
HER2-Specific Peptide (LTVSPWY) and Antibody (Herceptin) Targeted Core Cross-Linked Micelles for Breast Cancer: A Comparative Study

 ,
,  ,
,
Abstract
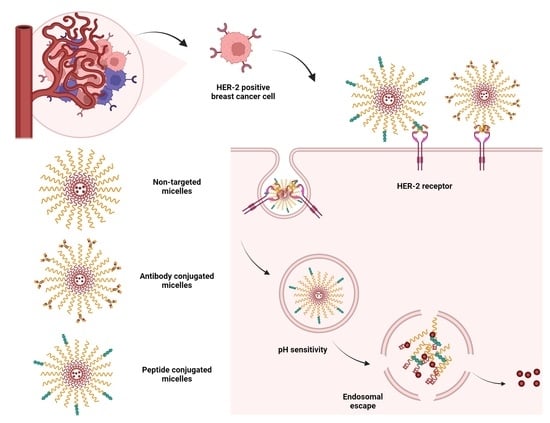
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
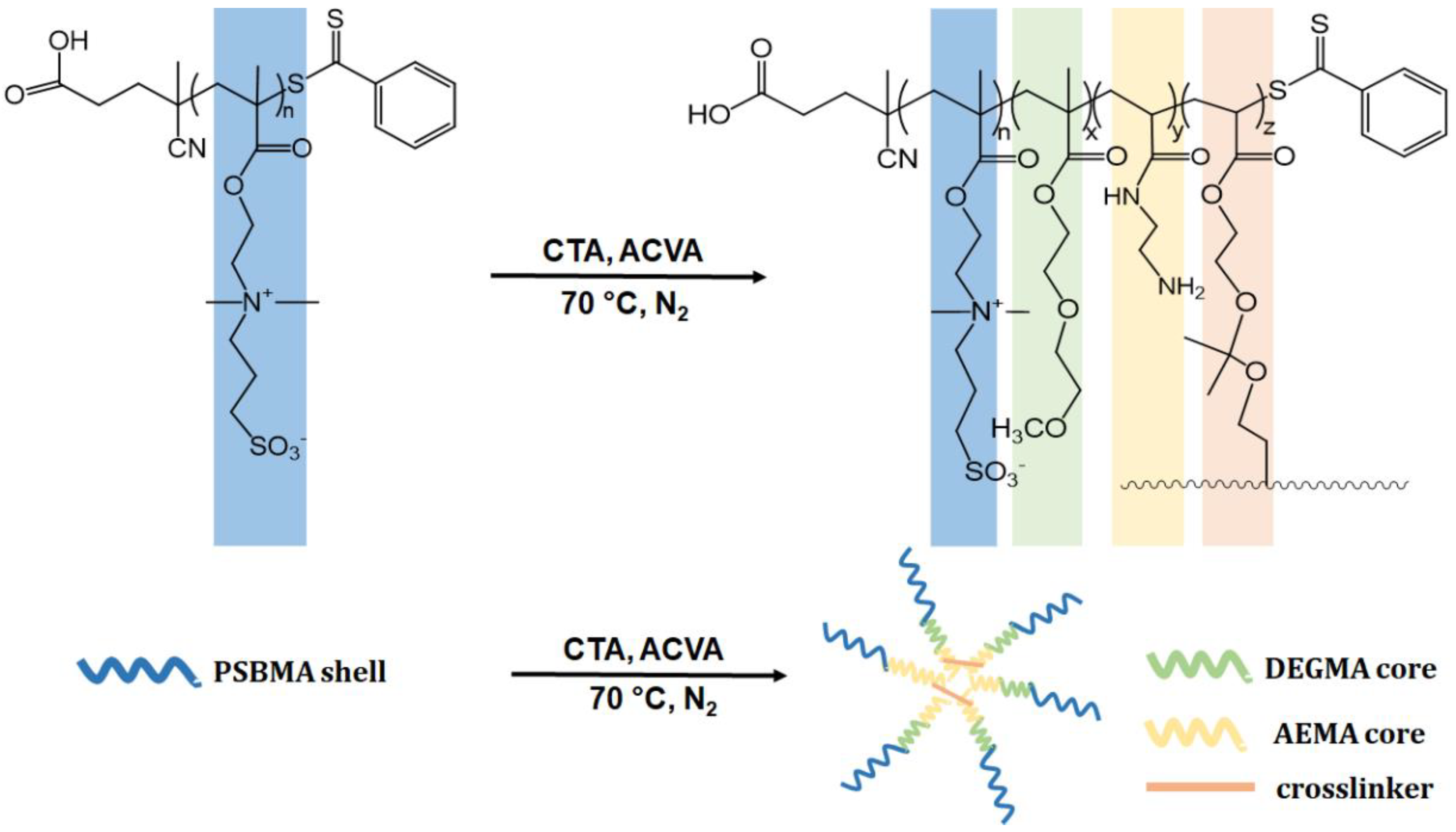
2.2.1. Synthesis of Homopolymer and Core Cross-Linked Micelles (CCMs) by RAFT Polymerization
2.2.2. Preparation of Targeted CCMs
2.2.3. Drug Loading Study
2.2.4. Drug Release Study
2.2.5. Preparation of Cell Lines
2.2.6. Cell Proliferation
2.2.7. Intracellular Uptake of CCMs
2.2.8. Apoptotic Effects
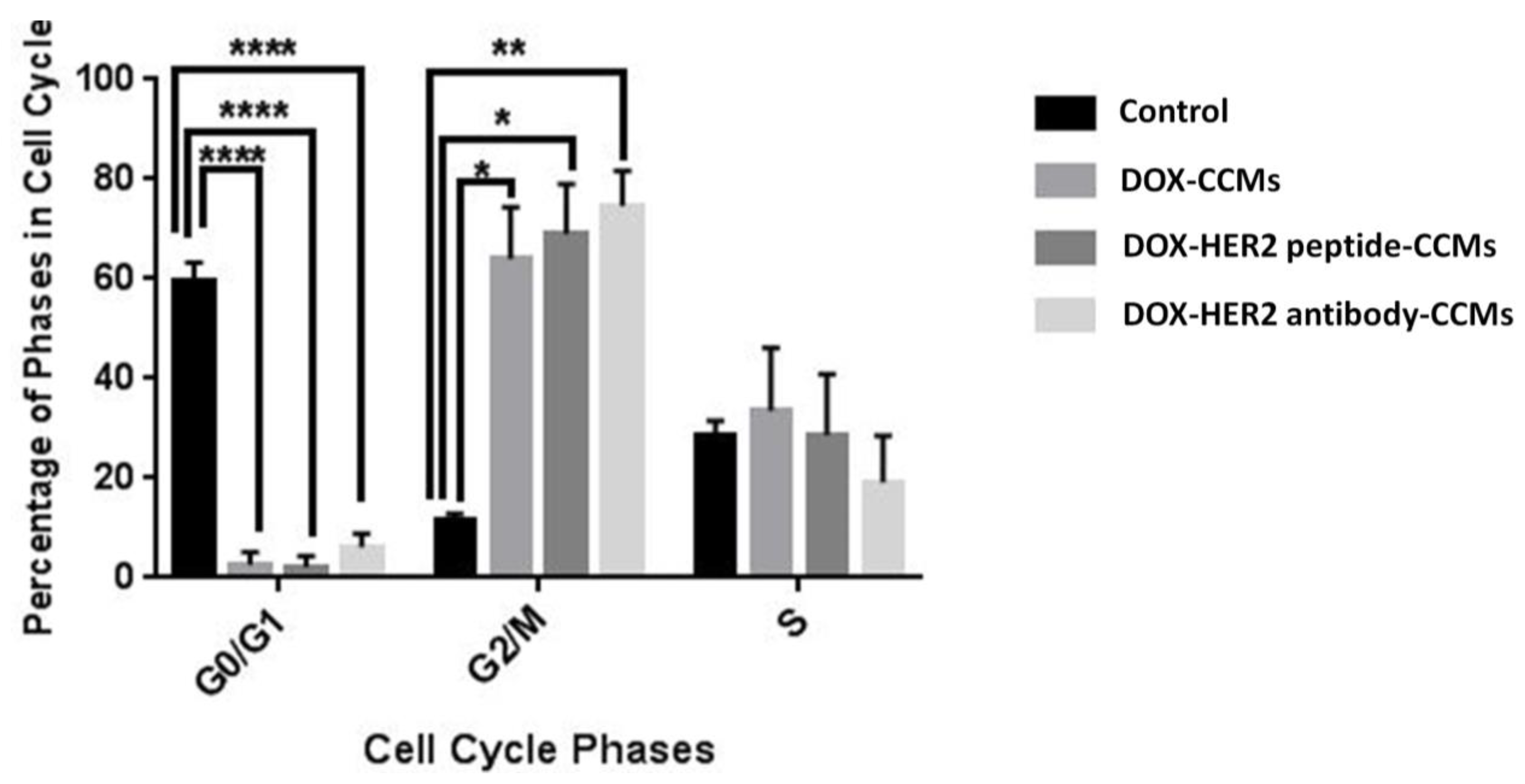
2.2.9. Cytostatic Effects
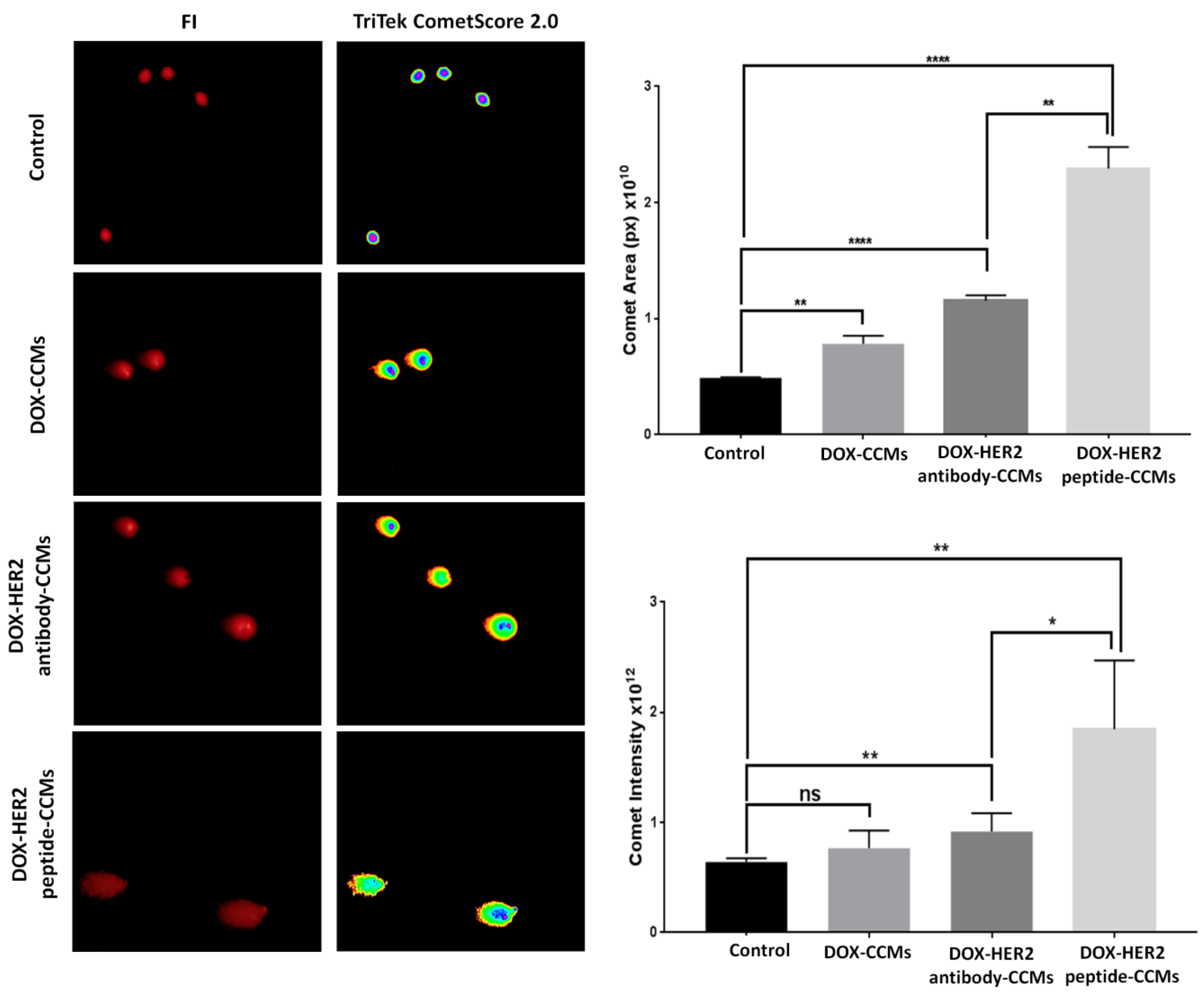
2.2.10. Genotoxic Effects
3. Results
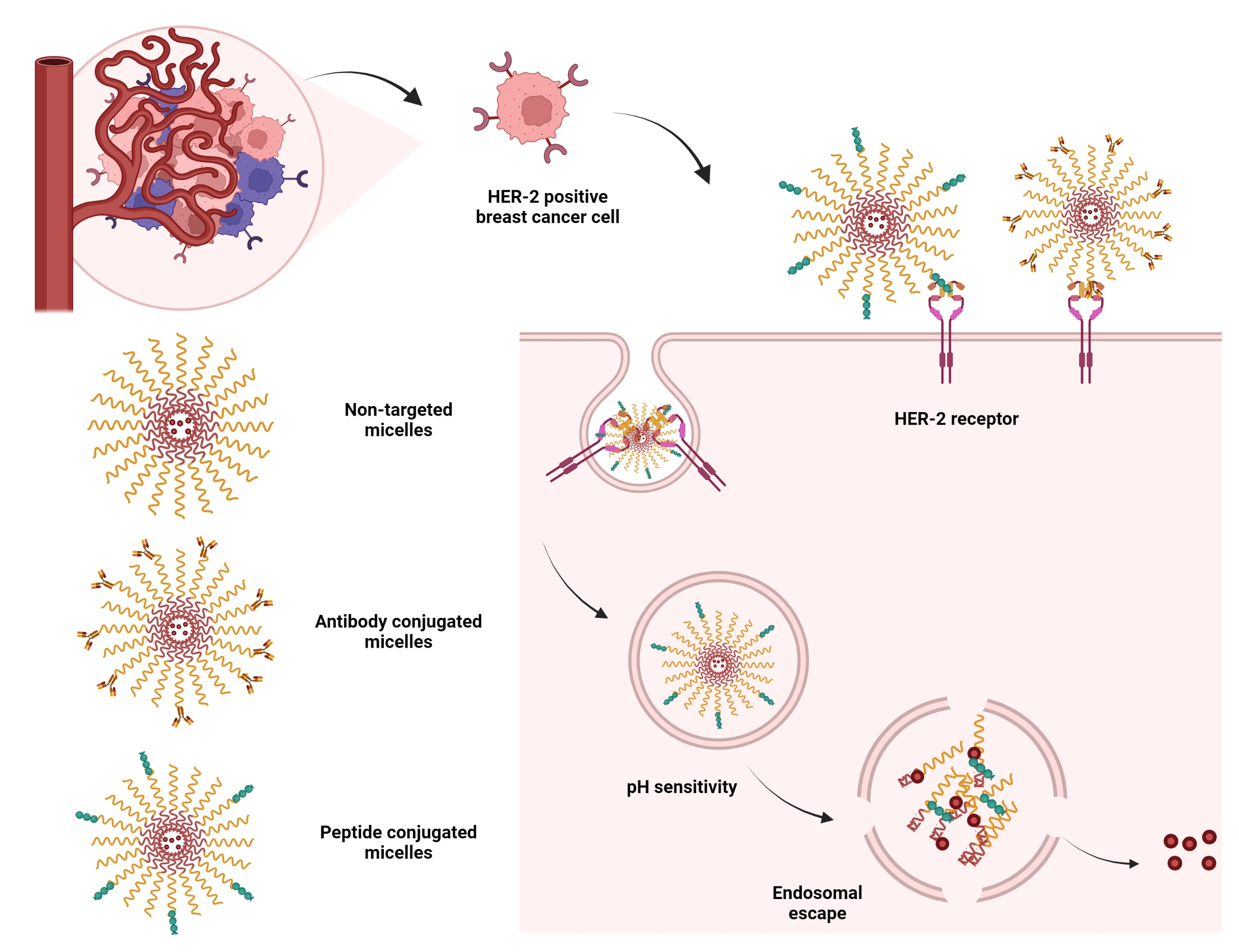
3.1. Synthesis and Characterization of Homopolymers, Micelles, and Targeted CCMs
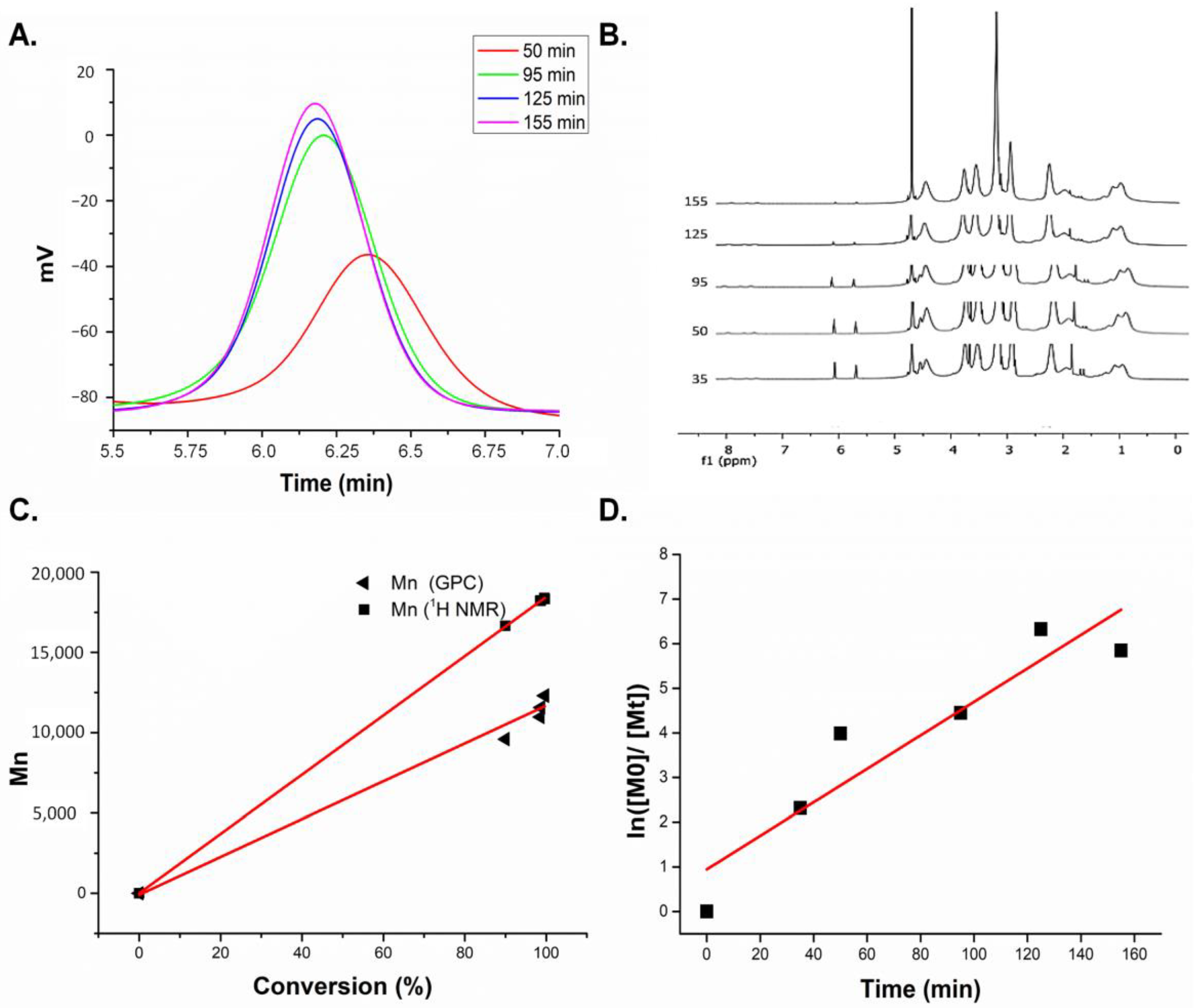
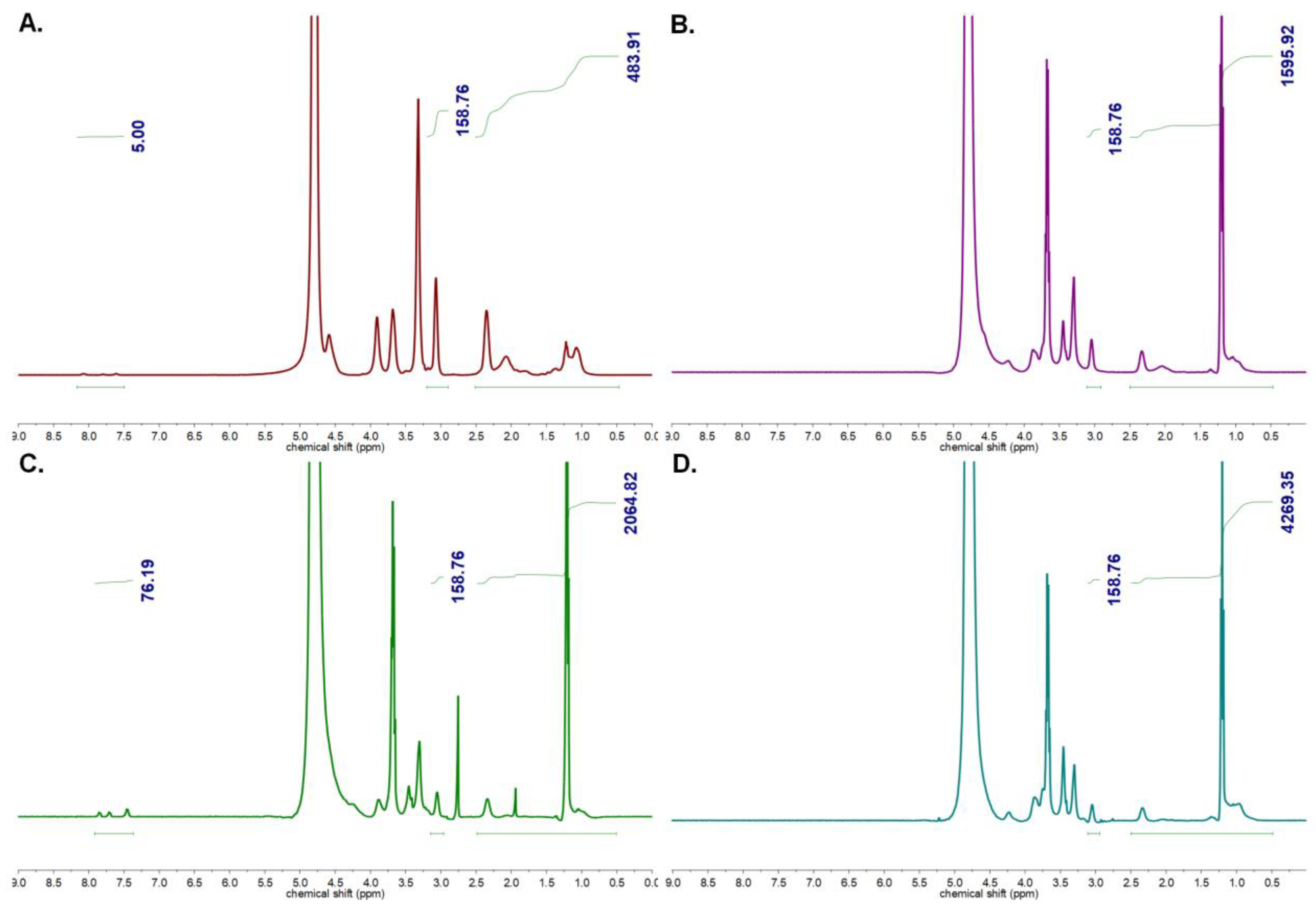
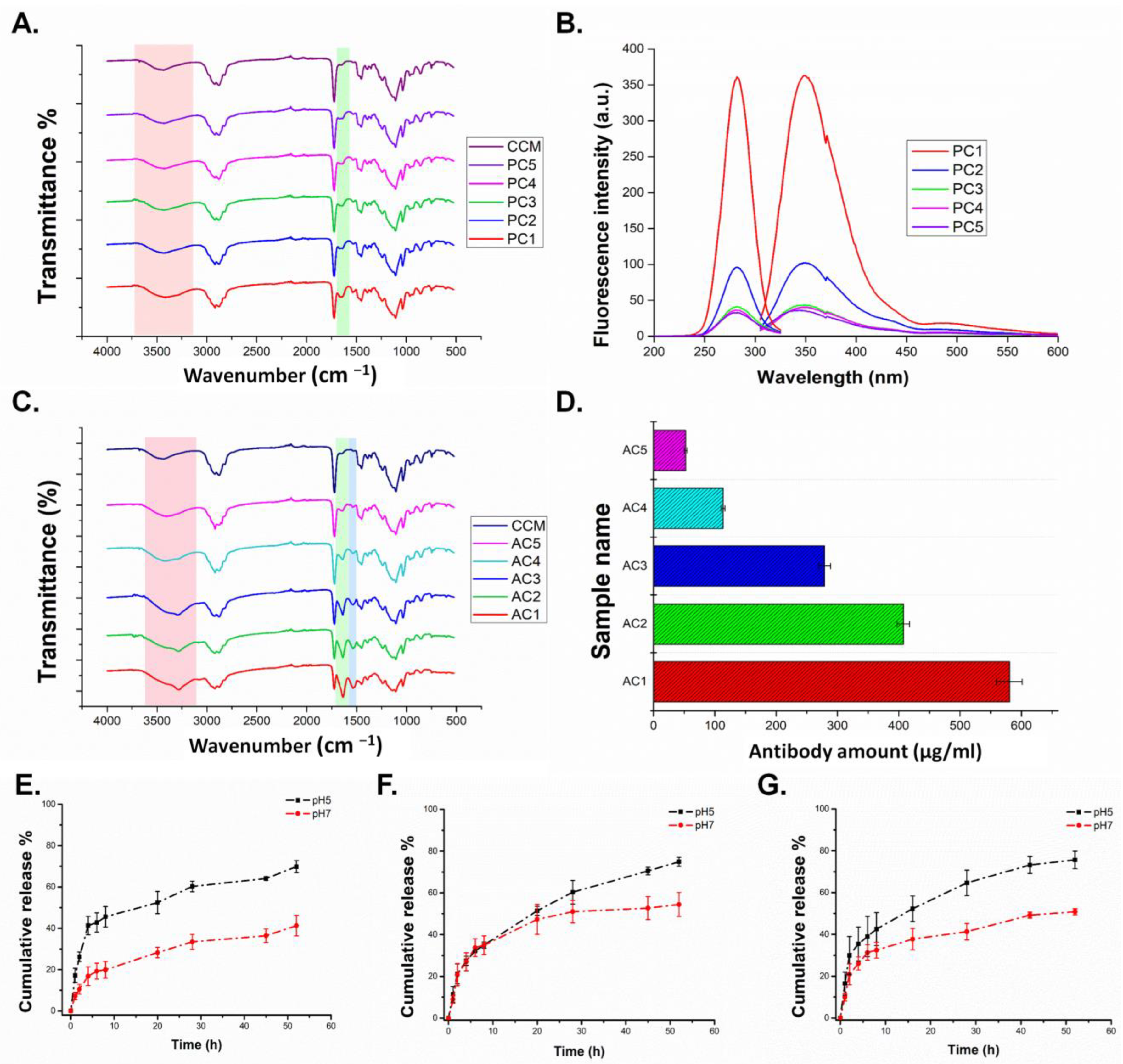
3.1.1. Characterization of Homopolymers and CCMs
3.1.2. Characterization of Peptide and Antibody Conjugated CCMs
3.1.3. Release Study
3.2. Cell Studies
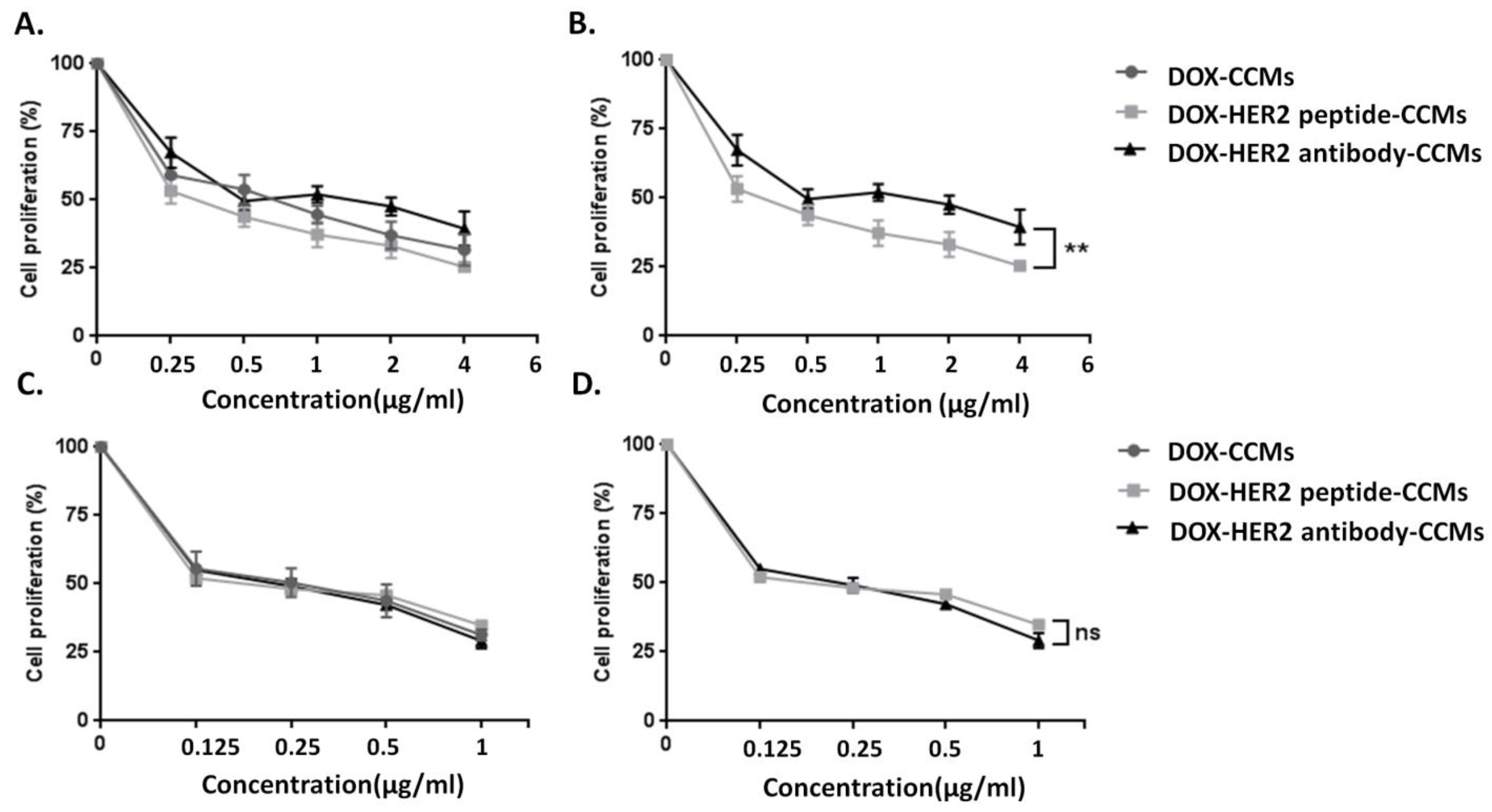
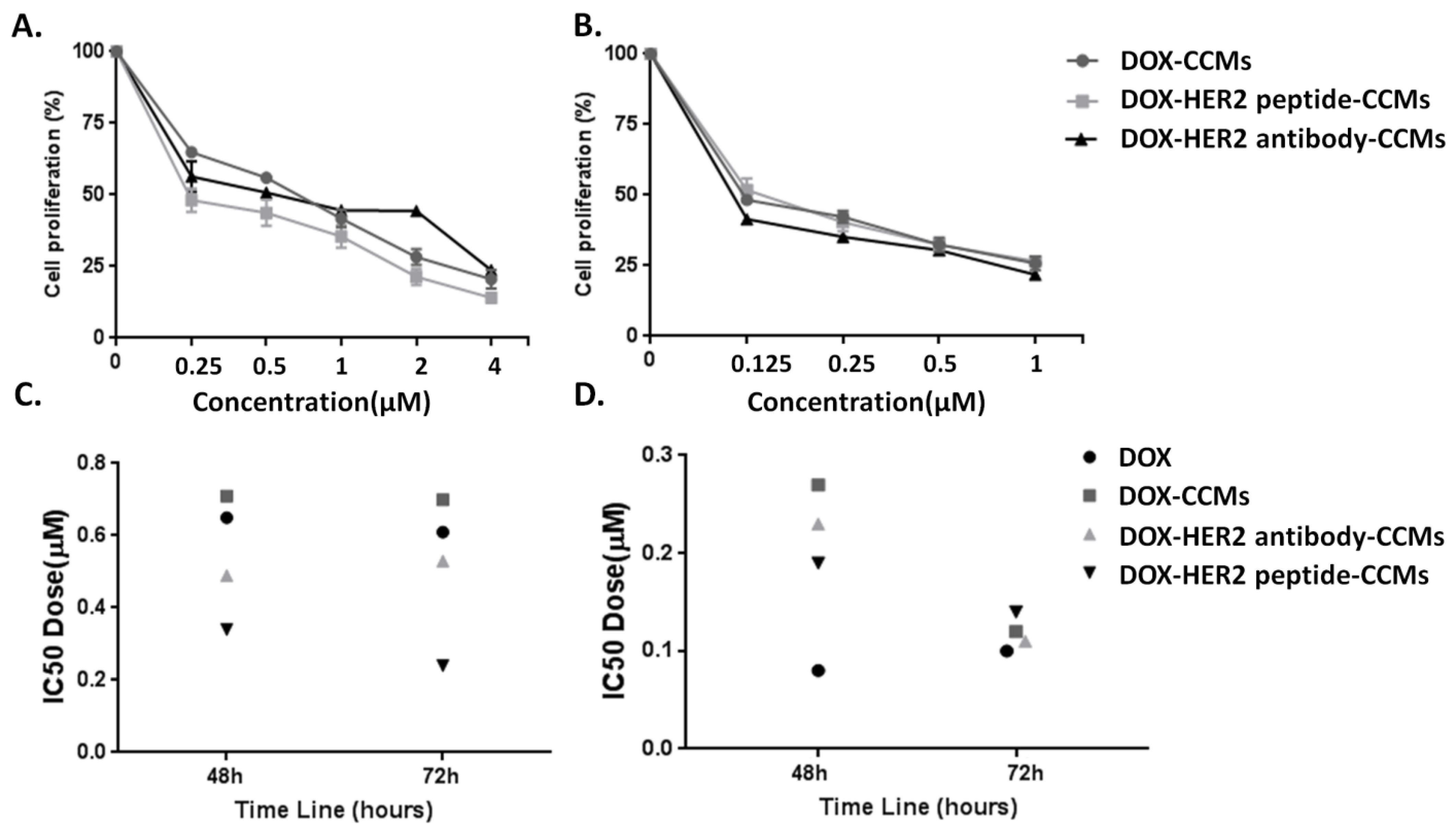
3.2.1. Determination of Cytotoxic Effects of CCMs on Cells by MTT Assay
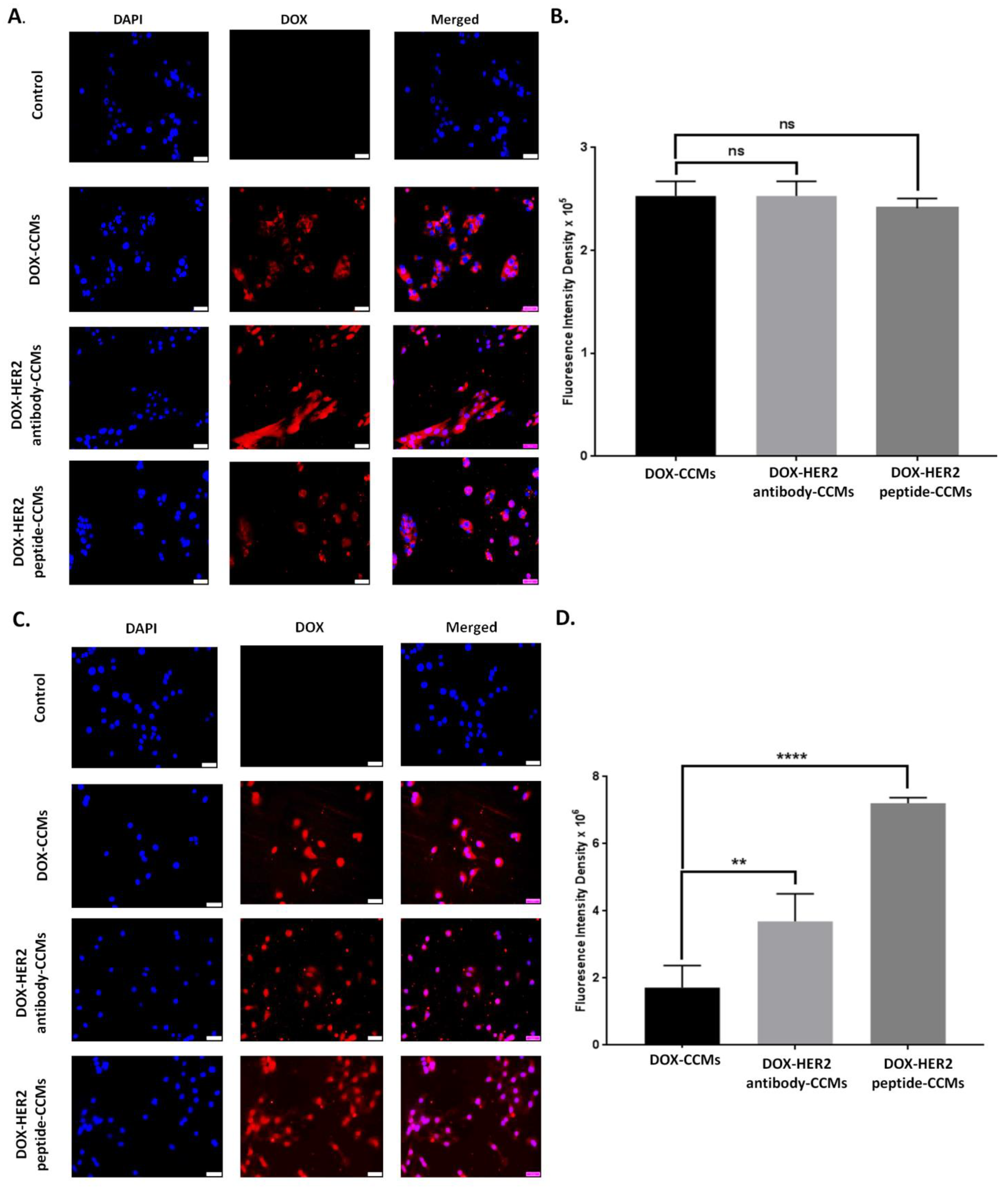
3.2.2. Determination of Uptake Amount of CCMs by Cells with Fluorescence Imaging
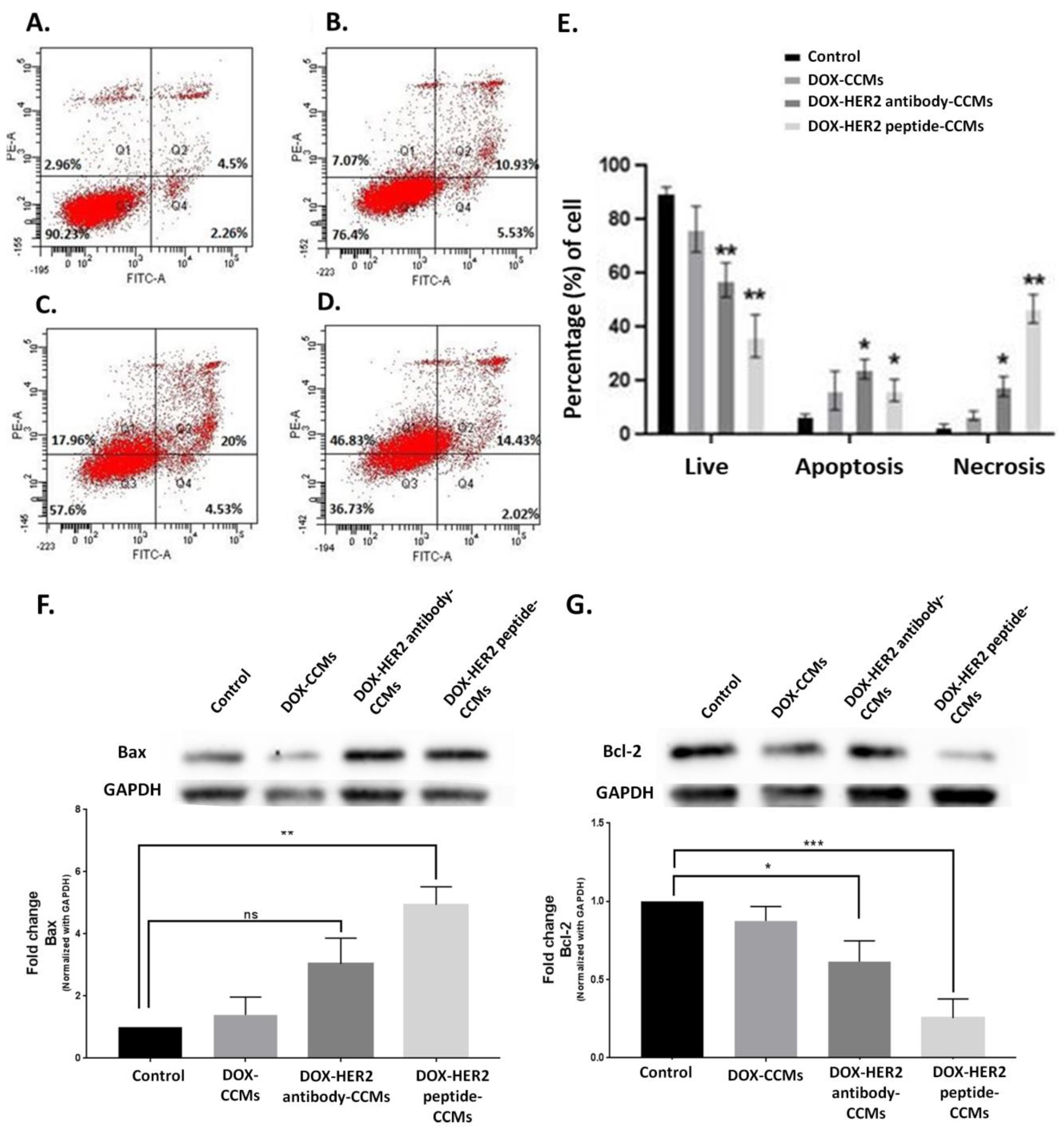
3.2.3. Determination of Apoptotic Effects of CCMs on Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Haggag, Y.; Abu Ras, B.; El-Tanani, Y.; Tambuwala, M.M.; McCarron, P.; Isreb, M.; El-Tanani, M. Co-delivery of a RanGTP inhibitory peptide and doxorubicin using dual-loaded liposomal carriers to combat chemotherapeutic resistance in breast cancer cells. Expert Opin. Drug Deliv. 2020, 17, 1655–1669. [Google Scholar] [CrossRef] [PubMed]
- Haggag, Y.A.; Yasser, M.; Tambuwala, M.M.; El Tokhy, S.S.; Isreb, M.; Donia, A.A. Repurposing of Guanabenz acetate by encapsulation into long-circulating nanopolymersomes for treatment of triple-negative breast cancer. Int. J. Pharm. 2021, 600, 120532. [Google Scholar] [CrossRef] [PubMed]
- Schottler, S.; Becker, G.; Winzen, S.; Steinbach, T.; Mohr, K.; Landfester, K.; Mailander, V.; Wurm, F.R. Protein adsorption is required for stealth effect of poly(ethylene glycol)- and poly(phosphoester)-coated nanocarriers. Nat. Nanotechnol. 2016, 11, 372–377. [Google Scholar] [CrossRef]
