1. Introduction
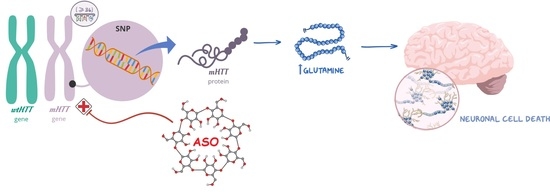
Huntington’s disease (HD) is a progressive, inherited neurodegenerative disorder caused by a CAG repeat expansion in the huntingtin gene, which results in the production of mutant huntingtin (mHTT) protein with an abnormally long polyglutamine (polyQ) expansion (≥36 CAG repeats). This mutant form of the HTT protein acquires a toxic gain-of-function by forming self-aggregates, leading to neuronal dysfunction and, ultimately, cell death [
1]. Given that the pathology is driven by the toxic mHTT protein, therapeutic strategies focused on lowering mHTT levels have been developed through the years. These strategies mainly include antisense oligonucleotides (ASO) and RNA interference (RNAi) based technologies, which target the RNA product in an allele-selective or non-selective way [
2,
3].
Ubiquitous at the tissue and subcellular levels, wild-type HTT (wtHTT) plays an essential role in embryonic neurodevelopment and has numerous functions in the adult brain, including transcriptional regulation, vesicular transport, regulation of endocytosis and vesicular trafficking, cellular stress responses (survival and cell death), and DNA repair [
4]. The long-term consequences of wtHTT reduction in the adult brain remain unclear. For this reason, selective reduction of mHTT while maintaining wtHTT levels may be a safer approach in the longer term than the unselective knockdown of both HTT alleles [
5,
6]. Although not covering all HD patients, approximately two-thirds of HD patients with European ancestry are expected to possess single-nucleotide polymorphism (SNP) rs362307 (SNP1), rs362331 (SNP2), or both SNPs in association with the CAG repeat expansion, making these SNP variants promising therapeutic targets for the majority of HD patients [
7].
Despite initial promising outcomes, three clinical trials using HTT-lowering ASOs were terminated in 2021: the non-allele selective GENERATION HD1 (NCT03761849) trial, and the two distinct allele-selective PRECISION-HD1 (NCT03225833) and PRECISION-HD2 (NCT03225846) trials. The GENERATION HD1 trial aimed to evaluate the efficacy and safety of intrathecal tominersen in adults. Although there were significant decreases in cerebrospinal fluid (CSF) mHTT, patients treated with tominersen fared worse than those in the placebo group in Phase III. Wave Life Sciences’ PRECISION-HD trials aimed to evaluate the efficacy and safety of intrathecal WVE-120201 and WVE-120202, which carried SNPs rs362307 and rs362331, respectively. Preliminary results of both trials did not show significant decreases in mHTT or total HTT at the doses tested [
8]. Given these failures, the exploration of alternative ways to optimize ASO drug delivery has become a research hotspot.
Different strategies have been developed to improve ASO stability, targeting, and delivery, including chemical modifications, bioconjugation to different moieties, and the use of delivery vehicles [
9]. Among the delivery vehicles, cyclodextrins (CDs) are known as favorable natural nanocarriers for oligonucleotide therapy [
10,
11,
12,
13]. Cyclodextrins are non-toxic cyclic oligosaccharides constituted by six (α-CD), seven (β-CD), and eight (γ-CD)
d-glucopyranosyl units. They can be synthesized in relatively high quantities and exhibit a remarkable versatility for chemical modification, allowing for the addition of unique functional groups for cell and tissue-specific targeting [
14]. The conjugation of small peptides is especially interesting in this respect.
In previous studies, we developed new CD nanoplatforms based on PEGylated amphiphilic CDs conjugated with the brain-targeting peptide rabies virus glycoprotein (RVG) covalently via thiol–maleimide click chemistry [
15] and by CD–adamantyl inclusion complex formation [
16]. Regardless of the chemistry approach, both CDs successfully increased the delivery of siRNA into neuronal cells and induced gene knockdown. RVG can specifically interact with neuronal nicotinic acetylcholine receptors (nAChR) or γ-aminobutyric acid (GABA) receptors on the surface of endothelial cells and transport a variety of nanocarriers into the central nervous system (CNS) in a non-invasive way [
17].
In the present work, we designed and characterized three CDs with distinct chemical structures and properties as potential platforms for the delivery of ASO targeting total HTT. These CDs:ASO nanocomplexes were screened for cellular uptake and their potential to induce HTT protein knockdown. The lead formulation was then conjugated to RVG by the post-insertion method in order to improve brain uptake and silencing efficacy. The efficiency of the RVG-targeted CDs:ASO nanocomplexes was evaluated in an in vitro co-culture model of the blood–brain barrier (BBB), consisting of the human cerebral microvascular endothelial cell line (hCMEC/D3) and the rat striatal embryonic neuronal cell line, ST14A, which expresses human mHTT (residues 1-548) with 120 CAG repeats and has been established as a reliable model of HD [
18,
19]. Finally, RVG-targeted CDs:ASO nanocomplexes were validated in a patient-derived cellular model of HD containing clinically relevant CAG expansion-associated SNPs.
2. Materials and Methods
2.1. Oligonucleotide
The human HTT-targeting ASO was purchased from Eurogentec (Seraing, Belgium) and was 20 nucleotides in length with five 2′-O-methoxyethyl (2′-O-MOE) modified nucleosides at the 5′ and 3′ends (upper case letters) and ten unmodified nucleotides in the central part (lower case letters). The internucleoside bonds (marked with an asterisk) were phosphorothioated to improve nuclease resistance. ASO sequence was as follows: C*TCAG*t*a*a*c*a*t*t*g*a*c*ACCA*C*. This sequence was previously described by [
20,
21].
