lee, Y.-h.; Cho, Y.; Lee, B.-W.; Park, C.-Y.; Lee, D.H.; Cha, B.-S.; Rhee, E.-J. Nonalcoholic fatty liver disease in diabetes. Part I: Epidemiology and diagnosis. Diabetes Metab. J. 2019, 43, 31–45. [Google Scholar] [CrossRef]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef]
- Treeprasertsuk, S.; Björnsson, E.; Enders, F.; Suwanwalaikorn, S.; Lindor, K.D. NAFLD fibrosis score: A prognostic predictor for mortality and liver complications among NAFLD patients. World J. Gastroenterol. 2013, 19, 1219. [Google Scholar] [CrossRef]
- Eren, F.; Kaya, E.; Yilmaz, Y. Accuracy of Fibrosis-4 index and non-alcoholic fatty liver disease fibrosis scores in metabolic (dysfunction) associated fatty liver disease according to body mass index: Failure in the prediction of advanced fibrosis in lean and morbidly obese individuals. Eur. J. Gastroenterol. Hepatol. 2022, 34, 98–103. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Harrison, S.A.; Ratziu, V.; Abdelmalek, M.F.; Diehl, A.M.; Caldwell, S.; Shiffman, M.L.; Aguilar Schall, R.; Jia, C.; McColgan, B. The natural history of advanced fibrosis due to nonalcoholic steatohepatitis: Data from the simtuzumab trials. Hepatology 2019, 70, 1913–1927. [Google Scholar] [CrossRef]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef] [PubMed]
- Kawano, Y.; Cohen, D.E. Mechanisms of hepatic triglyceride accumulation in non-alcoholic fatty liver disease. J. Gastroenterol. 2013, 48, 434–441. [Google Scholar] [CrossRef]
- Kumar, S.; Duan, Q.; Wu, R.; Harris, E.N.; Su, Q. Pathophysiological communication between hepatocytes and non-parenchymal cells in liver injury from NAFLD to liver fibrosis. Adv. Drug Deliv. Rev. 2021, 176, 113869. [Google Scholar] [CrossRef] [PubMed]
- Hammoutene, A.; Rautou, P.-E. Role of liver sinusoidal endothelial cells in non-alcoholic fatty liver disease. J. Hepatol. 2019, 70, 1278–1291. [Google Scholar] [CrossRef] [PubMed]
- Lang, A.; Schoonhoven, R.; Tuvia, S.; Brenner, D.A.; Rippe, R.A. Nuclear factor κB in proliferation, activation, and apoptosis in rat hepatic stellate cells. J. Hepatol. 2000, 33, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Hassan, N.; Schwartz, R.E. Organelle stress and alterations in inter-organelle crosstalk during liver fibrosis. Hepatology 2023. [Google Scholar] [CrossRef]
- Solaiman, A.; Sawires, S.K.S. The Ameliorative Potential of Alda-1 on Experimentally Induced Liver Fibrosis in Adult Male Mice. A Histological, Immunohistochemical and Biochemical Study. Egypt. J. Histol. 2022, 45, 949–968. [Google Scholar] [CrossRef]
- Pierantonelli, I.; Rychlicki, C.; Agostinelli, L.; Giordano, D.M.; Gaggini, M.; Fraumene, C.; Saponaro, C.; Manghina, V.; Sartini, L.; Mingarelli, E. Lack of NLRP3-inflammasome leads to gut-liver axis derangement, gut dysbiosis and a worsened phenotype in a mouse model of NAFLD. Sci. Rep. 2017, 7, 12200. [Google Scholar] [CrossRef]
- Tilg, H.; Effenberger, M. From NAFLD to MAFLD: When pathophysiology succeeds. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 387–388. [Google Scholar] [CrossRef]
- Finotti, M.; Romano, M.; Auricchio, P.; Scopelliti, M.; Brizzolari, M.; Grossi, U.; Piccino, M.; Benvenuti, S.; Morana, G.; Cillo, U. Target therapies for NASH/NAFLD: From the molecular aspect to the pharmacological and surgical alternatives. J. Pers. Med. 2021, 11, 499. [Google Scholar] [CrossRef]
- Sumida, Y.; Naito, Y.; Tanaka, S.; Sakai, K.; Inada, Y.; Taketani, H.; Kanemasa, K.; Yasui, K.; Itoh, Y.; Okanoue, T. Long-term (≥2 yr) efficacy of vitamin E for non-alcoholic steatohepatitis. Hepatogastroenterology 2013, 60, 1445–1450. [Google Scholar]
- Sasaki, A.; Nitta, H.; Otsuka, K.; Umemura, A.; Baba, S.; Obuchi, T.; Wakabayashi, G. Bariatric surgery and non-alcoholic Fatty liver disease: Current and potential future treatments. Front. Endocrinol. 2014, 5, 164. [Google Scholar] [CrossRef]
- Saeed, N.; Glass, L.; Sharma, P.; Shannon, C.; Sonnenday, C.J.; Tincopa, M.A. Incidence and risks for nonalcoholic fatty liver disease and steatohepatitis post-liver transplant: Systematic review and meta-analysis. Transplantation 2019, 103, e345–e354. [Google Scholar] [CrossRef]
- Cardoso, A.C.; de Figueiredo-Mendes, C.; Villela-Nogueira, C.A. Current management of NAFLD/NASH. Liver Int. 2021, 41, 89–94. [Google Scholar] [CrossRef]
- Androutsakos, T.; Nasiri-Ansari, N.; Bakasis, A.-D.; Kyrou, I.; Efstathopoulos, E.; Randeva, H.S.; Kassi, E. SGLT-2 inhibitors in NAFLD: Expanding their role beyond diabetes and cardioprotection. Int. J. Mol. Sci. 2022, 23, 3107. [Google Scholar] [CrossRef]
- Jojima, T.; Tomotsune, T.; Iijima, T.; Akimoto, K.; Suzuki, K.; Aso, Y. Empagliflozin (an SGLT2 inhibitor), alone or in combination with linagliptin (a DPP-4 inhibitor), prevents steatohepatitis in a novel mouse model of non-alcoholic steatohepatitis and diabetes. Diabetol. Metab. Syndr. 2016, 8, 45. [Google Scholar] [CrossRef] [PubMed]
- Wallenius, K.; Kroon, T.; Hagstedt, T.; Löfgren, L.; Sörhede-Winzell, M.; Boucher, J.; Lindén, D.; Oakes, N.D. The SGLT2 inhibitor dapagliflozin promotes systemic FFA mobilization, enhances hepatic β-oxidation, and induces ketosis. J. Lipid Res. 2022, 63, 100176. [Google Scholar] [CrossRef]
- Komiya, C.; Tsuchiya, K.; Shiba, K.; Miyachi, Y.; Furuke, S.; Shimazu, N.; Yamaguchi, S.; Kanno, K.; Ogawa, Y. Ipragliflozin improves hepatic steatosis in obese mice and liver dysfunction in type 2 diabetic patients irrespective of body weight reduction. PLoS ONE 2016, 11, e0151511. [Google Scholar] [CrossRef]
- Takahashi, H.; Kessoku, T.; Kawanaka, M.; Nonaka, M.; Hyogo, H.; Fujii, H.; Nakajima, T.; Imajo, K.; Tanaka, K.; Kubotsu, Y. Ipragliflozin improves the hepatic outcomes of patients with diabetes with NAFLD. Hepatol. Commun. 2022, 6, 120–132. [Google Scholar] [CrossRef] [PubMed]
- Han, E.; Lee, Y.-H.; Lee, B.-W.; Kang, E.S.; Cha, B.-S. Ipragliflozin additively ameliorates non-alcoholic fatty liver disease in patients with type 2 diabetes controlled with metformin and pioglitazone: A 24-week randomized controlled trial. J. Clin. Med. 2020, 9, 259. [Google Scholar] [CrossRef] [PubMed]
- He, K.; Li, J.; Xi, W.; Ge, J.; Sun, J.; Jing, Z. Dapagliflozin for nonalcoholic fatty liver disease: A systematic review and meta-analysis. Diabetes Res. Clin. Pract. 2022, 185, 109791. [Google Scholar] [CrossRef]
