

Resensitisation of Methicillin-Resistant Staphylococcus aureus to Conventional Antibiotics in the Presence of an Engineered Enzybiotic

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains and Growth Conditions
2.2. Software Used for Protein Structure Prediction
2.3. PCR Amplification of Genes
2.4. Gene Cloning and Synthesis of Recombinant Protein
2.5. Purification and Refolding of the Synthesized Proteins
2.6. Cell-Binding Activity of BAC100
2.7. Live/Dead Staining and Confocal Microscopy
2.8. Software Used for Graph Preparation and Statistical Analysis of the Data
3. Results
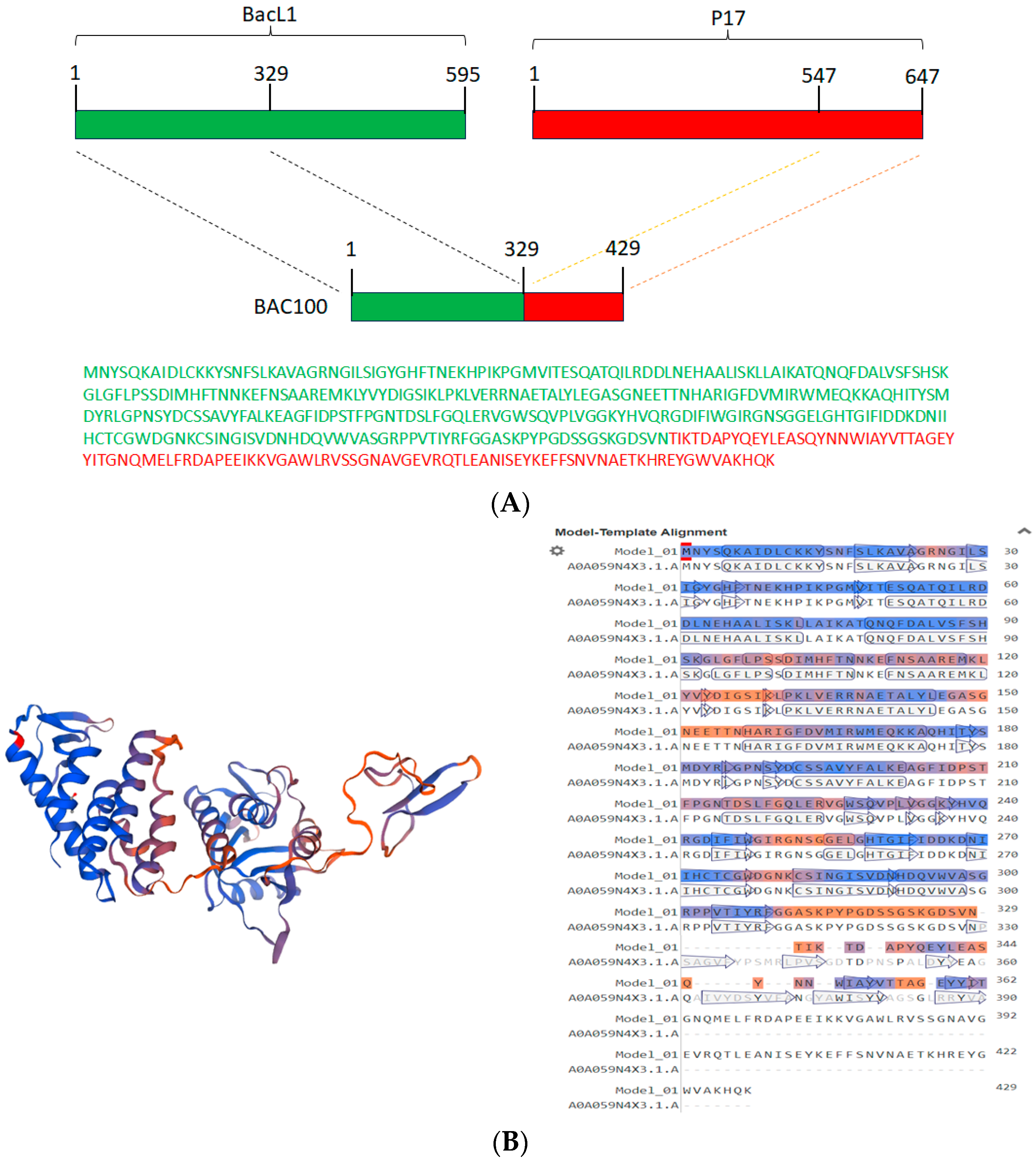
3.1. Construction of BAC100 Protein
3.2. Synthesis and Purification of BAC100 Protein
3.3. BAC100 Displays Cell Wall Binding to MRSA Clinical Isolates
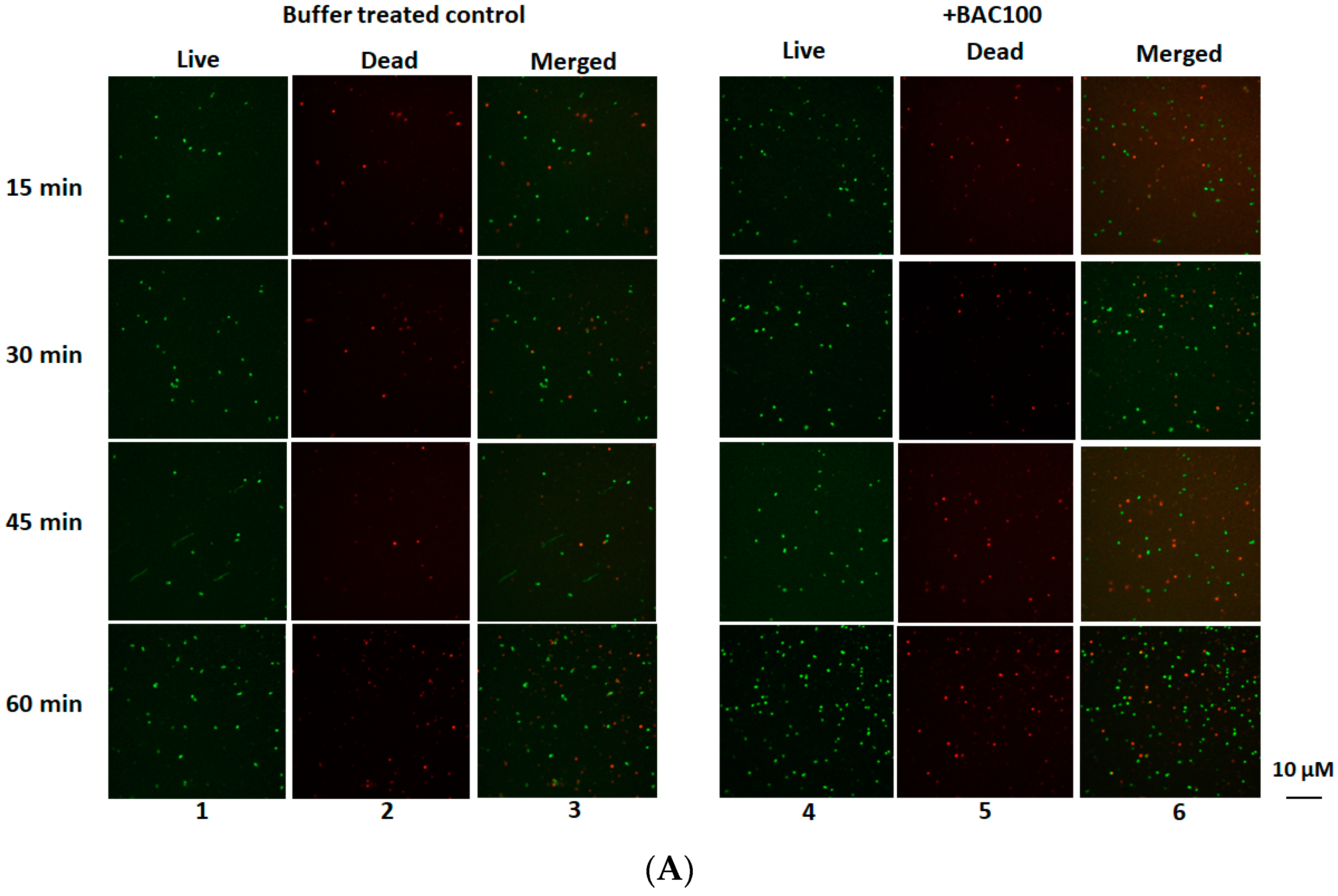
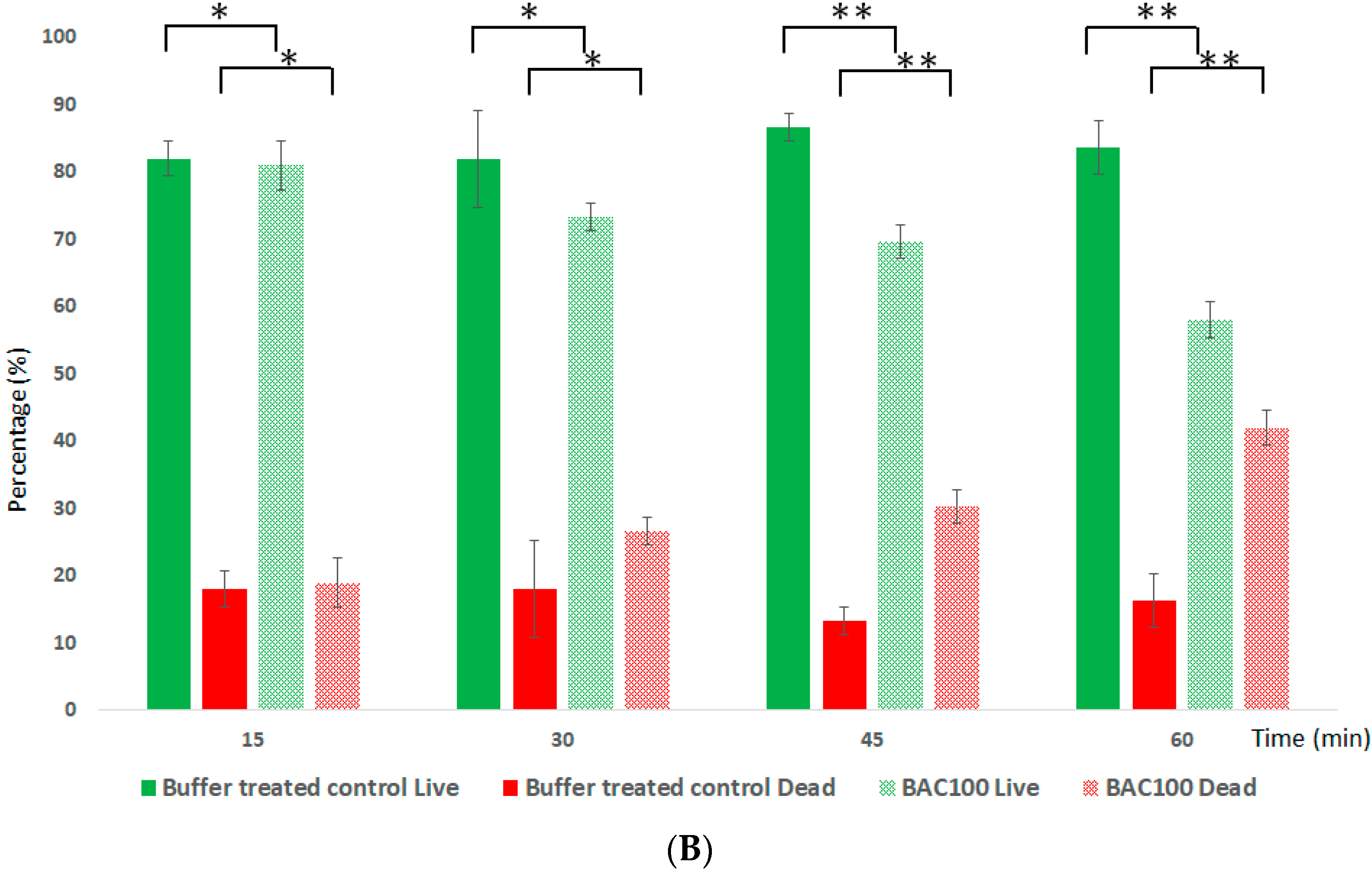
3.4. BAC100 Displays Activity against MRSA Clinical Isolate
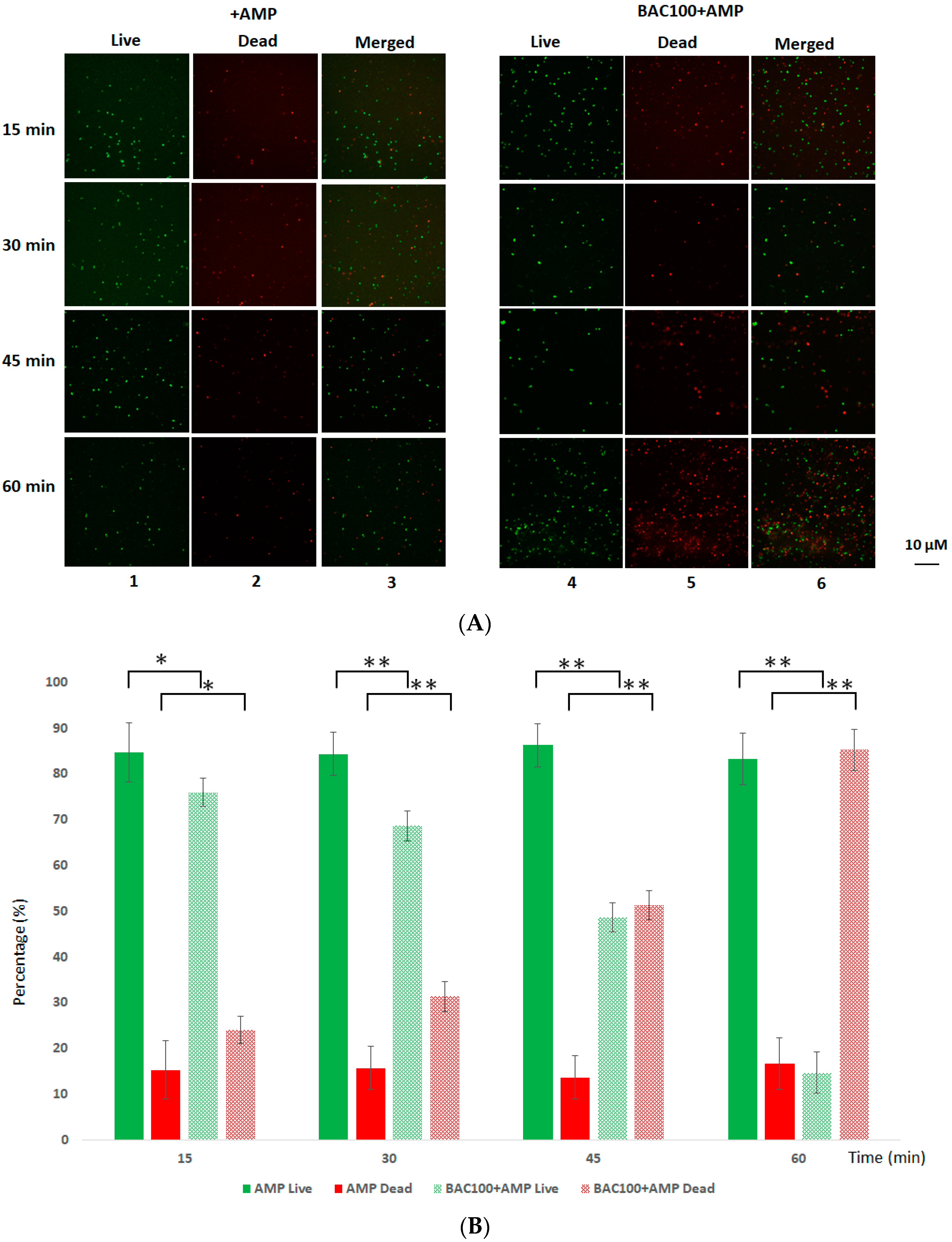
3.5. BAC100 Resensitises MRSA to Ampicillin
3.6. Tetracycline Actively Annihilates MRSA in the Presence of BAC100
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mulani, M.S.; Kamble, E.E.; Kumkar, S.N.; Tawre, M.S.; Pardesi, K.R. Emerging Strategies to Combat ESKAPE Pathogens in the Era of Antimicrobial Resistance: A Review. Front. Microbiol. 2019, 10, 539. [Google Scholar] [CrossRef]
- Tacconelli, E.; Carrara, E.; Savoldi, A.; Harbarth, S.; Mendelson, M.; Monnet, D.L.; Pulcini, C.; Kahlmeter, G.; Kluytmans, J.; Carmeli, Y.; et al. Discovery, research, and development of new antibiotics: The WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis. 2018, 18, 318–327. [Google Scholar] [CrossRef]