2.2. Cyclodextrins-Based NPs
2.2.1. Cyclodextrins:ASO Nanocomplexes
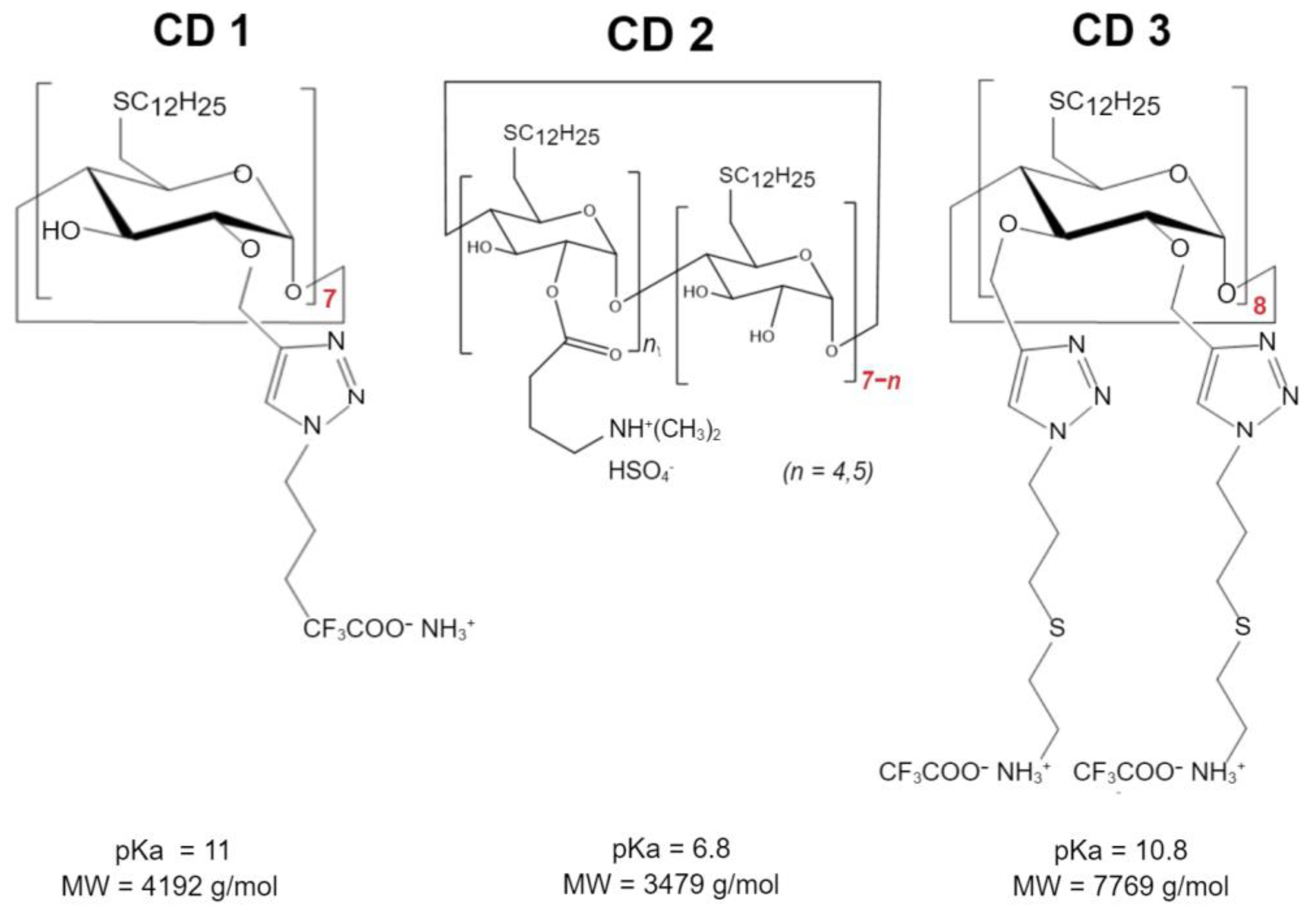
Three different CDs were investigated (
Scheme 1): Cationic amphiphilic β-CDs synthesized as previously described [
22] using the “click chemistry” strategy, herein referred to as CD1; a second cationic amphiphilic β-CDs, previously described [
16] and designed with an amino group to decrease the pKa relative to CD1, herein referred to as CD2; and a γ-CD with a double-charged chain synthesized and kindly provided by Cyclolab, R&D Laboratory (Hungary), herein referred to as CD3. These structures were selected to compare the type of CDs-β-CDs (CD1 and CD2) vs. γ-CDs (CD3), the cationic nature of the headgroup—primary amines (CD1) vs. tertiary amines (CD2), and the presence of a second charged chain (CD3, primary amines).
The CDs were reconstituted in RNAse-free water to obtain a 0.25 mg/mL (CD1 and CD2) or 1 mg/mL (CD3) solution. These solutions were mixed with ASO (500 nM) at different weight ratios (WR) of CDs:ASO (5:1; 7.5:1 and 10:1) and incubated at 65 °C and 450 rpm for 30 min in a thermomixer.
2.2.2. Synthesis of DSPE-PEG-RVG
For surface modification of the CDs:ASO nanocomplexes, DSPE-PEG
2000-Maleimide (DSPE-PEG-Mal) was conjugated to RVG by the Michael reaction. Firstly, the -SH group in RVG peptide (N’-CYTIWMPENPRPGTPCDIFTNSRG KRASNGGGG-C′, TAG Copenhagen, Denmark, Belgium) was activated with 2-Mercaptoethanol (2-ME, M3148, Sigma-Aldrich, Wicklow, Ireland) by mixing RVG (1 Equiv) and 2-ME (15 Equiv) in deionized water (DIW) for 4 h with continuous stirring at room temperature and under nitrogen. The final product was then freeze-dried. The activated RVG peptide (1 Equiv) was dissolved in a solvent mixture of chloroform, methanol, and DIW [CHCl3:MeOH:DIW = 65:35:8 (
v/
v/
v)] and added to the DSPE-PEG-Mal (1.5 Equiv). Using triethylamine as a catalyst, the solution was incubated at 41 °C for 48 h with stirring, avoiding oxygen and light. Post incubation, the solution was dialyzed against 96% ethanol in a dialysis cassette (3.5k MWCO, 66,332, Thermo Fisher Scientific, Bremen, Germany) for 2 days, replacing 96% ethanol every 12 h to remove triethylamine and any unconjugated DSPE-PEG-Mal. The product, named DSPE-PEG-RVG, was freeze-dried and stored at −20 °C until used. The DSPE-PEG-Mal-RVG product was identified by
1H NMR (
Figure S1).
To prepare non-targeted and RVG-targeted formulations, DSPE-PEG/DSPE-PEG-RVG was dissolved in water (1 mg/mL) and sonicated for 30 min in an ultrasonic bath at 60 °C. Subsequently, DSPE-PEG/DSPE-PEG-RVG was added to the CDs:ASO nanocomplexes (WR 10:1, 750 nM) in order to achieve CDs:ASO:DSPE-PEG (0.35 molar ratio of DSPE-PEG to CD) or CDs:ASO:DSPE-PEG-RVG (60 mol% to DSPE-PEG). The non-targeted and target formulations were incubated at 65 °C and 450 rpm for 30 min in a thermomixer.
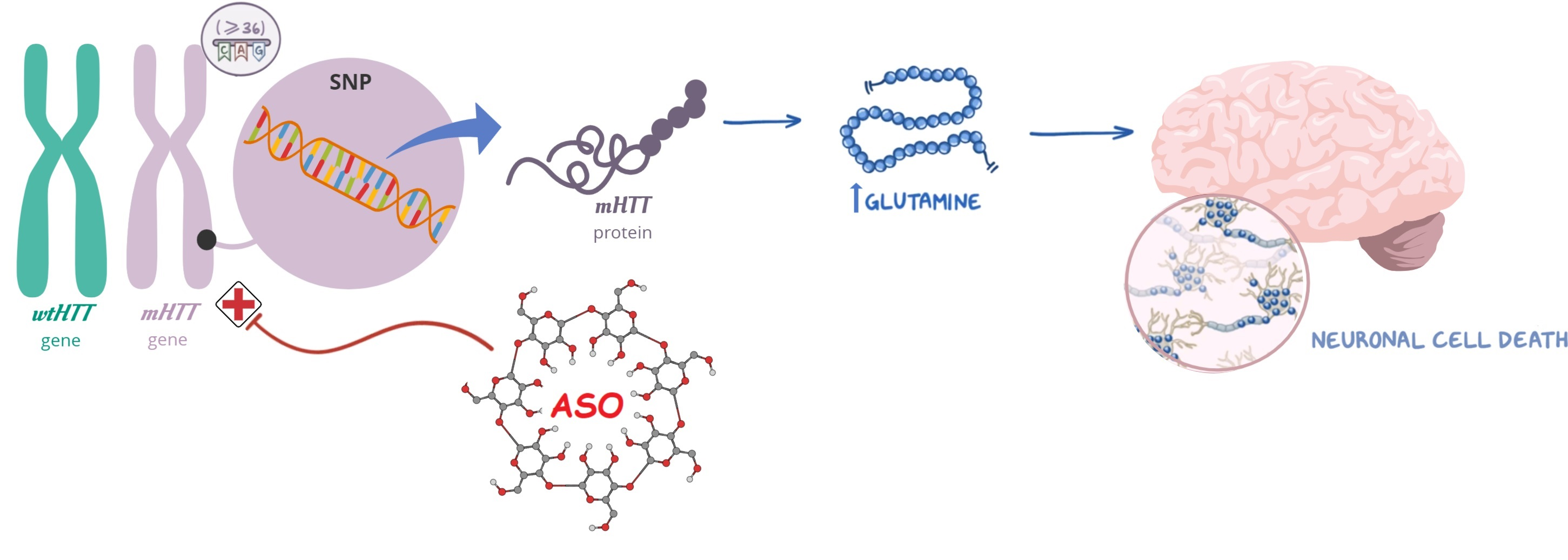
2.3. Physicochemical Profiles
Hydrodynamic size, polydispersity index (PDI), and zeta potential were determined by dynamic light scattering using a Zetasizer NanoZS
® instrument (Malvern Instruments, Worcestershire, UK). The size and morphology were verified by transmission electron microscope (TEM). The samples were deposited to a 400-mesh carbon film copper grid, stained with 2% (
w/
v) phosphotungstic acid, and imaged at a JEOL operated at 200 kV. The binding of ASO to CDs was confirmed by agarose gel electrophoresis, as previously described [
16].