- Eriksson, J.W.; Lundkvist, P.; Jansson, P.-A.; Johansson, L.; Kvarnström, M.; Moris, L.; Miliotis, T.; Forsberg, G.-B.; Risérus, U.; Lind, L. Effects of dapagliflozin and n-3 carboxylic acids on non-alcoholic fatty liver disease in people with type 2 diabetes: A double-blind randomised placebo-controlled study. Diabetologia 2018, 61, 1923–1934. [Google Scholar] [CrossRef]
- Sumida, Y.; Murotani, K.; Saito, M.; Tamasawa, A.; Osonoi, Y.; Yoneda, M.; Osonoi, T. Effect of luseogliflozin on hepatic fat content in type 2 diabetes patients with non-alcoholic fatty liver disease: A prospective, single-arm trial (LEAD trial). Hepatol. Res. 2019, 49, 64–71. [Google Scholar] [CrossRef]
- Kuchay, M.S.; Krishan, S.; Mishra, S.K.; Farooqui, K.J.; Singh, M.K.; Wasir, J.S.; Bansal, B.; Kaur, P.; Jevalikar, G.; Gill, H.K. Effect of empagliflozin on liver fat in patients with type 2 diabetes and nonalcoholic fatty liver disease: A randomized controlled trial (E-LIFT Trial). Diabetes Care 2018, 41, 1801–1808. [Google Scholar] [CrossRef]
- Itani, T.; Ishihara, T. Efficacy of canagliflozin against nonalcoholic fatty liver disease: A prospective cohort study. Obes. Sci. Pract. 2018, 4, 477–482. [Google Scholar] [CrossRef]
- Andersen, A.; Lund, A.; Knop, F.K.; Vilsbøll, T. Glucagon-like peptide 1 in health and disease. Nat. Rev. Endocrinol. 2018, 14, 390–403. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Liang, Y.; Luo, Y.; Li, Z.; Wen, Y.; Shen, J.; Li, R.; Zheng, H.; Gu, H.F.; Xia, N. Liraglutide ameliorates nonalcoholic fatty liver disease in diabetic mice via the IRS2/PI3K/Akt signaling pathway. Diabetes Metab. Syndr. Obes. Targets Ther. 2019, 12, 1013. [Google Scholar] [CrossRef] [PubMed]
- Ao, N.; Ma, Z.; Yang, J.; Jin, S.; Zhang, K.; Luo, E.; Du, J. Liraglutide ameliorates lipotoxicity-induced inflammation through the mTORC1 signalling pathway. Peptides 2020, 133, 170375. [Google Scholar] [CrossRef] [PubMed]
- Ali, E.S.; Girard, D.; Petrovsky, N. Impaired Ca2+ signaling due to hepatic steatosis mediates hepatic insulin resistance in Alström syndrome mice that is reversed by GLP-1 analog treatment. Am. J. Physiol. Cell Physiol. 2021, 321, C187–C198. [Google Scholar] [CrossRef]
- Chen, Y.; Xu, Y.-N.; Ye, C.-Y.; Feng, W.-B.; Zhou, Q.-T.; Yang, D.-H.; Wang, M.-W. GLP-1 mimetics as a potential therapy for nonalcoholic steatohepatitis. Acta Pharmacol. Sin. 2022, 43, 1156–1166. [Google Scholar] [CrossRef]
- Duparc, T.; Briand, F.; Trenteseaux, C.; Merian, J.; Combes, G.; Najib, S.; Sulpice, T.; Martinez, L.O. Liraglutide improves hepatic steatosis and metabolic dysfunctions in a 3-week dietary mouse model of nonalcoholic steatohepatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2019, 317, G508–G517. [Google Scholar] [CrossRef] [PubMed]
- Somm, E.; Montandon, S.A.; Loizides-Mangold, U.; Gaïa, N.; Lazarevic, V.; De Vito, C.; Perroud, E.; Bochaton-Piallat, M.-L.; Dibner, C.; Schrenzel, J. The GLP-1R agonist liraglutide limits hepatic lipotoxicity and inflammatory response in mice fed a methionine-choline deficient diet. Transl. Res. 2021, 227, 75–88. [Google Scholar] [CrossRef]
- Pontes-da-Silva, R.M.; de Souza Marinho, T.; de Macedo Cardoso, L.E.; Mandarim-de-Lacerda, C.A.; Aguila, M.B. Obese mice weight loss role on nonalcoholic fatty liver disease and endoplasmic reticulum stress treated by a GLP-1 receptor agonist. Int. J. Obes. 2022, 46, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, M.J.; Hull, D.; Guo, K.; Barton, D.; Hazlehurst, J.M.; Gathercole, L.L.; Nasiri, M.; Yu, J.; Gough, S.C.; Newsome, P.N. Glucagon-like peptide 1 decreases lipotoxicity in non-alcoholic steatohepatitis. J. Hepatol. 2016, 64, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, M.J.; Gaunt, P.; Aithal, G.P.; Barton, D.; Hull, D.; Parker, R.; Hazlehurst, J.M.; Guo, K.; Abouda, G.; Aldersley, M.A. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): A multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet 2016, 387, 679–690. [Google Scholar] [CrossRef]
- Song, T.; Jia, Y.; Li, Z.; Wang, F.; Ren, L.; Chen, S. Effects of liraglutide on nonalcoholic fatty liver disease in patients with type 2 diabetes mellitus: A systematic review and meta-analysis. Diabetes Ther. 2021, 12, 1735–1749. [Google Scholar] [CrossRef]
- Newsome, P.; Francque, S.; Harrison, S.; Ratziu, V.; Van Gaal, L.; Calanna, S.; Hansen, M.; Linder, M.; Sanyal, A. Effect of semaglutide on liver enzymes and markers of inflammation in subjects with type 2 diabetes and/or obesity. Aliment. Pharmacol. Ther. 2019, 50, 193–203. [Google Scholar] [CrossRef]
- Mantovani, A.; Petracca, G.; Beatrice, G.; Csermely, A.; Lonardo, A.; Targher, G. Glucagon-like peptide-1 receptor agonists for treatment of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis: An updated meta-analysis of randomized controlled trials. Metabolites 2021, 11, 73. [Google Scholar] [CrossRef]
- Rubino, D.M.; Greenway, F.L.; Khalid, U.; O’Neil, P.M.; Rosenstock, J.; Sørrig, R.; Wadden, T.A.; Wizert, A.; Garvey, W.T.; Arauz-Pacheco, C. Effect of weekly subcutaneous semaglutide vs daily liraglutide on body weight in adults with overweight or obesity without diabetes: The STEP 8 randomized clinical trial. JAMA 2022, 327, 138–150. [Google Scholar] [CrossRef]
- Choudhary, N.S.; Kumar, N.; Duseja, A. Peroxisome proliferator-activated receptors and their agonists in nonalcoholic fatty liver disease. J. Clin. Exp. Hepatol. 2019, 9, 731–739. [Google Scholar] [CrossRef]
- Gross, B.; Pawlak, M.; Lefebvre, P.; Staels, B. PPARs in obesity-induced T2DM, dyslipidaemia and NAFLD. Nat. Rev. Endocrinol. 2017, 13, 36–49. [Google Scholar] [CrossRef]
- Grygiel-Górniak, B. Peroxisome proliferator-activated receptors and their ligands: Nutritional and clinical implications–A review. Nutr. J. 2014, 13, 17. [Google Scholar] [CrossRef]
- Lian, J.; Fu, J. Pioglitazone for NAFLD patients with prediabetes or type 2 diabetes mellitus: A meta-analysis. Front. Endocrinol. 2021, 12, 615409. [Google Scholar] [CrossRef]
- Ratziu, V.; Charlotte, F.; Bernhardt, C.; Giral, P.; Halbron, M.; LeNaour, G.; Hartmann-Heurtier, A.; Bruckert, E.; Poynard, T.; Group, L.S. Long-term efficacy of rosiglitazone in nonalcoholic steatohepatitis: Results of the fatty liver improvement by rosiglitazone therapy (FLIRT 2) extension trial. Hepatology 2010, 51, 445–453. [Google Scholar] [CrossRef]