- Turner, N.A.; Sharma-Kuinkel, B.K.; Maskarinec, S.A.; Eichenberger, E.M.; Shah, P.P.; Carugati, M.; Holland, T.L.; Fowler, V.G., Jr. Methicillin-resistant Staphylococcus aureus: An overview of basic and clinical research. Nat. Rev. Microbiol. 2019, 4, 203–218. [Google Scholar] [CrossRef]
- Malachowa, N.; DeLeo, F.R. Mobile genetic elements of Staphylococcus aureus. Cell. Mol. Life Sci. 2010, 67, 3057–3071. [Google Scholar] [CrossRef] [PubMed]
- Mandal, S.M.; Roy, A.; Ghosh, A.K.; Hazra, T.K.; Basak, A.; Franco, O.L. Challenges and future prospects of antibiotic therapy: From peptides to phages utilization. Front. Pharmacol. 2014, 5, 105. [Google Scholar] [CrossRef]
- Kaur, I. Novel strategies to combat antimicrobial resistance. J. Infect. Dis. Ther. 2016, 4, 292. [Google Scholar] [CrossRef]
- Coronado-Álvarez, N.M.; Parra, D.; Parra-Ruiz, J. Clinical efficacy of fosfomycin combinations against a variety of gram-positive cocci. Enferm. Infecc. Microbiol. Clin. 2018, 37, 4–10. [Google Scholar] [CrossRef]
- Vestergaard, M.; Paulander, W.; Marvig, R.L.; Clasen, J.; Jochumsen, N.; Molin, S.; Jelsbak, L.; Ingmer, H.; Folkesson, A. Antibiotic combination therapy can select for broad-spectrum multidrug resistance in Pseudomonas aeruginosa. Int. J. Antimicrob. Agents 2016, 47, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Durante-Mangoni, E.; Signoriello, G.; Andini, R.; Mattei, A.; De Cristoforo, M.; Murino, P.; Bassetti, M.; Malacarne, P.; Petrosillo, N.; Galdieri, N.; et al. Colistin and