
Glycomimetic Peptides as Therapeutic Tools



Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
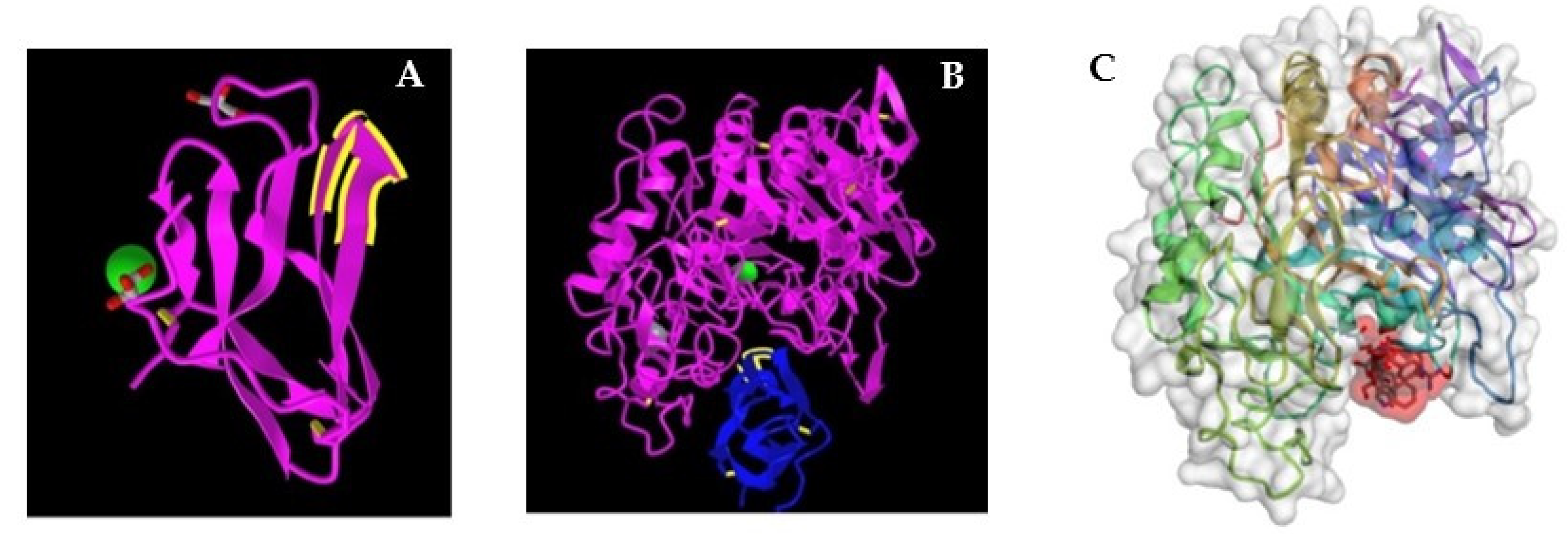
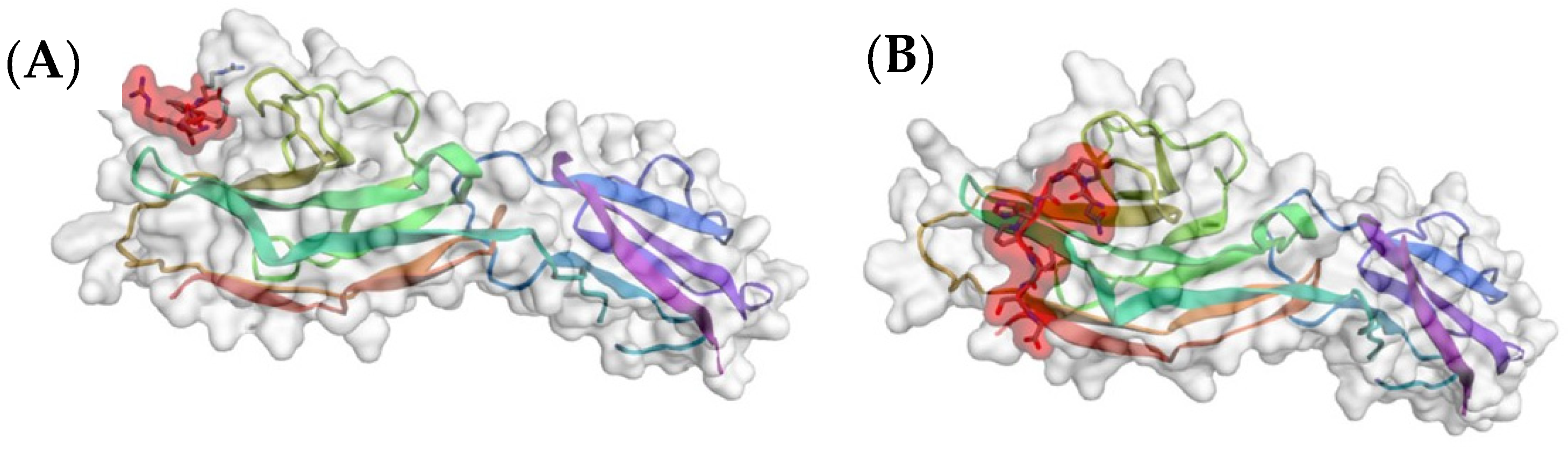
2. Tendamistat—An Inhibitor of α-Amylase
3. Major Families of Receptors on Immune Cells and Their Ligands
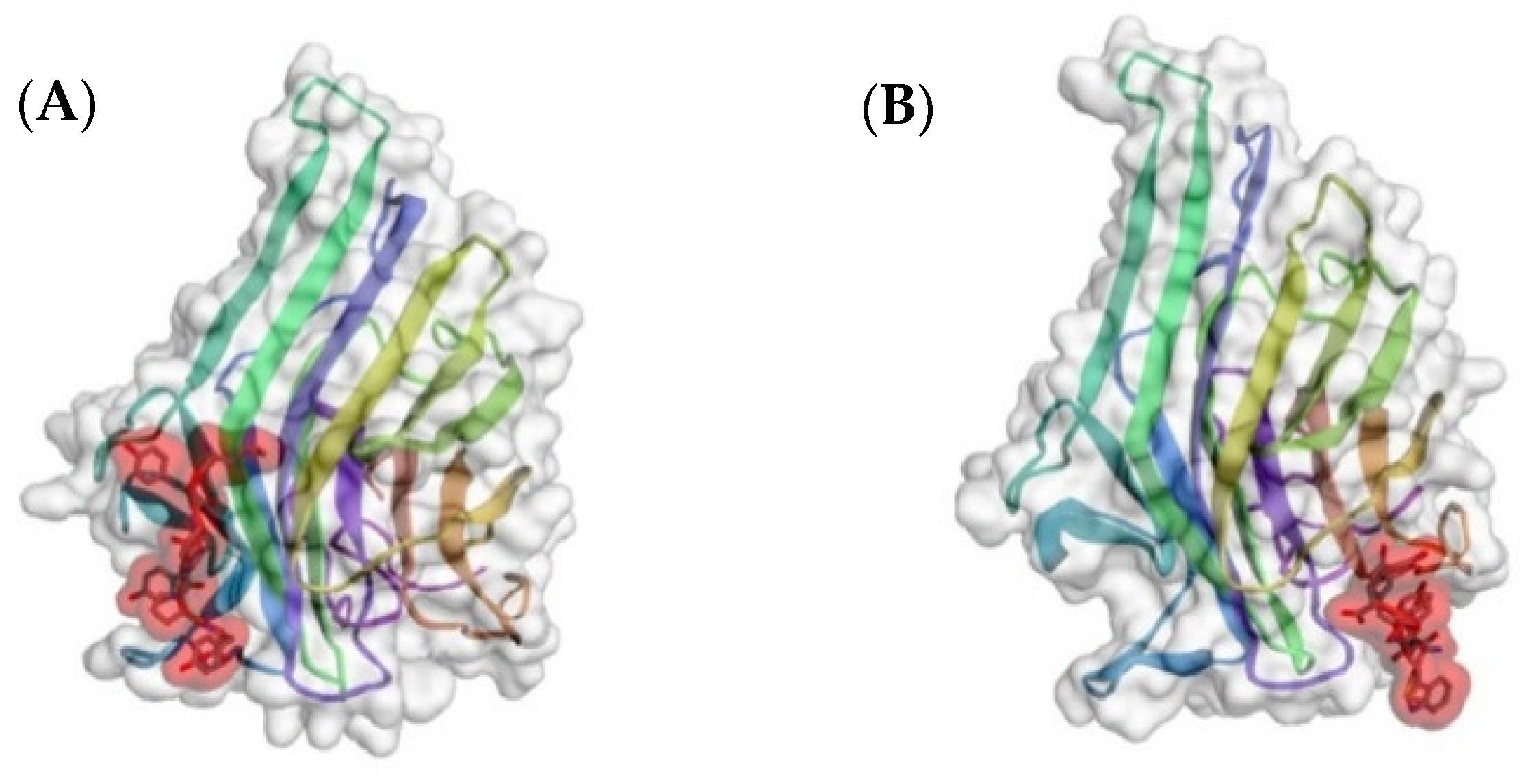
3.1. A Peptide Mimetic of Mannose
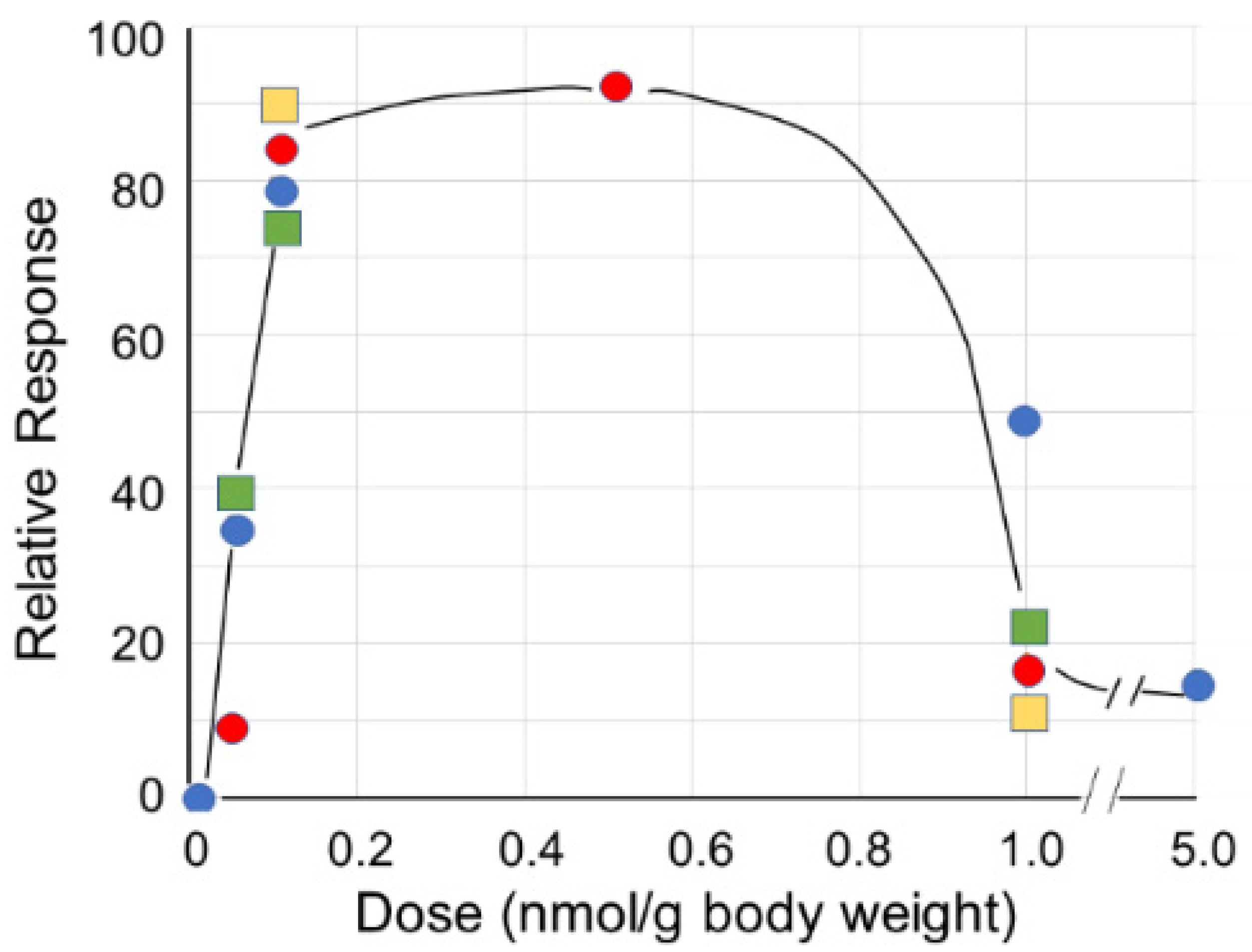
3.2. Peptide Mimetics of Sialic Acid

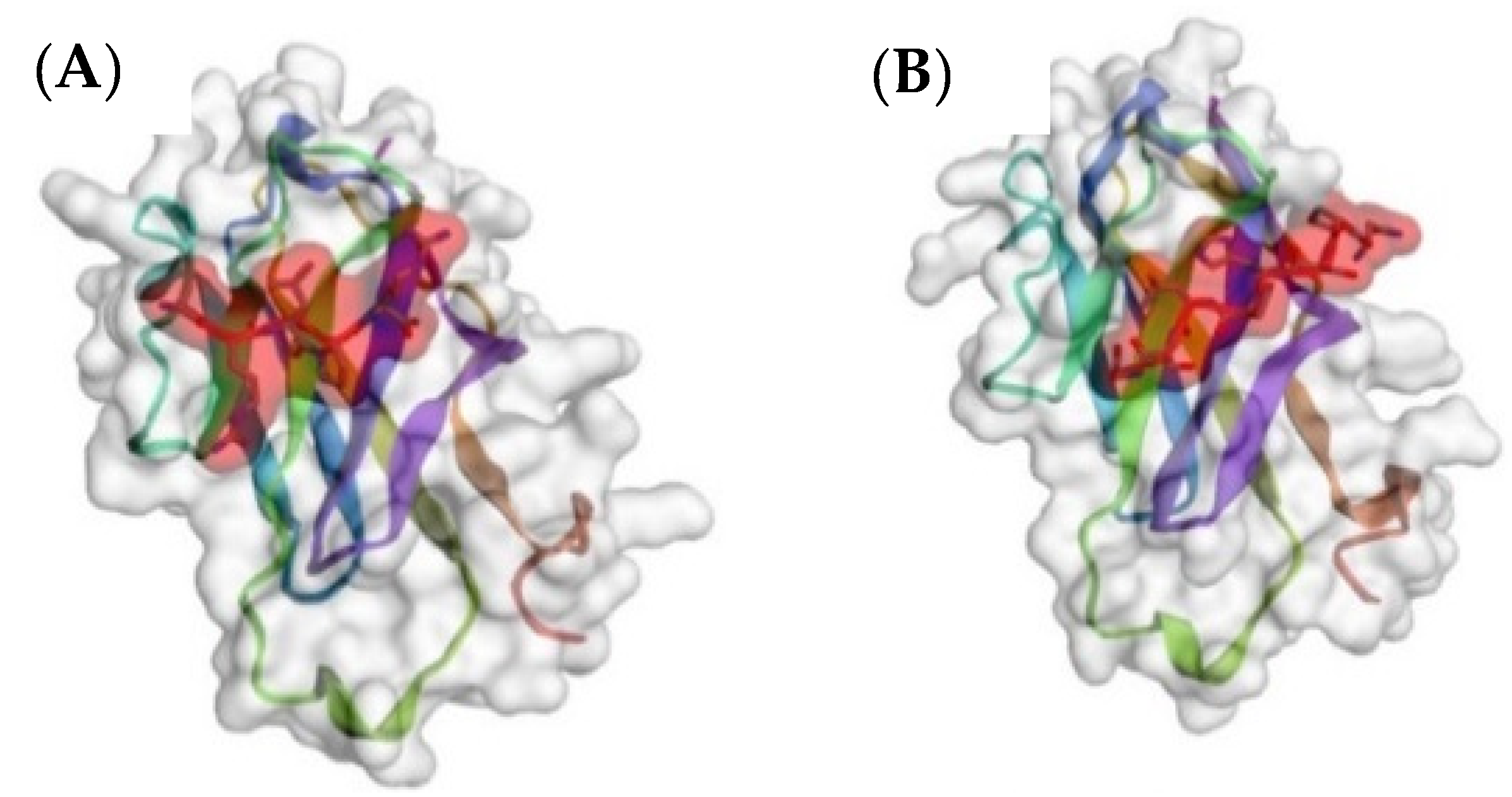
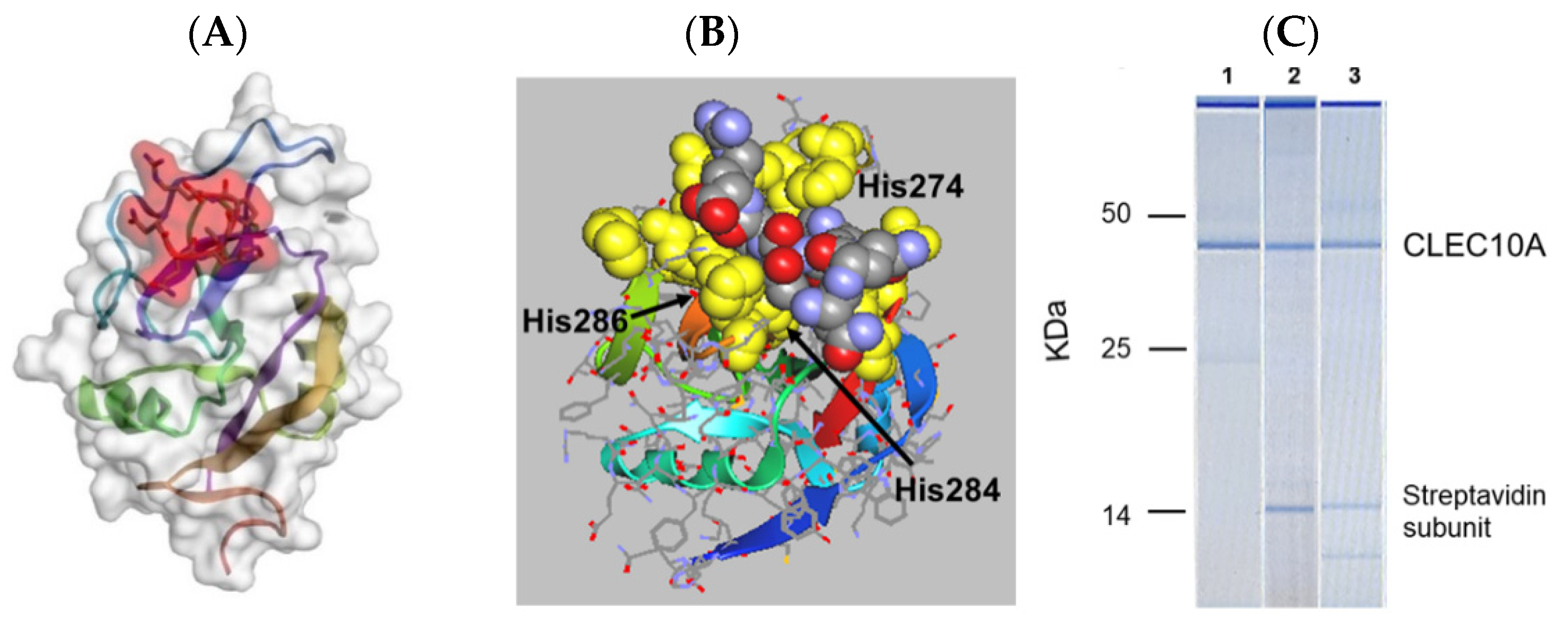
3.3. Peptide Mimetics of N-Acetylgalactosamine
3.4. E-Selectin and HNK-1
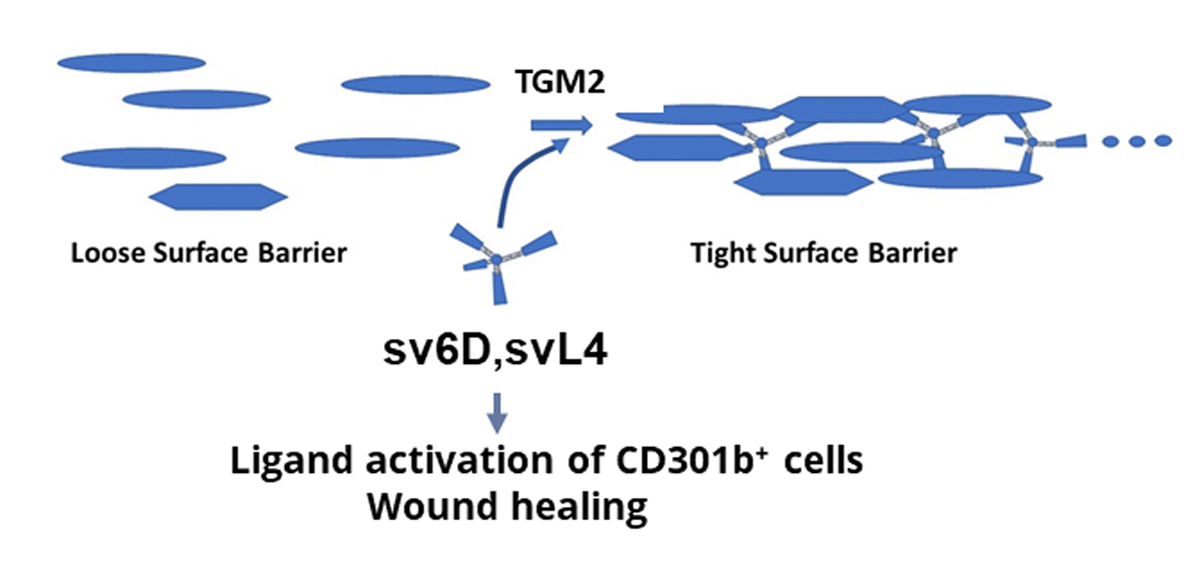
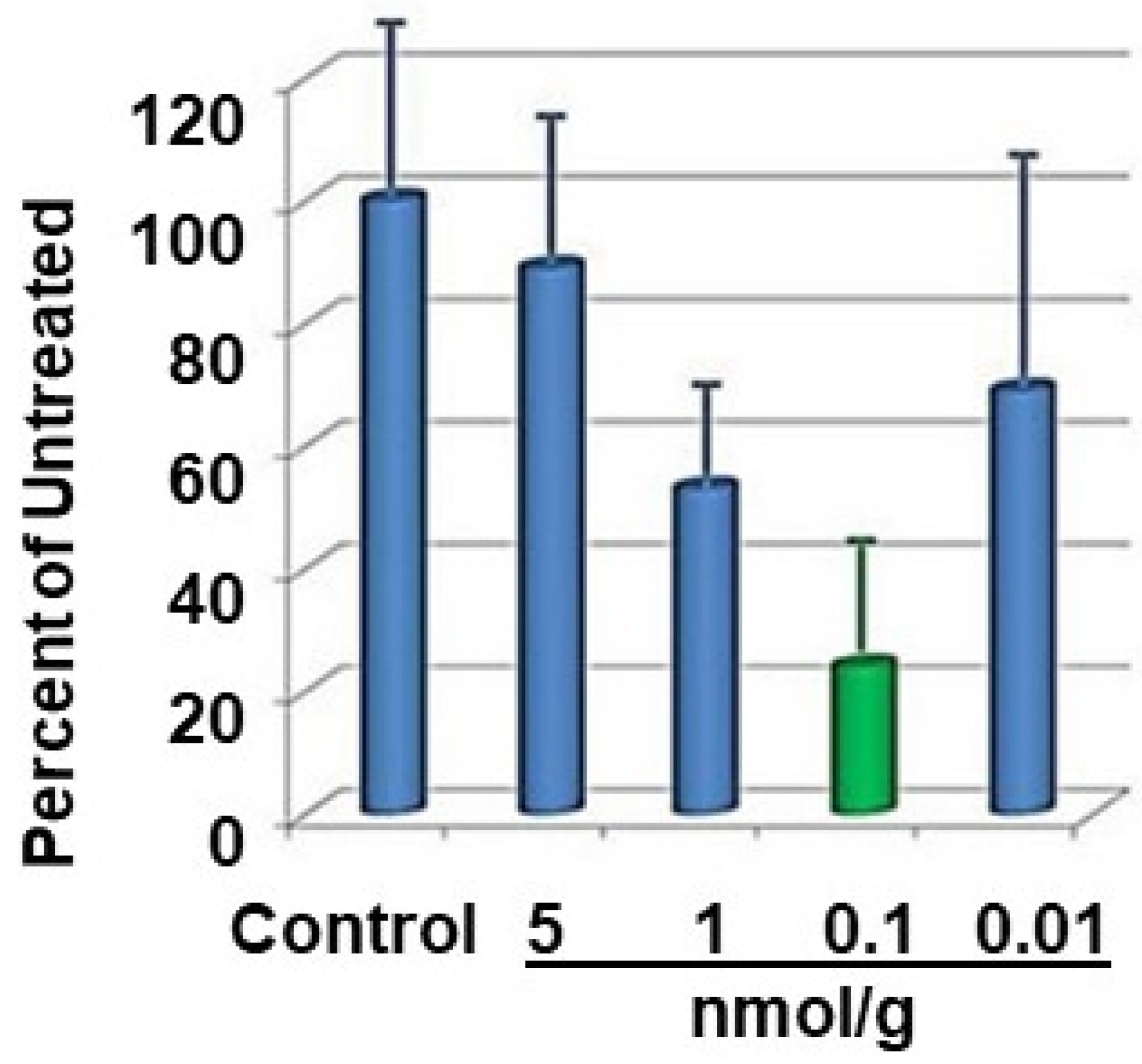
4. Peptides in Treatment of Neutrophilic Skin Diseases
5. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Khorev, O.; Stokmaier, D.; Schwardt, O.; Cutting, B.; Ernst, B. Trivalent, Gal/GalNAc-containing ligands designed for asialoglycoprotein receptor. Bioorg. Med. Chem. 2008, 16, 5216–5231. [Google Scholar] [CrossRef]
- Mūller, C.; Despras, G.; Lindhorst, T.K. Organizing multivalency in carbohydrate recognition. Chem. Soc. Rev. 2016, 45, 3275–3302. [Google Scholar] [CrossRef]
- Mende, M.; Tsouka, A.; Heidepriem, J.; Paris, G.; Mattes, D.S.; Eickelmann, S.; Bordoni, V.; Wawrzinek, R.; Fuchsberger, F.F.; Seeberger, P.H. On-chip neo-glycopeptide synthesis for multivalent glycan presentation. Chem. Eur. J. 2020, 26, 9954–9963. [Google Scholar] [CrossRef]
- Nair, J.K.; Willoughby, J.L.S.; Chan, A.; Charisse, K.; Alam, M.R.; Wang, Q.; Hoekstra, M.; Kandasamy, P.; Kel’In, A.V.; Milstein, S.; et al. Multivalent N-acetylgalactosamine-conjugated siRNA localizes in hepatocytes and elicits robust RNAi-mediated gene silencing. J. Am. Chem. Soc. 2014, 136, 16958–16961. [Google Scholar] [CrossRef]
- Prakash, T.P.; Yu, J.; Migawa, M.T.; Kinberger, G.A.; Wan, W.B.; Østergaard, M.E.; Vasquez, G.; Low, A.; Chappell, A.; Schmidt, K.; et al. Comprehensive structure-activity relationship of triantennary N-acetylgalactosamine conjugated antisense oligonucleotides for targeted delivery to hepatocytes. J. Med. Chem. 2016, 59, 2718–2733. [Google Scholar] [CrossRef] [PubMed]
- Duinkerken, S.; Horrevorts, S.K.; Kalay, H.; Ambrosini, M.; Rutte, L.; de Gruijl, T.D.; Garcia-Vallejo, J.J.; van Kooyk, Y. Glyco-dendrimers as intradermal anti-tumor vaccine targeting multiple skin DC subsets. Theranostics 2019, 9, 5797–5809. [Google Scholar] [CrossRef] [PubMed]
- Gao, T.; Yan, J.; Liu, C.-C.; Palma, A.S.; Guo, Z.; Xiao, M.; Chen, X.; Liang, X.; Chai, W.; Cao, H. Chemoenzymatic synthesis of O-mannose glycans containing sulfated or nonsulfated HNK-1 epitope. J. Am. Chem. Soc. 2019, 141, 19351–19359. [Google Scholar] [CrossRef] [PubMed]
- Ernst, B.; Magnani, J.L. From carbohydrate leads to glycomimetic drugs. Nat. Rev. Drug Discov. 2009, 8, 661–677. [Google Scholar] [CrossRef]
- Hevey, R. Strategies for the development of glycomimetic drug candidates. Pharmaceuticsls 2019, 12, 55. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.A.; Pinto, B.M. Structural and functional studies of peptide-carbohydrate mimicry. Top. Curr. Chem. 2008, 273, 55–116. [Google Scholar] [PubMed]