- Psaty, B.M.; Furberg, C.D. Rosiglitazone and cardiovascular risk. N. Engl. J. Med. 2007, 356, 2522–2524. [Google Scholar] [CrossRef] [PubMed]
- Aaldijk, A.S.; Verzijl, C.R.; Jonker, J.W.; Struik, D. Biological and pharmacological functions of the FGF19-and FGF21-coreceptor beta klotho. Front. Endocrinol. 2023, 14, 1150222. [Google Scholar] [CrossRef]
- Velingkar, A.; Vuree, S.; Prabhakar, P.K.; Kalashikam, R.R.; Banerjee, A.; Kondeti, S. Fibroblast growth factor 21 as a potential master regulator in metabolic disorders. Am. J. Physiol. Endocrinol. Metab. 2023, 324, E409–E424. [Google Scholar] [CrossRef] [PubMed]
- Kharitonenkov, A.; Shiyanova, T.L.; Koester, A.; Ford, A.M.; Micanovic, R.; Galbreath, E.J.; Sandusky, G.E.; Hammond, L.J.; Moyers, J.S.; Owens, R.A. FGF-21 as a novel metabolic regulator. J. Clin. Investig. 2005, 115, 1627–1635. [Google Scholar] [CrossRef]
- Wang, W.-F.; Li, S.-M.; Ren, G.-P.; Zheng, W.; Lu, Y.-J.; Yu, Y.-H.; Xu, W.-J.; Li, T.-H.; Zhou, L.-H.; Liu, Y. Recombinant murine fibroblast growth factor 21 ameliorates obesity-related inflammation in monosodium glutamate-induced obesity rats. Endocrinoligy 2015, 49, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Keinicke, H.; Sun, G.; Mentzel, C.M.J.; Fredholm, M.; John, L.M.; Andersen, B.; Raun, K.; Kjaergaard, M. FGF21 regulates hepatic metabolic pathways to improve steatosis and inflammation. Endocr. Connect. 2020, 9, 755–768. [Google Scholar] [CrossRef]
- Kolumam, G.; Chen, M.Z.; Tong, R.; Zavala-Solorio, J.; Kates, L.; van Bruggen, N.; Ross, J.; Wyatt, S.K.; Gandham, V.D.; Carano, R.A. Sustained brown fat stimulation and insulin sensitization by a humanized bispecific antibody agonist for fibroblast growth factor receptor 1/βKlotho complex. EBioMedicine 2015, 2, 730–743. [Google Scholar] [CrossRef]
- Wong, C.; Dash, A.; Fredrickson, J.; Lewin-Koh, N.; Chen, S.; Yoshida, K.; Liu, Y.; Gutierrez, J.; Kunder, R. Fibroblast growth factor receptor 1/Klothoβ agonist BFKB8488A improves lipids and liver health markers in patients with diabetes or NAFLD: A phase 1b randomized trial. Hepatology, 2022; online ahead of print. [Google Scholar] [CrossRef]
- Depaoli, A.; Phung, V.; Bashir, M.R.; Morrow, L.; Beysen, C.; Yan, A.; Ling, L.; Baxter, B.; Luskey, K.L.; Olefsky, J.M. 140-LB: NGM313, a novel activator of b-Klotho/FGFR1c, improves insulin resistance and reduces hepatic fat in obese, nondiabetic subjects. Diabetes 2019, 68, 140-LB. [Google Scholar] [CrossRef]
- Kurosu, H.; Choi, M.; Ogawa, Y.; Dickson, A.S.; Goetz, R.; Eliseenkova, A.V.; Mohammadi, M.; Rosenblatt, K.P.; Kliewer, S.A.; Kuro-o, M. Tissue-specific expression of βKlotho and fibroblast growth factor (FGF) receptor isoforms determines metabolic activity of FGF19 and FGF21. J. Biol. Chem. 2007, 282, 26687–26695. [Google Scholar] [CrossRef]
- Wunsch, E.; Milkiewicz, M.; Wasik, U.; Trottier, J.; Kempińska-Podhorodecka, A.; Elias, E.; Barbier, O.; Milkiewicz, P. Expression of hepatic fibroblast growth factor 19 is enhanced in primary biliary cirrhosis and correlates with severity of the disease. Sci. Rep. 2015, 5, 13462. [Google Scholar] [CrossRef] [PubMed]
- Wojcik, M.; Janus, D.; Dolezal-Oltarzewska, K.; Kalicka-Kasperczyk, A.; Poplawska, K.; Drozdz, D.; Sztefko, K.; Starzyk, J.B. A decrease in fasting FGF19 levels is associated with the development of non-alcoholic fatty liver disease in obese adolescents. J. Pediatr. Endocrinol. Metab. 2012, 25, 1089–1093. [Google Scholar] [CrossRef] [PubMed]
- Gadaleta, R.M.; Scialpi, N.; Peres, C.; Cariello, M.; Ko, B.; Luo, J.; Porru, E.; Roda, A.; Sabbà, C.; Moschetta, A. Suppression of hepatic bile acid synthesis by a non-tumorigenic FGF19 analogue protects mice from fibrosis and hepatocarcinogenesis. Sci. Rep. 2018, 8, 17210. [Google Scholar] [CrossRef]
- Harrison, S.A.; Rossi, S.J.; Paredes, A.H.; Trotter, J.F.; Bashir, M.R.; Guy, C.D.; Banerjee, R.; Jaros, M.J.; Owers, S.; Baxter, B.A. NGM282 improves liver fibrosis and histology in 12 weeks in patients with nonalcoholic steatohepatitis. Hepatology 2020, 71, 1198–1212. [Google Scholar] [CrossRef] [PubMed]
- Rinella, M.E.; Trotter, J.F.; Abdelmalek, M.F.; Paredes, A.H.; Connelly, M.A.; Jaros, M.J.; Ling, L.; Rossi, S.J.; DePaoli, A.M.; Harrison, S.A. Rosuvastatin improves the FGF19 analogue NGM282-associated lipid changes in patients with non-alcoholic steatohepatitis. J. Hepatol. 2019, 70, 735–744. [Google Scholar] [CrossRef]
- Zhu, Y.; Li, F.; Guo, G.L. Tissue-specific function of farnesoid X receptor in liver and intestine. Pharmacol. Res. 2011, 63, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Bjursell, M.; Wedin, M.; Admyre, T.; Hermansson, M.; Böttcher, G.; Göransson, M.; Lindén, D.; Bamberg, K.; Oscarsson, J.; Bohlooly-Y, M. Ageing Fxr deficient mice develop increased energy expenditure, improved glucose control and liver damage resembling NASH. PLoS ONE 2013, 8, e64721. [Google Scholar] [CrossRef] [PubMed]
- Cariou, B.; Staels, B. FXR: A promising target for the metabolic syndrome? Trends Pharmacol. Sci. 2007, 28, 236–243. [Google Scholar] [CrossRef]
- Zhou, S.; You, H.; Qiu, S.; Yu, D.; Bai, Y.; He, J.; Cao, H.; Che, Q.; Guo, J.; Su, Z. A new perspective on NAFLD: Focusing on the crosstalk between peroxisome proliferator-activated receptor alpha (PPARα) and farnesoid X receptor (FXR). Biomed. Pharmacother. 2022, 154, 113577. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, T.; Honda, A.; Ikegami, T.; Iida, T.; Matsuzaki, Y. Human-specific dual regulations of FXR-activation for reduction of fatty liver using in vitro cell culture model. J. Clin. Biochem. Nutr. 2019, 64, 112–123. [Google Scholar] [CrossRef]
- Fiorucci, S.; Mencarelli, A.; Palladino, G.; Cipriani, S. Bile-acid-activated receptors: Targeting TGR5 and farnesoid-X-receptor in lipid and glucose disorders. Trends Pharmacol. Sci. 2009, 30, 570–580. [Google Scholar] [CrossRef]
- Alemi, F.; Kwon, E.; Poole, D.P.; Lieu, T.; Lyo, V.; Cattaruzza, F.; Cevikbas, F.; Steinhoff, M.; Nassini, R.; Materazzi, S. The TGR5 receptor mediates bile acid–induced itch and analgesia. J. Clin. Investig. 2013, 123, 1513–1530. [Google Scholar] [CrossRef]