rifampicin compared with colistin alone for the treatment of serious infections due to extensively drug-resistant Acinetobacter baumannii: A multicenter, randomized clinical trial. Clin. Infect. Dis. 2013, 57, 349–358. [Google Scholar] [CrossRef]
- Wang, G.; Li, X.; Zasloff, M. A database view of naturally occurring antimicrobial peptides: Nomenclature, classification and amino acid sequence analysis. In Antimicrobial Peptides: Discovery, Design and Novel Therapeutic Strategies; Wang, G., Ed.; CABI: Oxfordshire, UK, 2010; pp. 1–21. [Google Scholar]
- Berglund, N.A.; Piggot, T.J.; Jefferies, D.; Sessions, R.B.; Bond, P.J.; Khalid, S. Interaction of the antimicrobial peptide polymyxin B1 with both membranes of E. coli: A molecular dynamics study. PLoS Comput. Biol. 2015, 11, e1004180. [Google Scholar] [CrossRef]
- Pfalzgraff, A.; Brandenburg, K.; Weindl, G. Antimicrobial peptides and their therapeutic potential for bacterial skin infections and wounds. Front. Pharmacol. 2018, 9, 281. [Google Scholar] [CrossRef] [PubMed]
- Duong, L.; Gross, S.P.; Siryaporn, A. Developing antimicrobial synergy with AMPs. Front. Med. Technol. 2021, 3, 640981. [Google Scholar] [CrossRef]
- Zhu, Y.; Hao, W.; Wang, X.; Ouyang, J.; Deng, X.; Yu, H.; Wang, Y. Antimicrobial peptides, conventional antibiotics, and their synergistic utility for the treatment of drug-resistant infections. Med. Res. Rev. 2022, 42, 1377–1422. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.; Rolff, J.; Friedman, J.; Hayouka, Z. Antimicrobial peptide combination can hinder resistance evolution. Microbiol. Spectr. 2022, 10, e00973-22. [Google Scholar] [CrossRef]
