
Novel Regioselective Synthesis of 1,3,4,5-Tetrasubstituted Pyrazoles and Biochemical Valuation on F1FO-ATPase and Mitochondrial Permeability Transition Pore Formation

 ,
,  ,
,  ,
,  ,
,  ,
,  , , ,
, , , 
 and
and 
Abstract
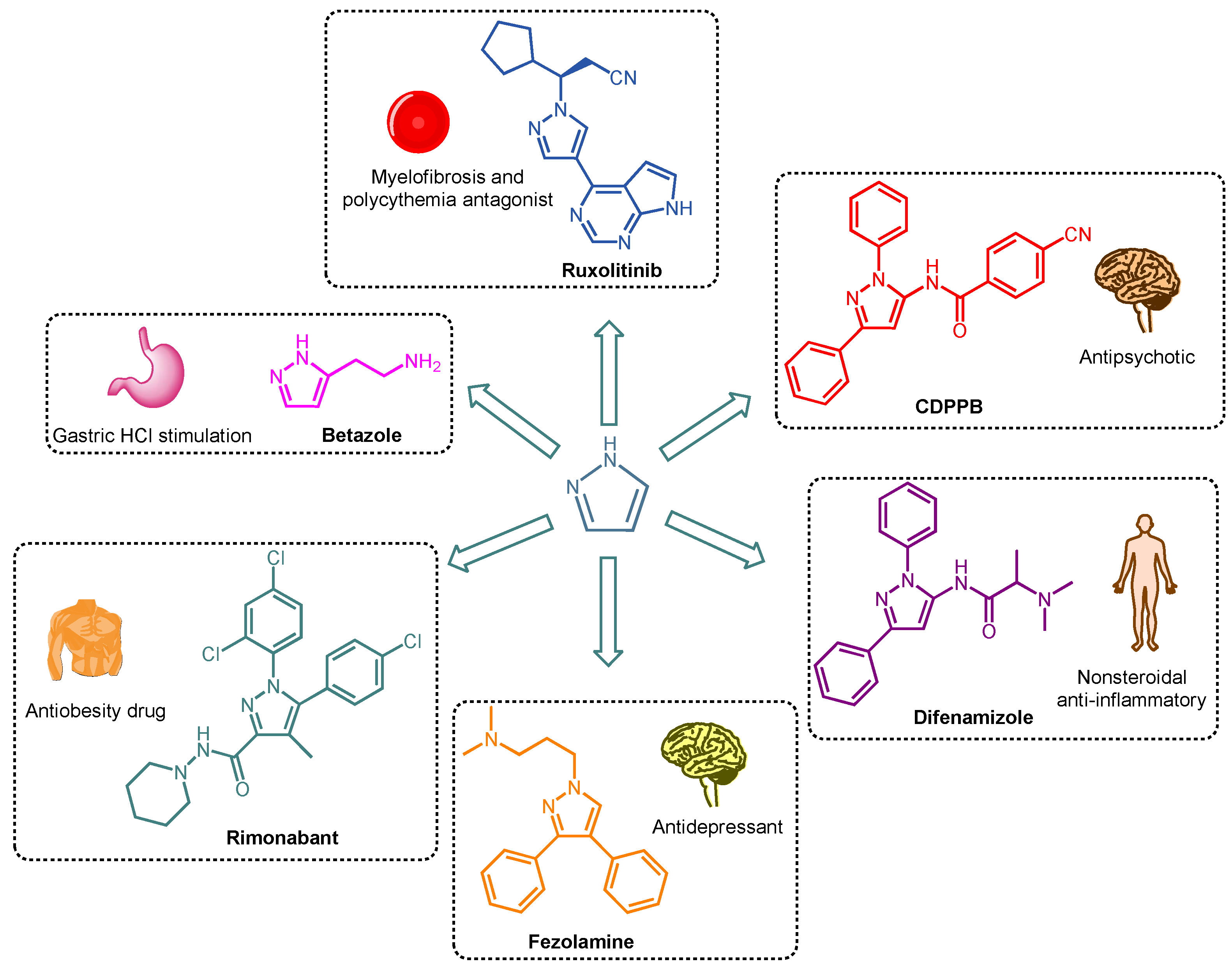
1. Introduction
2. Materials and Methods
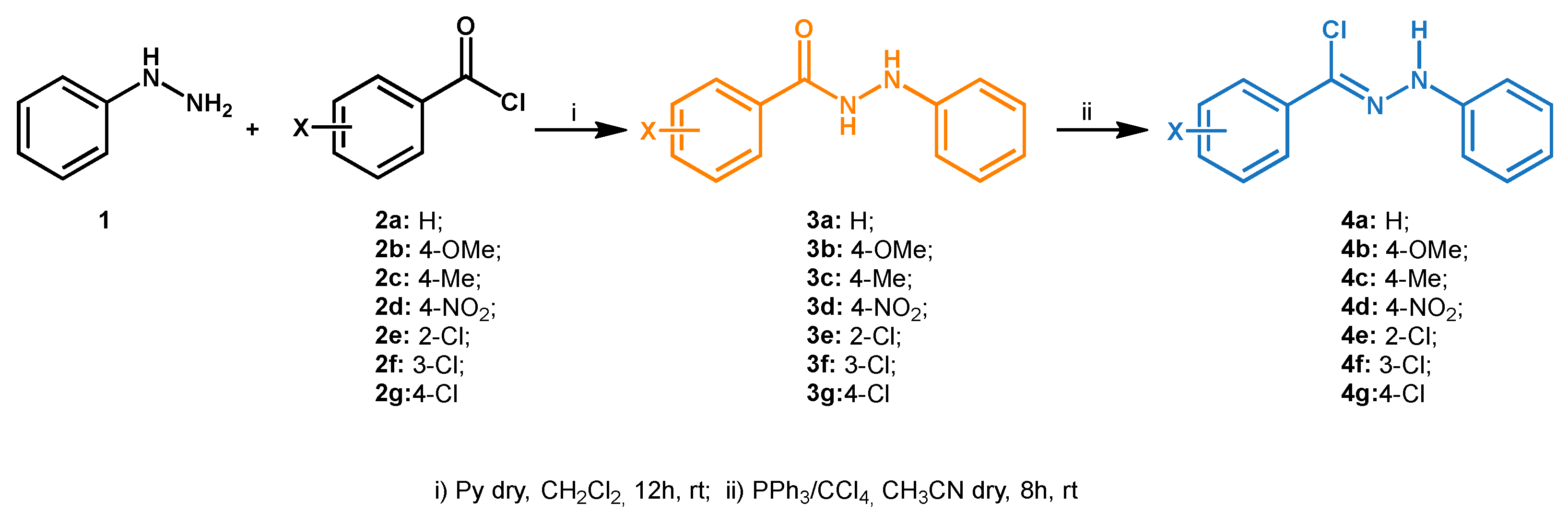
2.1. General Procedure for Synthesis of 1,3,4,5-Tetrasubstituted Pyrazoles 6a–6g
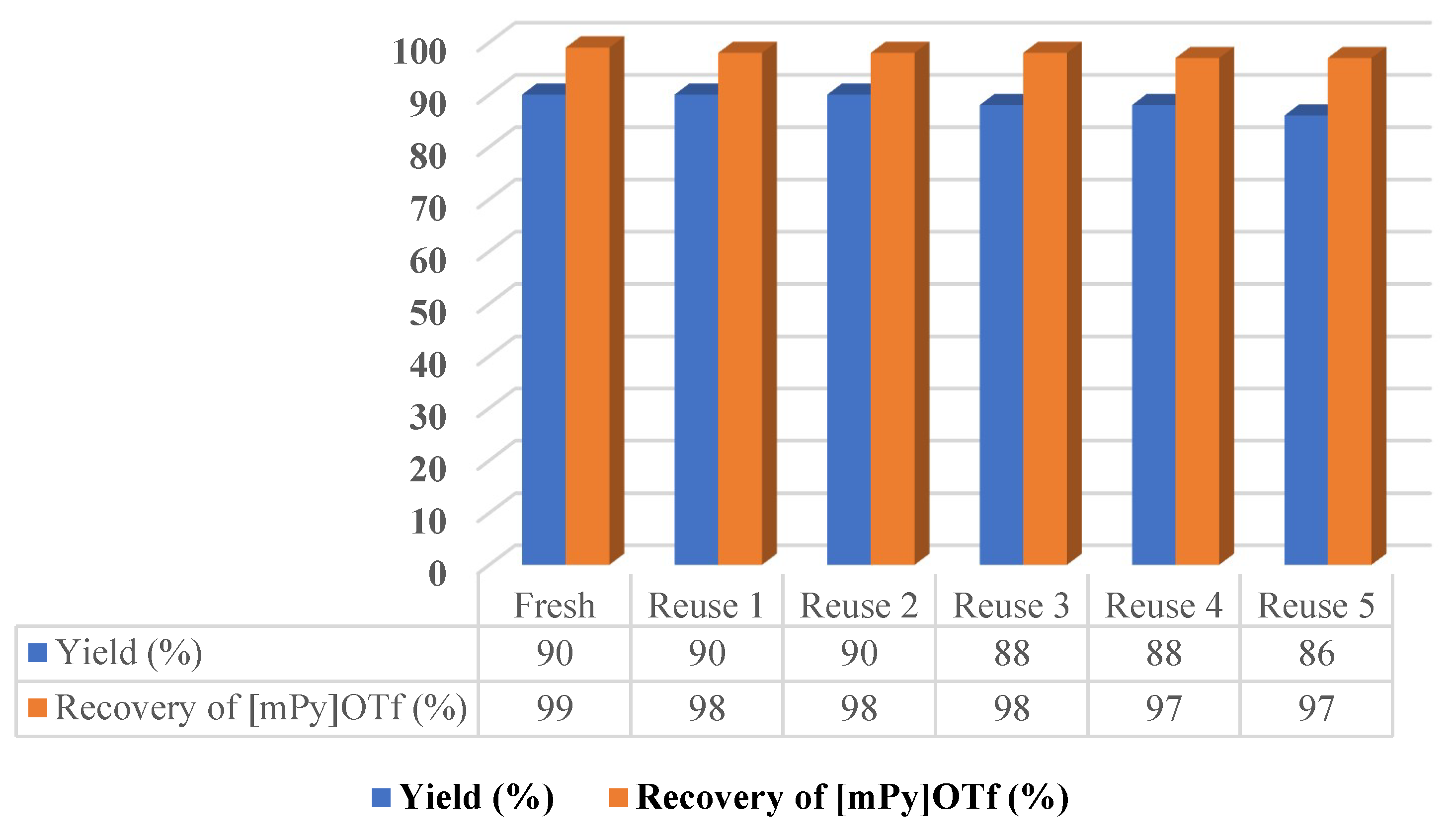
2.2. Recovery and Recycling Procedure of Ionic Liquid [mPy](OTf)
2.3. Preparation of Mitochondrial Fraction
2.4. Mitochondrial F-ATPase Activity Assay
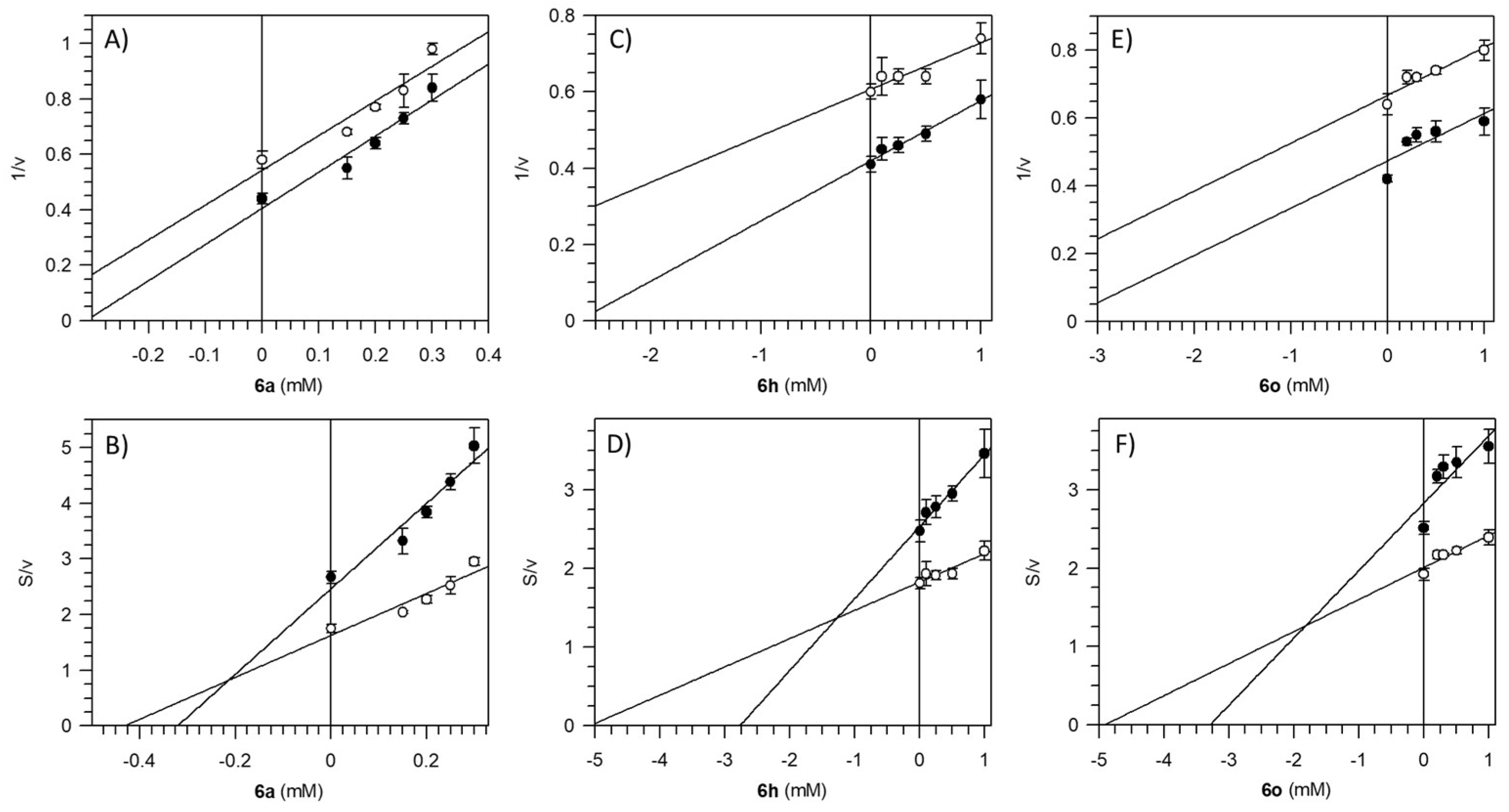
2.5. Kinetic Analysis
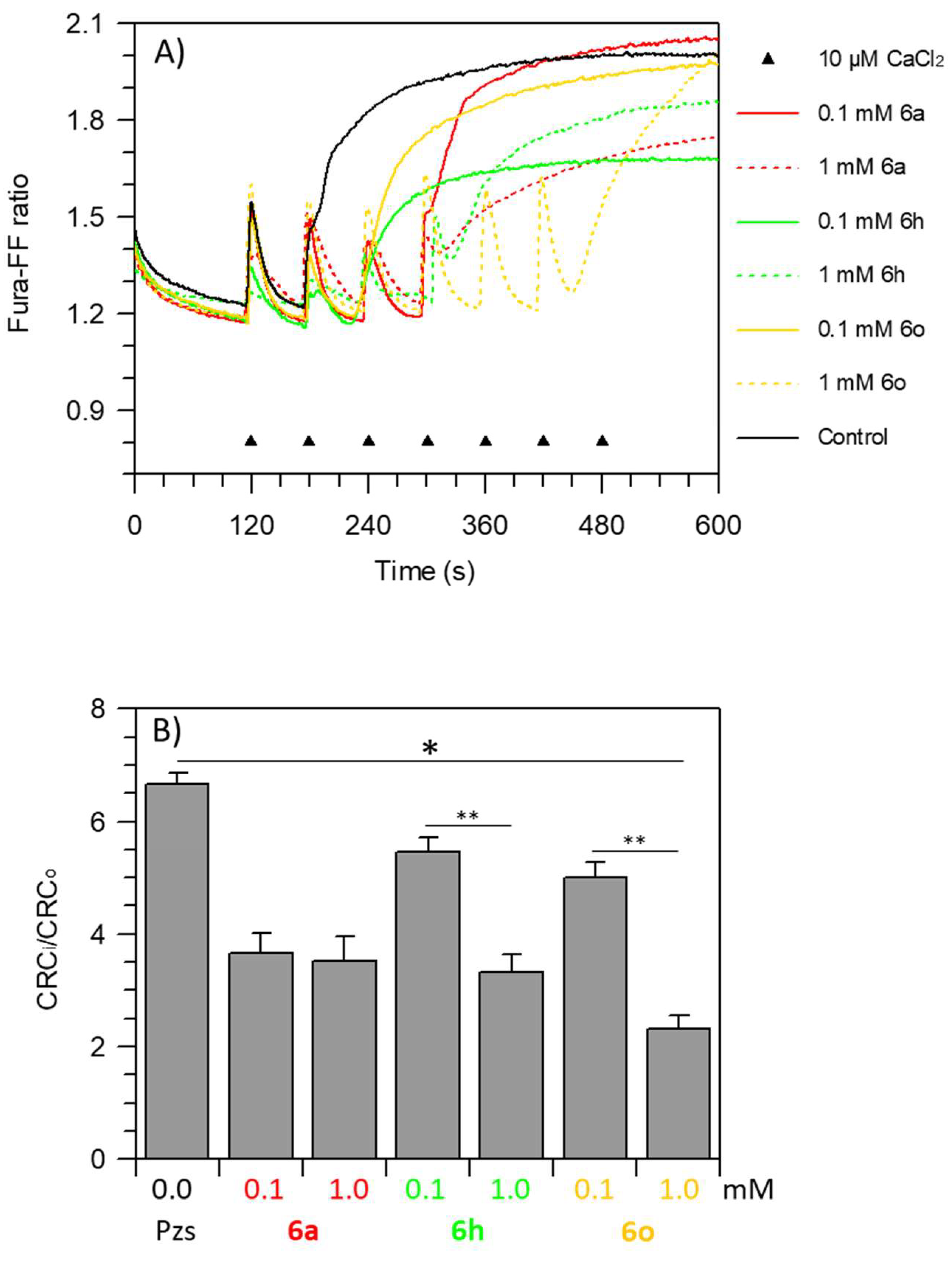
2.6. mPTP Assay
2.7. Statistical Analysis
3. Results and Discussion
3.1. Synthesis
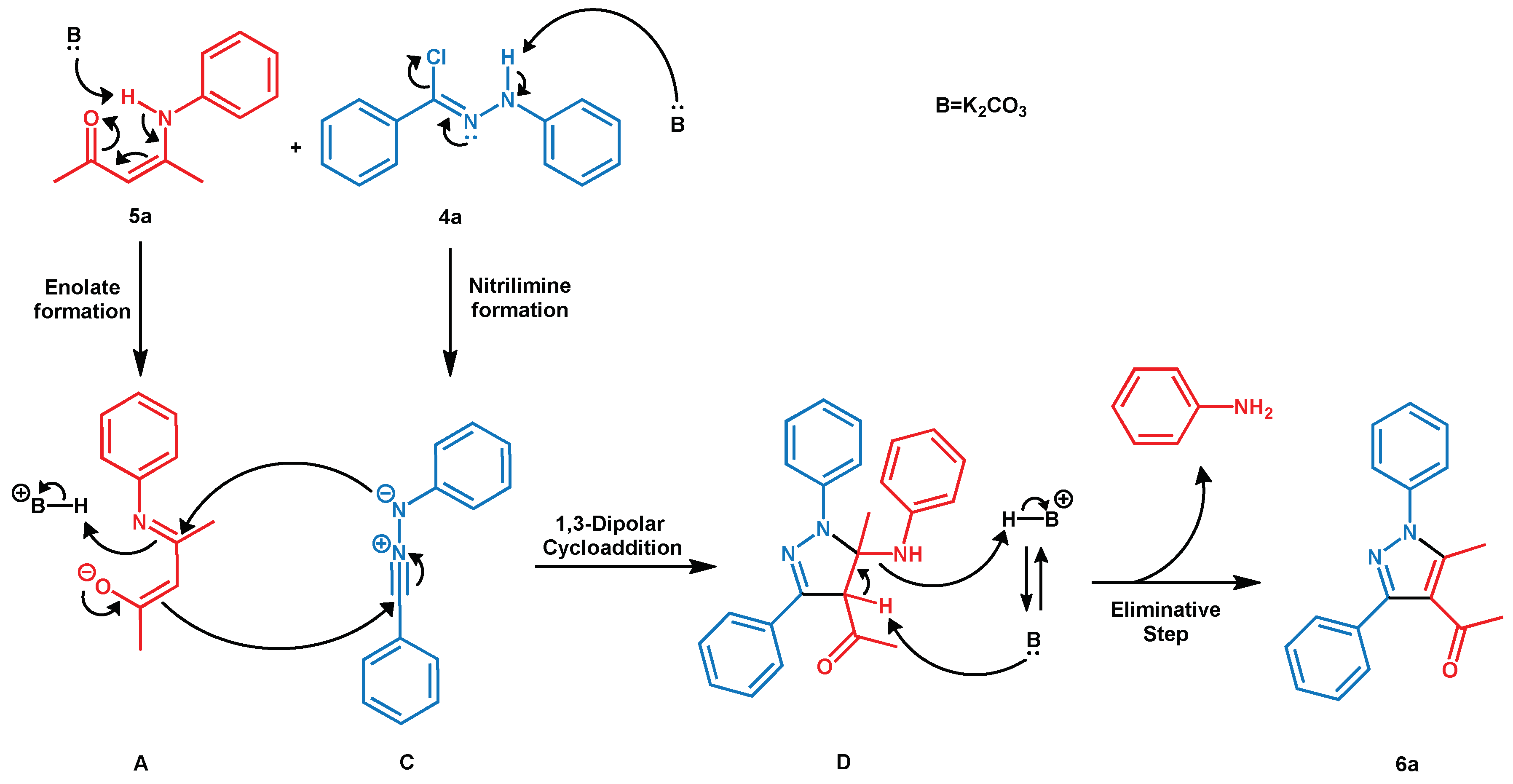
3.2. Reaction Mechanism
3.3. Recycling Ionic Liquid
3.4. Pzs’ Effect on F1FO-ATPase
3.5. Pzs Effect on mPTP
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Gomes, P.M.O.; Ouro, P.M.S.; Silva, A.M.S.; Silva, V.L.M. Styrylpyrazoles: Properties, synthesis and transformations. Molecules 2020, 25, 5886. [Google Scholar] [CrossRef] [PubMed]
- Kumar, H.; Bansal, K.K.; Goyal, A. Synthetic methods and antimicrobial perspective of pyrazole derivatives: An insight. Anti-Infect. Agents 2020, 18, 207–223. [Google Scholar] [CrossRef]
- Giornal, F.; Pazenok, S.; Rodefeld, L.; Lui, N.; Vors, J.-P.; Leroux, F.R. Synthesis of diversely fluorinated pyrazoles as novel active agrochemical ingredients. J. Fluorine Chem. 2013, 152, 2–11. [Google Scholar] [CrossRef]
- Judge, N.R.; Chacktas, G.; Ma, L.; Schink, A.; Buckpesch, R.; Schmutzler, D.; Machettira, A.B.; Dietrich, H.; Asmus, E.; Bierer, D.; et al. Flexible synthesis and herbicidal activity of fully substituted 3-hydroxypyrazoles. Eur. J. Org. Chem. 2021, 2021, 5677–5684. [Google Scholar] [CrossRef]
- Catalan, J.; Fabero, F.; Claramunt, R.M.; Santa Maria, M.D.; Foces-Foces, M.C.; Cano, F.H.; Martinez-Ripoll, M.; Elguero, J.; Sastre, R. New ultraviolet stabilizers: 3- and 5-(20 -hydroxyphenyl)pyrazoles. J. Am. Chem. Soc. 1992, 114, 5039–5048. [Google Scholar] [CrossRef]
- Garcia, H.; Iborra, S.; Miranda, M.A.; Morera, I.M.; Primo, J. Pyrazoles and isoxazoles derived from 2-hydroxyaryl phenylethynyl ketones: Synthesis and spectrophotometric evaluation of their potential applicability as sunscreens. Heterocycles 1991, 32, 1745–1748. [Google Scholar] [CrossRef]
- Busev, A.I.; Akimov, V.K.; Gusev, S.I. Pyrazolone Derivatives as Analytical Reagents. Russ. Chem. Rev. 1965, 34, 237. [Google Scholar] [CrossRef]
- Trofimenko, S. Coordination chemistry of pyrazole-derived ligands. Chem. Rev. 1972, 72, 497–509. [Google Scholar]
- Maeda, H.; Ito, Y.; Kusunose, Y.; Nakanishi, T. Dipyrrolylpyrazoles: Anion receptors in protonated form and efficient building blocks for organized structures. Chem. Commun. 2007, 1136–1138. [Google Scholar] [CrossRef]
- Massah, M.; Balmeh, N.; Goodarzi, K.; Fard, N.A. Molecular, docking analysis of H1 and H2 antihistamines groups with L-asparaginase II for reducing allergenicity; an in silico approach. Inform. Med. Unlocked 2022, 28, 100865. [Google Scholar] [CrossRef]
