
Unraveling the Crosstalk between Lipids and NADPH Oxidases in Diabetic Kidney Disease

Abstract
1. Introduction
2. The Role of Renal Lipids in the Pathogenesis of Diabetic Kidney Disease
2.1. Cholesterol
2.2. Fatty Acids and Triglycerides
2.3. Sphingolipids
3. The Role of NADPH Oxidases in the Pathogenesis of Diabetic Kidney Disease
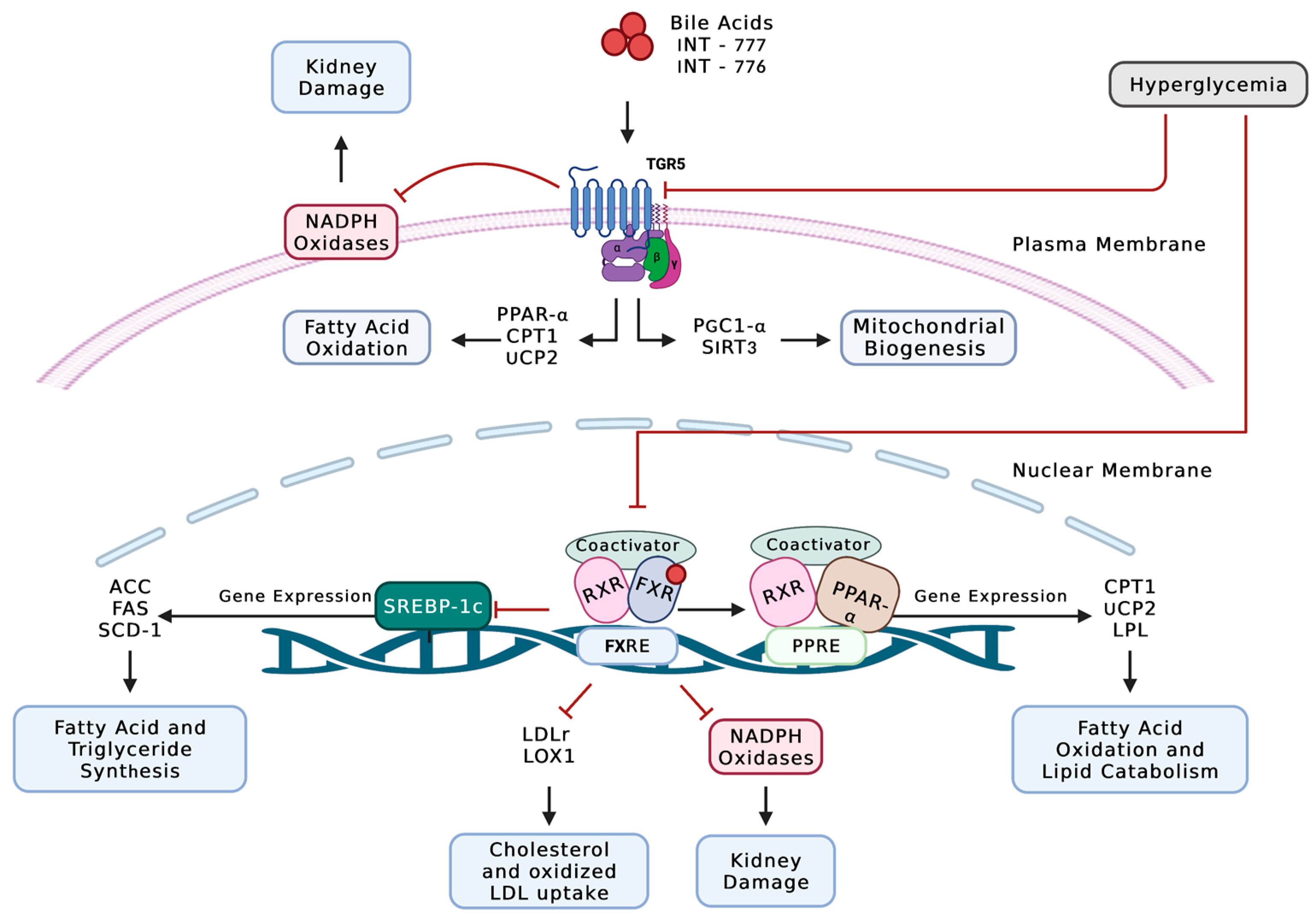
4. The Crosstalk between Lipids and NADPH Oxidases in Diabetic Kidney Disease
4.1. Cholesterol, Phospholipids, and NADPH Oxidase Signaling in DKD
4.2. Fatty Acids, Triglycerides, and NADPH Oxidase Signaling in DKD
4.3. Sphingolipids and NADPH Oxidase Signaling in DKD
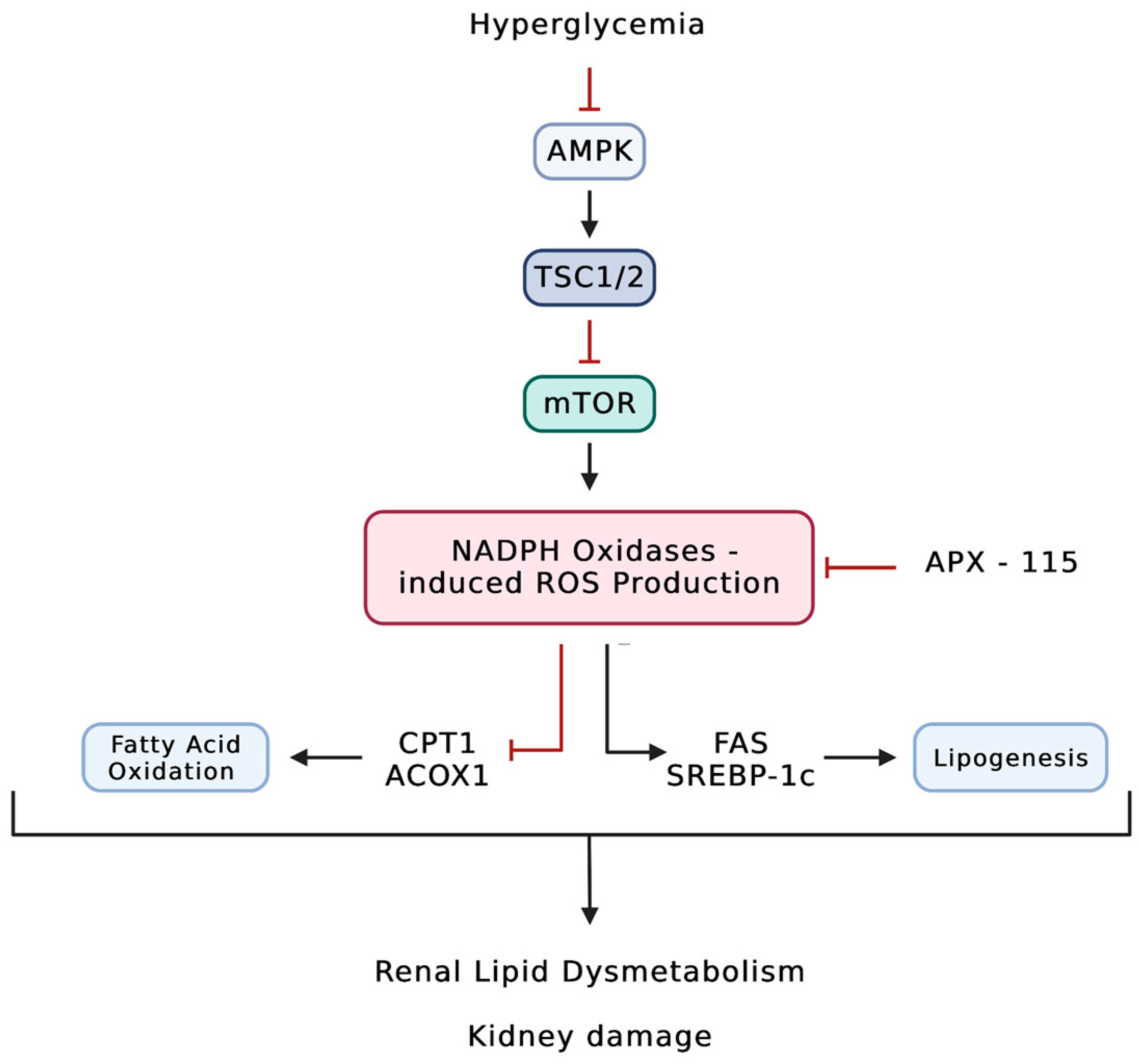
5. A Two-Way Crosstalk: Increased ROS Production May Cause Lipid Dysmetabolism
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Saran, R.; Robinson, B.; Abbott, K.C.; Agodoa, L.Y.C.; Bragg-Gresham, J.; Balkrishnan, R.; Bhave, N.; Dietrich, X.; Ding, Z.; Eggers, P.W.; et al. US Renal Data System 2018 Annual Data Report: Epidemiology of Kidney Disease in the United States. Am. J. Kidney Dis. 2019, 73, A7–A8. [Google Scholar] [CrossRef] [PubMed]
- Persson, F.; Rossing, P. Diagnosis of diabetic kidney disease: State of the art and future perspective. Kidney Int. Suppl. 2018, 8, 2–7. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, D.; Tuncel, M.; Levi, M. Diabetic nephropathy—A multifaceted target of new therapies. Discov. Med. 2010, 10, 406–415. [Google Scholar] [PubMed]
- Abboud, H.E. Mesangial cell biology. Exp. Cell Res. 2012, 318, 979–985. [Google Scholar] [CrossRef] [PubMed]
- Meyer, T.W.; Bennett, P.H.; Nelson, R.G. Podocyte number predicts long-term urinary albumin excretion in Pima Indians with Type II diabetes and microalbuminuria. Diabetologia 1999, 42, 1341–1344. [Google Scholar] [CrossRef]
- Pagtalunan, M.E.; Miller, P.L.; Jumping-Eagle, S.; Nelson, R.G.; Myers, B.D.; Rennke, H.G.; Coplon, N.S.; Sun, L.; Meyer, T.W. Podocyte loss and progressive glomerular injury in type II diabetes. J. Clin. Investig. 1997, 99, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Toyoda, M.; Najafian, B.; Kim, Y.; Caramori, M.L.; Mauer, M. Podocyte detachment and reduced glomerular capillary endothelial fenestration in human type 1 diabetic nephropathy. Diabetes 2007, 56, 2155–2160. [Google Scholar] [CrossRef]
- Weil, E.J.; Lemley, K.V.; Mason, C.C.; Yee, B.; Jones, L.I.; Blouch, K.; Lovato, T.; Richardson, M.; Myers, B.D.; Nelson, R.G. Podocyte detachment and reduced glomerular capillary endothelial fenestration promote kidney disease in type 2 diabetic nephropathy. Kidney Int. 2012, 82, 1010–1017. [Google Scholar] [CrossRef]
- White, K.E.; Bilous, R.W.; Marshall, S.M.; El Nahas, M.; Remuzzi, G.; Piras, G.; De Cosmo, S.; Viberti, G. Podocyte number in normotensive type 1 diabetic patients with albuminuria. Diabetes 2002, 51, 3083–3089. [Google Scholar] [CrossRef] [PubMed]
- Herman-Edelstein, M.; Scherzer, P.; Tobar, A.; Levi, M.; Gafter, U. Altered renal lipid metabolism and renal lipid accumulation in human diabetic nephropathy. J. Lipid Res. 2014, 55, 561–572. [Google Scholar] [CrossRef]
- Proctor, G.; Jiang, T.; Iwahashi, M.; Wang, Z.; Li, J.; Levi, M. Regulation of renal fatty acid and cholesterol metabolism, inflammation, and fibrosis in Akita and OVE26 mice with type 1 diabetes. Diabetes 2006, 55, 2502–2509. [Google Scholar] [CrossRef]
- Wang, X.X.; Jiang, T.; Shen, Y.; Adorini, L.; Pruzanski, M.; Gonzalez, F.J.; Scherzer, P.; Lewis, L.; Miyazaki-Anzai, S.; Levi, M. The farnesoid X receptor modulates renal lipid metabolism and diet-induced renal inflammation, fibrosis, and proteinuria. Am. J. Physiol. Renal Physiol. 2009, 297, F1587–F1596. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Jiang, T.; Li, J.; Proctor, G.; McManaman, J.L.; Lucia, S.; Chua, S.; Levi, M. Regulation of renal lipid metabolism, lipid accumulation, and glomerulosclerosis in FVBdb/db mice with type 2 diabetes. Diabetes 2005, 54, 2328–2335. [Google Scholar] [CrossRef]
- Merscher-Gomez, S.; Guzman, J.; Pedigo, C.E.; Lehto, M.; Aguillon-Prada, R.; Mendez, A.; Lassenius, M.I.; Forsblom, C.; Yoo, T.; Villarreal, R.; et al. Cyclodextrin protects podocytes in diabetic kidney disease. Diabetes 2013, 62, 3817–3827. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Wang, Z.; Proctor, G.; Moskowitz, S.; Liebman, S.E.; Rogers, T.; Lucia, M.S.; Li, J.; Levi, M. Diet-induced obesity in C57BL/6J mice causes increased renal lipid accumulation and glomerulosclerosis via a sterol regulatory element-binding protein-1c-dependent pathway. J. Biol. Chem. 2005, 280, 32317–32325. [Google Scholar] [CrossRef]
- Kimmelstiel, P.; Wilson, C. Intercapillary Lesions in the Glomeruli of the Kidney. Am. J. Pathol. 1936, 12, 83–98.7. [Google Scholar] [PubMed]
- Brownlee, M. The pathobiology of diabetic complications: A unifying mechanism. Diabetes 2005, 54, 1615–1625. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Eid, A.A.; Ford, B.M.; Bhandary, B.; de Cassia Cavaglieri, R.; Block, K.; Barnes, J.L.; Gorin, Y.; Choudhury, G.G.; Abboud, H.E. Mammalian target of rapamycin regulates Nox4-mediated podocyte depletion in diabetic renal injury. Diabetes 2013, 62, 2935–2947. [Google Scholar] [CrossRef]
- Eid, S.; Boutary, S.; Braych, K.; Sabra, R.; Massaad, C.; Hamdy, A.; Rashid, A.; Moodad, S.; Block, K.; Gorin, Y.; et al. mTORC2 Signaling Regulates Nox4-Induced Podocyte Depletion in Diabetes. Antioxid. Redox Signal. 2016, 25, 703–719. [Google Scholar] [CrossRef]
- Ford, B.M.; Eid, A.A.; Gooz, M.; Barnes, J.L.; Gorin, Y.C.; Abboud, H.E. ADAM17 mediates Nox4 expression and NADPH oxidase activity in the kidney cortex of OVE26 mice. Am. J. Physiol. Renal Physiol. 2013, 305, F323–F332. [Google Scholar] [CrossRef]
- Gorin, Y.; Block, K.; Hernandez, J.; Bhandari, B.; Wagner, B.; Barnes, J.L.; Abboud, H.E. Nox4 NAD(P)H oxidase mediates hypertrophy and fibronectin expression in the diabetic kidney. J. Biol. Chem. 2005, 280, 39616–39626. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.H.; Rincon-Choles, H.; Bhandari, B.; Choudhury, G.G.; Abboud, H.E.; Gorin, Y. Redox dependence of glomerular epithelial cell hypertrophy in response to glucose. Am. J. Physiol. Renal Physiol. 2006, 290, F741–F751. [Google Scholar] [CrossRef] [PubMed]