- Pellicciari, R.; Fiorucci, S.; Camaioni, E.; Clerici, C.; Costantino, G.; Maloney, P.R.; Morelli, A.; Parks, D.J.; Willson, T.M. 6α-ethyl-chenodeoxycholic acid (6-ECDCA), a potent and selective FXR agonist endowed with anticholestatic activity. J. Med. Chem. 2002, 45, 3569–3572. [Google Scholar] [CrossRef]
- Zhang, Y.; Jackson, J.P.; St. Claire, R.L., III; Freeman, K.; Brouwer, K.R.; Edwards, J.E. Obeticholic acid, a selective farnesoid X receptor agonist, regulates bile acid homeostasis in sandwich-cultured human hepatocytes. Pharmacol. Res. Perspect. 2017, 5, e00329. [Google Scholar] [CrossRef]
- Goto, T.; Itoh, M.; Suganami, T.; Kanai, S.; Shirakawa, I.; Sakai, T.; Asakawa, M.; Yoneyama, T.; Kai, T.; Ogawa, Y. Obeticholic acid protects against hepatocyte death and liver fibrosis in a murine model of nonalcoholic steatohepatitis. Sci. Rep. 2018, 8, 8157. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Wang, X.; Chung, T.-Y.; Ye, W.; Hodge, L.; Zhang, L.; Chng, K.; Xiao, Y.-F.; Wang, Y.J. Carbon tetrachloride (CCl4) accelerated development of non-alcoholic fatty liver disease (NAFLD)/steatohepatitis (NASH) in MS-NASH mice fed western diet supplemented with fructose (WDF). BMC Gastroenterol. 2020, 20, 339. [Google Scholar] [CrossRef]
- Rizzo, G.; Passeri, D.; De Franco, F.; Ciaccioli, G.; Donadio, L.; Rizzo, G.; Orlandi, S.; Sadeghpour, B.; Wang, X.X.; Jiang, T. Functional characterization of the semisynthetic bile acid derivative INT-767, a dual farnesoid X receptor and TGR5 agonist. Mol. Pharmacol. 2010, 78, 617–630. [Google Scholar] [CrossRef]
- Roth, J.D.; Feigh, M.; Veidal, S.S.; Fensholdt, L.K.; Rigbolt, K.T.; Hansen, H.H.; Chen, L.C.; Petitjean, M.; Friley, W.; Vrang, N. INT-767 improves histopathological features in a diet-induced ob/ob mouse model of biopsy-confirmed non-alcoholic steatohepatitis. World J. Gastroenterol. 2018, 24, 195. [Google Scholar] [CrossRef] [PubMed]
- Comeglio, P.; Cellai, I.; Mello, T.; Filippi, S.; Maneschi, E.; Corcetto, F.; Corno, C.; Sarchielli, E.; Morelli, A.; Rapizzi, E. INT-767 prevents NASH and promotes visceral fat brown adipogenesis and mitochondrial function. J. Endocrinol. 2018, 238, 107–127. [Google Scholar] [CrossRef]
- Chau, M.; Li, Y.; Roqueta-Rivera, M.; Garlick, K.; Shen, R.; Wang, G.; Or, Y.S.; Jiang, L.-J. Characterization of EDP-305, a highly potent and selective farnesoid X receptor agonist, for the treatment of non-alcoholic steatohepatitis. Int. J. Gastroenterol. 2019, 3, 4–16. [Google Scholar] [CrossRef]
- Schwabl, P.; Hambruch, E.; Budas, G.R.; Supper, P.; Burnet, M.; Liles, J.T.; Birkel, M.; Brusilovskaya, K.; Königshofer, P.; Peck-Radosavljevic, M. The non-steroidal FXR agonist cilofexor improves portal hypertension and reduces hepatic fibrosis in a rat NASH model. Biomedicines 2021, 9, 60. [Google Scholar] [CrossRef]
- Chianelli, D.; Rucker, P.V.; Roland, J.; Tully, D.C.; Nelson, J.; Liu, X.; Bursulaya, B.; Hernandez, E.D.; Wu, J.; Prashad, M. Nidufexor (LMB763), a novel FXR modulator for the treatment of nonalcoholic steatohepatitis. J. Med. Chem. 2020, 63, 3868–3880. [Google Scholar] [CrossRef]
- Kang, J.; Kim, J.-H.; Yi, J.; Han, O.; Kim, Y.; Baek, E.; Jung, S.Y.; Kwon, S.C.; Trautmann, M.E.; Hompesch, M. The ultra-long acting LAPS GLP/GCG dual agonist, HM12525A, demonstrated safety and prolonged pharmacokinetics in healthy volunteers: A phase 1 first-in-human study. Diabetologia 2015, 58, PS-069-79. [Google Scholar]
- Francque, S.M.; Bedossa, P.; Ratziu, V.; Anstee, Q.M.; Bugianesi, E.; Sanyal, A.J.; Loomba, R.; Harrison, S.A.; Balabanska, R.; Mateva, L. A randomized, controlled trial of the pan-PPAR agonist lanifibranor in NASH. N. Engl. J. Med. 2021, 385, 1547–1558. [Google Scholar] [CrossRef]
- Gawrieh, S.; Noureddin, M.; Loo, N.; Mohseni, R.; Awasty, V.; Cusi, K.S.; Kowdley, K.; Lai, M.; Schiff, E.; Parmar, D. A PPAR-α/γ Agonist, for Treatment of Nonalcoholic Fatty Liver Disease: A Randomized Controlled Double-Blind Phase 2 Trial. Hepatology 2021, 74, 1809–1824. [Google Scholar] [CrossRef] [PubMed]
- Gabe, M.B.N.; van der Velden, W.J.; Smit, F.X.; Gasbjerg, L.S.; Rosenkilde, M.M. Molecular interactions of full-length and truncated GIP peptides with the GIP receptor–A comprehensive review. Peptides 2020, 125, 170224. [Google Scholar] [CrossRef]
- Bastin, M.; Andreelli, F. Dual GIP–GLP1-receptor agonists in the treatment of type 2 diabetes: A short review on emerging data and therapeutic potential. Diabetes Metab. Syndr. Obes. Targets Ther. 2019, 12, 1973–1985. [Google Scholar] [CrossRef] [PubMed]
- Seino, Y.; Fukushima, M.; Yabe, D. GIP and GLP-1, the two incretin hormones: Similarities and differences. J. Diabetes Investig. 2010, 1, 8–23. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Seino, Y. Physiology of GIP-a lesson from GIP receptor knockout mice. Horm. Metab. Res. 2004, 36, 771–774. [Google Scholar] [CrossRef]
- Nauck, M.A.; Meier, J.J. The incretin effect in healthy individuals and those with type 2 diabetes: Physiology, pathophysiology, and response to therapeutic interventions. Lancet Diabetes Endocrinol. 2016, 4, 525–536. [Google Scholar] [CrossRef]
- Hartman, M.L.; Sanyal, A.J.; Loomba, R.; Wilson, J.M.; Nikooienejad, A.; Bray, R.; Karanikas, C.A.; Duffin, K.L.; Robins, D.A.; Haupt, A. Effects of novel dual GIP and GLP-1 receptor agonist tirzepatide on biomarkers of nonalcoholic steatohepatitis in patients with type 2 diabetes. Diabetes Care 2020, 43, 1352–1355. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Zhou, Q.; Cong, Z.; Hang, K.; Zou, X.; Zhang, C.; Chen, Y.; Dai, A.; Liang, A.; Ming, Q. Structural insights into multiplexed pharmacological actions of tirzepatide and peptide 20 at the GIP, GLP-1 or glucagon receptors. Nat. Commun. 2022, 13, 1057. [Google Scholar] [CrossRef] [PubMed]
- Foreman, R.E.; George, A.L.; Reimann, F.; Gribble, F.M.; Kay, R.G. Peptidomics: A review of clinical applications and methodologies. J. Proteome Res. 2021, 20, 3782–3797. [Google Scholar] [CrossRef]
- Sirhan, W.; Piran, R. Therapeutic peptidomimetics in metabolic diseases. In Peptide and Peptidomimetic Therapeutics; Elsevier: Amsterdam, The Netherlands, 2022; pp. 521–550. [Google Scholar]
- Zhang, X.-X.; Pan, Y.-H.; Huang, Y.-M.; Zhao, H.-L. Neuroendocrine hormone amylin in diabetes. World J. Diabetes 2016, 7, 189. [Google Scholar] [CrossRef]
- Habegger, K.M.; Heppner, K.M.; Geary, N.; Bartness, T.J.; DiMarchi, R.; Tschöp, M.H. The metabolic actions of glucagon revisited. Nat. Rev. Endocrinol. 2010, 6, 689–697. [Google Scholar] [CrossRef]