- Alam, M.A. Antibacterial Pyrazoles: Tackling resistant bacteria. Future Med. Chem. 2022, 14, 343–362. [Google Scholar] [CrossRef] [PubMed]
- Karrouchi, K.; Radi, S.; Ramli, Y.; Taoufik, J.; Mabkhot, Y.N.; Al-aizari, F.; Ansar, M. Synthesis and pharmacological activities of pyrazole derivatives: A review. Molecules 2018, 23, 134. [Google Scholar] [CrossRef] [PubMed]
- Verma, K.; Lahariya, A.K.; Dubey, S.; Verma, A.K.; Das, A.; Schneider, K.A.; Bharti, P.K. An integrated virtual screening and drug repurposing strategy for the discovery of new antimalarial drugs against plasmodium falciparum phosphatidylinositol 3-kinase. J. Cell. Biochem. 2021, 122, 1326–1336. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.-T.; Wang, M.; Galvin, V.; Yang, Y.; Arnsten, A.F.T. Effects of blocking mGluR5 on primate dorsolateral prefrontal cortical neuronal filing and working memory performance. Psychopharmacology 2021, 238, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Bennani, F.E.; Doudach, L.; Cherrah, Y.; Ramli, Y.; Karrouchi, K.; Ansar, M.; Faouzi, M.E.A. Overview of recent developments of pyrazole derivatives as an anticancer agent in different cell line. Bioorganic Chem. 2020, 97, 103470. [Google Scholar] [CrossRef] [PubMed]
- Chuang, H.; Huang, L.-C.S.; Kapoor, M.; Liao, Y.-J.; Yang, C.-L.; Chang, C.-C.; Wu, C.-Y.; Hwu, J.R.; Huang, T.-J.; Hsu, M.-H. Design and synthesis of pyridine-pyrazole-sulfonate derivatives as potential anti-HBV agents. Med. Chem. Commun. 2016, 7, 832–836. [Google Scholar] [CrossRef]
- Priya, D.; Gopinath, P.; Dhivya, L.S.; Vijaybabu, A.; Haritha, M.; Palaniappan, S.; Kathiravan, M.K. Structural insights into pyrazoles as agents against anti-inflammatory and related disorders. Chem. Sel. 2022, 7, e202104429. [Google Scholar] [CrossRef]
- Wang, Y.M.; Teusink-Cross, A.; Elborai, Y.; Krupski, M.C.; Nelson, A.S.; Grimley, M.S.; Flannery, A.; Mehta, P.A.; Bleesing, J.J.; Chandra, S.; et al. Ruxolitinib for the treatment of chronic GVHD and overlap syndrome in children and young adults. Transplantation 2022, 106, 412–419. [Google Scholar] [CrossRef]
- Niknam, Z.; Jafari, A.; Golchin, A.; Pouya, F.D.; Nemati, M.; Rezaei-Tavirani, M.; Rasmi, Y. Potential therapeutic options for COVID-19: An update on current evidence. Eur. J. Med. Res. 2022, 27, 6. [Google Scholar] [CrossRef]
- Dadiboyena, S.; Nefzi, A. Synthesis of functionalized tetrasubstituted pyrazolyl heterocycles—A review. Eur. J. Med. Chem. 2011, 46, 5258–5275. [Google Scholar] [CrossRef]
- Naoum, F.; Kasiotis, K.M.; Magiatis, P.; Haroutounian, S.A. Synthesis of novel nitro-substituted triaryl pyrazole derivatives as potential estrogen receptor ligands. Molecules 2007, 12, 1259–1273. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Mani, N.S. Reaction of N-Monosubstituted hydrazones with nitroolefins: A novel regioselective pyrazole synthesis. Org. Lett. 2006, 8, 3505–3508. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.S.M.; Kobayashi, K.; Mori, A. One-pot construction of pyrazoles and isoxazoles with palladium-catalyzed four-component coupling. Org. Lett. 2005, 7, 4487–4489. [Google Scholar] [CrossRef]