- Lu, A.J.; Wu, Z.Z.; Luo, X.L.; Li, S.M. Protein adsorption and macrophage uptake of zwitterionic sulfobetaine containing micelles. Colloid Surf. B 2018, 167, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Taghipour, Y.D.; Zarebkohan, A.; Salehi, R.; Rahimi, F.; Torchilin, V.P.; Hamblin, M.R.; Seifalian, A. An update on dual targeting strategy for cancer treatment. J. Control. Release 2022, 349, 67–96. [Google Scholar] [CrossRef] [PubMed]
- Phan, Q.T.; Patil, M.P.; Tu, T.T.K.; Kim, G.D.; Lim, K.T. Synthesis of zwitterionic redox-responsive nanogels by one-pot amine-thiol-ene reaction for anticancer drug release application. React. Funct. Polym. 2020, 147, 104463. [Google Scholar] [CrossRef]
- Garcia, K.P.; Zarschler, K.; Barbaro, L.; Barreto, J.A.; O’Malley, W.; Spiccia, L.; Stephan, H.; Graham, B. Zwitterionic-Coated “Stealth” Nanoparticles for Biomedical Applications: Recent Advances in Countering Biomolecular Corona Formation and Uptake by the Mononuclear Phagocyte System. Small 2014, 10, 2516–2529. [Google Scholar] [CrossRef]
- Jin, Q.; Chen, Y.J.; Wang, Y.; Ji, J. Zwitterionic drug nanocarriers: A biomimetic strategy for drug delivery. Colloid Surf. B 2014, 124, 80–86. [Google Scholar] [CrossRef]
- Kim, D.; Matsuoka, H.; Saruwatari, Y. Formation of Sulfobetaine-Containing Entirely Ionic PIC (Polyion Complex) Micelles and Their Temperature Responsivity. Langmuir 2020, 36, 10130–10137. [Google Scholar] [CrossRef]
- Fujii, S.; Takano, S.; Nakazawa, K.; Sakurai, K. Impact of Zwitterionic Polymers on the Tumor Permeability of Molecular Bottlebrush-Based Nanoparticles. Biomacromolecules 2022, 23, 2846–2855. [Google Scholar] [CrossRef]
- Alves, C.G.; de Melo-Diogo, D.; Lima-Sousa, R.; Correia, I.J. IR780 loaded sulfobetaine methacrylate-functionalized albumin nanoparticles aimed for enhanced breast cancer phototherapy. Int. J. Pharm. 2020, 582, 119346. [Google Scholar] [CrossRef] [PubMed]
- Leitao, M.M.; Alves, C.G.; de Melo-Diogo, D.; Lima-Sousa, R.; Moreira, A.F.; Correia, I.J. Sulfobetaine methacrylate-functionalized graphene oxide-IR780 nanohybrids aimed at improving breast cancer phototherapy. RSC Adv. 2020, 10, 38621–38630. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.J.; Ouyang, B.S.; Men, Y.Z.; Du, Y.; Cao, Y.B.; Xie, R.H.; Pang, Z.Q.; Shen, S.; Yang, W.L. Biodegradable zwitterionic polymer membrane coating endowing nanoparticles with ultra-long circulation and enhanced tumor photothermal therapy (Biomaterials 231 (2020) 119680). Biomaterials 2021, 275, 120920. [Google Scholar] [CrossRef] [PubMed]
- Men, Y.Z.; Peng, S.J.; Yang, P.; Jiang, Q.; Zhang, Y.H.; Shen, B.; Dong, P.; Pang, Z.Q.; Yang, W.L. Biodegradable Zwitterionic Nanogels with Long Circulation for Antitumor Drug Delivery. ACS Appl. Mater. Interfaces 2018, 10, 23509–23521. [Google Scholar] [CrossRef] [PubMed]