- Agostino, M.; Sandrin, M.; Thompson, P.; Farrugia, W.; Ramsland, P.; Yuriev, E. Carbohydrate-mimetic peptides: Structural aspects of mimicry and therapeutic implications. Exp. Op. Biol. Ther. 2011, 11, 211–224. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, T. Potential of peptides as inhibitors and mimotopes: Selection of carbohydrate-mimetic peptides from phage display libraries. J. Nuleic Acids 2012, 2012, 740982. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Yu, P.S.; Mui, E.Y.Y.; McKelvie, J.C.; Pham, T.P.T.; Yap, Y.W.; Wong, W.Q.; Wu, J.; Deng, W.; Orner, B.P. Phage display screening against a set of targets to establish peptide-based sugar mimetics and molecular docking to predict binding site. Bioorg. Med. Chem. 2009, 17, 4825–4832. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.K.; Smith, G.P. Searching for peptide ligands with an epitope library. Science 1990, 249, 386–390. [Google Scholar] [CrossRef]
- Devlin, J.J.; Panganiban, L.C.; Devlin, P.E. Random peptide libraries: A souce of specific protein binding molecules. Science 1990, 249, 404–406. [Google Scholar] [CrossRef]
- Bächle, D.; Loers, G.; Guthöhrlein, E.W.; Schachner, M.; Sewald, N. Glycomimetic cyclic peptides stimulate neurite outgrowth. Angew. Chem. Int. Ed. 2006, 45, 6582–6585. [Google Scholar] [CrossRef]
- Bhunia, A.; Vivekanandan, S.; Eckert, T.; Bung-Roderfeld, M.; Wechselberger, R.; Romanuka, J.; Bächle, D.; Kornilov, A.V.; von der Lieth, C.-W.; Jiménez-Barbero, J.; et al. Why structurally different cyclic peptides can be glycomimetics of the HNK-1 carbohydrate antigen. J. Am. Chem. Soc. 2010, 132, 96–105. [Google Scholar] [CrossRef]
- Pashov, A.D.; Plaxco, J.; Kaveri, S.Y.; Monzavi-Karbassi, B.; Harn, D.; Kieber-Emmons, T. Multiple antigenic mimotopes of HIV carbohydrate antigens. Relating structure and antigenicity. J. Biol. Chem. 2006, 281, 29675–29683. [Google Scholar] [CrossRef]
- Kieber-Emmons, T.; Saha, S.; Pashov, A.; Monzavi-Karbassi, B.; Murali, R. Carbohydrate-mimetic peptides for pan anti-tumor responses. Front. Immunol. 2014, 5, 308. [Google Scholar] [CrossRef]
- Muttenthaler, M.; King, G.F.; Adams, D.J.; Alewood, P.F. Trends in peptide drug discovery. Nat. Rev. Drug Discov. 2021, 20, 309–325. [Google Scholar] [CrossRef]
- Furukawa, N.; Popel, A.S. Peptides that immunoactivate the tumor environment. Biochim. Biophys. Acta-Rev. Cancer 2021, 1875, 188486. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, T. Peptide mimotopes to emulate carbohydrates. Chem. Soc. Rev. 2022, 51, 8160–8173. [Google Scholar] [CrossRef] [PubMed]
- Vértesy, L.; Oeding, V.; Bender, R.; Zepf, K.; Nesemann, G. Tendamistat (HOE 467), a tight-binding α-amylase inhibitor from Streptomyces tendae 4158. Isolation, biochemical properties. Eur. J. Biochem. 1984, 141, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Wiegand, G.; Epp, O.; Huber, R. The crystal structure of porcine pancreatic α-amylase in complex with the microbial inhibitor tendamistat. J. Mol. Biol. 1995, 247, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Blaszczyk, M.; Kurcinski, M.; Kouza, M.; Wieteska, L.; Debinski, A.; Kolinski, A.; Kmiecik, S. Modeling of protein-peptide interactions using the CABS-dock web server for binding site search and flexible docking. Methods 2016, 93, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Kurcinski, M.; Badaczewska-Dawid, A.; Kolinski, M.; Kolinski, A.; Kmiecik, S. Flexible docking of peptides to proteins using CABS-dock. Prot. Sci. 2020, 29, 211–222. [Google Scholar] [CrossRef]
- König, V.; Vértesy, L.; Schneider, T.R. Structure of the alpha-amylase inhibitor tendamistat at 0.93 A. Acta Crystallogr. D Biol. Crystallogr. 2003, 59, 1737–1743. [Google Scholar] [CrossRef]