- Bonini, B.F.; Franchini, M.C.; Gentili, D.; Locatelli, E.; Ricci, A. 1,3-dipolar cycloaddition of nitrile imines with functionalized acetylenes: Regiocontrolled Sc(OTf)3-catalyzed synthesis of 4- and 5- substituted pyrazoles. Synlett 2009, 14, 2328–2332. [Google Scholar] [CrossRef]
- Conti, P.; Pinto, A.; Tamborini, L.; Rizzo, V.; De Micheli, C. A regioselective route to 5-substituted pyrazole- and pyrazoline-3-phosphonic acids and esters. Tetrahedron 2007, 63, 5554–5560. [Google Scholar] [CrossRef]
- Maiuolo, L.; Algieri, V.; Russo, B.; Tallarida, M.A.; Nardi, M.; Di Gioia, M.L.; Merchant, Z.; Merino, P.; Delso, I.; De Nino, A. Synthesis, Biological and In Silico Evaluation of Pure Nucleobase-Containing Spiro (Indane-Isoxazolidine) Derivatives as Potential Inhibitors of MDM2-p53 Interaction. Molecules 2019, 24, 2909. [Google Scholar] [CrossRef]
- Maiuolo, L.; Feriotto, G.; Algieri, V.; Nardi, M.; Russo, B.; Di Gioia, M.L.; Furia, E.; Tallarida, M.A.; Mischiati, C.; De Nino, A. Antiproliferative Activity of Novel Isatinyl/Indanyl Nitrones (INs) as Potential Spin Trapping of Free Radical Intermediates. MedChemComm 2018, 9, 299–304. [Google Scholar] [CrossRef]
- Maiuolo, L.; De Nino, A.; Algieri, V.; Nardi, M. Microwave-Assisted 1,3-Dipolar Cyclo-addition: Recent Advances In Synthesis of Isoxazolidines. Mini-Rev. Org. Chem. 2017, 14, 136–142. [Google Scholar] [CrossRef]
- Costanzo, P.; Calandruccio, C.; Di Gioia, M.L.; Nardi, M.; Oliverio, M.; Procopio, A. First multicomponent reaction exploiting glycerol carbonate synthesis. J. Clean. Prod. 2018, 202, 504–509. [Google Scholar] [CrossRef]
- De Nino, A.; Merino, P.; Algieri, V.; Nardi, M.; Di Gioia, M.L.; Russo, B.; Tallarida, M.A.; Maiuolo, L. Synthesis of 1,5-functionalized 1,2,3-triazoles using ionic liquid/iron(III) chloride as an efficient and reusable homogeneous catalyst. Catalysts 2018, 8, 364–376. [Google Scholar] [CrossRef]
- De Nino, A.; MAiuolo, L.; Costanzo, P.; Algieri, V.; Jiritano, A.; Olivito, F.; Tallarida, M.A. Recent progress in catalytic synthesis of 1,2,3-triazoles. Catalysts 2021, 11, 1120. [Google Scholar] [CrossRef]
- Shi, C.; Ma, C.; Ma, H.; Zhou, X.; Cao, J.; Fan, Y.; Huang, G. Copper-catalyzed synthesis of 1,3,4-trisubstituted and 1,3,4,5-tetrasubstituted pyrazoles via [3+2] cycloadditions of hydrazones and nitroolefins. Tetrahedron 2016, 72, 4055–4058. [Google Scholar] [CrossRef]
- Molteni, G.; Ponti, A. The nitrilimine-alkene cycloaddition regioselectivity rationalized by density functional theory reactivity indices. Molecules 2017, 22, 202–213. [Google Scholar] [CrossRef]
- Kowalczyk, A.; Utecht-Jarzyńska, G.; Mlostoń, G.; Jasiński, M. Trifluoromethylated Pyrazoles via Sequential (3 + 2)-Cycloaddition of Fluorinated Nitrile Imines with Chalcones and Solvent-Dependent Deacylative Oxidation Reactions. Org. Lett. 2022, 24, 2499–2503. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Gao, C.-F.; Ma, H.; Nie, J.; Ma, J.-A.; Zhang, F.-G. Quadruple Functionalized Pyrazole Pharmacophores by One-pot Regioselective [3+2] Cycloaddition of Fluorinated Nitrile Imines and Dicyanoalkenes. Chem. Asian J. 2022, 17, e202200436. [Google Scholar] [CrossRef]