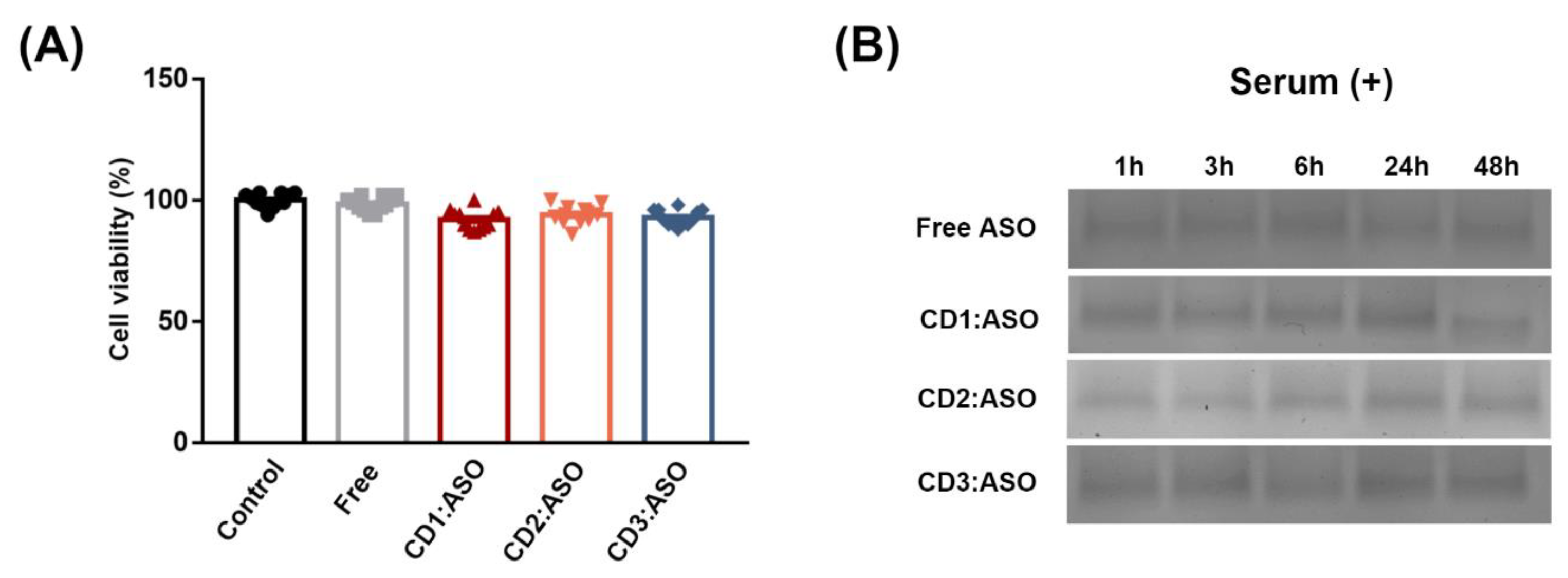
Heparin displacement was conducted to study the release of ASO from CDs. Heparin (10 mg/mL) was added to the complexes (WR of 1:5) for 30 min, 6 h, 24 h, 48 h, and 72 h, and agarose gel electrophoresis was performed at 90 V for 60 min. Finally, the stability profile in a simulated biological environment was evaluated by incubating the CDs:ASO nanocomplexes with an equal volume of 25% heat-inactivated serum over 48 h at 37 °C. At defined time intervals, i.e., 1, 3, 6, 24, and 48 h, aliquots were collected and stored at −20 °C till gel electrophoresis was executed.
2.4. Cells
The immortalized rat striatal neuronal cell line ST14A was purchased from Coriell Institute (Camden, NJ, USA) and cultured in DMEM with 4.5 g/L glucose, L-glutamine, and sodium pyruvate with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. Cells were maintained at 33 °C, with a 5% CO2 atmosphere, and the cell culture medium was changed every 2–3 days. The ST14A cell line expresses human mHTT (residues 1-548) with 120 polyglutamines/CAG repeats (Q120).
The immortalized human cerebral microvascular endothelial cell line (hCMEC/D3) was purchased from Cedarlane Laboratories (Burlington, Ontario, Canada) and cultured on rat collagen I-coated flasks (CELLCOAT®, Greiner Bio-One, Kremsmünster, Austria) and maintained in Endothelial Growth Medium (EBM-2, #CC-3156, Lonza, Basel, Switzerland) supplemented with SingleQuots™ Bullet-Kit (#CC-4176, Lonza, Basel, Switzerland) at 37 °C in 5% CO2. The growth medium was changed every 2–3 days until the cells reached approximately 80% confluency. The cells used were between passages 27 and 35.
Human HD patient-derived fibroblasts GM04723 (19-year-old, female, CAG15/67) were purchased from Coriell Institute and maintained in complete minimal essential medium with 15% FBS, 1% L-glutamine, and 1% penicillin/streptomycin at 37 °C in 5% CO2. The cell culture medium was changed every 2–3 days. The cells were cultured for approximately 20 passages.