- Chang, C.A.; Chen, W.; Gilson, M.K. Ligand configurational entropy and protein binding. Proc. Nat. Acad. Sci. USA 2007, 104, 1534–1539. [Google Scholar] [CrossRef]
- Zhou, H.X.; Gilson, M.K. Theory of free energy and entropy in noncovalent binding. Chem. Rev. 2008, 109, 4092–4107. [Google Scholar] [CrossRef]
- Gimeno, A.; Delgado, S.; Valverde, P.; Bertuzzi, S.; Berbis, M.A.; Echavarren, J.; Lacetera, A.; Martín-Santamaría, S.; Surolia, A.; Cañada, F.J.; et al. Minimizing the entropy penalty for ligand binding: Lessons from the molecular recognition of the histo blood-group antigens by human Galectin-3. Angew. Chem. Int. Ed. 2019, 58, 7268–7272. [Google Scholar] [CrossRef]
- Changeux, J.-P. Allostery and the Monod-Wyman-Changeux model after 50 years. Annu. Rev. Biophys. 2012, 41, 103–133. [Google Scholar] [CrossRef] [PubMed]
- Mammen, M.; Choi, S.-K.; Whitesides, G.M. Polyvalent interactions in biological systems: Implications for design and use of multivalent ligands and inhibitors. Angew. Chem. Internat. Ed. 1998, 37, 2754–2794. [Google Scholar] [CrossRef]
- Dimick, S.M.; Powell, S.C.; McMahon, S.A.; Moothoo, D.N.; Naismith, J.H.; Toone, E.J. On the meaning of affinity: Cluster glycoside effects and concanavalin A. J. Am. Chem. Soc. 1999, 121, 10286–10296. [Google Scholar] [CrossRef]
- Cairo, C.W.; Gestwicki, J.E.; Kanai, M.; Kiessling, L.L. Control of multivalent interactions by binding epitope density. J. Am. Chem. Soc. 2002, 124, 1615–1619. [Google Scholar] [CrossRef]
- Dam, T.K.; Gerken, T.A.; Brewer, C.F. Thermodynamics of multivalent carbohydrate-lectin cross-linking interactions: Importance of entropy in the bind and jump mechanism. Biochemistry 2009, 48, 3822–3827. [Google Scholar] [CrossRef]
- Eggink, L.L.; Salas, M.; Hanson, C.V.; Hoober, J.K. Peptide sugar mimetics prevent HIV-1 replication in peripheral blood mononuclear cells in the presence of HIV-positive antiserum. AIDS Res. Hum. Retrovir. 2010, 26, 149–160. [Google Scholar] [CrossRef]
- Eggink, L.L.; Hoober, J.K. Peptide mimetics of terminal sugars of complex glycans. Glycobiol. Insights 2010, 2, 63–74. [Google Scholar]
- Cote, R.; Eggink, L.L.; Hoober, J.K. CLEC receptors, endocytosis, and calcium signaling. AIMS Aller. Immunol. 2017, 1, 207–231. [Google Scholar] [CrossRef]
- Oldenburg, K.R.; Loganathan, D.; Goldstein, I.W.; Schultz, P.G.; Gallop, M.A. Peptide ligands for a sugar-binding protein isolated from a random peptide library. Proc. Natl. Acad. Sci. USA 1992, 89, 5393–5397. [Google Scholar] [CrossRef]
- Scott, J.K.; Loganathan, D.; Easley, R.B.; Gong, X.; Goldstein, I.J. A family of concanavalin A-binding peptides from a hexapeptide epitope library. Proc. Natl. Acad. Sci. USA 1992, 89, 5398–5402. [Google Scholar] [CrossRef]
- Kaur, K.J.; Khurana, S.; Salunke, D.M. Topological analysis of the functional mimicry between a peptide and a carbohydrate moiety. J. Biol. Chem. 1997, 272, 5539–5543. [Google Scholar] [CrossRef] [PubMed]
- Kaur, K.J.; Jain, D.; Goel, M.; Salunke, D.M. Immunological implications of structural mimicry between a dodecapeptide and a carbohydrate moiety. Vaccine 2001, 19, 3124–3130. [Google Scholar] [CrossRef] [PubMed]
- Jain, D.; Kaur, K.; Sundaravandivel, B.; Salunke, D.M. Structural and functional consequences of peptide-carbohydrate mimicry. J. Biol. Chem. 2000, 275, 16098–16102. [Google Scholar] [CrossRef] [PubMed]