- Tian, Y.-C.; Li, J.-K.; Zhang, F.-G.; Ma, J.-A. Regioselective Decarboxylative Cycloaddition Route to Fully Substituted 3-CF3-Pyrazoles from Nitrilimines and Isoxazolidinediones. Adv. Synth. Catal. 2021, 363, 2093–2097. [Google Scholar] [CrossRef]
- Ghasempour, L.; Asghari, S.; Tajbakhsh, M.; Mohseni, M. Preparation of New Spiropyrazole, Pyrazole and Hydantoin Derivatives and Investigation of Their Antioxidant and Antibacterial Activities. Chem. Biodivers. 2021, 18, e21001. [Google Scholar] [CrossRef]
- Yavari, I.; Taheri, Z.; Naeimabadi, M.; Bahemmat, S.; Halvag, M.R. A Convenient Synthesis of Tetrasubstituted Pyrazoles from Nitrile Imines and 2-(Thioxothiazolidin-5-ylidene)acetates. Synlett 2018, 29, 918–921. [Google Scholar] [CrossRef]
- Filkina, M.E.; Baray, D.N.; Beloglazkina, E.K.; Grishin, Y.K.; Roznyatovsky, V.A.; Kukushkin, M.E. Regioselective Cycloaddition of Nitrile Imines to 5-Methylidene-3-phenyl-hydantoin: Synthesis and DFT Calculations. Int. J. Mol. Sci. 2023, 24, 1289–1307. [Google Scholar] [CrossRef]
- De Nino, A.; Algieri, V.; Tallarida, M.A.; Costanzo, P.; Pedròn, M.; Tejero, T.; Merino, P.; Maiuolo, L. Regioselective synthesis of 1,4,5,-trisubstituted-1,2,3-triazoles from ary azides and enaminones. Eur. J. Org. Chem. 2019, 2019, 5725–5731. [Google Scholar] [CrossRef]
- Duan, L.; Zhou, H.; Gu, Y.; Gong, P.; Qin, M. The use of enaminones and enamines as effective synthons for MSA-catalyzed regioselective synthesis of 1,3,4-tri- and 1,3,4,5-tetrasubstituted pyrazoles. New. J. Chem. 2019, 43, 16131–16137. [Google Scholar] [CrossRef]
- Maiuolo, L.; Algieri, V.; Olivito, F.; De Nino, A. Recent developments on 1,3-dipolar cycloaddition reactions by catalysis in green solvents. Catalysts 2020, 10, 65–91. [Google Scholar] [CrossRef]
- Maiuolo, L.; Russo, B.; Algieri, V.; Nardi, M.; Di Gioia, M.L.; Tallarida, M.A.; De Nino, A. Regioselective synthesis of 1,5-disubstituted 1,2,3-triazoles by 1,3-dipolar cycloaddition: Role of Er(OTf)3, ionic liquid and water. Tetrahedron Lett. 2019, 60, 672–674. [Google Scholar] [CrossRef]
- De Nino, A.; Maiuolo, L.; Merino, P.; Nardi, M.; Procopio, A.; Roca-López, D.; Russo, B.; Algieri, V. Efficient Organocatalyst Supported on a Simple Ionic Liquid as a Recoverable System for the Asymmetric Diels-Alder Reaction in the Presence of Water. ChemCatChem. 2015, 7, 830–835. [Google Scholar] [CrossRef]
- Nesci, S.; Pagliarani, A. Incoming News on the F-Type ATPase Structure and Functions in Mammalian Mitochondria. BBA Advances 2021, 1, 100001–100003. [Google Scholar] [CrossRef]
- Mnatsakanyan, N.; Llaguno, M.C.; Yang, Y.; Yan, Y.; Weber, J.; Sigworth, F.J.; Jonas, E.A. A Mitochondrial Megachannel Resides in Monomeric F1FO ATP Synthase. Nat. Commun. 2019, 10, 5823–5833. [Google Scholar] [CrossRef]