- Isoglu, I.A.; Ozsoy, Y.; Isoglu, S.D. Advances in Micelle-based Drug Delivery: Cross-linked Systems. Curr. Top. Med. Chem. 2017, 17, 1469–1489. [Google Scholar] [CrossRef] [PubMed]
- Ozdemir, Z.; Topuzogullari, M.; Isoglu, I.A.; Dincer, S. RAFT-mediated synthesis of poly(N-(2-hydroxypropyl)methacrylamide-b-4-vinylpyridine) by conventional and microwave heating. Polym. Bull. 2013, 70, 2857–2872. [Google Scholar] [CrossRef]
- Topuzogullari, M.; Bulmus, V.; Dalgakiran, E.; Dincer, S. pH- and temperature-responsive amphiphilic diblock copolymers of 4-vinylpyridine and oligoethyleneglycol methacrylate synthesized by RAFT polymerization. Polymer 2014, 55, 525–534. [Google Scholar] [CrossRef]
- Bayram, N.N.; Topuzogullari, M.; Isoglu, I.A.; Isoglu, S.D. RAFT-synthesized POEGMA-b-P4VP block copolymers: Preparation of nanosized micelles for anticancer drug release. Polym. Bull. 2022, 79, 9575–9588. [Google Scholar] [CrossRef]
- Bayram, N.N.; Ulu, G.T.; Topuzogullari, M.; Baran, Y.; Isoglu, S.D. HER2-Targeted, Degradable Core Cross-Linked Micelles for Specific and Dual pH-Sensitive DOX Release. Macromol. Biosci. 2022, 22, 2100375. [Google Scholar] [CrossRef]
- Peng, J.; Chen, J.; Xie, F.; Bao, W.; Xu, H.; Wang, H.; Xu, Y.; Du, Z. Herceptin-conjugated paclitaxel loaded PCL-PEG worm-like nanocrystal micelles for the combinatorial treatment of HER2-positive breast cancer. Biomaterials 2019, 222, 119420. [Google Scholar] [CrossRef]
- Furman, O.; Zaporozhets, A.; Tobi, D.; Bazylevich, A.; Firer, M.A.; Patsenker, L.; Gellerman, G.; Lubin, B.C.R. Novel Cyclic Peptides for Targeting EGFR and EGRvIII Mutation for Drug Delivery. Pharmaceutics 2022, 14, 1505. [Google Scholar] [CrossRef] [PubMed]
- Yogi, A.; Hussack, G.; van Faassen, H.; Haqqani, A.S.; Delaney, C.E.; Brunette, E.; Sandhu, J.K.; Hewitt, M.; Sulea, T.; Kemmerich, K.; et al. Brain Delivery of IGF1R5, a Single-Domain Antibody Targeting Insulin-like Growth Factor-1 Receptor. Pharmaceutics 2022, 14, 1452. [Google Scholar] [CrossRef] [PubMed]
- Oh, D.Y.; Bang, Y.J. HER2-targeted therapies—A role beyond breast cancer. Nat. Rev. Clin. Oncol. 2020, 17, 33–48. [Google Scholar] [CrossRef]
- Shien, T.; Iwata, H. Adjuvant and neoadjuvant therapy for breast cancer. Jpn. J. Clin. Oncol. 2020, 50, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Park, J.W.; Hong, K.L.; Kirpotin, D.B.; Colbern, G.; Shalaby, R.; Baselga, J.; Shao, Y.; Nielsen, U.B.; Marks, J.D.; Moore, D.; et al. Anti-HER2 immunoliposomes: Enhanced efficacy attributable to targeted delivery. Clin. Cancer Res. 2002, 8, 1172–1181. [Google Scholar]