- Latz, S.; Wahida, A.; Arif, A.; Häfner, H.; Hoß, M.; Ritter, K.; Horz, H.P. Preliminary survey of local bacteriophages with lytic activity against multi-drug resistant bacteria. J. Basic Microbiol. 2016, 56, 1117–1123. [Google Scholar] [CrossRef]
- Domingo-Calap, P.; Delgado-Martínez, J. Bacteriophages: Protagonists of a post-antibiotic era. Antibiotics 2018, 7, 66. [Google Scholar] [CrossRef]
- Dvořáčková, M.; Růžička, F.; Benešík, M.; Pantůček, R.; Dvoráková-Heroldová, M. Antimicrobial effect of commercial phage preparation Stafal® on biofilm and planktonic forms of methicillin-resistant Staphylococcus aureus. Folia Microbiol. 2018, 64, 121–126. [Google Scholar] [CrossRef]
- Chen, J.; Novick, R.P. Phage-mediated intergeneric transfer of toxin genes. Science 2009, 323, 139–141. [Google Scholar] [CrossRef]
- Malik, D.J.; Sokolov, I.J.; Vinner, G.K.; Mancuso, F.; Cinquerrui, S.; Vladisavljevic, G.T.; Clokie, M.R.J.; Garton, N.J.; Stapley, A.G.F.; Kirpichnikova, A. Formulation, stabilisation and encapsulation of bacteriophage for phage therapy. Adv. Colloid Interface Sci. 2017, 249, 100–133. [Google Scholar] [CrossRef]
- Cooper, C.J.; Koonjan, S.; Nilsson, A.S. Enhancing whole phage therapy and their derived antimicrobial enzymes through complex formulation. Pharmaceuticals 2018, 11, 34. [Google Scholar] [CrossRef]
- Dams, D.; Briers, Y. Enzybiotics: Enzyme-Based Antibacterials as Therapeutics. Adv. Exp. Med. Biol. 2019, 1148, 233–253. [Google Scholar] [PubMed]