- Urbani, A.; Giorgio, V.; Carrer, A.; Franchin, C.; Arrigoni, G.; Jiko, C.; Abe, K.; Maeda, S.; Shinzawa-Itoh, K.; Bogers, J.F.M.; et al. Purified F-ATP Synthase Forms a Ca2+-Dependent High-Conductance Channel Matching the Mitochondrial Permeability Transition Pore. Nat. Commun. 2019, 10, 4341–4351. [Google Scholar] [CrossRef] [PubMed]
- Nesci, S.; Pagliarani, A.; Algieri, C.; Trombetti, F. Mitochondrial F-Type ATP Synthase: Multiple Enzyme Functions Revealed by the Membrane-Embedded FO Structure. Crit. Rev. Biochem. Mol. Biol. 2020, 55, 309–321. [Google Scholar] [CrossRef]
- Bonora, M.; Giorgi, C.; Pinton, P. Molecular Mechanisms and Consequences of Mitochondrial Permeability Transition. Nat. Rev. Mol. Cell Biol. 2021, 23, 266–285. [Google Scholar]
- Bernardi, P. Looking Back to the Future of Mitochondrial Research. Front. Physiol. 2021, 12, 682467–682475. [Google Scholar]
- Izzo, V.; Bravo-San Pedro, J.M.; Sica, V.; Kroemer, G.; Galluzzi, L. Mitochondrial Permeability Transition: New Findings and Persisting Uncertainties. Trends Cell Biol. 2016, 26, 655–667. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, P.; Di Lisa, F.; Fogolari, F.; Lippe, G. From ATP to PTP and Back: A Dual Function for the Mitochondrial ATP Synthase. Circ. Res. 2015, 116, 1850–1862. [Google Scholar] [CrossRef]
- Nesci, S. The Mitochondrial Permeability Transition Pore in Cell Death: A Promising Drug Binding Bioarchitecture. Med. Res. Rev. 2020, 40, 811–817. [Google Scholar] [CrossRef] [PubMed]
- Nesci, S.; Trombetti, F.; Algieri, C.; Pagliarani, A. A Therapeutic Role for the F1FO-ATP Synthase. SLAS Discov. 2019, 24, 893–903. [Google Scholar] [CrossRef] [PubMed]
- Pagliarani, A.; Nesci, S.; Ventrella, V. Novel Drugs Targeting the C-Ring of the F1FO-ATP Synthase. Mini Rev. Med. Chem. 2016, 16, 815–824. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, P.; Rasola, A.; Forte, M.; Lippe, G. The Mitochondrial Permeability Transition Pore: Channel Formation by F-ATP Synthase, Integration in Signal Transduction, and Role in Pathophysiology. Physiol. Rev. 2015, 95, 1111–1155. [Google Scholar] [CrossRef]
- Gallagher, N.; Zangh, H.; Junghoefer, T.; Giangrisostomi, E.; Ovsyannikov, R.; Pink, M.; Rajca, S.; Casu, M.B.; Rajca, A. Termally and magnetically robust triplet ground state diradical. J. Am. Chem. Soc. 2019, 141, 4764–4774. [Google Scholar] [CrossRef]
- Zhang, C.-Y.; Liu, X.-H.; Wang, B.-L.; Wang, S.-H.; Li, Z.-M. Synthesis and antifungal activities of new pyrazole derivatives via 1,3-dipolar cycloaddition reaction. Chem. Biol. Drug. Des. 2010, 75, 489–493. [Google Scholar] [CrossRef]
- Dalpozzo, R.; De Nino, A.; Nardi, M.; Russo, B.; Procopio, A. Erbium(III) Triflate: A Valuable Catalyst for the Synthesis of Aldimines, Ketimines, and Enaminones. Synthesis 2006, 7, 1127–1132. [Google Scholar] [CrossRef]
- Xie, C.; Feng, L.; Li, W.; Ma, X.; Ma, X.; Liu, Y.; Ma, C. Efficient synthesis of pyrrolo[1,2-a]quinoxalines catalyzed by a bronsted acid through cleavage of C-C bonds. Org. Biomol. Chem. 2016, 14, 8529–8535. [Google Scholar] [CrossRef]