- Satoh, M.; Fujimoto, S.; Haruna, Y.; Arakawa, S.; Horike, H.; Komai, N.; Sasaki, T.; Tsujioka, K.; Makino, H.; Kashihara, N. NAD(P)H oxidase and uncoupled nitric oxide synthase are major sources of glomerular superoxide in rats with experimental diabetic nephropathy. Am. J. Physiol. Renal Physiol. 2005, 288, F1144–F1152. [Google Scholar] [CrossRef]
- Eid, A.A.; Gorin, Y.; Fagg, B.M.; Maalouf, R.; Barnes, J.L.; Block, K.; Abboud, H.E. Mechanisms of podocyte injury in diabetes: Role of cytochrome P450 and NADPH oxidases. Diabetes 2009, 58, 1201–1211. [Google Scholar] [CrossRef]
- Jha, J.C.; Banal, C.; Okabe, J.; Gray, S.P.; Hettige, T.; Chow, B.S.M.; Thallas-Bonke, V.; De Vos, L.; Holterman, C.E.; Coughlan, M.T.; et al. NADPH Oxidase Nox5 Accelerates Renal Injury in Diabetic Nephropathy. Diabetes 2017, 66, 2691–2703. [Google Scholar] [CrossRef] [PubMed]
- Ducasa, G.M.; Mitrofanova, A.; Mallela, S.K.; Liu, X.; Molina, J.; Sloan, A.; Pedigo, C.E.; Ge, M.; Santos, J.V.; Hernandez, Y.; et al. ATP-binding cassette A1 deficiency causes cardiolipin-driven mitochondrial dysfunction in podocytes. J. Clin. Investig. 2019, 129, 3387–3400. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, I.J.; Trent, C.M.; Schulze, P.C. Lipid metabolism and toxicity in the heart. Cell Metab. 2012, 15, 805–812. [Google Scholar] [CrossRef]
- Lara-Castro, C.; Garvey, W.T. Intracellular lipid accumulation in liver and muscle and the insulin resistance syndrome. Endocrinol. Metab. Clin. N. Am. 2008, 37, 841–856. [Google Scholar] [CrossRef]
- Perry, R.J.; Samuel, V.T.; Petersen, K.F.; Shulman, G.I. The role of hepatic lipids in hepatic insulin resistance and type 2 diabetes. Nature 2014, 510, 84–91. [Google Scholar] [CrossRef]
- Ruan, X.Z.; Varghese, Z.; Powis, S.H.; Moorhead, J.F. Dysregulation of LDL receptor under the influence of inflammatory cytokines: A new pathway for foam cell formation. Kidney Int. 2001, 60, 1716–1725. [Google Scholar] [CrossRef]
- Yuan, Y.; Zhao, L.; Chen, Y.; Moorhead, J.F.; Varghese, Z.; Powis, S.H.; Minogue, S.; Sun, Z.; Ruan, X.Z. Advanced glycation end products (AGEs) increase human mesangial foam cell formation by increasing Golgi SCAP glycosylation in vitro. Am. J. Physiol. Renal Physiol. 2011, 301, F236–F243. [Google Scholar] [CrossRef] [PubMed]
- Nohturfft, A.; DeBose-Boyd, R.A.; Scheek, S.; Goldstein, J.L.; Brown, M.S. Sterols regulate cycling of SREBP cleavage-activating protein (SCAP) between endoplasmic reticulum and Golgi. Proc. Natl. Acad. Sci. USA 1999, 96, 11235–11240. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, J.L.; DeBose-Boyd, R.A.; Brown, M.S. Protein sensors for membrane sterols. Cell 2006, 124, 35–46. [Google Scholar] [CrossRef]
- Sun, H.; Yuan, Y.; Sun, Z.L. Cholesterol Contributes to Diabetic Nephropathy through SCAP-SREBP-2 Pathway. Int. J. Endocrinol. 2013, 2013, 592576. [Google Scholar] [CrossRef]
- Zhang, Y.; Ma, K.L.; Liu, J.; Wu, Y.; Hu, Z.B.; Liu, L.; Lu, J.; Zhang, X.L.; Liu, B.C. Inflammatory stress exacerbates lipid accumulation and podocyte injuries in diabetic nephropathy. Acta Diabetol. 2015, 52, 1045–1056. [Google Scholar] [CrossRef]
- Yang, Y.; Yang, Q.; Yang, J.; Ma, Y.; Ding, G. Angiotensin II induces cholesterol accumulation and injury in podocytes. Sci. Rep. 2017, 7, 10672. [Google Scholar] [CrossRef]
- Wiciński, M.; Żak, J.; Malinowski, B.; Popek, G.; Grześk, G. PCSK9 signaling pathways and their potential importance in clinical practice. EPMA J. 2017, 8, 391–402. [Google Scholar] [CrossRef] [PubMed]
- Lo Surdo, P.; Bottomley, M.J.; Calzetta, A.; Settembre, E.C.; Cirillo, A.; Pandit, S.; Ni, Y.G.; Hubbard, B.; Sitlani, A.; Carfí, A. Mechanistic implications for LDL receptor degradation from the PCSK9/LDLR structure at neutral pH. EMBO Rep. 2011, 12, 1300–1305. [Google Scholar] [CrossRef]
- Zhang, D.W.; Lagace, T.A.; Garuti, R.; Zhao, Z.; McDonald, M.; Horton, J.D.; Cohen, J.C.; Hobbs, H.H. Binding of proprotein convertase subtilisin/kexin type 9 to epidermal growth factor-like repeat A of low density lipoprotein receptor decreases receptor recycling and increases degradation. J. Biol. Chem. 2007, 282, 18602–18612. [Google Scholar] [CrossRef]
- Lambert, G.; Sjouke, B.; Choque, B.; Kastelein, J.J.; Hovingh, G.K. The PCSK9 decade. J. Lipid Res. 2012, 53, 2515–2524. [Google Scholar] [CrossRef] [PubMed]
- Seidah, N.G. The PCSK9 revolution and the potential of PCSK9-based therapies to reduce LDL-cholesterol. Glob. Cardiol. Sci. Pract. 2017, 2017, e201702. [Google Scholar] [CrossRef] [PubMed]
- Haas, M.E.; Levenson, A.E.; Sun, X.; Liao, W.-H.; Rutkowski, J.M.; Ferranti, S.D.d.; Schumacher, V.A.; Scherer, P.E.; Salant, D.J.; Biddinger, S.B. The Role of Proprotein Convertase Subtilisin/Kexin Type 9 in Nephrotic Syndrome-Associated Hypercholesterolemia. Circulation 2016, 134, 61–72. [Google Scholar] [CrossRef]
- Pavlakou, P.; Liberopoulos, E.; Dounousi, E.; Elisaf, M. PCSK9 in chronic kidney disease. Int. Urol. Nephrol. 2017, 49, 1015–1024. [Google Scholar] [CrossRef] [PubMed]
- Igweonu-Nwakile, E.O.; Ali, S.; Paul, S.; Yakkali, S.; Teresa Selvin, S.; Thomas, S.; Bikeyeva, V.; Abdullah, A.; Radivojevic, A.; Abu Jad, A.A.; et al. A Systematic Review on the Safety and Efficacy of PCSK9 Inhibitors in Lowering Cardiovascular Risks in Patients with Chronic Kidney Disease. Cureus 2022, 14, e29140. [Google Scholar] [CrossRef]
- Leiter, L.A.; Cariou, B.; Müller-Wieland, D.; Colhoun, H.M.; Del Prato, S.; Tinahones, F.J.; Ray, K.K.; Bujas-Bobanovic, M.; Domenger, C.; Mandel, J.; et al. Efficacy and safety of alirocumab in insulin-treated individuals with type 1 or type 2 diabetes and high cardiovascular risk: The ODYSSEY DM-INSULIN randomized trial. Diabetes Obes. Metab. 2017, 19, 1781–1792. [Google Scholar] [CrossRef]