- Siwak, D.R.; Tari, A.M.; Lopez-Berestein, G. The potential of drug-carrying immunoliposomes as anticancer agents—Commentary re: J. W. Park et al., anti-HER2 immunoliposomes: Enhanced efficacy due to targeted delivery. Clin. Cancer Res., 8: 1172–1181, 2002. Clin. Cancer Res. 2002, 8, 955–956. [Google Scholar] [PubMed]
- Goldstein, D.; Gofrit, O.; Nyska, A.; Benita, S. Anti-HER2 cationic immunoemulsion as a potential targeted drug delivery system for the treatment of prostate cancer. Cancer Res. 2007, 67, 269–275. [Google Scholar] [CrossRef]
- Shadidi, M.; Sioud, M. Identification of novel carrier peptides for the specific delivery of therapeutics into cancer cells. FASEB J. 2002, 16, 256–258. [Google Scholar] [CrossRef]
- Chopra, A. LTVSPWY peptide-modified PEGylated chitosan magnetic nanoparticles. In Molecular Imaging and Contrast Agent Database (MICAD); National Center for Biotechnology Information (US) Publisher: Bethesda, MD, USA, 2004. [Google Scholar]
- Abbineni, G.; Modali, S.; Safiejko-Mroczka, B.; Petrenko, V.A.; Mao, C.B. Evolutionary Selection of New Breast Cancer Cell-Targeting Peptides and Phages with the Cell-Targeting Peptides Fully Displayed on the Major Coat and Their Effects on Actin Dynamics during Cell Internalization (vol 7, pg 1629, 2010). Mol. Pharm. 2010, 7, 2369. [Google Scholar] [CrossRef]
- Gandra, N.; Abbineni, G.; Qu, X.W.; Huai, Y.Y.; Wang, L.; Mao, C.B. Bacteriophage Bionanowire as a Carrier for Both Cancer-Targeting Peptides and Photosensitizers and its use in Selective Cancer Cell Killing by Photodynamic Therapy. Small 2013, 9, 215–221. [Google Scholar] [CrossRef]
- Meschenmoser, K.; Kim, Y.; Franken, S.; Nowak, M.; Feldmann, G.; Bendas, G.; Wolfgarten, M.; Messmer, D.; Schmidt-Wolf, I.G. Targeting cancer with a bi-functional peptide: In vitro and in vivo results. In Vivo 2013, 27, 431–442. [Google Scholar]
- Newton, J.; Deutscher, S.L. Phage Peptide Display. In Molecular Imaging II; Semmler, W., Schwaiger, M., Eds.; Springer: Berlin/Heidelberg, Germany, 2008; pp. 145–163. [Google Scholar]
- Tai, W.Y.; Mahato, R.; Cheng, K. The role of HER2 in cancer therapy and targeted drug delivery. J. Control. Release 2010, 146, 264–275. [Google Scholar] [CrossRef]
- Wang, X.F.; Birringer, M.; Dong, L.F.; Veprek, P.; Low, P.; Swettenham, E.; Stantic, M.; Yuan, L.H.; Zobalova, R.; Vu, K.; et al. A peptide conjugate of vitamin E succinate targets breast cancer cells with high ErbB2 expression. Cancer Res. 2007, 67, 3337–3344. [Google Scholar] [CrossRef]