- Manoharadas, S.; Altaf, M.; Alrefaei, A.F.; Ahmad, N.; Althaf Hussain, S.; Al-Rayes, B.F. An Engineered Multimodular Enzybiotic against Methicillin-Resistant Staphylococcus aureus. Life 2021, 11, 1384. [Google Scholar] [CrossRef] [PubMed]
- Manoharadas, S.; Altaf, M.; Ahmad, N.; Alrefaei, A.F.; Al-Rayes, B.F. Construction and Activity Testing of a Modular Fusion Peptide against Enterococcus faecalis. Antibiotics 2023, 12, 388. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Tomita, H.; Kamei, E.; Ike, Y. Cloning and Genetic Analyses of the Bacteriocin 41 Determinant Encoded on the Enterococcus faecalis Pheromone-Responsive Conjugative Plasmid pYI14: A Novel Bacteriocin Complemented by Two Extracellular Components (Lysin and Activator). J. Bacteriol. 2008, 190, 2075–2085. [Google Scholar] [CrossRef] [PubMed]
- Vybiral, D.; Takác, M.; Loessner, M.; Witte, A.; von Ahsen, U.; Bläsi, U. Complete nucleotide sequence and molecular characterization of two lytic Staphylococcus aureus phages: 44AHJD and P68. FEMS Microbiol Lett. 2003, 219, 275–283. [Google Scholar] [CrossRef]
- Manoharadas, S.; Altaf, M.; Alrefaei, A.W.F.; Devasia, R.M.; Hadj, A.Y.M.B.; Abuhasil, M.S. Concerted dispersion of Staphylococcus aureus biofilm by bacteriophage and ‘green synthesized’ silver nanoparticles. RSC Adv. 2021, 11, 1420–1429. [Google Scholar] [CrossRef]
- Manoharadas, S.; Witte, A.; Bläsi, U. Antimicrobial activity of a chimeric enzybiotic towards Staphylococcus aureus. J. Biotechnol. 2009, 139, 118–123. [Google Scholar] [CrossRef]