- Galsgaard, K.D. The vicious circle of hepatic glucagon resistance in non-alcoholic fatty liver disease. J. Clin. Med. 2020, 9, 4049. [Google Scholar] [CrossRef]
- Cohen, M.A.; Ellis, S.M.; Le Roux, C.W.; Batterham, R.L.; Park, A.; Patterson, M.; Frost, G.S.; Ghatei, M.A.; Bloom, S.R. Oxyntomodulin suppresses appetite and reduces food intake in humans. J. Clin. Endocrinol. Metab. 2003, 88, 4696–4701. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.; Huo, S.; Xu, B.; Li, F.; Wang, P.; Liu, Y.; Lei, H. A novel long-acting oxyntomodulin analogue eliminates diabetes and obesity in mice. Eur. J. Med. Chem. 2020, 203, 112496. [Google Scholar] [CrossRef] [PubMed]
- Pocai, A.; Carrington, P.E.; Adams, J.R.; Wright, M.; Eiermann, G.; Zhu, L.; Du, X.; Petrov, A.; Lassman, M.E.; Jiang, G. Glucagon-like peptide 1/glucagon receptor dual agonism reverses obesity in mice. Diabetes 2009, 58, 2258–2266. [Google Scholar] [CrossRef] [PubMed]
- Henderson, S.; Konkar, A.; Hornigold, D.; Trevaskis, J.; Jackson, R.; Fritsch Fredin, M.; Jansson-Löfmark, R.; Naylor, J.; Rossi, A.; Bednarek, M. Robust anti-obesity and metabolic effects of a dual GLP-1/glucagon receptor peptide agonist in rodents and non-human primates. Diabetes Obes. Metab. 2016, 18, 1176–1190. [Google Scholar] [CrossRef]
- Boland, M.L.; Laker, R.C.; Mather, K.; Nawrocki, A.; Oldham, S.; Boland, B.B.; Lewis, H.; Conway, J.; Naylor, J.; Guionaud, S. Resolution of NASH and hepatic fibrosis by the GLP-1R and GCGR dual-agonist cotadutide via modulating mitochondrial function and lipogenesis. Nat. Metab. 2020, 2, 413–431. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.K.; Lee, J.S.; Kim, D.; Park, E.; Kim, Y.H.; Choi, I.Y. 990-P: Therapeutic Effect of a Novel Long-Acting GLP-1/GIP/Glucagon Triple Agonist (HM15211) in a NASH and Fibrosis Animal Model. Diabetes 2019, 68, 990-P. [Google Scholar] [CrossRef]
- Choi, I.Y.; Lee, J.S.; Kim, J.K.; Park, Y.J.; Jung, S.Y.; Kim, Y.H.; Kwon, S.C. Potent body weight loss and efficacy in a NASH animal model by a novel long-acting GLP-1/Glucagon/GIP triple-agonist (HM15211). In Proceedings of the American Diabetes Association’s 77th Scientific Session, San Diego, CA, USA, 9–13 June 2017. [Google Scholar]
- Choi, J.; Jo, H.; Kim, J.K.; Lee, S.D.; Lee, S.H.; Choi, I.Y. 996-P: Therapeutic Effect of a Novel Long-Acting GLP-1/GIP/Glucagon Triple Agonist (HM15211) in a Dyslipidemia Animal Model. Diabetes 2019, 68, 996-P. [Google Scholar] [CrossRef]
- Kim, J.K.; Lee, J.S.; Kwon, H.; Lee, J.; Kim, D.; Bae, S.; Lee, S.H.; Choi, I.Y. 1803-P: Antifibrotic Effect of a Novel Long-Acting GLP-1/GIP/Glucagon Triple Agonist (HM15211) in BDL-Induced Liver Fibrosis Mice. Diabetes 2020, 69, 1803-P. [Google Scholar] [CrossRef]
- Zarei, M.; Aguilar-Recarte, D.; Palomer, X.; Vázquez-Carrera, M. Revealing the role of peroxisome proliferator-activated receptor β/δ in nonalcoholic fatty liver disease. Metabolism 2021, 114, 154342. [Google Scholar] [CrossRef]
- Nabavi, S.M.; Devi, K.P.; Sathya, S.; Sanches-Silva, A.; Joanna, L.; Talarek, S.; Xu, S.; Daglia, M.; Nabavi, S.F.; Shirooie, S. New trends in the pharmacological intervention of PPARs in obesity: Role of natural and synthetic compounds. Curr. Med. Chem. 2021, 28, 4004–4022. [Google Scholar] [CrossRef]
- Kalliora, C.; Drosatos, K. The glitazars paradox: Cardiotoxicity of the metabolically beneficial dual PPARα and PPARγ activation. J. Cardiovasc. Pharmacol. 2020, 76, 514. [Google Scholar] [CrossRef]
- Agrawal, R. The first approved agent in the Glitazar’s class: Saroglitazar. Curr. Drug Targets 2014, 15, 151–155. [Google Scholar] [CrossRef]
- Munigoti, S.P.; Harinarayan, C. Role of Glitazars in atherogenic dyslipidemia and diabetes: Two birds with one stone? Indian J. Endocrinol. Metab. 2014, 18, 283. [Google Scholar] [CrossRef]
- Kumar, D.P.; Caffrey, R.; Marioneaux, J.; Santhekadur, P.K.; Bhat, M.; Alonso, C.; Koduru, S.V.; Philip, B.; Jain, M.R.; Giri, S.R. The PPAR α/γ agonist saroglitazar improves insulin resistance and steatohepatitis in a diet induced animal model of nonalcoholic fatty liver disease. Sci. Rep. 2020, 10, 9330. [Google Scholar] [CrossRef]
- Staels, B.; Rubenstrunk, A.; Noel, B.; Rigou, G.; Delataille, P.; Millatt, L.J.; Baron, M.; Lucas, A.; Tailleux, A.; Hum, D.W. Hepatoprotective effects of the dual peroxisome proliferator-activated receptor alpha/delta agonist, GFT505, in rodent models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Hepatology 2013, 58, 1941–1952. [Google Scholar] [CrossRef] [PubMed]
- Sumida, Y.; Okanoue, T.; Nakajima, A.; NAFLD, J.S.G.o. Phase 3 drug pipelines in the treatment of non-alcoholic steatohepatitis. Hepatol. Res. 2019, 49, 1256–1262. [Google Scholar] [CrossRef]
- Lefere, S.; Puengel, T.; Hundertmark, J.; Penners, C.; Frank, A.K.; Guillot, A.; De Muynck, K.; Heymann, F.; Adarbes, V.; Defrêne, E. Differential effects of selective-and pan-PPAR agonists on experimental steatohepatitis and hepatic macrophages. J. Hepatol. 2020, 73, 757–770. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yang, K.; Yang, J.; Xiao, W.; Le, Y.; Yu, F.; Gu, L.; Lang, S.; Tian, Q.; Jin, T. Liver-derived fibroblast growth factor 21 mediates effects of glucagon-like peptide-1 in attenuating hepatic glucose output. EBioMedicine 2019, 41, 73–84. [Google Scholar] [CrossRef]
- Hong, H.; Kim, J.; Choi, H.; Kim, D.; Kim, T.; Tølbøl, K.; Feigh, M.; Illemann, M.; Rigbolt, K.; Vrang, N. YH25724, a novel long-acting GLP-1/FGF21 dual agonist lowers both non-alcoholic fatty liver disease activity score and fibrosis stage in a diet-induced obese mouse model of biopsy-confirmed non-alcoholic steatohepatitis. J. Hepatol. 2017, 1, S16–S17. [Google Scholar] [CrossRef]
- Pan, Q.; Lin, S.; Li, Y.; Liu, L.; Li, X.; Gao, X.; Yan, J.; Gu, B.; Chen, X.; Li, W. A novel GLP-1 and FGF21 dual agonist has therapeutic potential for diabetes and non-alcoholic steatohepatitis. EBioMedicine 2021, 63, 103202. [Google Scholar] [CrossRef]
- Zaman, R.; Islam, R.A.; Ibnat, N.; Othman, I.; Zaini, A.; Lee, C.Y.; Chowdhury, E.H. Current strategies in extending half-lives of therapeutic proteins. J. Control. Release 2019, 301, 176–189. [Google Scholar] [CrossRef] [PubMed]