- Jain, D.; Kaur, K.J.; Salunke, D. Plasticity in protein-peptide recognition: Crystal structures of two different peptides bound to concanavalin A. Biochem. J. 2001, 80, 2912–2921. [Google Scholar] [CrossRef] [PubMed]
- Jain, D.; Kaur, K.J.; Salunke, D.M. Enhanced binding of a rationally designed peptide ligand of concanavalin A arises from improved geometrical complementarity. Biochemistry 2001, 40, 12059–12066. [Google Scholar] [CrossRef]
- de Isla, N.G.; Riquelme, B.D.; Rasia, R.J.; Valverde, J.R.; Stoltz, J.F. Quantification of glycophorin A and glycophorin B on normal human RBCs by flow cytometry. Transfusion 2003, 43, 1145–1152. [Google Scholar] [CrossRef]
- Morell, A.G.; Gregoriadis, G.; Scheinberg, I.H.; Hickman, J.; Ashwell, G. The role of sialic acid in determining the survival of glycoproteins in the circulation. J. Biol. Chem. 1971, 246, 1461–1467. [Google Scholar] [CrossRef]
- Hoober, J.K. ASGR1 and its enigmatic relative CLEC10A. Int. J. Mol. Sci. 2020, 21, 4818. [Google Scholar] [CrossRef]
- Macauley, M.S.; Crocker, P.R.; Paulson, J.C. Siglec-mediated regulation of immune cell function in disease. Nat. Rev. Immunol. 2014, 14, 653–666. [Google Scholar] [CrossRef]
- Duan, S.; Paulson, J.C. Siglecs as immune cell checkpoints in disease. Annu. Rev. Immunol. 2020, 38, 365–395. [Google Scholar] [CrossRef]
- Gonzalez-Gil, A.; Schnaar, R.L. Siglec ligands. Cells 2021, 10, 1260. [Google Scholar] [CrossRef] [PubMed]
- Rabinovich, G.A.; Toscano, M.A. Turning ‘sweet’ on immunity: Galectin-glycan interactions in immune tolerance and inflammation. Nat. Rev. Immunol. 2009, 9, 338–352. [Google Scholar] [CrossRef] [PubMed]
- Di Lella, S.; Sundblad, V.; Cerliani, J.P.; Guardia, C.M.; Estrin, D.A.; Vasta, G.R.; Rabinovich, G.A. When galectins recognize glycans: From biochemistry to physiology and back again. Biochemistry 2011, 50, 7842–7857. [Google Scholar] [CrossRef] [PubMed]
- Johannes, L.; Jacob, R.; Leffler, H. Galecins at a glance. J. Cell Sci. 2018, 131, jcs208884. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.E.; Drickamer, K. Mammalian sugar-binding receptors: Known functions and unexplored roles. FEBS J. 2019, 286, 1800–1814. [Google Scholar] [CrossRef]
- Cummings, R.D.; Chiffoleau, E.; van Kooyk, Y.; McEver, R.P. C-Type lectins. In Essentials of Glycobiology, 4th ed.; Varki, A., Cummings, R.D., Esko, J.K., Stanley, P., Hart, G.W., Aebi, M., Darvill, A.G., Kinoshita, T., Packer, N.H., Prestegard, J.H., et al., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2022. [Google Scholar] [CrossRef]
- Zheng, J.; Xiao, H.; Wu, R. Specific identification of glycoproteins bearing the Tn antigen in human cells. Angew. Chem. Int. Ed. 2017, 56, 7107–7111. [Google Scholar] [CrossRef]
- Pirro, M.; Schoof, E.; van Vliet, S.J.; Rombouts, Y.; Stella, A.; de Ru, A.; Mohammed, Y.; Wuhrer, M.; Van Veelen, P.A.; Hensbergen, P.J. Glycoproteomic analysis of MGL-binding proteins on acute T-cell leukemia cells. J. Proteome Res. 2019, 18, 1125–1132. [Google Scholar] [CrossRef]
- Seki, I.; Suzuki, M.; Miyasaka, N.; Kohsaka, H. Expression of CD45 isoforms correlates with differential proliferative responses of peripheral CD4+ and CD8+ T cells. Immunol. Lett. 2010, 129, 39–46. [Google Scholar] [CrossRef]
- Xu, Z.; Weiss, A. Negative regulation of CD45 by differential homodimerization of the alternatively spliced isoforms. Nat. Immunol. 2002, 3, 764–771. [Google Scholar] [CrossRef]