- Liu, K.; Shang, X.; Cheng, Y.; Chang, X.; Li, P.; Li, W. Regioselective [3+2]-annulation of hydrazonyl chlorides with 1,3-dicarbonyl compounds for assembling of polysubstituted pyrazoles. Org. Biomol. Chem. 2018, 16, 7811–7814. [Google Scholar] [CrossRef] [PubMed]
- Nesci, S.; Ventrella, V.; Trombetti, F.; Pirini, M.; Pagliarani, A. The mitochondrial F1FO-ATPase desensitization to oligomycin by tributyltin is due to thiol oxidation. Biochimie 2014, 97, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Ventrella, V.; Nesci, S.; Trombetti, F.; Bandiera, P.; Pirini, M.; Borgatti, A.R.; Pagliarani, A. Tributyltin inhibits the oligomycin-sensitive Mg-ATPase activity in Mytilus galloprovincialis digestive gland mitochondria. Comp. Biochem. Physiol. Toxicol. Pharmacol. 2011, 153, 75–81. [Google Scholar] [CrossRef]
- Algieri, C.; Trombetti, F.; Pagliarani, A.; Ventrella, V.; Bernardini, C.; Fabbri, M.; Forni, M.; Nesci, S. Mitochondrial Ca2+ -activated F1FO -ATPase hydrolyzes ATP and promotes the permeability transition pore. Ann. N. Y. Acad. Sci. 2019, 1457, 142–157. [Google Scholar] [CrossRef] [PubMed]
- Fiske, C.H.; Subbarow, Y. The colorimetric determination of phosphorus. J. Biol. Chem. 1925, 66, 375–400. [Google Scholar] [CrossRef]
- Cornish-Bowden, A. A simple graphical method for determining the inhibition constants of mixed, uncompetitive and non-competitive inhibitors. Biochem. J. 1974, 137, 143–144. [Google Scholar] [CrossRef]
- Nesci, S.; Algieri, C.; Trombetti, F.; Ventrella, V.; Fabbri, M.; Pagliarani, A. Sulfide affects the mitochondrial respiration, the Ca2+-activated F1FO-ATPase activity and the permeability transition pore but does not change the Mg2+-activated F1FO-ATPase activity in swine heart mitochondria. Pharmacol. Res. 2021, 166, 105495–105504. [Google Scholar] [CrossRef]
- Liu, H.; Jia, H.; Wang, B.; Xiao, Y.; Guo, H. Synthesis of spirobidihydropyrazole through double 1,3-dipolar cycloaddition of nitrilimines with allenoates. Org. Lett. 2017, 19, 4714–4717. [Google Scholar] [CrossRef] [PubMed]
- Jamieson, C.; Livingstone, K. The Nitrile Imine 1,3-Dipole–Properties, Reactivity and Applications; Springer: Berlin, Germany, 2020; pp. 1–152. [Google Scholar]
- Bakavoli, M.; Moeinpour, F.; Davoodnia, A.; Morsali, A. 1,3-dipolar cycloaddition of N-[4-nitrophenyl]-C-[2-furyl] nitrilimine with some dipolarophiles: A combined experimental and theoretical study. J. Mol. Struct. 2010, 969, 139–144. [Google Scholar] [CrossRef]
- Gaspar, F.V.; Azevedo, M.F.M.F.; Carneiro, L.S.A.; Ribeiro, S.B.; Esteves, P.M.; Buarque, C.D. 1,3-Dipolar cycloaddition reactions of enaminones and azides: Synthesis of 4-acyl-1,2,3-triazoles and mechanistic studies. Tetrahedron 2022, 120, 132856–132866. [Google Scholar] [CrossRef]
- Goossens, K.; Lava, K.; Bielawski, C.W.; Binnemans, K. Ionic liquid crystals: Versatile materials. Chem. Rev. 2016, 116, 4643–4807. [Google Scholar] [PubMed]
- Bini, R.; Chiappe, C.; Mestre, V.L.; Pomelli, C.S.; Welton, T. A theoretical study of the solvent effect on Diels-Alder reaction in room temperature ionic liquids using a supramolecular approach. Theor. Chem. Acc. 2009, 123, 347–352. [Google Scholar] [CrossRef]