- Zhihong, Y.; Chen, W.; Qianqian, Z.; Lidan, S.; Qiang, Z.; Jing, H.; Wenxi, W.; Bhawal, R. Emerging roles of oxyntomodulin-based glucagon-like peptide-1/glucagon co-agonist analogs in diabetes and obesity. Peptides 2023, 162, 170955. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Qi, J.; Sun, G.; Ren, G.; Zhu, S.; Wu, Y.; Yu, D.; Zhao, J.; Liu, M.; Li, D. Enhancement of the pharmacological efficacy of FGF-21 by genetic modification and PEGylation. Curr. Pharm. Biotechnol. 2013, 14, 1287–1298. [Google Scholar] [CrossRef]
- Cui, A.; Li, J.; Ji, S.; Ma, F.; Wang, G.; Xue, Y.; Liu, Z.; Gao, J.; Han, J.; Tai, P. The effects of B1344, a novel fibroblast growth factor 21 analog, on nonalcoholic steatohepatitis in nonhuman primates. Diabetes 2020, 69, 1611–1623. [Google Scholar] [CrossRef]
- Xu, J.; Bussiere, J.; Yie, J.; Sickmier, A.; An, P.; Belouski, E.; Stanislaus, S.; Walker, K.W. Polyethylene glycol modified FGF21 engineered to maximize potency and minimize vacuole formation. Bioconjugate Chem. 2013, 24, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Mu, J.; Pinkstaff, J.; Li, Z.; Skidmore, L.; Li, N.; Myler, H.; Dallas-Yang, Q.; Putnam, A.-M.; Yao, J.; Bussell, S. FGF21 analogs of sustained action enabled by orthogonal biosynthesis demonstrate enhanced antidiabetic pharmacology in rodents. Diabetes 2012, 61, 505–512. [Google Scholar] [CrossRef]
- Verzijl, C.R.; Van De Peppel, I.P.; Struik, D.; Jonker, J.W. Pegbelfermin (BMS-986036): An investigational PEGylated fibroblast growth factor 21 analogue for the treatment of nonalcoholic steatohepatitis. Expert Opin. Investig. Drugs 2020, 29, 125–133. [Google Scholar] [CrossRef]
- Sanyal, A.; Charles, E.D.; Neuschwander-Tetri, B.A.; Loomba, R.; Harrison, S.A.; Abdelmalek, M.F.; Lawitz, E.J.; Halegoua-DeMarzio, D.; Kundu, S.; Noviello, S. Pegbelfermin (BMS-986036), a PEGylated fibroblast growth factor 21 analogue, in patients with non-alcoholic steatohepatitis: A randomised, double-blind, placebo-controlled, phase 2a trial. Lancet 2018, 392, 2705–2717. [Google Scholar] [CrossRef]
- Charles, E.D.; Neuschwander-Tetri, B.A.; Pablo Frias, J.; Kundu, S.; Luo, Y.; Tirucherai, G.S.; Christian, R. Pegbelfermin (BMS-986036), PEGylated FGF21, in patients with obesity and type 2 diabetes: Results from a randomized phase 2 study. Obesity 2019, 27, 41–49. [Google Scholar] [CrossRef]
- Abdelmalek, M.F.; Charles, E.D.; Sanyal, A.J.; Harrison, S.A.; Neuschwander-Tetri, B.A.; Goodman, Z.; Ehman, R.A.; Karsdal, M.; Nakajima, A.; Du, S. The FALCON program: Two phase 2b randomized, double-blind, placebo-controlled studies to assess the efficacy and safety of pegbelfermin in the treatment of patients with nonalcoholic steatohepatitis and bridging fibrosis or compensated cirrhosis. Contemp. Clin. Trials 2021, 104, 106335. [Google Scholar] [CrossRef]
- Pierce, A.; Rosenstock, M.; Margalit, M.; Mansbach, H. BIO89-100, a novel glycoPEGylated FGF21 Analog, Demonstrates Triglyceride Reduction and Broad Metabolic Effects in Spontaneously Diabetic Obese Cynomolgus Monkeys. J. Clin. Lipidol. 2020, 14, 584–585. [Google Scholar] [CrossRef]
- Rosenstock, M.; Ayalon, M.; Mansbach, H.; Liu, Y.; Margalit, M. LBP-29-BIO89-100, a novel PEG-FGF21 analogue, is efficacious following weekly and every 2-week subcutaneous dosing in spontaneous diabetic cynomolgus monkeys. J. Hepatol. 2019, 70, e155. [Google Scholar] [CrossRef]
- Loomba, R.; Lawitz, E.J.; Frias, J.P.; Ortiz-Lasanta, G.; Johansson, L.; Franey, B.B.; Morrow, L.; Rosenstock, M.; Hartsfield, C.L.; Chen, C.-Y. Safety, pharmacokinetics, and pharmacodynamics of pegozafermin in patients with non-alcoholic steatohepatitis: A randomised, double-blind, placebo-controlled, phase 1b/2a multiple-ascending-dose study. Lancet Gastroenterol. Hepatol. 2023, 8, 120–132. [Google Scholar] [CrossRef]
- Saxena, R.; Nanjan, M.J. Elastin-like polypeptides and their applications in anticancer drug delivery systems: A review. Drug Deliv. 2015, 22, 156–167. [Google Scholar] [CrossRef]
- Gilroy, C.; Capozzi, M.; Varanko, A.; Tong, J.; D–Alessio, D.; Campbell, J.; Chilkoti, A. Sustained release of a GLP-1 and FGF21 dual agonist from an injectable depot protects mice from obesity and hyperglycemia. Sci. Adv. 2020, 6, eaaz9890. [Google Scholar] [CrossRef] [PubMed]
- Roopenian, D.C.; Akilesh, S. FcRn: The neonatal Fc receptor comes of age. Nat. Rev. Immunol. 2007, 7, 715–725. [Google Scholar] [CrossRef]
- Kontermann, R.E. Strategies for extended serum half-life of protein therapeutics. Curr. Opin. Biotechnol. 2011, 22, 868–876. [Google Scholar] [CrossRef]
- Fanali, G.; Di Masi, A.; Trezza, V.; Marino, M.; Fasano, M.; Ascenzi, P. Human serum albumin: From bench to bedside. Mol. Asp. Med. 2012, 33, 209–290. [Google Scholar] [CrossRef]
- Chaudhury, C.; Brooks, C.L.; Carter, D.C.; Robinson, J.M.; Anderson, C.L. Albumin binding to FcRn: Distinct from the FcRn− IgG interaction. Biochemistry 2006, 45, 4983–4990. [Google Scholar] [CrossRef]
- Bhattacharya, A.A.; Grüne, T.; Curry, S. Crystallographic analysis reveals common modes of binding of medium and long-chain fatty acids to human serum albumin. J. Mol. Biol. 2000, 303, 721–732. [Google Scholar] [CrossRef]
- Brønden, A.; Knop, F.K.; Christensen, M.B. Clinical pharmacokinetics and pharmacodynamics of albiglutide. Clin. Pharmacokinet. 2017, 56, 719–731. [Google Scholar] [CrossRef]
- Knudsen, L.B.; Nielsen, P.F.; Huusfeldt, P.O.; Johansen, N.L.; Madsen, K.; Pedersen, F.Z.; Thøgersen, H.; Wilken, M.; Agersø, H. Potent derivatives of glucagon-like peptide-1 with pharmacokinetic properties suitable for once daily administration. J. Med. Chem. 2000, 43, 1664–1669. [Google Scholar] [CrossRef]
- Lau, J.; Bloch, P.; Schäffer, L.; Pettersson, I.; Spetzler, J.; Kofoed, J.; Madsen, K.; Knudsen, L.B.; McGuire, J.; Steensgaard, D.B. Discovery of the once-weekly glucagon-like peptide-1 (GLP-1) analogue semaglutide. J. Med. Chem. 2015, 58, 7370–7380. [Google Scholar] [CrossRef]
- Elbrønd, B.; Jakobsen, G.; Larsen, S.; Agersø, H.; Jensen, L.B.; Rolan, P.; Sturis, J.; Hatorp, V.; Zdravkovic, M. Pharmacokinetics, pharmacodynamics, safety, and tolerability of a single-dose of NN2211, a long-acting glucagon-like peptide 1 derivative, in healthy male subjects. Diabetes Care 2002, 25, 1398–1404. [Google Scholar] [CrossRef] [PubMed]