- Elewa, U.; Fernández-Fernández, B.; Mahillo-Fernández, I.; Martin-Cleary, C.; Sanz, A.B.; Sanchez-Niño, M.D.; Ortiz, A. PCSK9 in diabetic kidney disease. Eur. J. Clin. Investig. 2016, 46, 779–786. [Google Scholar] [CrossRef]
- Morena, M.; Le May, C.; Chenine, L.; Arnaud, L.; Dupuy, A.M.; Pichelin, M.; Leray-Moragues, H.; Chalabi, L.; Canaud, B.; Cristol, J.P.; et al. Plasma PCSK9 concentrations during the course of nondiabetic chronic kidney disease: Relationship with glomerular filtration rate and lipid metabolism. J. Clin. Lipidol. 2017, 11, 87–93. [Google Scholar] [CrossRef]
- Konarzewski, M.; Szolkiewicz, M.; Sucajtys-Szulc, E.; Blaszak, J.; Lizakowski, S.; Swierczynski, J.; Rutkowski, B. Elevated circulating PCSK-9 concentration in renal failure patients is corrected by renal replacement therapy. Am. J. Nephrol. 2014, 40, 157–163. [Google Scholar] [CrossRef]
- Oram, J.F.; Lawn, R.M. ABCA1. The gatekeeper for eliminating excess tissue cholesterol. J. Lipid Res. 2001, 42, 1173–1179. [Google Scholar] [CrossRef]
- Wang, N.; Silver, D.L.; Thiele, C.; Tall, A.R. ATP-binding cassette transporter A1 (ABCA1) functions as a cholesterol efflux regulatory protein. J. Biol. Chem. 2001, 276, 23742–23747. [Google Scholar] [CrossRef]
- Tang, C.; Kanter, J.E.; Bornfeldt, K.E.; Leboeuf, R.C.; Oram, J.F. Diabetes reduces the cholesterol exporter ABCA1 in mouse macrophages and kidneys. J. Lipid Res. 2010, 51, 1719–1728. [Google Scholar] [CrossRef] [PubMed]
- Pedigo, C.E.; Ducasa, G.M.; Leclercq, F.; Sloan, A.; Mitrofanova, A.; Hashmi, T.; Molina-David, J.; Ge, M.; Lassenius, M.I.; Forsblom, C.; et al. Local TNF causes NFATc1-dependent cholesterol-mediated podocyte injury. J. Clin. Investig. 2016, 126, 3336–3350. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Ducasa, G.M.; Mallela, S.K.; Kim, J.J.; Molina, J.; Mitrofanova, A.; Wilbon, S.S.; Ge, M.; Fontanella, A.; Pedigo, C.; et al. Sterol-O-acyltransferase-1 has a role in kidney disease associated with diabetes and Alport syndrome. Kidney Int. 2020, 98, 1275–1285. [Google Scholar] [CrossRef]
- Kim, J.J.; Wilbon, S.S.; Fornoni, A. Podocyte Lipotoxicity in CKD. Kidney360 2021, 2, 755–762. [Google Scholar] [CrossRef]
- Mitrofanova, A.; Burke, G.; Merscher, S.; Fornoni, A. New insights into renal lipid dysmetabolism in diabetic kidney disease. World J. Diabetes 2021, 12, 524–540. [Google Scholar] [CrossRef] [PubMed]
- Bhat, O.M.; Yuan, X.; Li, G.; Lee, R.; Li, P.L. Sphingolipids and Redox Signaling in Renal Regulation and Chronic Kidney Diseases. Antioxid. Redox Signal. 2018, 28, 1008–1026. [Google Scholar] [CrossRef]
- Chalfant, C.E.; Spiegel, S. Sphingosine 1-phosphate and ceramide 1-phosphate: Expanding roles in cell signaling. J. Cell Sci. 2005, 118, 4605–4612. [Google Scholar] [CrossRef]
- Geoffroy, K.; Troncy, L.; Wiernsperger, N.; Lagarde, M.; El Bawab, S. Glomerular proliferation during early stages of diabetic nephropathy is associated with local increase of sphingosine-1-phosphate levels. FEBS Lett. 2005, 579, 1249–1254. [Google Scholar] [CrossRef]
- Lan, T.; Liu, W.; Xie, X.; Xu, S.; Huang, K.; Peng, J.; Shen, X.; Liu, P.; Wang, L.; Xia, P.; et al. Sphingosine kinase-1 pathway mediates high glucose-induced fibronectin expression in glomerular mesangial cells. Mol. Endocrinol. 2011, 25, 2094–2105. [Google Scholar] [CrossRef]
- Nojiri, T.; Kurano, M.; Tokuhara, Y.; Ohkubo, S.; Hara, M.; Ikeda, H.; Tsukamoto, K.; Yatomi, Y. Modulation of sphingosine-1-phosphate and apolipoprotein M levels in the plasma, liver and kidneys in streptozotocin-induced diabetic mice. J. Diabetes Investig. 2014, 5, 639–648. [Google Scholar] [CrossRef]
- Lovric, S.; Goncalves, S.; Gee, H.Y.; Oskouian, B.; Srinivas, H.; Choi, W.I.; Shril, S.; Ashraf, S.; Tan, W.; Rao, J.; et al. Mutations in sphingosine-1-phosphate lyase cause nephrosis with ichthyosis and adrenal insufficiency. J. Clin. Investig. 2017, 127, 912–928. [Google Scholar] [CrossRef]
- Ren, S.; Babelova, A.; Moreth, K.; Xin, C.; Eberhardt, W.; Doller, A.; Pavenstadt, H.; Schaefer, L.; Pfeilschifter, J.; Huwiler, A. Transforming growth factor-beta2 upregulates sphingosine kinase-1 activity, which in turn attenuates the fibrotic response to TGF-beta2 by impeding CTGF expression. Kidney Int. 2009, 76, 857–867. [Google Scholar] [CrossRef] [PubMed]
- Awad, A.S.; Rouse, M.D.; Khutsishvili, K.; Huang, L.; Bolton, W.K.; Lynch, K.R.; Okusa, M.D. Chronic sphingosine 1-phosphate 1 receptor activation attenuates early-stage diabetic nephropathy independent of lymphocytes. Kidney Int. 2011, 79, 1090–1098. [Google Scholar] [CrossRef] [PubMed]
- Bekpinar, S.; Yenidunya, G.; Gurdol, F.; Unlucerci, Y.; Aycan-Ustyol, E.; Dinccag, N. The effect of nephropathy on plasma sphingosine 1-phosphate concentrations in patients with type 2 diabetes. Clin. Biochem. 2015, 48, 1264–1267. [Google Scholar] [CrossRef]
- Merrill, A.H., Jr. De novo sphingolipid biosynthesis: A necessary, but dangerous, pathway. J. Biol. Chem. 2002, 277, 25843–25846. [Google Scholar] [CrossRef] [PubMed]
- Haus, J.M.; Kashyap, S.R.; Kasumov, T.; Zhang, R.; Kelly, K.R.; Defronzo, R.A.; Kirwan, J.P. Plasma ceramides are elevated in obese subjects with type 2 diabetes and correlate with the severity of insulin resistance. Diabetes 2009, 58, 337–343. [Google Scholar] [CrossRef]
- Liu, G.; Han, F.; Yang, Y.; Xie, Y.; Jiang, H.; Mao, Y.; Wang, H.; Wang, M.; Chen, R.; Yang, J.; et al. Evaluation of sphingolipid metabolism in renal cortex of rats with streptozotocin-induced diabetes and the effects of rapamycin. Nephrol. Dial. Transplant. 2011, 26, 1493–1502. [Google Scholar] [CrossRef]
- Fornoni, A.; Sageshima, J.; Wei, C.; Merscher-Gomez, S.; Aguillon-Prada, R.; Jauregui, A.N.; Li, J.; Mattiazzi, A.; Ciancio, G.; Chen, L.; et al. Rituximab targets podocytes in recurrent focal segmental glomerulosclerosis. Sci. Transl. Med. 2011, 3, 85ra46. [Google Scholar] [CrossRef]