- Manoharadas, S.; Ahmad, N.; Altaf, M.; Alrefaei, A.F.; Al-Rayes, B.F. An Enzybiotic Cocktail Effectively Disrupts Preformed Dual Biofilm of Staphylococcus aureus and Enterococcus faecalis. Pharmaceuticals 2023, 16, 564. [Google Scholar] [CrossRef]
- Foxley, M.A.; Friedline, A.W.; Jensen, J.M.; Nimmo, S.L.; Scull, E.M.; King, J.B.; Strange, S.; Xiao, M.T.; Smith, B.E.; Thomas, K.J., III; et al. Efficacy of ampicillin against methicillin-resistant Staphylococcus aureus restored through synergy with branched poly(ethylenimine). J. Antibiot. 2016, 69, 871–878. [Google Scholar] [CrossRef]
- Watkins, R.R.; Holubar, M.; David, M.Z. Antimicrobial resistance in methicillin-resistant Staphylococcus aureus to newer antimicrobial agents. Antimicrob. Agents Chemother. 2019, 63, e01216-19. [Google Scholar] [CrossRef]
- World Health Organization. Antimicrobial Resistance: Global Report on Surveillance. 2014. Available online: https://apps.who.int/iris/handle/10665/112642 (accessed on 17 August 2023).
- Lowy, F.D. Antimicrobial resistance: The example of Staphylococcus aureus. J. Clin. Invest. 2003, 111, 1265–1273. [Google Scholar] [CrossRef]
- Barber, M. Methicillin-resistant staphylococci. J. Clin. Pathol. 1961, 14, 385–393. [Google Scholar] [CrossRef]
- Musser, J.M.; Kapur, V. Clonal analysis of methicillin-resistant Staphylococcus aureus strains from intercontinental sources: Association of the mec gene with divergent phylogenetic lineages implies dissemination by horizontal transfer and recombination. J. Clin. Microbiol. 1992, 30, 2058–2063. [Google Scholar] [CrossRef]
- Berger-Bächi, B.; Tschierske, M. Role of fem factors in methicillin resistance. Drug Resist. Updates 1998, 1, 325–335. [Google Scholar] [CrossRef]