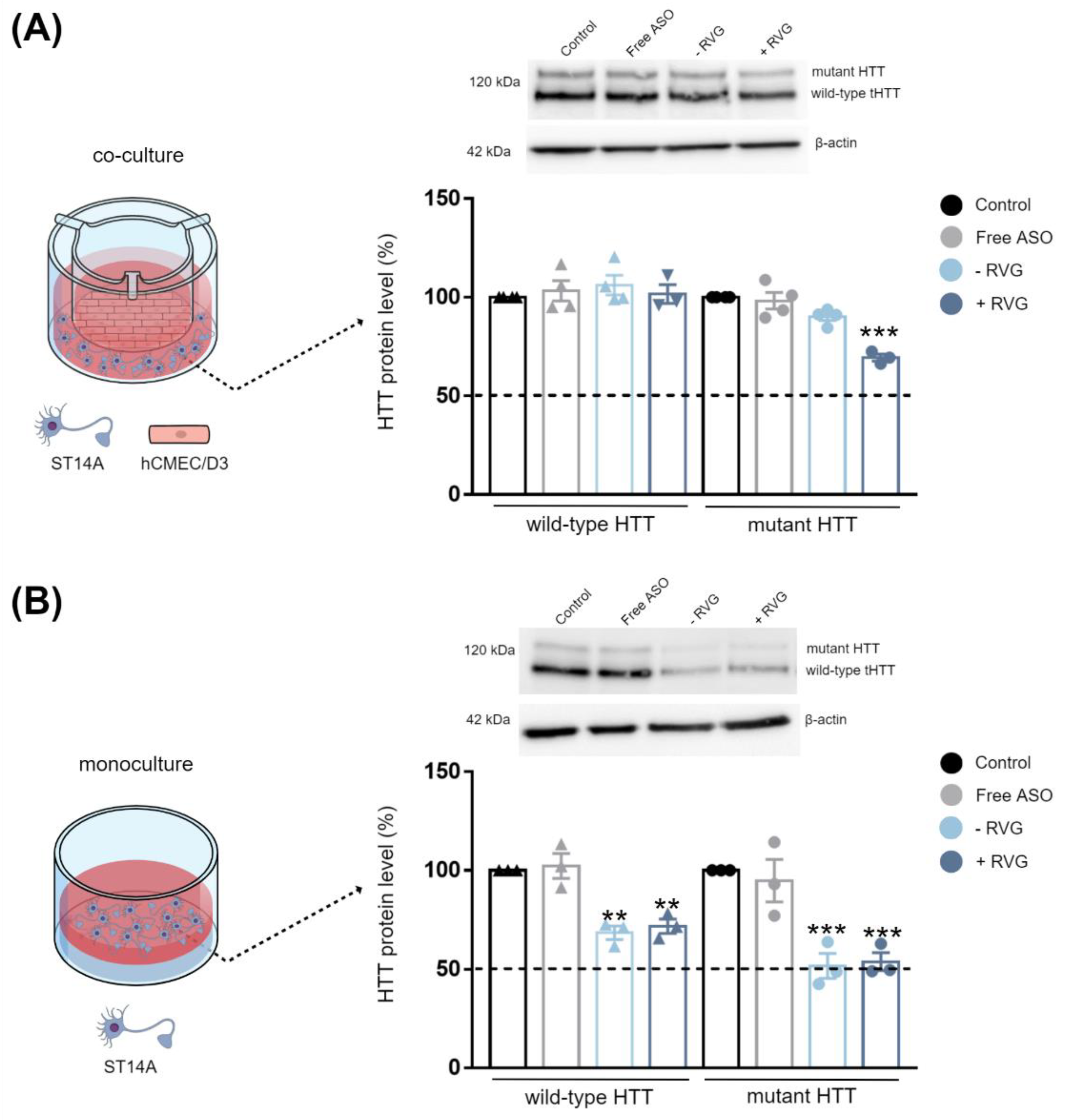
In Vitro Blood–Brain Barrier (BBB) and Co-Culture Model
The hCMEC/D3 BBB cell model was previously established and characterized in our laboratory [
16]. Briefly, hCMEC/D3 cells were seeded on coated transwell inserts (ThinCert #662641, Greiner Bio-One, Kremsmünster, Austria) at a density of 3.0 × 10
4 cells/cm
2 in 150 µL complete medium and grown for 8 days to achieve confluence (TEER plateau of 20 Ω × cm
2). After the establishment of the hCMEC/D3 monolayer, the upper chamber was transferred to another bottom chamber seeded with ST14A cells (7.5 × 10
4 cells per well in 600 µL of DMEM media) or GM04723 (1.5 × 10
5 cells per well in 600 µL of DMEM media) the day before. Then, free ASO or CD3:ASO conjugated to DSPE-PEG or co-formulated with RVG in EBM-2 medium were added to the upper compartment. After incubation for 72 h, ST14A and GM04723 cells in the lower chamber were collected for western blot analysis and RT-qPCR, respectively.
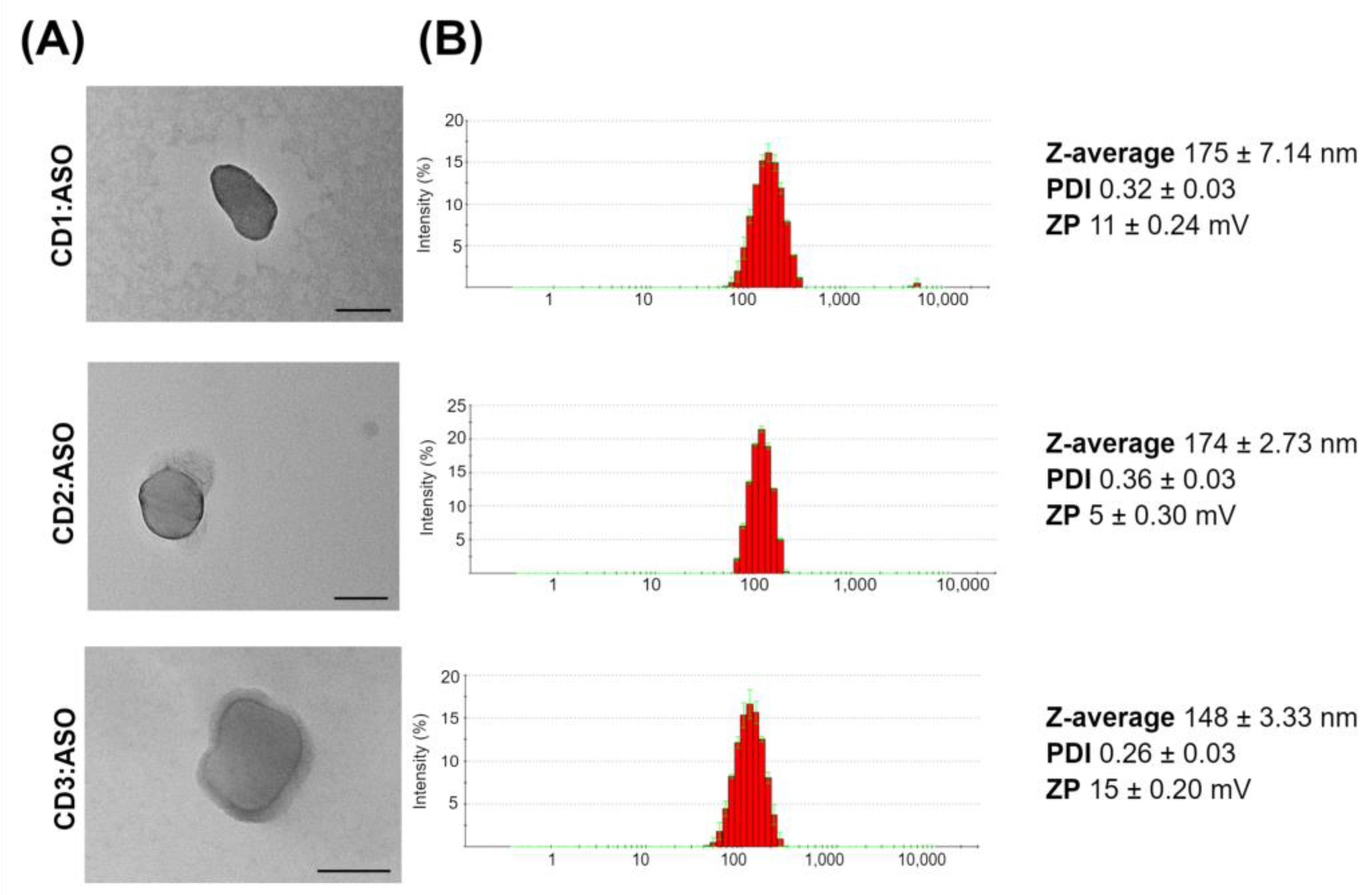
2.5. Viability Assay
The cell viability was assessed by ATP content using the CellTiter-Fluor™ cell viability assay (Promega, Madison, WI, USA). ST14A cells (7.5 × 104 cells per well) and GM04723 (1.5 × 105 cells) were seeded in 96-well plates and cultured for 24 h, whereas hCMEC/D3 cells (3.0 × 104 cells per well) were seeded in coated 96-well plates and cultured for 8 days. After this period, cells were treated with CDs:ASO nanocomplexes for 72 h, and the cell viability from each treated sample was measured in triplicate according to the manufacturer’s instructions.