- Jie, L.Y.; Cai, L.L.; Wang, L.J.; Ying, X.Y.; Yu, R.S.; Zhang, M.M.; Du, Y.Z. Actively-targeted LTVSPWY peptide-modified magnetic nanoparticles for tumor imaging. Int. J. Nanomed. 2012, 7, 3981–3989. [Google Scholar] [CrossRef]
- Gurdap, S.; Bayram, N.N.; Isoglu, I.A.; Isoglu, S.D. Sulfobetaine-Based Homo- and Copolymers by RAFT: Cross-Linked Micelles and Aqueous Solution Properties. ACS Appl. Polym. Mater. 2022, 4, 6303–6311. [Google Scholar] [CrossRef]
- Bhuchar, N.; Sunasee, R.; Ishihara, K.; Thundat, T.; Narain, R. Degradable Thermoresponsive Nanogels for Protein Encapsulation and Controlled Release. Bioconjug. Chem. 2012, 23, 75–83. [Google Scholar] [CrossRef]
- van der Vlies, A.J.; O’Neil, C.P.; Hasegawa, U.; Hammond, N.; Hubbell, J.A. Synthesis of Pyridyl Disulfide-Functionalized Nanoparticles for Conjugating Thiol-Containing Small Molecules, Peptides, and Proteins. Bioconjug. Chem. 2010, 21, 653–662. [Google Scholar] [CrossRef] [PubMed]
- Kulhari, H.; Pooja, D.; Shrivastava, S.; Naidu, V.G.M.; Sistla, R. Peptide conjugated polymeric nanoparticles as a carrier for targeted delivery of docetaxel. Colloid Surf. B 2014, 117, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.R.; Mezghrani, O.; Zhang, L.; Chen, Y.; Ke, X.; Ci, T.Y. GE11 peptide modified and reduction-responsive hyaluronic acid-based nanoparticles induced higher efficacy of doxorubicin for breast carcinoma therapy. Int. J. Nanomed. 2016, 11, 5125–5147. [Google Scholar] [CrossRef] [PubMed]
- Son, S.; Shin, S.; Rao, N.V.; Um, W.; Jeon, J.; Ko, H.; Deepagan, V.G.; Kwon, S.; Lee, J.Y.; Park, J.H. Anti-Trop2 antibody-conjugated bioreducible nanoparticles for targeted triple negative breast cancer therapy. Int. J. Biol. Macromol. 2018, 110, 406–415. [Google Scholar] [CrossRef]
- Marques, A.C.; Costa, P.J.; Velho, S.; Amaral, M.H. Functionalizing nanoparticles with cancer-targeting antibodies: A comparison of strategies. J. Control. Release 2020, 320, 180–200. [Google Scholar] [CrossRef] [PubMed]
- Liao, Z.S.; Huang, S.Y.; Huang, J.J.; Chen, J.K.; Lee, A.W.; Lai, J.Y.; Lee, D.J.; Cheng, C.C. Self-Assembled pH-Responsive Polymeric Micelles for Highly Efficient, Noncytotoxic Delivery of Doxorubicin Chemotherapy To Inhibit Macrophage Activation: In Vitro Investigation. Biomacromolecules 2018, 19, 2772–2781. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Yi, M.J.; Zhang, M.C.; Shi, Z.Y.; Zhang, S.P. Folate-Conjugated Cell Membrane Mimetic Polymer Micelles for Tumor-Cell-Targeted Delivery of Doxorubicin. Langmuir 2019, 35, 504–512. [Google Scholar] [CrossRef] [PubMed]