- Yoo, T.H.; Pedigo, C.E.; Guzman, J.; Correa-Medina, M.; Wei, C.; Villarreal, R.; Mitrofanova, A.; Leclercq, F.; Faul, C.; Li, J.; et al. Sphingomyelinase-like phosphodiesterase 3b expression levels determine podocyte injury phenotypes in glomerular disease. J. Am. Soc. Nephrol. 2015, 26, 133–147. [Google Scholar] [CrossRef]
- Mitrofanova, A.; Mallela, S.K.; Ducasa, G.M.; Yoo, T.H.; Rosenfeld-Gur, E.; Zelnik, I.D.; Molina, J.; Varona Santos, J.; Ge, M.; Sloan, A.; et al. SMPDL3b modulates insulin receptor signaling in diabetic kidney disease. Nat. Commun. 2019, 10, 2692. [Google Scholar] [CrossRef] [PubMed]
- Zador, I.Z.; Deshmukh, G.D.; Kunkel, R.; Johnson, K.; Radin, N.S.; Shayman, J.A. A role for glycosphingolipid accumulation in the renal hypertrophy of streptozotocin-induced diabetes mellitus. J. Clin. Investig. 1993, 91, 797–803. [Google Scholar] [CrossRef] [PubMed]
- Shayman, J.A. Targeting Glycosphingolipid Metabolism to Treat Kidney Disease. Nephron 2016, 134, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Shayman, J.A. Targeting Glucosylceramide Synthesis in the Treatment of Rare and Common Renal Disease. Semin. Nephrol. 2018, 38, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Subathra, M.; Korrapati, M.; Howell, L.A.; Arthur, J.M.; Shayman, J.A.; Schnellmann, R.G.; Siskind, L.J. Kidney glycosphingolipids are elevated early in diabetic nephropathy and mediate hypertrophy of mesangial cells. Am. J. Physiol. Renal Physiol. 2015, 309, F204–F215. [Google Scholar] [CrossRef]
- Ozbek, E. Induction of oxidative stress in kidney. Int. J. Nephrol. 2012, 2012, 465897. [Google Scholar] [CrossRef]
- Gill, P.S.; Wilcox, C.S. NADPH oxidases in the kidney. Antioxid. Redox Signal. 2006, 8, 1597–1607. [Google Scholar] [CrossRef]
- Kaneto, H.; Katakami, N.; Kawamori, D.; Miyatsuka, T.; Sakamoto, K.; Matsuoka, T.A.; Matsuhisa, M.; Yamasaki, Y. Involvement of oxidative stress in the pathogenesis of diabetes. Antioxid. Redox Signal. 2007, 9, 355–366. [Google Scholar] [CrossRef]
- Gooch, J.L.; Barnes, J.L.; Garcia, S.; Abboud, H.E. Calcineurin is activated in diabetes and is required for glomerular hypertrophy and ECM accumulation. Am. J. Physiol. Renal Physiol. 2003, 284, F144–F154. [Google Scholar] [CrossRef]
- Gooch, J.L.; Gorin, Y.; Zhang, B.X.; Abboud, H.E. Involvement of calcineurin in transforming growth factor-beta-mediated regulation of extracellular matrix accumulation. J. Biol. Chem. 2004, 279, 15561–15570. [Google Scholar] [CrossRef]
- Gooch, J.L.; Pergola, P.E.; Guler, R.L.; Abboud, H.E.; Barnes, J.L. Differential expression of calcineurin A isoforms in the diabetic kidney. J. Am. Soc. Nephrol. 2004, 15, 1421–1429. [Google Scholar] [CrossRef] [PubMed]
- Gooch, J.L.; Tang, Y.; Ricono, J.M.; Abboud, H.E. Insulin-like growth factor-I induces renal cell hypertrophy via a calcineurin-dependent mechanism. J. Biol. Chem. 2001, 276, 42492–42500. [Google Scholar] [CrossRef] [PubMed]
- Reddy, R.N.; Knotts, T.L.; Roberts, B.R.; Molkentin, J.D.; Price, S.R.; Gooch, J.L. Calcineurin A-beta is required for hypertrophy but not matrix expansion in the diabetic kidney. J. Cell. Mol. Med. 2011, 15, 414–422. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Chang, J.H.; Paik, S.Y.; Tang, Y.; Eisner, W.; Spurney, R.F. Calcineurin (CN) activation promotes apoptosis of glomerular podocytes both in vitro and in vivo. Mol. Endocrinol. 2011, 25, 1376–1386. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.M.; Wu, Y.G.; Liang, C.; Zhang, P.; Dong, J.; Ren, K.J.; Zhang, W.; Fang, F.; Shen, J.J. FK506 ameliorates renal injury in early experimental diabetic rats induced by streptozotocin. Int. Immunopharmacol. 2011, 11, 1613–1619. [Google Scholar] [CrossRef]
- Gooch, J.L.; Roberts, B.R.; Cobbs, S.L.; Tumlin, J.A. Loss of the alpha-isoform of calcineurin is sufficient to induce nephrotoxicity and altered expression of transforming growth factor-beta. Transplantation 2007, 83, 439–447. [Google Scholar] [CrossRef]
- Williams, C.R.; Gooch, J.L. Calcineurin Aβ regulates NADPH oxidase (Nox) expression and activity via nuclear factor of activated T cells (NFAT) in response to high glucose. J. Biol. Chem. 2014, 289, 4896–4905. [Google Scholar] [CrossRef]
- Ferrans, V.J.; Fredrickson, D.S. The pathology of Tangier disease. A light and electron microscopic study. Am. J. Pathol. 1975, 78, 101–158. [Google Scholar]
- Fobker, M.; Voss, R.; Reinecke, H.; Crone, C.; Assmann, G.; Walter, M. Accumulation of cardiolipin and lysocardiolipin in fibroblasts from Tangier disease subjects. FEBS Lett. 2001, 500, 157–162. [Google Scholar] [CrossRef]
- Paradies, G.; Paradies, V.; De Benedictis, V.; Ruggiero, F.M.; Petrosillo, G. Functional role of cardiolipin in mitochondrial bioenergetics. Biochim. Biophys. Acta 2014, 1837, 408–417. [Google Scholar] [CrossRef]
- Forbes, J.M.; Thorburn, D.R. Mitochondrial dysfunction in diabetic kidney disease. Nat. Rev. Nephrol. 2018, 14, 291–312. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.; Karl, B.; Mathew, A.V.; Gangoiti, J.A.; Wassel, C.L.; Saito, R.; Pu, M.; Sharma, S.; You, Y.H.; Wang, L.; et al. Metabolomics reveals signature of mitochondrial dysfunction in diabetic kidney disease. J. Am. Soc. Nephrol. 2013, 24, 1901–1912. [Google Scholar] [CrossRef] [PubMed]
- Sirtori, C.R. The pharmacology of statins. Pharmacol. Res. 2014, 88, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Athyros, V.G.; Mikhailidis, D.P.; Liberopoulos, E.N.; Kakafika, A.I.; Karagiannis, A.; Papageorgiou, A.A.; Tziomalos, K.; Ganotakis, E.S.; Elisaf, M. Effect of statin treatment on renal function and serum uric acid levels and their relation to vascular events in patients with coronary heart disease and metabolic syndrome: A subgroup analysis of the GREek Atorvastatin and Coronary heart disease Evaluation (GREACE) Study. Nephrol. Dial. Transplant. 2007, 22, 118–127. [Google Scholar] [CrossRef]
- Kolavennu, V.; Zeng, L.; Peng, H.; Wang, Y.; Danesh, F.R. Targeting of RhoA/ROCK signaling ameliorates progression of diabetic nephropathy independent of glucose control. Diabetes 2008, 57, 714–723. [Google Scholar] [CrossRef] [PubMed]