2.6. Cellular Uptake
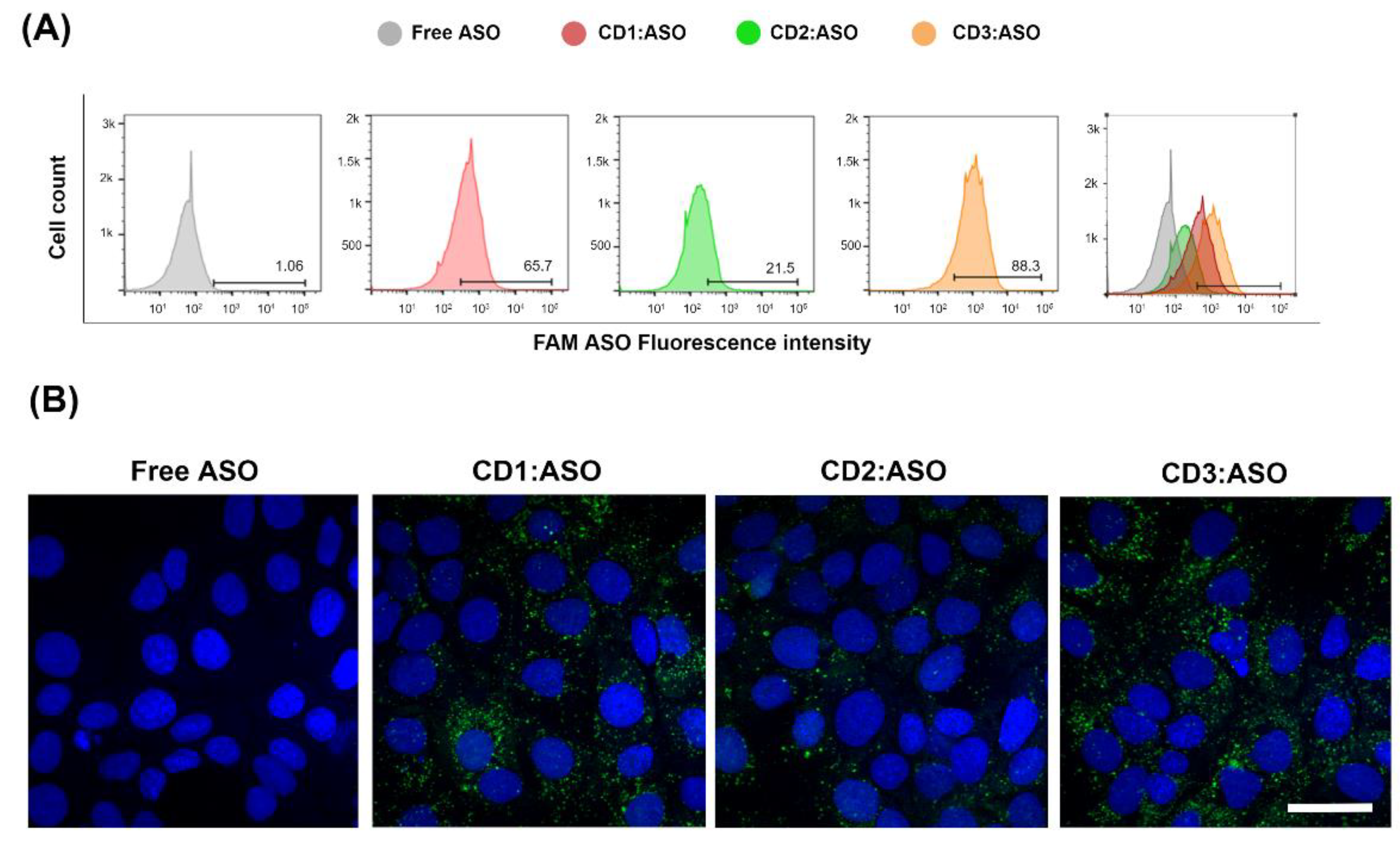
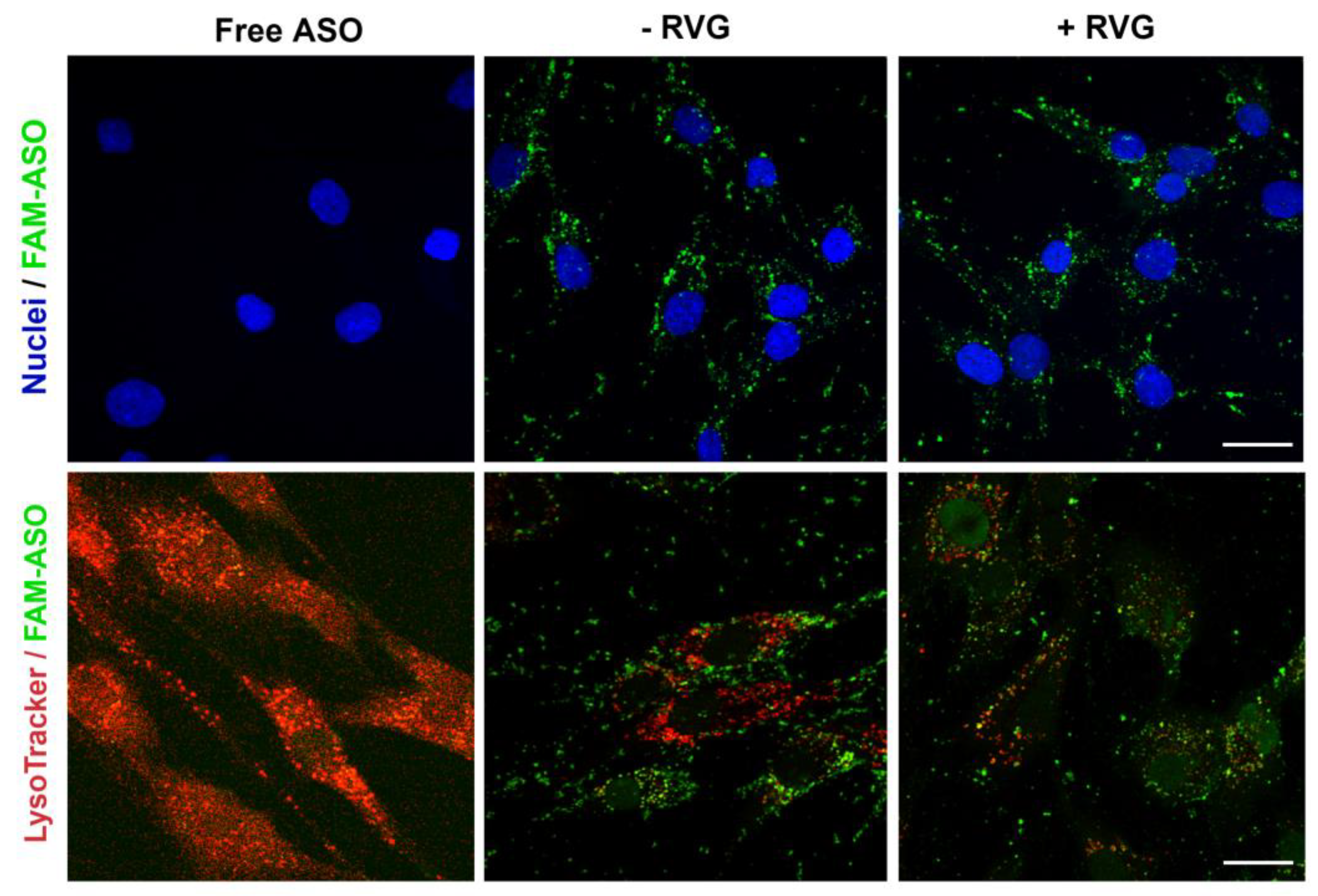
The amount of fluorescently labeled 6-FAM ASO taken up by cells was measured by flow cytometry and confocal microscopy. ST14A cells were plated in 24-well tissue culture plates at 1.5 × 105 cells/well 24 h prior to transfection. After 24 h, cells were washed twice with D-PBS and CDs:ASO nanocomplexes were added to cells in serum-reduced medium (2% FBS) at a final concentration of 100 nM 6-FAM labeled ASO. After 6 h of incubation, cells were washed with D-PBS, trypsinized, and collected into a complete DMEM medium. Samples were centrifuged for 5 min at 1500 rpm at 4 °C and resuspended in 400 μL of D-PBS. All data were acquired on a BD FACSCelesta™ flow cytometer (FACSCalibur, BD Biosciences, San Jose, CA, USA) and analyzed using FlowJo™ Software (version 10).