- Jain, P.S.; Chaudhari, A.J.; Patel, S.A.; Patel, Z.N.; Patel, D.T. Development and validation of the UV-spectrophotometric method for determination of terbinafine hydrochloride in bulk and in formulation. Pharm. Methods 2011, 2, 198–202. [Google Scholar] [CrossRef]
- Sharma, P.K.; Singh, R.; Novakovic, K.R.; Eaton, J.W.; Grizzle, W.E.; Singh, S. CCR9 mediates PI3K/AKT-dependent antiapoptotic signals in prostate cancer cells and inhibition of CCR9-CCL25 interaction enhances the cytotoxic effects of etoposide. Int. J. Cancer 2010, 127, 2020–2030. [Google Scholar] [CrossRef]
- Sonar, M.V.; Wampole, M.E.; Jin, Y.Y.; Chen, C.P.; Thakur, M.L.; Wickstrom, E. Fluorescence Detection of KRAS2 mRNA Hybridization in Lung Cancer Cells with PNA-Peptides Containing an Internal Thiazole Orange. Bioconjug. Chem. 2014, 25, 1697–1708. [Google Scholar] [CrossRef]
- Donovan, M.S.; Sumerlin, B.S.; Lowe, A.B.; McCormick, C.L. Controlled/“living” polymerization of sulfobetaine monomers directly in aqueous media via RAFT. Macromolecules 2002, 35, 8663–8666. [Google Scholar] [CrossRef]
- Doncom, K.E.B.; Willcock, H.; O’Reilly, R.K. The direct synthesis of sulfobetaine-containing amphiphilic block copolymers and their self-assembly behavior. Eur. Polym. J. 2017, 87, 497–507. [Google Scholar] [CrossRef]
- Nam, J.P.; Lee, K.J.; Choi, J.W.; Yun, C.O.; Nah, J.W. Targeting delivery of tocopherol and doxorubicin grafted-chitosan polymeric micelles for cancer therapy: In vitro and in vivo evaluation. Colloid Surf. B 2015, 133, 254–262. [Google Scholar] [CrossRef]
- Kim, Y.J.; Ha, J.H.; Kim, Y.J. Self-assembled polymeric micelles for targeted photodynamic therapy of human epidermal growth factor receptor 2 overexpressing breast cancer. Nanotechnology 2021, 32, 275101. [Google Scholar] [CrossRef]
- Fiandra, L.; Mazzucchelli, S.; De Palma, C.; Colombo, M.; Allevi, R.; Sommaruga, S.; Clementi, E.; Bellini, M.; Prosperi, D.; Corsi, F. Assessing the In Vivo Targeting Efficiency of Multifunctional Nanoconstructs Bearing Antibody-Derived Ligands. ACS Nano 2013, 7, 6092–6102. [Google Scholar] [CrossRef] [PubMed]
- Ghisaidoobe, A.B.T.; Chung, S.J. Intrinsic Tryptophan Fluorescence in the Detection and Analysis of Proteins: A Focus on Forster Resonance Energy Transfer Techniques. Int. J. Mol. Sci. 2014, 15, 22518–22538. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Mi, Y.; Feng, S.S. Targeted co-delivery of docetaxel and siPlk1 by herceptin-conjugated vitamin E TPGS based immunomicelles. Biomaterials 2013, 34, 3411–3421. [Google Scholar] [CrossRef] [PubMed]