- Fujii, M.; Inoguchi, T.; Maeda, Y.; Sasaki, S.; Sawada, F.; Saito, R.; Kobayashi, K.; Sumimoto, H.; Takayanagi, R. Pitavastatin ameliorates albuminuria and renal mesangial expansion by downregulating NOX4 in db/db mice. Kidney Int. 2007, 72, 473–480. [Google Scholar] [CrossRef]
- Bruder-Nascimento, T.; Callera, G.; Montezano, A.C.; Antunes, T.T.; He, Y.; Cat, A.N.; Ferreira, N.S.; Barreto, P.A.; Olivon, V.C.; Tostes, R.C.; et al. Renoprotective Effects of Atorvastatin in Diabetic Mice: Downregulation of RhoA and Upregulation of Akt/GSK3. PLoS ONE 2016, 11, e0162731. [Google Scholar] [CrossRef]
- Wang, A.; Lin, Y.; Liang, B.; Zhao, X.; Qiu, M.; Huang, H.; Li, C.; Wang, W.; Kong, Y. Statins attenuate cholesterol-induced ROS via inhibiting NOX2/NOX4 and mitochondrial pathway in collecting ducts of the kidney. BMC Nephrol. 2022, 23, 184. [Google Scholar] [CrossRef]
- Lu, L.; Peng, W.H.; Wang, W.; Wang, L.J.; Chen, Q.J.; Shen, W.F. Effects of atorvastatin on progression of diabetic nephropathy and local RAGE and soluble RAGE expressions in rats. J. Zhejiang Univ. Sci. B 2011, 12, 652–659. [Google Scholar] [CrossRef]
- Wang, L.; Yao, X.; Li, Q.; Sun, S. Effect of Simvastatin on Lipid Accumulation and the Expression of CXCL16 and Nephrin in Podocyte Induced by Oxidized LDL. J. Investig. Surg. 2018, 31, 69–74. [Google Scholar] [CrossRef]
- Wei, P.; Grimm, P.R.; Settles, D.C.; Balwanz, C.R.; Padanilam, B.J.; Sansom, S.C. Simvastatin reverses podocyte injury but not mesangial expansion in early stage type 2 diabetes mellitus. Ren. Fail. 2009, 31, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Wang, Y.; He, P.; Li, D. Probucol Inhibited Nox2 Expression and Attenuated Podocyte Injury in Type 2 Diabetic Nephropathy of db/db Mice. Biol. Pharm. Bull. 2013, 36, 1883–1890. [Google Scholar] [CrossRef]
- Lu, Q.; Yin, X.X.; Wang, J.Y.; Gao, Y.Y.; Pan, Y.M. Effects of Ginkgo biloba on prevention of development of experimental diabetic nephropathy in rats. Acta Pharmacol. Sin. 2007, 28, 818–828. [Google Scholar] [CrossRef] [PubMed]
- Silla, A.; Fogacci, F.; Punzo, A.; Hrelia, S.; Simoni, P.; Caliceti, C.; Cicero, A.F.G. Treatment with PCSK9 Inhibitor Evolocumab Improves Vascular Oxidative Stress and Arterial Stiffness in Hypercholesterolemic Patients with High Cardiovascular Risk. Antioxidants 2023, 12, 578. [Google Scholar] [CrossRef]
- Wang, G.; Liu, Z.; Li, M.; Li, Y.; Alvi, S.S.; Ansari, I.A.; Khan, M.S. Ginkgolide B Mediated Alleviation of Inflammatory Cascades and Altered Lipid Metabolism in HUVECs via Targeting PCSK-9 Expression and Functionality. Biomed. Res. Int. 2019, 2019, 7284767. [Google Scholar] [CrossRef] [PubMed]
- Keating, G.M.; Croom, K.F. Fenofibrate. Drugs 2007, 67, 121–153. [Google Scholar] [CrossRef]
- Yaribeygi, H.; Mohammadi, M.T.; Rezaee, R.; Sahebkar, A. Fenofibrate improves renal function by amelioration of NOX-4, IL-18, and p53 expression in an experimental model of diabetic nephropathy. J. Cell. Biochem. 2018, 119, 7458–7469. [Google Scholar] [CrossRef]
- Yvan-Charvet, L.; Pagler, T.A.; Seimon, T.A.; Thorp, E.; Welch, C.L.; Witztum, J.L.; Tabas, I.; Tall, A.R. ABCA1 and ABCG1 protect against oxidative stress-induced macrophage apoptosis during efferocytosis. Circ. Res. 2010, 106, 1861–1869. [Google Scholar] [CrossRef]
- Yagi, S.; Akaike, M.; Aihara, K.; Iwase, T.; Ishikawa, K.; Yoshida, S.; Sumitomo-Ueda, Y.; Kusunose, K.; Niki, T.; Yamaguchi, K.; et al. Ezetimibe ameliorates metabolic disorders and microalbuminuria in patients with hypercholesterolemia. J. Atheroscler. Thromb. 2010, 17, 173–180. [Google Scholar] [CrossRef]
- Cho, K.H.; Kim, H.J.; Rodriguez-Iturbe, B.; Vaziri, N.D. Niacin ameliorates oxidative stress, inflammation, proteinuria, and hypertension in rats with chronic renal failure. Am. J. Physiol. Renal Physiol. 2009, 297, F106–F113. [Google Scholar] [CrossRef]
- Park, F.; Miller, D.D. Role of lysophosphatidic acid and its receptors in the kidney. Physiol. Genom. 2017, 49, 659–666. [Google Scholar] [CrossRef] [PubMed]
- Yung, Y.C.; Stoddard, N.C.; Chun, J. LPA receptor signaling: Pharmacology, physiology, and pathophysiology. J. Lipid Res. 2014, 55, 1192–1214. [Google Scholar] [CrossRef]
- Aikawa, S.; Hashimoto, T.; Kano, K.; Aoki, J. Lysophosphatidic acid as a lipid mediator with multiple biological actions. J. Biochem. 2015, 157, 81–89. [Google Scholar] [CrossRef]
- Budd, D.C.; Qian, Y. Development of lysophosphatidic acid pathway modulators as therapies for fibrosis. Future Med. Chem. 2013, 5, 1935–1952. [Google Scholar] [CrossRef]
- Lee, J.H.; Kim, D.; Oh, Y.S.; Jun, H.S. Lysophosphatidic Acid Signaling in Diabetic Nephropathy. Int. J. Mol. Sci. 2019, 20, 2850. [Google Scholar] [CrossRef]
- Grove, K.J.; Voziyan, P.A.; Spraggins, J.M.; Wang, S.; Paueksakon, P.; Harris, R.C.; Hudson, B.G.; Caprioli, R.M. Diabetic nephropathy induces alterations in the glomerular and tubule lipid profiles. J. Lipid Res. 2014, 55, 1375–1385. [Google Scholar] [CrossRef]
- Rancoule, C.; Attane, C.; Gres, S.; Fournel, A.; Dusaulcy, R.; Bertrand, C.; Vinel, C.; Treguer, K.; Prentki, M.; Valet, P.; et al. Lysophosphatidic acid impairs glucose homeostasis and inhibits insulin secretion in high-fat diet obese mice. Diabetologia 2013, 56, 1394–1402. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Sarker, M.K.; Choi, H.; Shin, D.; Kim, D.; Jun, H.S. Lysophosphatidic acid receptor 1 inhibitor, AM095, attenuates diabetic nephropathy in mice by downregulation of TLR4/NF-kappaB signaling and NADPH oxidase. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1332–1340. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Halaihel, N.; Zhang, W.; Rogers, T.; Levi, M. Role of sterol regulatory element-binding protein 1 in regulation of renal lipid metabolism and glomerulosclerosis in diabetes mellitus. J. Biol. Chem. 2002, 277, 18919–18927. [Google Scholar] [CrossRef]
- Eberle, D.; Hegarty, B.; Bossard, P.; Ferre, P.; Foufelle, F. SREBP transcription factors: Master regulators of lipid homeostasis. Biochimie 2004, 86, 839–848. [Google Scholar] [CrossRef]