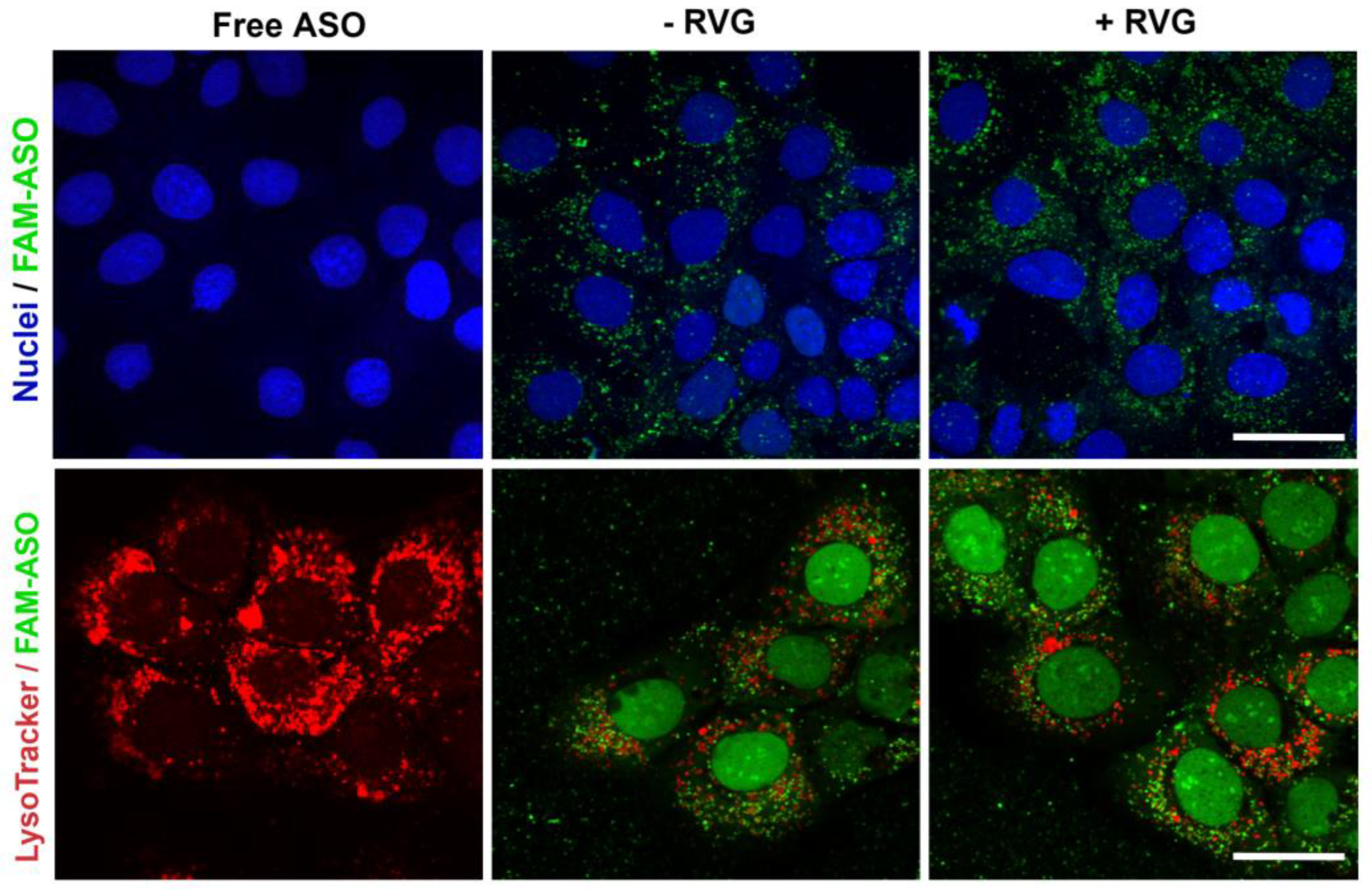
For confocal imaging, ST14A cells (1.25 × 105 cells per well) and GM04723 (1.0 × 105 cells) were seeded on round coverslips, at per coverslip in a 24-well plate and 500 uL medium. After 24 h, the medium was replaced with 400 uL of serum-reduced medium (2% FBS) plus 100 uL of 6-FAM labeled ASO (at a final concentration of 100 nM ASO). For staining of lysosomes, 50 nM LysoTracker Red DND-99 (ThermoFisher, Bremen, Germany) was added into the medium 1 h prior to fixation with 4% PFA (6 h of incubation in total). Coverslips were mounted in Mowiol, and confocal images were acquired on a Zeiss LSM510 META confocal microscope (Carl Zeiss, Jena, Germany) using a 63× objective.
2.7. Protein Quantification
ST14A cells were seeded in 24-well plates at a density of 7.5 × 104 cells per well and incubated for 24 h. After growing overnight, the medium was replaced with 400 uL serum-reduced medium (2% FBS) plus 100 µL of CDs containing ASO (at a final concentration of 100 nM ASO). For co-culture experiments, ST14A cells were treated as described above.
2.7.1. Western Blotting
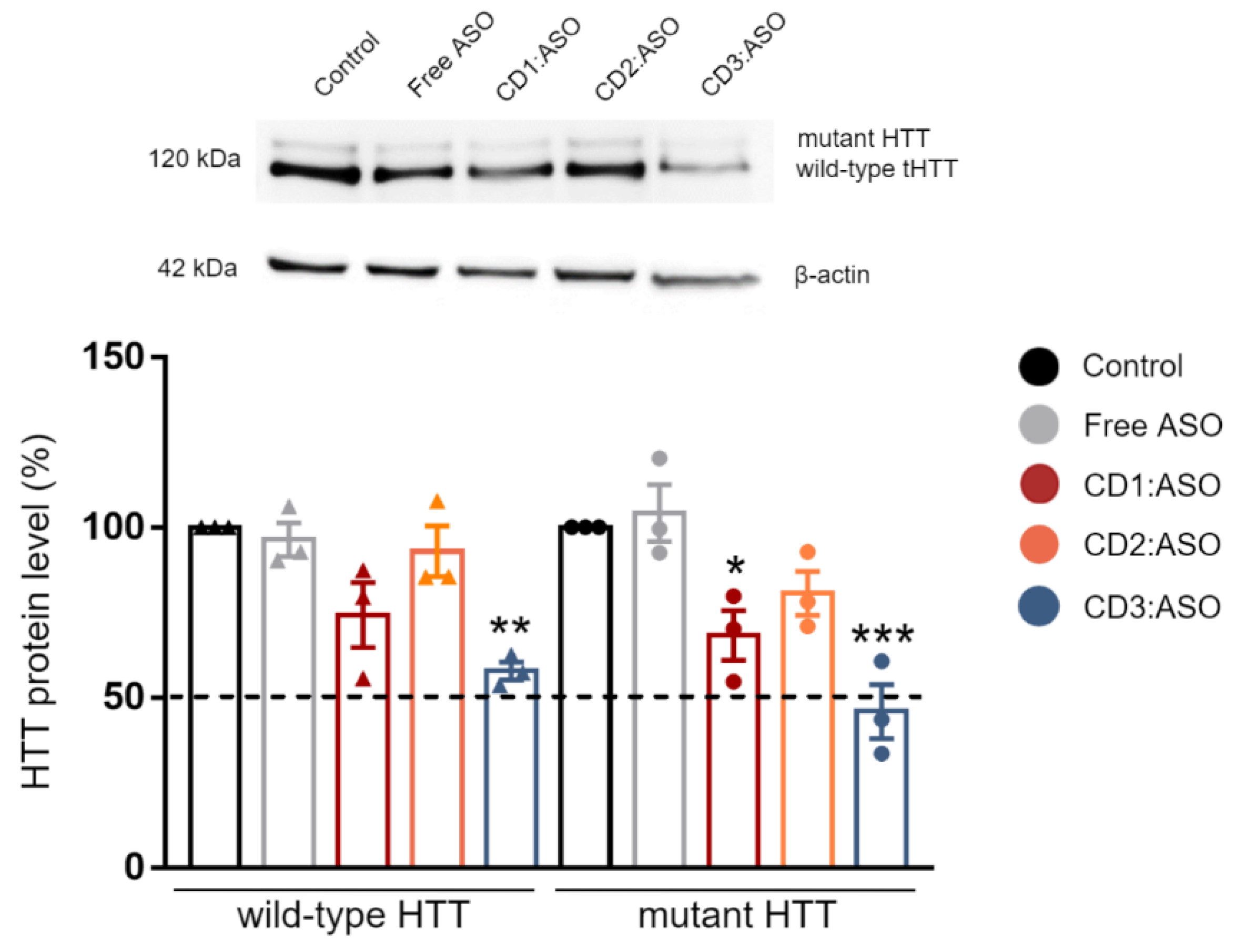
Cells were lysed 72 h post-transfection in RIPA buffer (Sigma Aldrich, Wicklow, Ireland) containing 200 mM PMSF, 100 mM sodium orthovanadate, and cOmplete™ EDTA-free protease inhibitor cocktail solution (Roche, San Francisco, CA, USA) for 30 min on ice. The lysate was centrifuged at 14,000 rpm at 4 °C for 10 min and protein concentration was quantified using Pierce™ BCA Protein Assay kit (ThermoFisher, Bremen, Germany). The samples were mixed with an equal volume of Laemli buffer (Sigma Aldrich) and denatured by boiling at 95 °C for 6 min. Equal amounts of protein (15 µg) were loaded into a 6–12% SDS-PAGE gel and then transferred to PVDF membranes (Bio-Rad, Hercules, CA, USA). Membranes were blocked with 5% milk in TBST buffer (150 mM NaCl; Tris, pH 7.4 and 0.1% Tween-20) for 