- Kumar, V.; Cheng, P.; Condamine, T.; Mony, S.; Languino, L.R.; McCaffrey, J.C.; Hockstein, N.; Guarino, M.; Masters, G.; Penman, E.; et al. CD45 phosphatase inhibits STAT3 transcription factor activity in myeloid cells and promotes tumor-associated macrophage differentiation. Immunity 2016, 44, 303–315. [Google Scholar] [CrossRef]
- Posnett, D.N.; McGrath, H.; Tam, J.P. A novel method for producing anti-peptide antibodies. J. Biol. Chem. 1988, 263, 1719–1725. [Google Scholar] [CrossRef] [PubMed]
- Eggink, L.L.; Spyroulias, G.A.; Jones, N.G.; Hanson, C.V.; Hoober, J.K. A peptide mimetic of 5-acetylneuraminic acid-galactose binds with high avidity to siglecs and NKG2D. PLoS ONE 2015, 10, e0130532. [Google Scholar] [CrossRef] [PubMed]
- Zhuravleva, M.A.; Trandem, K.; Sun, P.D. Structural implications of Siglec-5-mediated sialoglycan recognition. J. Mol. Biol. 2008, 375, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Alphey, M.S.; Attrill, H.; Crocker, P.R.; van Aalten, D.M. High resolution crystal structures of Siglec-7. Insights into ligand specificity in the Siglec family. J. Biol. Chem. 2003, 278, 3372–3377. [Google Scholar] [CrossRef] [PubMed]
- Eggink, L.L.; Hoober, J.K. A biologically active peptide mimetic of N-acetylgalactosamine/galactose. BMC Res. Notes 2009, 2, 23. [Google Scholar] [CrossRef]
- Eggink, L.L.; Roby, K.F.; Cote, R.; Hoober, J.K. An innovative immunotherapeutic strategy for ovarian cancer: CLEC10A and glycomimetic peptides. J. ImmunoTher. Cancer 2018, 6, 28. [Google Scholar] [CrossRef]
- Gabba, A.; Bogucka, A.; Luz, J.G.; Diniz, A.; Coelho, H.; Corzana, F.; Cañada, F.J.; Marcelo, F.; Murphy, P.V.; Birrane, G. Crystal structure of the carbohydrate recognition domain of the human macrophage galactose C-type lectin bound to GalNAc and the tumor-associated antigen. Biochemistry 2021, 60, 1327–1336. [Google Scholar] [CrossRef]
- Biasini, M.; Bienert, S.; Waterhouse, A.; Arnold, K.; Studer, G.; Schmidt, T.; Kiefer, F.; Cassarino, T.G.; Bertoni, M.; Bordoli, L.; et al. SWISS-MODEL: Modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014, 42, W252–W258. [Google Scholar] [CrossRef]
- Napoletano, C.; Rughetti, A.; Tarp, M.P.A.; Coleman, J.; Bennett, E.P.; Picco, G. Tumor-associated Tn-MUC1 glycform is internalized through the macrophage galactose-type C-type lectin and delivered to the HLA I and II compartments in dendritic cells. Cancer Res. 2007, 67, 8358–8367. [Google Scholar] [CrossRef]
- Van Vliet, S.J.; Aarnoudse, C.A.; Broks-van den Berg, V.C.M.; Boks, M.; Geijtenbeek, T.B.H.; van Kooyk, Y. MGL-mediated internalization and antigen presentation by dendritic cellsf: A role for tyrosine 5. Eur. J. Immunol. 2007, 37, 2075–2081. [Google Scholar] [CrossRef]
- Heger, L.; Balk, S.; Lühr, J.J.; Heidkamp, G.F.; Lehmann, C.H.K.; Hatscher, L.; Purbojo, A.; Hartmann, A.; Garcia-Martin, F.; Nishimura, S.-I.; et al. CLEC10A is a specific marker for human CD1c+ dendritic cells and enhances their toll-like receptor 7/8-induced cytokine secretion. Front. Immunol. 2018, 9, 744. [Google Scholar] [CrossRef] [PubMed]
- Nunes, P.; Demaurex, N. The role of calcium signaling in phagocytosis. J. Leukoc. Biol. 2010, 88, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Ilarregui, J.M.; Kooij, G.; Rodriguez, E.; van der Pol, S.M.A.; Koning, N.; Kalay, H.; van der Horst, J.C.; van Vliet, S.J.; García-Vallejo, J.J.; de Vries, H.E.; et al. Macrophage galactose-type lectin (MGL) is induced on M2 microglia and participates in the resolution phase of autoimmune neuroinflammation. J. Neuroinflamm. 2019, 16, 130. [Google Scholar] [CrossRef] [PubMed]