- Ishigaki, N.; Yamamoto, T.; Shimizu, Y.; Kobayashi, K.; Yatoh, S.; Sone, H.; Takahashi, A.; Suzuki, H.; Yamagata, K.; Yamada, N.; et al. Involvement of glomerular SREBP-1c in diabetic nephropathy. Biochem. Biophys. Res. Commun. 2007, 364, 502–508. [Google Scholar] [CrossRef]
- Van Krieken, R.; Marway, M.; Parthasarathy, P.; Mehta, N.; Ingram, A.J.; Gao, B.; Krepinsky, J.C. Inhibition of SREBP With Fatostatin Does Not Attenuate Early Diabetic Nephropathy in Male Mice. Endocrinology 2018, 159, 1479–1495. [Google Scholar] [CrossRef] [PubMed]
- Desvergne, B.; Wahli, W. Peroxisome proliferator-activated receptors: Nuclear control of metabolism. Endocr. Rev. 1999, 20, 649–688. [Google Scholar] [CrossRef] [PubMed]
- Yoon, M. The role of PPARalpha in lipid metabolism and obesity: Focusing on the effects of estrogen on PPARalpha actions. Pharmacol. Res. 2009, 60, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Braissant, O.; Foufelle, F.; Scotto, C.; Dauca, M.; Wahli, W. Differential expression of peroxisome proliferator-activated receptors (PPARs): Tissue distribution of PPAR-alpha, -beta, and -gamma in the adult rat. Endocrinology 1996, 137, 354–366. [Google Scholar] [CrossRef]
- Hong, Y.A.; Lim, J.H.; Kim, M.Y.; Kim, T.W.; Kim, Y.; Yang, K.S.; Park, H.S.; Choi, S.R.; Chung, S.; Kim, H.W.; et al. Fenofibrate improves renal lipotoxicity through activation of AMPK-PGC-1alpha in db/db mice. PLoS ONE 2014, 9, e96147. [Google Scholar] [CrossRef]
- Park, C.W.; Zhang, Y.; Zhang, X.; Wu, J.; Chen, L.; Cha, D.R.; Su, D.; Hwang, M.T.; Fan, X.; Davis, L.; et al. PPARalpha agonist fenofibrate improves diabetic nephropathy in db/db mice. Kidney Int. 2006, 69, 1511–1517. [Google Scholar] [CrossRef]
- Park, C.W.; Kim, H.W.; Ko, S.H.; Chung, H.W.; Lim, S.W.; Yang, C.W.; Chang, Y.S.; Sugawara, A.; Guan, Y.; Breyer, M.D. Accelerated diabetic nephropathy in mice lacking the peroxisome proliferator-activated receptor alpha. Diabetes 2006, 55, 885–893. [Google Scholar] [CrossRef]
- Cheng, R.; Ding, L.; He, X.; Takahashi, Y.; Ma, J.X. Interaction of PPARalpha With the Canonic Wnt Pathway in the Regulation of Renal Fibrosis. Diabetes 2016, 65, 3730–3743. [Google Scholar] [CrossRef]
- Maki, T.; Maeda, Y.; Sonoda, N.; Makimura, H.; Kimura, S.; Maeno, S.; Takayanagi, R.; Inoguchi, T. Renoprotective effect of a novel selective PPARalpha modulator K-877 in db/db mice: A role of diacylglycerol-protein kinase C-NAD(P)H oxidase pathway. Metabolism 2017, 71, 33–45. [Google Scholar] [CrossRef]
- Inoguchi, T.; Li, P.; Umeda, F.; Yu, H.Y.; Kakimoto, M.; Imamura, M.; Aoki, T.; Etoh, T.; Hashimoto, T.; Naruse, M.; et al. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C—Dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes 2000, 49, 1939–1945. [Google Scholar] [CrossRef] [PubMed]
- Inoguchi, T.; Sonta, T.; Tsubouchi, H.; Etoh, T.; Kakimoto, M.; Sonoda, N.; Sato, N.; Sekiguchi, N.; Kobayashi, K.; Sumimoto, H.; et al. Protein kinase C-dependent increase in reactive oxygen species (ROS) production in vascular tissues of diabetes: Role of vascular NAD(P)H oxidase. J. Am. Soc. Nephrol. 2003, 14, S227–S232. [Google Scholar] [CrossRef] [PubMed]
- Sheetz, M.J.; King, G.L. Molecular understanding of hyperglycemia’s adverse effects for diabetic complications. JAMA 2002, 288, 2579–2588. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, B.; Jones, S.A.; Price, R.R.; Watson, M.A.; McKee, D.D.; Moore, L.B.; Galardi, C.; Wilson, J.G.; Lewis, M.C.; Roth, M.E.; et al. A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol. Cell 2000, 6, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Kalaany, N.Y.; Mangelsdorf, D.J. LXRS AND FXR: The Yin and Yang of Cholesterol and Fat Metabolism. Annu. Rev. Physiol. 2006, 68, 159–191. [Google Scholar] [CrossRef]
- Rizzo, G.; Renga, B.; Mencarelli, A.; Pellicciari, R.; Fiorucci, S. Role of FXR in regulating bile acid homeostasis and relevance for human diseases. Curr. Drug Targets Immune Endocr. Metabol. Disord. 2005, 5, 289–303. [Google Scholar] [CrossRef]
- Wang, H.; Chen, J.; Hollister, K.; Sowers, L.C.; Forman, B.M. Endogenous bile acids are ligands for the nuclear receptor FXR/BAR. Mol. Cell 1999, 3, 543–553. [Google Scholar] [CrossRef]
- Zhang, Y.; Kast-Woelbern, H.R.; Edwards, P.A. Natural structural variants of the nuclear receptor farnesoid X receptor affect transcriptional activation. J. Biol. Chem. 2003, 278, 104–110. [Google Scholar] [CrossRef]
- Bishop-Bailey, D.; Walsh, D.T.; Warner, T.D. Expression and activation of the farnesoid X receptor in the vasculature. Proc. Natl. Acad. Sci. USA 2004, 101, 3668–3673. [Google Scholar] [CrossRef]
- Forman, B.M.; Goode, E.; Chen, J.; Oro, A.E.; Bradley, D.J.; Perlmann, T.; Noonan, D.J.; Burka, L.T.; McMorris, T.; Lamph, W.W.; et al. Identification of a nuclear receptor that is activated by farnesol metabolites. Cell 1995, 81, 687–693. [Google Scholar] [CrossRef]
- Jiang, T.; Wang, X.X.; Scherzer, P.; Wilson, P.; Tallman, J.; Takahashi, H.; Li, J.; Iwahashi, M.; Sutherland, E.; Arend, L.; et al. Farnesoid X Receptor Modulates Renal Lipid Metabolism, Fibrosis, and Diabetic Nephropathy. Diabetes 2007, 56, 2485–2493. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.X.; Wang, D.; Luo, Y.; Myakala, K.; Dobrinskikh, E.; Rosenberg, A.Z.; Levi, J.; Kopp, J.B.; Field, A.; Hill, A.; et al. FXR/TGR5 Dual Agonist Prevents Progression of Nephropathy in Diabetes and Obesity. J. Am. Soc. Nephrol. 2018, 29, 118–137. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.X.; Jiang, T.; Shen, Y.; Caldas, Y.; Miyazaki-Anzai, S.; Santamaria, H.; Urbanek, C.; Solis, N.; Scherzer, P.; Lewis, L.; et al. Diabetic Nephropathy Is Accelerated by Farnesoid X Receptor Deficiency and Inhibited by Farnesoid X Receptor Activation in a Type 1 Diabetes Model. Diabetes 2010, 59, 2916–2927. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.; Pellicciari, R.; Pruzanski, M.; Auwerx, J.; Schoonjans, K. Targeting bile-acid signalling for metabolic diseases. Nat. Rev. Drug Discov. 2008, 7, 678–693. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, A.; Maiti, P. TGR5: An emerging bile acid G-protein-coupled receptor target for the potential treatment of metabolic disorders. Drug Discov. Today 2009, 14, 523–530. [Google Scholar] [CrossRef]