- Hui, H.; Farilla, L.; Merkel, P.; Perfetti, R. The short half-life of glucagon-like peptide-1 in plasma does not reflect its long-lasting beneficial effects. Eur. J Endocrinol. 2002, 146, 863–869. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Ly, N.; Li, J.; Arends, R.H. Population Pharmacokinetics of Cotadutide in Subjects with Type 2 Diabetes. Clin. Pharmacokinet. 2022, 61, 833–845. [Google Scholar] [CrossRef] [PubMed]
- Monnet, C.; Jorieux, S.; Urbain, R.; Fournier, N.; Bouayadi, K.; De Romeuf, C.; Behrens, C.K.; Fontayne, A.; Mondon, P. Selection of IgG variants with increased FcRn binding using random and directed mutagenesis: Impact on effector functions. Front. Immunol. 2015, 6, 39. [Google Scholar] [CrossRef]
- Liu, R.; Oldham, R.J.; Teal, E.; Beers, S.A.; Cragg, M.S. Fc-engineering for modulated effector functions—Improving antibodies for cancer treatment. Antibodies 2020, 9, 64. [Google Scholar] [CrossRef]
- Naver, S.V.; Jimenez-Solem, E.; Christensen, M.; Andersen, J.T.; Knop, F.K. Dulaglutide: A novel once-weekly glucagon-like peptide-1 receptor agonist. Clin. Investig. 2014, 4, 729–743. [Google Scholar] [CrossRef]
- Choi, J.D.; Baek, S.; Kim, Y.; Eun, K.; Kwon, S.C.; Morrow, L.; Hompesch, M.; Kang, J. 982-P: A Double-Blinded, Placebo Controlled, Single Ascending Dose Study for Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics after Subcutaneous Administration of Novel Long-Acting GLP-1/GIP/Glucagon Triple Agonist (HM15211) in Healthy Obese Subjects. Diabetes 2019, 68, 982. [Google Scholar]
- Stanislaus, S.; Hecht, R.; Yie, J.; Hager, T.; Hall, M.; Spahr, C.; Wang, W.; Weiszmann, J.; Li, Y.; Deng, L. A novel Fc-FGF21 with improved resistance to proteolysis, increased affinity toward β-klotho, and enhanced efficacy in mice and cynomolgus monkeys. Endocrinology 2017, 158, 1314–1327. [Google Scholar] [CrossRef]
- Kaufman, A.; Abuqayyas, L.; Denney, W.S.; Tillman, E.J.; Rolph, T. AKR-001, an Fc-FGF21 analog, showed sustained pharmacodynamic effects on insulin sensitivity and lipid metabolism in type 2 diabetes patients. Cell Rep. Med. 2020, 1, 100057. [Google Scholar] [CrossRef]
- Harrison, S.A.; Ruane, P.J.; Freilich, B.L.; Neff, G.; Patil, R.; Behling, C.A.; Hu, C.; Fong, E.; de Temple, B.; Tillman, E.J. Efruxifermin in non-alcoholic steatohepatitis: A randomized, double-blind, placebo-controlled, phase 2a trial. Nat. Med. 2021, 27, 1262–1271. [Google Scholar] [CrossRef]
- Boey, A.; Ho, H.K. All roads lead to the liver: Metal nanoparticles and their implications for liver health. Small 2020, 16, 2000153. [Google Scholar] [CrossRef]
- Sadauskas, E.; Wallin, H.; Stoltenberg, M.; Vogel, U.; Doering, P.; Larsen, A.; Danscher, G. Kupffer cells are central in the removal of nanoparticles from the organism. Part. Fibre Toxicol. 2007, 4, 10. [Google Scholar] [CrossRef]
- Alexis, F.; Pridgen, E.; Molnar, L.K.; Farokhzad, O.C. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol. Pharm. 2008, 5, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Xin, X.; Ma, J.; Tan, C.; Osna, N.; Mahato, R.I. Therapeutic targets, novel drugs, and delivery systems for diabetes associated NAFLD and liver fibrosis. Adv. Drug Deliv. Rev. 2021, 176, 113888. [Google Scholar] [CrossRef]
- Park, J.-K.; Utsumi, T.; Seo, Y.-E.; Deng, Y.; Satoh, A.; Saltzman, W.M.; Iwakiri, Y. Cellular distribution of injected PLGA-nanoparticles in the liver. Nanomed. Nanotechnol. Biol. Med. 2016, 12, 1365–1374. [Google Scholar] [CrossRef]
- Monestier, M.; Charbonnier, P.; Gateau, C.; Cuillel, M.; Robert, F.; Lebrun, C.; Mintz, E.; Renaudet, O.; Delangle, P. ASGPR-Mediated Uptake of Multivalent Glycoconjugates for Drug Delivery in Hepatocytes. ChemBioChem 2016, 17, 590–594. [Google Scholar] [CrossRef] [PubMed]
- Petrov, R.A.; Maklakova, S.Y.; Ivanenkov, Y.A.; Petrov, S.A.; Sergeeva, O.V.; Yamansarov, E.Y.; Saltykova, I.V.; Kireev, I.I.; Alieva, I.B.; Deyneka, E.V. Synthesis and biological evaluation of novel mono-and bivalent ASGP-R-targeted drug-conjugates. Bioorg. Med. Chem. Lett. 2018, 28, 382–387. [Google Scholar] [CrossRef] [PubMed]
- Bon, C.; Hofer, T.; Bousquet-Mélou, A.; Davies, M.R.; Krippendorff, B.-F. Capacity limits of asialoglycoprotein receptor-mediated liver targeting. MAbs 2017, 9, 1360–1369. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Porterfield, J.E.; An, H.-T.; Jimenez, A.S.; Lee, S.; Kannan, S.; Sharma, A.; Kannan, R.M. Rationally designed galactose dendrimer for hepatocyte-specific targeting and intracellular drug delivery for the treatment of liver disorders. Biomacromolecules 2021, 22, 3574–3589. [Google Scholar] [CrossRef] [PubMed]
- Harris, E.N.; Baker, E. Role of the hyaluronan receptor, stabilin-2/HARE, in health and disease. Int. J. Mol. Sci. 2020, 21, 3504. [Google Scholar] [CrossRef]
- Yang, J.-A.; Kong, W.H.; Sung, D.K.; Kim, H.; Kim, T.H.; Lee, K.C.; Hahn, S.K. Hyaluronic acid–tumor necrosis factor-related apoptosis-inducing ligand conjugate for targeted treatment of liver fibrosis. Acta Biomater. 2015, 12, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhou, C.; Fu, Y.; Chen, T.; Liu, X.; Zhang, Z.; Gong, T. Targeted delivery of hyaluronic acid nanomicelles to hepatic stellate cells in hepatic fibrosis rats. Acta Pharm. Sin. B 2020, 10, 693–710. [Google Scholar] [CrossRef]
- Hu, R.; Yang, X.; He, X.; Song, G. The relationship between NAFLD and retinol-binding protein 4-an updated systematic review and meta-analysis. Lipids Health Dis. 2023, 22, 8. [Google Scholar] [CrossRef]
- Uchio, K.; Tuchweber, B.; Manabe, N.; Gabbiani, G.; Rosenbaum, J.; Desmouliere, A. Cellular retinol-binding protein-1 expression and modulation during in vivo and in vitro myofibroblastic differentiation of rat hepatic stellate cells and portal fibroblasts. Lab. Investig. 2002, 82, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Jeong, W.I. Retinoic acids and hepatic stellate cells in liver disease. J. Gastroenterol. Hepatol. 2012, 27, 75–79. [Google Scholar] [CrossRef]
- Qiao, J.-B.; Fan, Q.-Q.; Xing, L.; Cui, P.-F.; He, Y.-J.; Zhu, J.-C.; Wang, L.; Pang, T.; Oh, Y.-K.; Zhang, C. Vitamin A-decorated biocompatible micelles for chemogene therapy of liver fibrosis. J. Control. Release 2018, 283, 113–125. [Google Scholar] [CrossRef]