1 h at room temperature, rinsed, and incubated overnight at 4 °C with primary antibody against HTT (1:1000; MAB2166; Sigma Aldrich, Wicklow, Ireland). Subsequently, the membranes were incubated with anti-mouse HRP-conjugated secondary antibodies for 2 h at room temperature (1:2000; A0545; Sigma Aldrich, Wicklow, Ireland). The chemiluminescent signal was revealed using SuperSignal™ West Pico PLUS Substrate (ThermoFisher, Bremen, Germany) and imaged with an Alliance Imaging system (Uvitec, Cambridge, UK). Band intensities were quantified by densitometry using the LabImage 1D program and normalized to β-actin control (1:1000; A5441; Sigma Aldrich, Wicklow, Ireland).
2.7.2. Immunohistochemistry
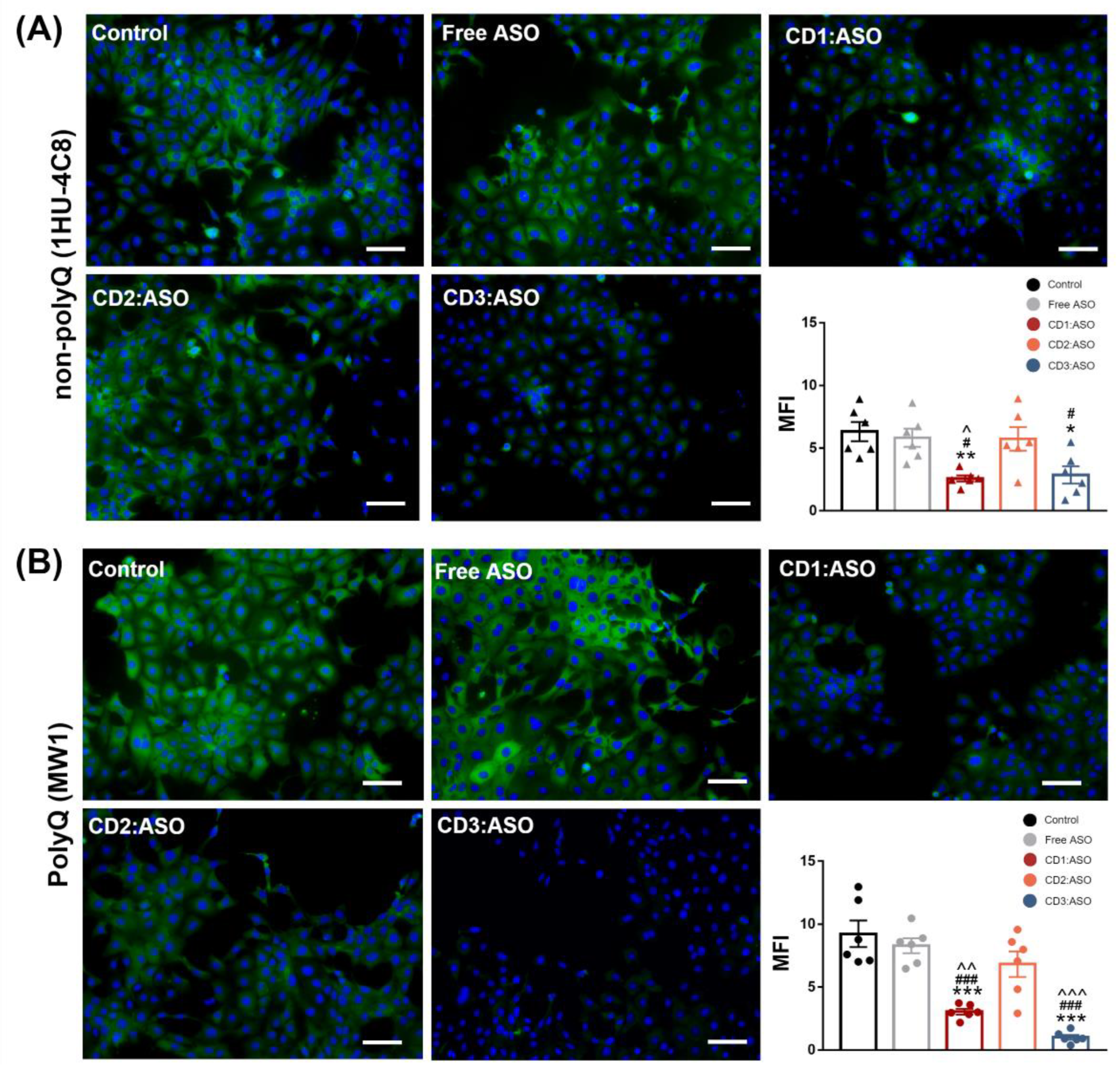
Cells were seeded in coverslips and transfected for 72 h. Then, the cells were washed with warm D-PBS, fixed with 4% paraformaldehyde (PFA) at room temperature for 15 min, and quenched with 50 mM ammonium chloride followed by permeabilization with 0.2% Triton X-100 for 5 min. Blocking was performed with 3% BSA for 30 min at room temperature. Afterward, the samples were incubated with anti-HTT antibody, clone 1 HU-4C8 (1:100; MAB2166; Sigma Aldrich, Wicklow, Ireland), and Anti-polyQ specific, clone MW1 (1:100; MABN2427; Sigma Aldric, Wicklow, Ireland) overnight at 4 °C. Following incubation, the samples were washed in D-PBS and incubated for 1 h in 1% BSA with Alexa 488 goat anti-mouse (1:200; #A11001; Invitrogen, Waltham, MA, USA) and DAPI (0.1 µg/mL, # D1306; Invitrogen, Waltham, MA, USA). Finally, samples were washed in D-PBS, mounted with ProLong™ Gold anti-fade reagent (ThermoScientific, Bremen, Germany), dried, and imaged using an Olympus IX73 microscope (Olympus Inc., Tokyo, Japan). Immunohistochemistry was carried out at the same time to allow for a comparison of the mean fluorescence intensity (MFI), which was quantified using Image J software (NIH) (version 1.53t).