- Guo, C.; Chen, W.-D.; Wang, Y.-D. TGR5, Not Only a Metabolic Regulator. Front. Physiol. 2016, 7, 646. [Google Scholar] [CrossRef]
- Wang, X.X.; Edelstein, M.H.; Gafter, U.; Qiu, L.; Luo, Y.; Dobrinskikh, E.; Lucia, S.; Adorini, L.; D’Agati, V.D.; Levi, J.; et al. G Protein-Coupled Bile Acid Receptor TGR5 Activation Inhibits Kidney Disease in Obesity and Diabetes. J. Am. Soc. Nephrol. 2016, 27, 1362–1378. [Google Scholar] [CrossRef]
- Corda, S.; Laplace, C.; Vicaut, E.; Duranteau, J. Rapid reactive oxygen species production by mitochondria in endothelial cells exposed to tumor necrosis factor-alpha is mediated by ceramide. Am. J. Respir. Cell Mol. Biol. 2001, 24, 762–768. [Google Scholar] [CrossRef]
- Lecour, S.; Van der Merwe, E.; Opie, L.H.; Sack, M.N. Ceramide attenuates hypoxic cell death via reactive oxygen species signaling. J. Cardiovasc. Pharmacol. 2006, 47, 158–163. [Google Scholar] [CrossRef]
- Bhunia, A.K.; Han, H.; Snowden, A.; Chatterjee, S. Redox-regulated signaling by lactosylceramide in the proliferation of human aortic smooth muscle cells. J. Biol. Chem. 1997, 272, 15642–15649. [Google Scholar] [CrossRef]
- Zhang, A.Y.; Yi, F.; Jin, S.; Xia, M.; Chen, Q.Z.; Gulbins, E.; Li, P.L. Acid sphingomyelinase and its redox amplification in formation of lipid raft redox signaling platforms in endothelial cells. Antioxid. Redox Signal. 2007, 9, 817–828. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, X.; Carpinteiro, A.; Gulbins, E. Acid sphingomyelinase amplifies redox signaling in Pseudomonas aeruginosa-induced macrophage apoptosis. J. Immunol. 2008, 181, 4247–4254. [Google Scholar] [CrossRef]
- Brown, D.A.; London, E. Structure and origin of ordered lipid domains in biological membranes. J. Membr. Biol. 1998, 164, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Zhang, Y.; Yi, F.; Li, P.L. Critical role of lipid raft redox signaling platforms in endostatin-induced coronary endothelial dysfunction. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 485–490. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.Y.; Yi, F.; Zhang, G.; Gulbins, E.; Li, P.L. Lipid raft clustering and redox signaling platform formation in coronary arterial endothelial cells. Hypertension 2006, 47, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Yi, F.; Zhang, A.Y.; Janscha, J.L.; Li, P.L.; Zou, A.P. Homocysteine activates NADH/NADPH oxidase through ceramide-stimulated Rac GTPase activity in rat mesangial cells. Kidney Int. 2004, 66, 1977–1987. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.T.; Schalinske, K.L. Homocysteine metabolism and its relation to health and disease. Biofactors 2010, 36, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Lippi, G.; Plebani, M. Hyperhomocysteinemia in health and disease: Where we are now, and where do we go from here? Clin. Chem. Lab. Med. 2012, 50, 2075–2080. [Google Scholar] [CrossRef]
- Rudy, A.; Kowalska, I.; Straczkowski, M.; Kinalska, I. Homocysteine concentrations and vascular complications in patients with type 2 diabetes. Diabetes Metab. 2005, 31, 112–117. [Google Scholar] [CrossRef]
- Mujumdar, V.S.; Aru, G.M.; Tyagi, S.C. Induction of oxidative stress by homocyst(e)ine impairs endothelial function. J. Cell. Biochem. 2001, 82, 491–500. [Google Scholar] [CrossRef]
- Tyagi, S.C. Homocyst(e)ine and heart disease: Pathophysiology of extracellular matrix. Clin. Exp. Hypertens. 1999, 21, 181–198. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Chen, Y.F.; Zou, A.P. Implications of hyperhomocysteinemia in glomerular sclerosis in hypertension. Hypertension 2002, 39, 443–448. [Google Scholar] [CrossRef]
- Ma, L.; Liu, Q.; Jiang, Y.; Zhao, H.; Zhao, T.; Cao, Y.; Li, P.; Niu, W. Genetically elevated circulating homocysteine concentrations increase the risk of diabetic kidney disease in Chinese diabetic patients. J. Cell. Mol. Med. 2019, 23, 2794–2800. [Google Scholar] [CrossRef]
- Yang, Z.Z.; Zou, A.P. Homocysteine enhances TIMP-1 expression and cell proliferation associated with NADH oxidase in rat mesangial cells. Kidney Int. 2003, 63, 1012–1020. [Google Scholar] [CrossRef] [PubMed]
- Li, P.L.; Zhang, Y. Cross talk between ceramide and redox signaling: Implications for endothelial dysfunction and renal disease. In Sphingolipids in Disease; Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2013; pp. 171–197. [Google Scholar] [CrossRef]
- Li, X.; Becker, K.A.; Zhang, Y. Ceramide in redox signaling and cardiovascular diseases. Cell. Physiol. Biochem. 2010, 26, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Yi, F.; Zhang, A.Y.; Li, N.; Muh, R.W.; Fillet, M.; Renert, A.F.; Li, P.L. Inhibition of ceramide-redox signaling pathway blocks glomerular injury in hyperhomocysteinemic rats. Kidney Int. 2006, 70, 88–96. [Google Scholar] [CrossRef]
- Yi, F.; Jin, S.; Zhang, F.; Xia, M.; Bao, J.X.; Hu, J.; Poklis, J.L.; Li, P.L. Formation of lipid raft redox signalling platforms in glomerular endothelial cells: An early event of homocysteine-induced glomerular injury. J. Cell. Mol. Med. 2009, 13, 3303–3314. [Google Scholar] [CrossRef]
- Yamanaka, N.; Shimizu, A. Role of glomerular endothelial damage in progressive renal disease. Kidney Blood Press. Res. 1999, 22, 13–20. [Google Scholar] [CrossRef]
- Boini, K.M.; Xia, M.; Li, C.; Zhang, C.; Payne, L.P.; Abais, J.M.; Poklis, J.L.; Hylemon, P.B.; Li, P.L. Acid sphingomyelinase gene deficiency ameliorates the hyperhomocysteinemia-induced glomerular injury in mice. Am. J. Pathol. 2011, 179, 2210–2219. [Google Scholar] [CrossRef]
- Zhang, C.; Hu, J.J.; Xia, M.; Boini, K.M.; Brimson, C.A.; Laperle, L.A.; Li, P.L. Protection of podocytes from hyperhomocysteinemia-induced injury by deletion of the gp91phox gene. Free Radic. Biol. Med. 2010, 48, 1109–1117. [Google Scholar] [CrossRef]