- Bolu, B.S.; Golba, B.; Sanyal, A.; Sanyal, R. Trastuzumab targeted micellar delivery of docetaxel using dendron-polymer conjugates. Biomater. Sci. 2020, 8, 2600–2610. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.T.; Li, K.; Liu, B.; Feng, S.S. A strategy for precision engineering of nanoparticles of biodegradable copolymers for quantitative control of targeted drug delivery. Biomaterials 2010, 31, 9145–9155. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Badkas, A.; Stevenson, M.; Lee, J.Y.; Leung, Y.K. Herceptin conjugated PLGA-PHis-PEG pH sensitive nanoparticles for targeted and controlled drug delivery. Int. J. Pharm. 2015, 487, 81–90. [Google Scholar] [CrossRef]
- Kumar, A.; Lale, S.V.; Alex, M.R.A.; Choudhary, V.; Koul, V. Folic acid and trastuzumab conjugated redox responsive random multiblock copolymeric nanocarriers for breast cancer therapy: In-vitro and in-vivo studies. Colloid Surf. B 2017, 149, 369–378. [Google Scholar] [CrossRef]
- Sheng, Y.; You, Y.W.; Chen, Y. Dual-targeting hybrid peptide-conjugated doxorubicin for drug resistance reversal in breast cancer. Int. J. Pharm. 2016, 512, 1–13. [Google Scholar] [CrossRef]
- Adams, J.M.; Cory, S. The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene 2007, 26, 1324–1337. [Google Scholar] [CrossRef]
- Kale, J.; Osterlund, E.J.; Andrews, D.W. BCL-2 family proteins: Changing partners in the dance towards death. Cell Death Differ. 2018, 25, 65–80. [Google Scholar] [CrossRef]
- Pillai-Kastoori, L.; Schutz-Geschwender, A.R.; Harford, J.A. A systematic approach to quantitative Western blot analysis. Anal. Biochem. 2020, 593, 113608. [Google Scholar] [CrossRef]
- Choi, J.Y.; Ramasamy, T.; Kim, S.Y.; Kim, J.; Ku, S.K.; Youn, Y.S.; Kim, J.R.; Jeong, J.H.; Choi, H.G.; Yong, C.S.; et al. PEGylated lipid bilayer-supported mesoporous silica nanoparticle composite for synergistic co-delivery of axitinib and celastrol in multi-targeted cancer therapy. Acta Biomater. 2016, 39, 94–105. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Q.H.; Ma, X.Y.; Song, Y.M.H.; Chen, Q.Q.; Jiao, Q.L.; Zhou, L.Q. Targeting regulated cell death in tumor nanomedicines. Theranostics 2022, 12, 817–841. [Google Scholar] [CrossRef] [PubMed]
- Ghanem, A.; Emara, H.A.; Muawia, S.; Abd El Maksoud, A.I.; Al-Karmalawy, A.A.; Elshal, M.F. Tanshinone IIA synergistically enhances the antitumor activity of doxorubicin by interfering with the PI3K/AKT/mTOR pathway and inhibition of topoisomerase II: In vitro and molecular docking studies. New J. Chem. 2020, 44, 17374–17381. [Google Scholar] [CrossRef]