- Algieri, V.; Algieri, C.; Maiuolo, L.; De Nino, A.; Pagliarani, A.; Tallarida, M.A.; Trombetti, F.; Nesci, S. 1,5-Disubstituted-1,2,3-triazoles as inhibitors of the mitochondrial Ca2+-activated F1Fo-ATP(hydrol)ase and the permeability transition pore. Ann. N. Y. Acad. Sci. 2021, 1485, 43–85. [Google Scholar] [CrossRef]
- Algieri, C.; Bernardini, C.; Marchi, S.; Forte, M.; Tallarida, M.A.; Bianchi, F.; La Mantia, D.; Algieri, V.; Stanzione, R.; Cotugno, M.; et al. 1,5-disubstituted-1,2,3-triazoles counteract mitochondrial dysfunction acting on F1FO-ATPase in models of cardiovascular diseases. Pharmacol. Res. 2023, 187, 106561. [Google Scholar] [CrossRef]
- Neginskaya, M.A.; Solesio, M.E.; Berezhnaya, E.V.; Amodeo, G.F.; Mnatsakanyan, N.; Jonas, E.A.; Pavlov, E.V. ATP Synthase C-Subunit-Deficient Mitochondria Have a Small Cyclosporine A-Sensitive Channel, but Lack the Permeability Transition Pore. Cell Rep. 2019, 26, 11–17. [Google Scholar] [CrossRef]
- Algieri, C.; Trombetti, F.; Pagliarani, A.; Ventrella, V.; Nesci, S. Phenylglyoxal Inhibition of the Mitochondrial F1FO-ATPase Activated by Mg2+ or by Ca2+ Provides Clues on the Mitochondrial Permeability Transition Pore. Arch. Biochem. Bioph. 2020, 681, 108258–108264. [Google Scholar] [CrossRef]
- Nesci, S. Protein Folding and Unfolding: Proline Cis-Trans Isomerization at the c Subunits of F1 FO -ATPase Might Open a High Conductance Ion Channel. Proteins 2022, 90, 2001–2005. [Google Scholar] [CrossRef]
- Nesci, S. What Happens When the Mitochondrial H+-Translocating F1FO-ATP(Hydrol)Ase Becomes a Molecular Target of Calcium? The Pore Opens. Biochimie 2022, 198, 92–95. [Google Scholar] [CrossRef]
- Mnatsakanyan, N.; Park, H.-A.; Wu, J.; He, X.; Llaguno, M.C.; Latta, M.; Miranda, P.; Murtishi, B.; Graham, M.; Weber, J.; et al. Mitochondrial ATP Synthase C-Subunit Leak Channel Triggers Cell Death upon Loss of Its F1 Subcomplex. Cell Death Differ. 2022, 29, 1874–1887. [Google Scholar] [CrossRef]
- Bernardi, P.; Carraro, M.; Lippe, G. The Mitochondrial Permeability Transition: Recent Progress and Open Questions. FEBS J. 2021, 289, 7051–7074. [Google Scholar] [CrossRef] [PubMed]
- Šileikytė, J.; Devereaux, J.; de Jong, J.; Schiavone, M.; Jones, K.; Nilsen, A.; Bernardi, P.; Forte, M.; Cohen, M.S. Second-Generation Inhibitors of the Mitochondrial Permeability Transition Pore with Improved Plasma Stability. Chem. Med. Chem 2019, 14, 1771–1782. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry a | Solvent b | Base | T (°C) | t (h) | Yield (%) c | 6a:7a e |
|---|---|---|---|---|---|---|
| 1 | [mPy](OTf)/H2O | Et3N | rt | 2 | 50 | 90:10 |
| 2 | [mPy](OTf)/H2O | Et3N | rt | 24 | 51 | 80:20 |
| 3 | [mPy](OTf)/H2O | Et3N | 50 | 2 | 67 | 96:4 |
| 4 | [mPy](OTf)/H2O | Et3N | 85 | 2 | 32 | 95:5 |
| 5 | [mPy](OTf)/H2O | DBU | 50 | 2 | 42 | 96:4 |
| 6 | [mPy](OTf)/H2O | DMAP | 50 | 2 | 38 | 96:4 |
| 7 | [mPy](OTf)/H2O | NaOH | 50 | 2 | traces | - |
| 8 | [mPy](OTf)/H2O | K2CO3 | rt | 2 | 72 | 96:4 |
| 9 | [mPy](OTf)/H2O | K2CO3 | 50 | 2 | 85 | 96:4 |