- Abais, J.M.; Xia, M.; Zhang, Y.; Boini, K.M.; Li, P.L. Redox regulation of NLRP3 inflammasomes: ROS as trigger or effector? Antioxid. Redox Signal. 2015, 22, 1111–1129. [Google Scholar] [CrossRef]
- Ding, W.; Guo, H.; Xu, C.; Wang, B.; Zhang, M.; Ding, F. Mitochondrial reactive oxygen species-mediated NLRP3 inflammasome activation contributes to aldosterone-induced renal tubular cells injury. Oncotarget 2016, 7, 17479–17491. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.V.; Anders, H.J. Glomerular disease: Limiting autoimmune tissue injury: ROS and the inflammasome. Nat. Rev. Nephrol. 2014, 10, 545–546. [Google Scholar] [CrossRef]
- Qiu, Y.Y.; Tang, L.Q. Roles of the NLRP3 inflammasome in the pathogenesis of diabetic nephropathy. Pharmacol. Res. 2016, 114, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Sakai, N.; Wada, T. Revisiting inflammation in diabetic nephropathy: The role of the Nlrp3 inflammasome in glomerular resident cells. Kidney Int. 2015, 87, 12–14. [Google Scholar] [CrossRef] [PubMed]
- Boini, K.M.; Zhang, C.; Xia, M.; Han, W.Q.; Brimson, C.; Poklis, J.L.; Li, P.L. Visfatin-induced lipid raft redox signaling platforms and dysfunction in glomerular endothelial cells. Biochim. Biophys. Acta 2010, 1801, 1294–1304. [Google Scholar] [CrossRef] [PubMed]
- Catarzi, S.; Romagnoli, C.; Marcucci, G.; Favilli, F.; Iantomasi, T.; Vincenzini, M.T. Redox regulation of ERK1/2 activation induced by sphingosine 1-phosphate in fibroblasts: Involvement of NADPH oxidase and platelet-derived growth factor receptor. Biochim. Biophys. Acta 2011, 1810, 446–456. [Google Scholar] [CrossRef]
- Yaghobian, D.; Don, A.S.; Yaghobian, S.; Chen, X.; Pollock, C.A.; Saad, S. Increased sphingosine 1-phosphate mediates inflammation and fibrosis in tubular injury in diabetic nephropathy. Clin. Exp. Pharmacol. Physiol. 2016, 43, 56–66. [Google Scholar] [CrossRef]
- Etoh, T.; Inoguchi, T.; Kakimoto, M.; Sonoda, N.; Kobayashi, K.; Kuroda, J.; Sumimoto, H.; Nawata, H. Increased expression of NAD(P)H oxidase subunits, NOX4 and p22phox, in the kidney of streptozotocin-induced diabetic rats and its reversibity by interventive insulin treatment. Diabetologia 2003, 46, 1428–1437. [Google Scholar] [CrossRef]
- Den Hartigh, L.J.; Omer, M.; Goodspeed, L.; Wang, S.; Wietecha, T.; O’Brien, K.D.; Han, C.Y. Adipocyte-Specific Deficiency of NADPH Oxidase 4 Delays the Onset of Insulin Resistance and Attenuates Adipose Tissue Inflammation in Obesity. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 466–475. [Google Scholar] [CrossRef]
- Furukawa, S.; Fujita, T.; Shimabukuro, M.; Iwaki, M.; Yamada, Y.; Nakajima, Y.; Nakayama, O.; Makishima, M.; Matsuda, M.; Shimomura, I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J. Clin. Investig. 2017, 114, 1752–1761. [Google Scholar] [CrossRef] [PubMed]
- Keaney, J.F., Jr.; Larson, M.G.; Vasan, R.S.; Wilson, P.W.; Lipinska, I.; Corey, D.; Massaro, J.M.; Sutherland, P.; Vita, J.A.; Benjamin, E.J. Obesity and systemic oxidative stress: Clinical correlates of oxidative stress in the Framingham Study. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 434–439. [Google Scholar] [CrossRef] [PubMed]
- Youn, J.Y.; Siu, K.L.; Lob, H.E.; Itani, H.; Harrison, D.G.; Cai, H. Role of vascular oxidative stress in obesity and metabolic syndrome. Diabetes 2014, 63, 2344–2355. [Google Scholar] [CrossRef]
- Cha, J.J.; Min, H.S.; Kim, K.T.; Kim, J.E.; Ghee, J.Y.; Kim, H.W.; Lee, J.E.; Han, J.Y.; Lee, G.; Ha, H.J.; et al. APX-115, a first-in-class pan-NADPH oxidase (Nox) inhibitor, protects db/db mice from renal injury. Lab. Investig. 2017, 97, 419–431. [Google Scholar] [CrossRef]
- Kwon, G.; Uddin, M.J.; Lee, G.; Jiang, S.; Cho, A.; Lee, J.H.; Lee, S.R.; Bae, Y.S.; Moon, S.H.; Lee, S.J.; et al. A novel pan-Nox inhibitor, APX-115, protects kidney injury in streptozotocin-induced diabetic mice: Possible role of peroxisomal and mitochondrial biogenesis. Oncotarget 2017, 8, 74217–74232. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.S.; Kim, H.M.; Lee, S.H.; Ha, K.B.; Bae, Y.S.; Lee, S.J.; Moon, S.H.; Lee, E.Y.; Lee, J.-H.; Chung, C.H. APX-115, a pan-NADPH oxidase inhibitor, protects development of diabetic nephropathy in podocyte specific NOX5 transgenic mice. Free Radic. Biol. Med. 2020, 161, 92–101. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Anti-Hyperlipidemic Drugs | Mechanism of Action | Effect on Oxidative Stress and NADPH Oxidases | Major Findings | Reference |
|---|---|---|---|---|
| Pitavastatin | Inhibits HMG-CoA reductase |
|
| [96] |
| Atorvastatin | Inhibits HMG-CoA reductase |
|
| [97,98] |
| Probucol | Increases the rate of LDL catabolism |
|
| [102] |
| Evolocumab | Monoclonal antibody against PCSK9 |
|
| [104] |
| Ginkgolide B | Downregulates PCSK-9 expression |
|
| [105] |
| Fenofibrate | Peroxisome proliferator-activated receptor-α (PPAR-α) agonist |
|
| [107] |
| Cyclodextrin | Removes cholesterol from cells |
|
| [108] |
| Ezetimibe | Inhibits of Niemann–Pick C1-like 1 protein |
|
| [109] |
| Niacin | Lipid-lowering drug |
|
| [110] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Njeim, R.; Alkhansa, S.; Fornoni, A. Unraveling the Crosstalk between Lipids and NADPH Oxidases in Diabetic Kidney Disease. Pharmaceutics 2023, 15, 1360. https://doi.org/10.3390/pharmaceutics15051360
Njeim R, Alkhansa S, Fornoni A. Unraveling the Crosstalk between Lipids and NADPH Oxidases in Diabetic Kidney Disease. Pharmaceutics. 2023; 15(5):1360. https://doi.org/10.3390/pharmaceutics15051360
Chicago/Turabian StyleNjeim, Rachel, Sahar Alkhansa, and Alessia Fornoni. 2023. "Unraveling the Crosstalk between Lipids and NADPH Oxidases in Diabetic Kidney Disease" Pharmaceutics 15, no. 5: 1360. https://doi.org/10.3390/pharmaceutics15051360
APA StyleNjeim, R., Alkhansa, S., & Fornoni, A. (2023). Unraveling the Crosstalk between Lipids and NADPH Oxidases in Diabetic Kidney Disease. Pharmaceutics, 15(5), 1360. https://doi.org/10.3390/pharmaceutics15051360