- Valverde, P.; Martinez, J.D.; Cañada, F.J.; Ardá, A.; Jiménez-Barbero, J. Molecular recognition in C-type lectins: The cases of DC-SIGN, MGL, and L-selectin. ChemBioChem. 2020, 21, 2999–3025. [Google Scholar] [CrossRef]
- Zaal, A.; Li, R.J.E.; Lūbbers, J.; Bruijns, S.C.M.; Kalau, H.; van Kooyk, Y.; van Vliet, S.J. Activation of the C-type lectin MGL by terminal GalNAc ligands reduces the glycolytic activity of human dendritic cells. Front. Immunol. 2020, 11, 305. [Google Scholar] [CrossRef]
- Kushchayev, S.V.; Sankar, T.; Eggink, L.L.; Kushchayeva, Y.S.; Wiener, P.C.; Hoober, J.K.; Eschbacher, J.; Liu, R.; Shi, F.-D.; Abdelwahab, M.G.; et al. Monocyte galactose/N-acetylgalactosamine-specific C-type lectin receptor stimulant immunotherapy of an experimental glioma. Part I: Stimulatory effects on blood monocytes and monocyte-derived cells of the brain. Cancer Manag. Res. 2012, 4, 309–323. [Google Scholar]
- Kushchayev, S.V.; Sankar, T.; Eggink, L.L.; Kushchayeva, Y.S.; Wiener, P.C.; Hoober, J.K.; Eschbacher, J.; Liu, R.; Shi, F.-D.; Abdelwahab, M.G.; et al. Monocyte galactose/N-acetylgalactosamine-specific C-type lectin receptor stimulant immunotherapy of an experimental glioma. Part II: Combination with external radiation improves survival. Cancer Manag. Res. 2012, 4, 325–334. [Google Scholar]
- Dusoswa, A.; Verhoeff, J.; Abels, E.; Méndez-Huergo, S.P.; Croci, D.O.; Kuijper, L.H.; de Miguel, E.; Wouters, V.M.C.J.; Best, M.G.; Rodriguez, E.; et al. Glioblastomas exploit truncated O-linked glycans for local and distant immune modulation via the macrophage galactose-type lectin. Proc. Natl. Acad. Sci. USA 2020, 117, 3693–3703. [Google Scholar] [CrossRef]
- Tang-Huau, T.L.; Gueguen, P.; Goudot, C.; Durand, M.; Bohec, M.; Baulande, S. Human in vivo-generated monocyte-derived dendritic cells and macrophages cross-present antigens through a vacuolar pathway. Nat. Commun. 2018, 9, 2570. [Google Scholar] [CrossRef]
- Fukuda, M.N.; Ohyama, C.; Lowitz, K.; Matsuo, O.; Pasqualini, R.; Ruoslahti, E.; Fukuda, M. A peptide mimic of E-selectin ligand inhibits sialyl Lewis X-dependent lung colonization of tumor cells. Cancer Res. 2000, 60, 450–456. [Google Scholar]
- Jiménez, V.A.; Navarrete, K.R.; Duque-Noreña, M.; Marrugo, K.P.; Contreras, M.A.; Campos, C.H.; Alderete, J.B. Rational design of novel glycomimetic peptides for E-selectin targeting. J. Chem. Inf. Model. 2021, 61, 2463–2474. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, Y.; Yumoto, K.; Kawakami, T. An improved mouse model of atopic dermatitis and suppression of skin lesions by an inhibitor of Tec family kinases. Allergol. Internatl. 2007, 56, 403–409. [Google Scholar] [CrossRef] [PubMed]
- Johannessen, B.R.; Skov, L.K.; Kastrup, J.S.; Kristensen, O.; Bolwig, C.; Larsen, J.N.; Spangfort, M.; Lund, K.; Gajhede, M. Structure of the house dust mite allergen Der f 2: Implications for function and molecular basis of IgE cross-reactivity. FEBS Lett. 2005, 579, 1208–1212. [Google Scholar] [CrossRef] [PubMed]
- Kanemaru, K.; Noguchi, E.; Tahara-Hanaoka, S.; Mizuno, S.; Tateno, H.; Denda-Nagai, K.; Fujisawa, Y.; Nakamura, Y.; Irimura, T.; Matsuda, H.; et al. Clec10a regulates mite-induced dermatitis. Sci. Immunol. 2019, 4, eaax6908. [Google Scholar] [CrossRef] [PubMed]