2.8. SNP Genotyping Assay
GM04723 (CAG15/67) cells were seeded in 24-well plates at a density of 1.5 × 105 cells per well and incubated for 24 h. After growing overnight, the medium was replaced with 400 uL serum-reduced medium (2% FBS) plus 100 µL of CDs containing ASO (at a final concentration of 100 nM ASO). RNA extractions were performed 72 h post-transfection using a GenElute™ Mammalian Total RNA kit according to the supplier’s instructions (Sigma Aldrich, Wicklow, Ireland). cDNA synthesis was further performed using the High-Capacity cDNA Reverse Transcription kit (Applied Biosystems, Foster City, CA, USA). Allele-specific detection of HTT mRNA levels was performed using the SNP rs362307 (C___2229302_10, Thermo Fisher Scientific, Bremen, Germany) genotyping assay. For each sample, 10-μL qPCR reactions were set up as follows: 5 µL SsoFast Probes Supermix (Bio-Rad, Basel, Switzerland), 0.25 µL of 40× TaqMan SNP Genotyping Assay, 2.75 µL H2O, and 2 µL of cDNA sample. All reactions were carried out using the CFX96 Real-Time PCR Detection System (Bio-Rad, Basel, Switzerland) following the steps: 95 °C for 30 s; 95 °C for 5 s; 60 °C for 25 s; 40 cycles of amplification. Each reaction was run in duplicate. Results were expressed as Ct values after normalization to GAPDH as the housekeeping gene.
2.9. Statistical Analysis
All statistical analyses were performed using GraphPad Prism 7. Analysis of protein knockdown was performed using two-way ANOVAs with Sidak’s post hoc multiple comparison testing. Immunohistochemistry and RT-qPCR experiments were analysed using one-way ANOVA with Tukey’s post hoc multiple comparison testing. All error bars represent the standard error of the mean. Statistical significance was considered when p < 0.05.
4. Discussion
Therapeutic strategies focused on lowering mHTT levels using non-allele and allele-selective ASOs delivered by intrathecal injections have been assessed in human clinical trials; however, the three most promising HTT-lowering trials were halted early [
8]. Although they have failed for very different reasons (reviewed elsewhere [
26]), a common potential challenge for this therapy is the long-term tolerability of the monthly intrathecal injections [
5]. Therefore, the exploration of alternative strategies that can deliver ASOs to the brain in an effective way and using lower doses has become an important aspect of the development of RNA-based therapeutics.
In this study, a different approach was used to lower total HTT expression using ASO delivered via CDs. The three CD-derivatives have been all modified on the primary side with linear dodecanthiol moieties in order to provide a common lipophilic motif utilizable for the self-assembly process. CD1 possesses alkyl chains on the secondary side terminating with primary amines. A suitable precursor of CD1 has been regioselectively modified on the hydroxy in position 2 with propargyl groups that have been reacted with BOC-protected 1-azidopropylamine under cycloaddition conditions. Acidic cleavage of the BOC protecting groups yielded the desired compound. The installation of the tertiary amines on the secondary side of CD2 was achieved through ester formation between the commercially available 3-aminobutanoyl chloride and the corresponding primary-side modified cyclodextrin precursor. The resulting isomeric mixture is primarily substituted on the hydroxy groups in position 2. Exhaustive secondary side propargylation for CD3 can be achieved as previously described [
27]. Subsequent cycloaddition with ad hoc designed azido-alkyl-amine derivatives generated the titled compound in good yield. CD3 possesses alkyl chains on the secondary side, terminating with primary amines as with CD1, but in a larger amount (more than double). These structure modifications of CDs can directly affect their interactions with cellular components and, consequently, their biological activity [
28,
29,
30]. The physicochemical properties of the three CDs evaluated were similar and with uniform distribution, showing desirable characteristics for brain delivery (<200 nm and positively charged) [
31], strong stability, and protecting ASO from nuclease degradation. In the cytotoxicity evaluation, it was proven that CD1:ASO, CD2:ASO, and CD3:ASO nanocomplexes were not toxic to striatal neuronal cells nor HD patient-derived fibroblasts under the conditions used.
Given the complexity of HD, it is important to validate the efficacy of the delivery system in different therapeutically relevant cell lines. Striatal neurons are particularly vulnerable at the early stages of HD progression [
32], while in human fibroblast lines, heterozygosity of HD-SNPs provides a convenient and physiologically relevant platform for testing potential allele-selective ASOs. In the current study, we showed greater cellular uptake and silencing efficacy by treatment with double-charged chain γ-CDs (CD3:ASO nanocomplexes) than β-CDs with primary amines (CD1) followed by β-CDs with tertiary amines (CD2). Although the ASO sequence was not designed to target specific polyQ repeats within mHTT mRNA, a higher inhibition of the expression of mHTT protein was achieved compared to total HTT.
We hypothesized that active targeting would increase nanocarrier internalization in the targeted cells, thereby increasing the ASO intracellular concentration and efficacy. For this reason, CDs were further modified with RVG, a cell-penetrating peptide that has been considered an attractive approach for the targeted transport of nanocarriers across the BBB [
17,
33]. After incorporating RVG into γ-CDs, clear differences were observed between the mono- and co-culture models. The presence of the targeting moiety did not result in preferential accumulation in the monoculture system compared to the co-culture model, probably because it cannot facilitate the critical step of extravasation through the BBB. Although nAChR is expressed in endothelial cells, neurons, and fibroblasts, nAChRs in the CNS comprise a combination of different α (α1–α9) and β (β2–β4) subunits, or even α subunits alone. Depending on the cell type involved, the diversity in expression, dominance, and subunit composition may have different effects on ligand affinity [
34,
35]. Considering that nAChR was responsible for RVG uptake, in order to cross the BBB, it would have to bind to either α3, α5, α7, β2, or β3 subunits, whereas striatal neurons would have to bind to either α3, α4, α7, or β2 [
36]. It is reasonable to assume that the synthesized RVG sequence selected was responsible for interaction mainly with the BBB nAChR subunits and the relatively similar size and positive zeta potential of both RVG-targeted and untargeted nanocomplexes were accountable for their comparable efficiency in the monoculture model.
Although this study has provided a foundation for the rational design and future application of CD platforms in the delivery of ASOs to treat HD, some limitations still need to be addressed in order to successfully translate these findings into the clinic. For example, preclinical studies of allele-specific targeting ASOs should be performed in animal models with the same SNP genotypes as the prospective patient population. However, there are no mouse models of HD that include SNP1 and SNP2 [
37]. This was one of the challenges encountered by Wave Life Sciences in their WVE0120101 and WVE-120102 preclinical trials, as they needed access to suitable animal models of HD that contained the SNPs it was targeting. Therefore, they were unable to confidently predict the pharmacokinetic and pharmacodynamic effects of the molecules [
38].
5. Conclusions
In this work, we have explored the influence of distinct chemical structures on the biological behavior of CDs-based nanoparticles for the delivery of ASO. The double-charged chain γ-CDs exhibited superior internalization and knockdown efficacy compared to both β-CDs. Additionally, all three CDs evaluated were found to be non-cytotoxic in striatal neurons and HD fibroblast cells. Although designed as a non-selective approach, our strategy provides an additional benefit of some allele selectivity as demonstrated by western blotting and immunohistochemistry against the mutant polyQ tract. After the incorporation of RVG into γ-CDs, clear differences were observed between the mono- and co-culture models. RVG-targeting did not result in preferential accumulation of the nanocomplexes in the neuronal/fibroblast monoculture but did so in the co-cultured BBB model. These results may be an indication that, by direct injection into the brain, the use of untargeted CDs:ASO nanocomplexes might be sufficient to induce gene/protein knockdown. However, if systemic administration is desired, a targeting approach should be considered to overcome the BBB. Further studies will now be performed in vivo by intrathecal delivery as well as by intravenous delivery to corroborate these data.


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}