- Hassan, R.; Tammam, S.N.; El Safy, S.; Abdel-Halim, M.; Asimakopoulou, A.; Weiskirchen, R.; Mansour, S. Prevention of hepatic stellate cell activation using JQ1-and atorvastatin-loaded chitosan nanoparticles as a promising approach in therapy of liver fibrosis. Eur. J. Pharm. Biopharm. 2019, 134, 96–106. [Google Scholar] [CrossRef]
- Magnusson, S.; Berg, T. Extremely rapid endocytosis mediated by the mannose receptor of sinusoidal endothelial rat liver cells. Biochem. J. 1989, 257, 651–656. [Google Scholar] [CrossRef] [PubMed]
- Mak, K.M.; Shin, D.W. Hepatic sinusoids versus central veins: Structures, markers, angiocrines, and roles in liver regeneration and homeostasis. Anat. Rec. 2021, 304, 1661–1691. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.K.; Pandey, M.; Dewangan, H.K.; Sl, N.; Sahoo, P.K. A Comprehensive Review on Liver Targeting: Emphasis on Nanotechnology-based Molecular Targets and Receptors Mediated Approaches. Curr. Drug Targets 2022, 23, 1381–1405. [Google Scholar] [PubMed]
- Kim, M.; Jeong, M.; Hur, S.; Cho, Y.; Park, J.; Jung, H.; Seo, Y.; Woo, H.; Nam, K.; Lee, K. Engineered ionizable lipid nanoparticles for targeted delivery of RNA therapeutics into different types of cells in the liver. Sci. Adv. 2021, 7, eabf4398. [Google Scholar] [CrossRef]
- Go, G.-W.; Mani, A. Low-density lipoprotein receptor (LDLR) family orchestrates cholesterol homeostasis. Yale J. Biol. Med. 2012, 85, 19. [Google Scholar]
- Wang, Z.; Duan, X.; Lv, Y.; Zhao, Y. Low density lipoprotein receptor (LDLR)-targeted lipid nanoparticles for the delivery of sorafenib and Dihydroartemisinin in liver cancers. Life Sci. 2019, 239, 117013. [Google Scholar] [CrossRef]
- Akinc, A.; Querbes, W.; De, S.; Qin, J.; Frank-Kamenetsky, M.; Jayaprakash, K.N.; Jayaraman, M.; Rajeev, K.G.; Cantley, W.L.; Dorkin, J.R. Targeted delivery of RNAi therapeutics with endogenous and exogenous ligand-based mechanisms. Mol. Ther. 2010, 18, 1357–1364. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| No. | Molecules | Development Stage | Clinical Trial Identification | Estimated Year of Completion |
|---|---|---|---|---|
| 1 | Canagliflozin | N/A | NCT05422092 | 2023 |
| N/A | NCT05513729 | 2024 | ||
| 2 | Dapagliflozin | Phase 3 | NCT03723252 | 2023 |
| NCT05308160 | 2024 | |||
| Phase 4 | NCT05459701 | 2023 | ||
| NCT05140694 | 2025 | |||
| NCT05254626 | 2025 | |||
| 3 | Empagliflozin | N/A | NCT05694923 | 2024 |
| Phase 2 | NCT03867487 | 2024 | ||
| NCT05140694 | 2025 | |||
| Phase 3 | NCT05605158 | 2024 | ||
| Phase 4 | NCT04642261 | 2023 | ||
| NCT04976283 | 2023 | |||
| NCT04639414 | 2023 | |||
| NCT05140694 | 2025 | |||
| 4 | Ertugliflozin | Phase 4 | NCT05644717 | 2024 |
| No. | Molecules | Development Stage | Clinical Trial Identification | Estimated Year of Completion |
|---|---|---|---|---|
| 1 | Dulaglutide | Phase 4 | NCT03648554 | 2024 |
| 2 | Liraglutide | N/A | NCT05779644 | 2025 |
| 3 | Semaglutide | Phase 2 | NCT04216589 | 2023 |
| NCT03884075 | 2024 | |||
| NCT04971785 | 2024 | |||
| NCT05016882 | 2024 | |||
| Phase 3 | NCT05067621 | 2027 | ||
| NCT04822181 | 2029 | |||
| Phase 4 | NCT04639414 | 2023 | ||
| NCT05813249 | 2024 |
| No. | Molecules | Target | Development Stage | Clinical Trial Identification | Estimated Year of Completion |
|---|---|---|---|---|---|
| 1 | Pioglitazone | PPAR-γ | Phase 2 | NCT04501406 | 2027 |
| Phase 3 | NCT05605158 | 2024 | |||
| Phase 4 | NCT04976283 | 2023 | |||
| NCT05305287 | 2027 |
| No. | Molecules | Structure | Development Stage | Clinical Trial Identification | Estimated Year of Completion |
|---|---|---|---|---|---|
| 1 | Aldafermin (NGM282) | FGF19 analog | Phase 2 | NCT04210245 | 2023 |
| 2 | bFKB1 (BFKB8488A) | FGFR1c/β-klotho bispecific antibody | Phase 2 | NCT04171765 | 2023 |
| 3 | Efruxifermin (AR001) | Fc-FGF21 analog | Phase 2 | NCT05039450 | 2024 |
| NCT04767529 | 2024 | ||||
| 4 | MK-3655 (NGM313) | FGFR1c/β-klotho bispecific antibody | Phase 2 | NCT04583423 | 2023 |
| 5 | Pegozafermin (BIO89–100) | glycoPEGylated FGF21 | Phase 2 | NCT04929483 | 2023 |
| No. | Molecules | Development Stage | Clinical Trial Identification | Estimated Year of Completion |
|---|---|---|---|---|
| 1 | Cilofexor + semaglutide and firsocostat | Phase 2 | NCT04971785 | 2024 |
| 2 | OCA, 6-α-ethyl-chenodeoxycholic acid (6-ECDCA, INT-747) | Phase 2 | NCT05573204 | 2024 |
| Phase 3 | NCT02548351 | 2025 | ||
| 3 | MET409 | Phase 2 | NCT04702490 | 2022 |
| 4 | Tropifexor + licogliflozin | Phase 2 | NCT04065841 | 2024 |
| No. | Molecules | Target | Development Stage | Clinical Trial Identification | Estimated Year of Completion |
|---|---|---|---|---|---|
| 1 | Cotadutide (MEDI0382) | GLP-1R/GCGR | Phase 2/3 | NCT05364931 | 2024 |
| 2 | DD01 | GLP-1R/GCGR | Phase 1 | NCT04812262 | 2022 |
| 3 | HM15211 | GLP-1R/GIPR/GCGR | Phase 2 | NCT04505436 | 2024 |
| 4 | Tirzepatide (LY3298176, TPZ) | GLP-1R/GIPR | Phase 1/2 | NCT05751720 | 2025 |
| Phase 2 | NCT04166773 | 2024 |
| No. | Molecules | Target | Development Stage | Clinical Trial Identification | Estimated Year of Completion |
|---|---|---|---|---|---|
| 1 | Lanifibranor | PPAR-α/γ/δ agonist | Phase 2 | NCT05232071 | 2023 |
| NCT03459079 | 2024 | ||||
| Phase 3 | NCT04849728 | 2026 | |||
| 2 | Saroglitazar | PPAR-α/γ agonist | Phase 2 | NCT03617263 | 2023 |
| NCT03639623 | 2023 | ||||
| NCT05011305 | 2023 | ||||
| NCT05211284 | 2025 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amatya, R.; Lee, D.; Min, K.A.; Shin, M.C. Pharmaceutical Strategies to Improve Druggability of Potential Drug Candidates in Nonalcoholic Fatty Liver Disease Therapy. Pharmaceutics 2023, 15, 1963. https://doi.org/10.3390/pharmaceutics15071963
Amatya R, Lee D, Min KA, Shin MC. Pharmaceutical Strategies to Improve Druggability of Potential Drug Candidates in Nonalcoholic Fatty Liver Disease Therapy. Pharmaceutics. 2023; 15(7):1963. https://doi.org/10.3390/pharmaceutics15071963
Chicago/Turabian StyleAmatya, Reeju, Donghee Lee, Kyoung Ah Min, and Meong Cheol Shin. 2023. "Pharmaceutical Strategies to Improve Druggability of Potential Drug Candidates in Nonalcoholic Fatty Liver Disease Therapy" Pharmaceutics 15, no. 7: 1963. https://doi.org/10.3390/pharmaceutics15071963
APA StyleAmatya, R., Lee, D., Min, K. A., & Shin, M. C. (2023). Pharmaceutical Strategies to Improve Druggability of Potential Drug Candidates in Nonalcoholic Fatty Liver Disease Therapy. Pharmaceutics, 15(7), 1963. https://doi.org/10.3390/pharmaceutics15071963