| 10 d | [mPy](OTf)/H2O | K2CO3 | 50 | 2 | 90 | 97:3 |
| 11 d | [Bmim]Cl/H2O | K2CO3 | 50 | 2 | 75 | 95:5 |
| 12 d | [Bmim][BF4]/H2O | K2CO3 | 50 | 2 | 78 | 96:4 |
| 13 d | DMF | K2CO3 | 50 | 2 | 52 | 76:24 |
| 14 d | DMSO | K2CO3 | 50 | 2 | 64 | 82:18 |

| Entry a | Hydrazonyl Chloride | X | Enaminone | R1 | R2 | Product | t (h) | Yield 6a–6o (%) b | 6:7 c |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 4a | H | 5a | CH3 | CH3 | 6a | 2 | 90 | 97:3 |
| 2 | 4b | 4-OCH3 | 5a | CH3 | CH3 | 6b | 2 | 95 | 97:3 |
| 3 | 4c | 4-CH3 | 5a | CH3 | CH3 | 6c | 2 | 94 | 96:4 |
| 4 | 4d | 4-NO2 | 5a | CH3 | CH3 | 6d | 2 | 90 | 95:5 |
| 5 | 4e | 2-Cl | 5a | CH3 | CH3 | 6e | 2 | 92 | 96:4 |
| 6 | 4f | 3-Cl | 5a | CH3 | CH3 | 6f | 2 | 90 | 94:6 |
| 7 | 4g | 4-Cl | 5a | CH3 | CH3 | 6g | 2 | 92 | 98:2 |
| 8 | 4a | H | 5b | Ph | CH3 | 6h | 4 | 81 | 93:7 |
| 9 | 4b | 4-OCH3 | 5b | Ph | CH3 | 6i | 4 | 83 | 96:4 |
| 10 | 4c | 4-CH3 | 5b | Ph | CH3 | 6j | 4 | 82 | 96:4 |
| 11 | 4d | 4-NO2 | 5b | Ph | CH3 | 6k | 4 | 80 | 95:5 |
| 12 | 4e | 2-Cl | 5b | Ph | CH3 | 6l | 4 | 82 | 95:5 |
| 13 | 4f | 3-Cl | 5b | Ph | CH3 | 6m | 4 | 84 | 96:4 |
| 14 | 4g | 4-Cl | 5b | Ph | CH3 | 6n | 4 | 81 | 96:4 |
| 15 | 4a | H | 5c | Ph | Ph | 6o | 4 | 76 | 94:6 |
| Entry a | Compound | IC50 (mM) | K’i (mM) |
|---|---|---|---|
| 1 | 6a | 0.25 ± 0.01 | 0.21 ± 0.03 |
| 2 | 6h | 1.62 ± 0.85 | 1.27 ± 0.25 |
| 3 | 6o | 0.21 ± 0.15 | 1.86 ± 0.33 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Algieri, V.; Algieri, C.; Costanzo, P.; Fiorani, G.; Jiritano, A.; Olivito, F.; Tallarida, M.A.; Trombetti, F.; Maiuolo, L.; De Nino, A.; et al. Novel Regioselective Synthesis of 1,3,4,5-Tetrasubstituted Pyrazoles and Biochemical Valuation on F1FO-ATPase and Mitochondrial Permeability Transition Pore Formation. Pharmaceutics 2023, 15, 498. https://doi.org/10.3390/pharmaceutics15020498
Algieri V, Algieri C, Costanzo P, Fiorani G, Jiritano A, Olivito F, Tallarida MA, Trombetti F, Maiuolo L, De Nino A, et al. Novel Regioselective Synthesis of 1,3,4,5-Tetrasubstituted Pyrazoles and Biochemical Valuation on F1FO-ATPase and Mitochondrial Permeability Transition Pore Formation. Pharmaceutics. 2023; 15(2):498. https://doi.org/10.3390/pharmaceutics15020498
Chicago/Turabian StyleAlgieri, Vincenzo, Cristina Algieri, Paola Costanzo, Giulia Fiorani, Antonio Jiritano, Fabrizio Olivito, Matteo Antonio Tallarida, Fabiana Trombetti, Loredana Maiuolo, Antonio De Nino, and et al. 2023. "Novel Regioselective Synthesis of 1,3,4,5-Tetrasubstituted Pyrazoles and Biochemical Valuation on F1FO-ATPase and Mitochondrial Permeability Transition Pore Formation" Pharmaceutics 15, no. 2: 498. https://doi.org/10.3390/pharmaceutics15020498
APA StyleAlgieri, V., Algieri, C., Costanzo, P., Fiorani, G., Jiritano, A., Olivito, F., Tallarida, M. A., Trombetti, F., Maiuolo, L., De Nino, A., & Nesci, S. (2023). Novel Regioselective Synthesis of 1,3,4,5-Tetrasubstituted Pyrazoles and Biochemical Valuation on F1FO-ATPase and Mitochondrial Permeability Transition Pore Formation. Pharmaceutics, 15(2), 498. https://doi.org/10.3390/pharmaceutics15020498