- Kim, C.K.; Milheirico, C.; de Lencastre, H.; Tomasz, A. Antibiotic resistance as a stress response: Recovery of high-level oxacillin resistance in methicillin-resistant Staphylococcus aureus “auxiliary” (fem) mutants by induction of the stringent stress response. Antimicrob. Agents Chemother. 2017, 30, e00313-17. [Google Scholar] [CrossRef]
- Murthy, M.H.; Olson, M.E.; Wickert, R.W.; Fey, P.D.; Jalali, Z. Daptomycin non-susceptible meticillin-resistant Staphylococcus aureus USA 300 isolate. J. Med. Microbiol. 2008, 57, 1036–1038. [Google Scholar] [CrossRef]
- Mammina, C.; Bonura, C.; di Carlo, P.; Cala, C.; Aleo, A.; Monastero, R.; Palma, D.M. Daptomycin non-susceptible, vancomycin intermediate methicillin-resistant Staphylococcus aureus ST398 from a chronic leg ulcer, Italy. Scand. J. Infect. Dis. 2010, 42, 955–957. [Google Scholar] [CrossRef]
- Nguyen, F.; Starosta, A.L.; Arenz, S.; Sohmen, D.; Donhofer, A.; Wilson, D.N. Tetracycline antibiotics and resistance mechanisms. Biol. Chem. 2014, 395, 559–575. [Google Scholar] [CrossRef]
- Schwartz, B.S.; Graber, C.J.; Diep, B.A.; Basuino, L.; Perdreau-Remington, F.; Chambers, H.F. Doxycycline, not minocycline, induces its own resistance in multidrug-resistant, community-associated methicillin-resistant Staphylococcus aureus clone USA300. Clin. Infect. Dis. 2009, 48, 1483–1484. [Google Scholar] [CrossRef]
- Peechakara, B.V.; Basit, H.; Gupta, M. Ampicillin. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2023. Available online: https://www.ncbi.nlm.nih.gov/books/NBK519569/ (accessed on 17 August 2023).