- Dupasquier, M.; Stoitzner, P.; van Oudenaren, A.; Romani, N.; Leenen, P.J.M. Macrophages and dendritic cells constitute a major subpopulation of cells in the mouse dermis. J. Investig. Dermatol. 2004, 123, 876–879. [Google Scholar] [CrossRef] [PubMed]
- Dupasquier, M.; Stoitzner, P.; Wan, H.; Cerqueira, D.; van Oudenaren, A.; Voerman, J.S.A.; Denda-Nagai, K.; Irimura, T.; Raes, G.; Romani, N.; et al. The dermal microenvironment induces the expression of the alternate activation marker CD301/mMGL in mononuclear phagocytes, independent of IL-4/IL-13 signaling. J. Leukoc. Biol. 2006, 8, 838–849. [Google Scholar] [CrossRef]
- Palma, A.; Jarrah, A.S.; Tieri, P.; Cesareni, G.; Castiglione, F. Gene regulatory network modeling of macrophage differentiation corroborates the continuum hypothesis of polarization states. Front. Physiol. 2018, 9, 1659. [Google Scholar] [CrossRef]
- Davies, L.C.; Jenkins, S.J.; Allen, J.E.; Taylor, P.R. Tissue-resident macrophages. Nat. Immunol. 2013, 14, 986–995. [Google Scholar] [CrossRef]
- Greenlee-Wacker, M.C. Clearance of apoptotic neutrophils and resolution of inflammation. Immunol. Rev. 2016, 273, 357–370. [Google Scholar] [CrossRef]
- Zhong, X.; Lee, H.-N.; Kim, S.H.; Park, S.-A.; Kim, W.; Cha, Y.-N.; Surh, Y.-J. Myc-nick promotes efferocytosis through M2 macrophage polarization during resolution of inflammation. FEBS J. 2018, 32, 5312–5325. [Google Scholar] [CrossRef]
- Filep, J.G. Targeting neutrophils for promoting the resolution of inflammation. Front. Immunol. 2022, 13, 866747. [Google Scholar] [CrossRef]
- Eggink, L.L.; Hoober, J.K. Resolution of eczema with multivalent peptides. J. Investig. Dermatol. Innovat. 2022, 2, 100142. [Google Scholar] [CrossRef]
- Shook, B.; Xiao, E.; Kumamoto, Y.; Iwasaki, A.; Horsley, V. CD301b+ macrophages are essential for effective skin wound healing. J. Investig. Dermatol. 2016, 136, 1885–1891. [Google Scholar] [CrossRef] [PubMed]
- Shook, B.A.; Wasko, R.R.; Rivera-Gonzalez, G.C.; Salazar-Gatzimas, E.; López-Giráldez, F.; Dash, B.C.; Muñoz-Rojas, A.R.; Aultman, K.D.; Zwick, R.K.; Lei, V.; et al. Myofibroblast proliferation and heterogeneity are supported by macrophages during skin repair. Science 2018, 362, 909. [Google Scholar] [CrossRef] [PubMed]
- Saraiva, M.; Vieira, P.; O’Garra, A. Biology and therapeutic potential of interleukin-10. J. Exp. Med. 2019, 217, 0418. [Google Scholar] [CrossRef] [PubMed]
- Adib, Y.; Bensussan, A.; Michel, L. Cutaneous wound healing: A review about innate immune response and current therapeutic applications. Med. Inflamm. 2022, 2022, 5344085. [Google Scholar] [CrossRef]
- Conaway, E.A.; de Oliveira, D.C.; McInnis, C.M.; Snapper, S.B.; Horwitz, B.H. Inhibition of inflammatory gene transcription by IL-10 is associated with rapid suppression of LPS-induced enhancer activation. J. Immunol. 2017, 198, 2906–2915. [Google Scholar] [CrossRef]
- Hutchins, A.P.; Takahashi, Y.; Miranda-Saavedra, D. Genomic analysis of LPS-stimulated myeloid cells identifies a common pro-inflammatory response but divergent IL-10 anti-inflammatory responses. Sci. Rep. 2015, 5, 9100. [Google Scholar] [CrossRef]
- Shemer, A.; Scheyltjens, I.; Frumer, G.R.; Kim, J.S.; Grozovski, J.; Ayanaw, S.; Dassa, B.; Van Hove, H.; Chappell-Maor, L.; Boura-Halfon, S.; et al. Interleukin-10 prevents pathological microglia hyperactivation following peripheral endotoxin challenge. Immunity 2020, 53, 1033–1049. [Google Scholar] [CrossRef]