- Shokrzadeh, M.; Etebari, M.; Ghassemi-Barghi, N. An engineered non-erythropoietic erythropoietin-derived peptide, ARA290, attenuates doxorubicin induced genotoxicity and oxidative stress. Toxicol. In Vitro 2020, 66, 104864. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Size (nm) | PDI | Zeta Potential (mV) | Peptide Amount (µg/mL) | |
| CCMs | 65.5 ± 6.2 | 0.269 | 14.5 ± 0.7 | - |
| PC1 | 235 ± 127 | 0.345 | 9.15 ± 3.3 | 15.47 ± 0.49 |
| PC2 | 141 ± 60 | 0.318 | 13.4 ± 3.8 | 3.19 ± 0.37 |
| PC3 | 141 ± 33 | 0.415 | 13.8 ± 3.8 | 0.66 ± 0.09 |
| PC4 | 118 + 43 | 0.482 | 14.7 ± 3.6 | 0.60 ± 0.27 |
| PC5 | 113 ± 17 | 0.486 | 14.5 ± 3.7 | 0.34 ± 0.04 |
| Size (nm) | PDI | Zeta Potential (mV) | Antibody amount (µg/mL) | |
| CCMs | 65.5 ± 6.2 | 0.269 | 14.5 ± 0.7 | - |
| AC1 | 428 ± 113 | 0.466 | 13.3 ± 5.4 | 580.4 ± 21 |
| AC2 | 316 ± 119 | 0.499 | 16.2 ± 3.8 | 407.6 ± 10.2 |
| AC3 | 90 ± 40 | 0.344 | 10.2 ± 4.3 | 279 ± 9.5 |
| AC4 | 79 ± 45 | 0.287 | 11.4 ± 4.1 | 113.7 ± 3.0 |
| AC5 | 78 ± 38 | 0.280 | 15.0 ± 4.7 | 52.33 ± 2.7 |
| IC50 Value for 48 h Incubation Time | IC50 value for 72 h Incubation Time | |
|---|---|---|
| SKBR-3 cells | ||
| DOX-CCMs | 0.71 µM | 0.70 µM |
| DOX-HER2 antibody-CCMs | 0.49 µM | 0.53 µM |
| DOX-HER2 peptide-CCMs | 0.34 µM | 0.24 µM |
| MCF-10A cells | ||
| DOX-CCMs | 0.27 µM | 0.12 µM |
| DOX-HER2 antibody-CCMs | 0.23 µM | 0.11 µM |
| DOX-HER2 peptide-CCMs | 0.19 µM | 0.14 µM |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bayram, N.N.; Ulu, G.T.; Abdulhadi, N.A.; Gürdap, S.; İşoğlu, İ.A.; Baran, Y.; İşoğlu, S.D. HER2-Specific Peptide (LTVSPWY) and Antibody (Herceptin) Targeted Core Cross-Linked Micelles for Breast Cancer: A Comparative Study. Pharmaceutics 2023, 15, 733. https://doi.org/10.3390/pharmaceutics15030733
Bayram NN, Ulu GT, Abdulhadi NA, Gürdap S, İşoğlu İA, Baran Y, İşoğlu SD. HER2-Specific Peptide (LTVSPWY) and Antibody (Herceptin) Targeted Core Cross-Linked Micelles for Breast Cancer: A Comparative Study. Pharmaceutics. 2023; 15(3):733. https://doi.org/10.3390/pharmaceutics15030733
Chicago/Turabian StyleBayram, Nazende Nur, Gizem Tuğçe Ulu, Nusaibah Abdulsalam Abdulhadi, Seda Gürdap, İsmail Alper İşoğlu, Yusuf Baran, and Sevil Dinçer İşoğlu. 2023. "HER2-Specific Peptide (LTVSPWY) and Antibody (Herceptin) Targeted Core Cross-Linked Micelles for Breast Cancer: A Comparative Study" Pharmaceutics 15, no. 3: 733. https://doi.org/10.3390/pharmaceutics15030733
APA StyleBayram, N. N., Ulu, G. T., Abdulhadi, N. A., Gürdap, S., İşoğlu, İ. A., Baran, Y., & İşoğlu, S. D. (2023). HER2-Specific Peptide (LTVSPWY) and Antibody (Herceptin) Targeted Core Cross-Linked Micelles for Breast Cancer: A Comparative Study. Pharmaceutics, 15(3), 733. https://doi.org/10.3390/pharmaceutics15030733