- Haroon, Z.A.; Hettasch, J.M.; Lai, T.-S.; Dewhirst, M.W.; Greenberg, C.S. Tissue transglutaminase is expressed, active, and directly involved in rat dermal wound healing and angiogenesis. FASEB J. 1999, 13, 1787–1795. [Google Scholar] [CrossRef]
- Griffin, M.; Casadio, R.; Bergamini, C.M. Transglutaminases: Natures biological glues. Biochem. J. 2002, 368, 377–396. [Google Scholar] [CrossRef] [PubMed]
- Telei, D.; Griffin, M. Tissue transaminase (TG2)—A wound response enzyme. Front. Biosci. 2006, 11, 867–882. [Google Scholar]
- Liedén, A.; Winge, M.C.G.; Sääf, A.; Kockum, I.; Ekelund, E.; Rodriguez, E.; Fölster-Holst, R.; Franke, A.; Illig, T.; Tengvall-Linder, M.; et al. Genetic variation in the epidermal transglutaminase genes is not associated with atopic dermatitis. PLoS ONE 2012, 7, e49694. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Luo, Y.; Sun, J.; Liu, X.; Ling, S.; Xu, B.; Zhang, Y.; Liu, J.; Li, W.; Wang, B.; et al. Transglutaminase 3 promotes skin inflammation in atopic dermatitis by activating monocyte-derived dendritic cells via DC-SIGN. J. Investig. Dermatol. 2020, 140, 170–179. [Google Scholar] [CrossRef]
- Chou, C.-Y.; Streets, A.J.; Watson, P.; Huang, L.; Verderio, E.A.M.; Johnson, T.S. A crucial sequence for transglutaminase type 2 extracellular trafficking in renal tubular epithelial cells lies in its N-terminal β-sandwich domain. J. Biol. Chem. 2011, 286, 27825–27835. [Google Scholar] [CrossRef]
- Pinkas, D.M.; Strop, P.; Brunger, A.T.; Khosla, C. Transglutaminase 2 undergoes a large conformational change upon activation. PLoS Biol. 2007, 5, e327. [Google Scholar] [CrossRef]
- Folk, J.E.; Mullooly, J.P.; Cole, P.W. Mechanism of action of guinea pig liver transglutaminase. II. The role of metal in enzyme activation. J. Biol. Chem. 1967, 242, 1838–1844. [Google Scholar] [CrossRef]
- Bos, J.D.; Meinardi, M.M.H.M. The 500 Dalton rule for the skin penetration of chemical compounds and drugs. Exp. Dermatol. 2000, 9, 165–169. [Google Scholar] [CrossRef]
- Sano, T.; Okada, A.; Kawasaki, K.; Kume, T.; Fukui, M.; Todo, H.; Sugibayashi, K. Self-assembled structure of α-isostearyl glyceryl ether affects skin permeability—A lamellar with 70-nm spaces and L3 phase enhanced the transdermal delivery of a hydrophilic model drug. AAPS PharmSciTech 2022, 23, 296. [Google Scholar] [CrossRef]
- Peña-Juárez, M.C.; Guadarrama-Escobar, O.R.; Escobar-Chávez, J.J. Transdermal delivery systems for biomolecules. J. Pharm. Innovat. 2022, 17, 319–332. [Google Scholar] [CrossRef]
- London, N.; Movshovitz-Attias, D.; Schueler-Furman, O. The structural basis of peptide-protein binding strategies. Structure 2010, 18, 188–199. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hoober, J.K.; Eggink, L.L. Glycomimetic Peptides as Therapeutic Tools. Pharmaceutics 2023, 15, 688. https://doi.org/10.3390/pharmaceutics15020688
Hoober JK, Eggink LL. Glycomimetic Peptides as Therapeutic Tools. Pharmaceutics. 2023; 15(2):688. https://doi.org/10.3390/pharmaceutics15020688
Chicago/Turabian StyleHoober, J. Kenneth, and Laura L. Eggink. 2023. "Glycomimetic Peptides as Therapeutic Tools" Pharmaceutics 15, no. 2: 688. https://doi.org/10.3390/pharmaceutics15020688
APA StyleHoober, J. K., & Eggink, L. L. (2023). Glycomimetic Peptides as Therapeutic Tools. Pharmaceutics, 15(2), 688. https://doi.org/10.3390/pharmaceutics15020688