- Fishovitz, J.; Hermoso, J.A.; Chang, M.; Mobashery, S. Penicillin-binding protein 2a of methicillin-resistant Staphylococcus aureus. IUBMB Life 2014, 66, 572–577. [Google Scholar] [CrossRef]
- Chopra, I.; Roberts, M. Tetracycline antibiotics: Mode of action, applications, molecular biology, and epidemiology of bacterial resistance. Microbiol. Mol. Biol. Rev. 2001, 65, 232–260. [Google Scholar] [CrossRef]
- Gillespie, M.T.; May, J.W.; Skurray, R. Detection of an integrated tetracycline resistance plasmid in the chromosome of methicillin-resistant Staphylococcus aureus. J. Gen. Microbiol. 1986, 132, 1723–1728. [Google Scholar] [CrossRef]
- Horaud, T.; de Cerspedes, G.; Clermont, D.; David, F.; Delbos, F. Variability of chromosomal genetic elements in streptococci. In Genetics and Molecular Biology of Streptococci, Lactococci and Enterococci; Dunny, G.M., Cleary, P.P., McKay, L.L., Eds.; American Society for Microbiology: Washington, DC, USA, 1991; pp. 16–20. [Google Scholar]
- Shang, D.; Liu, Y.; Jiang, F.; Ji, F.; Wang, H.; Han, X. Synergistic antibacterial activity of designed Trp-containing antibacterial peptides in combination with antibiotics against multidrug-resistant Staphylococcus epidermidis. Front. Microbiol. 2019, 10, 2719. [Google Scholar] [CrossRef]
- Sechet, E.; Telford, E.; Bonamy, C.; Sansonetti, P.J.; Sperandio, B. Natural molecules induce and synergize to boost expression of the human antimicrobial peptide β-defensin-3. Proc. Natl. Acad. Sci. USA 2018, 115, E9869–E9878. [Google Scholar] [CrossRef]
- Dos Santos, C.; Dos Santos, L.S.; Franco, O.L. Fosfomycin and nitrofurantoin: Classic antibiotics and perspectives. J. Antibiot. 2021, 74, 547–558. [Google Scholar] [CrossRef]
- Lin, D.; Koskella, B.; Lin, H.C. Phage therapy: An alternative to antibiotics in the age of multi-drug resistance. World J. Gastrointest. Pharmacol. Ther. 2017, 8, 162–173. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manoharadas, S.; Al-Rayes, B.F.; Almuzaini, M.A.M.; Almohammadi, Y.M. Resensitisation of Methicillin-Resistant Staphylococcus aureus to Conventional Antibiotics in the Presence of an Engineered Enzybiotic. Pharmaceutics 2023, 15, 2511. https://doi.org/10.3390/pharmaceutics15102511
Manoharadas S, Al-Rayes BF, Almuzaini MAM, Almohammadi YM. Resensitisation of Methicillin-Resistant Staphylococcus aureus to Conventional Antibiotics in the Presence of an Engineered Enzybiotic. Pharmaceutics. 2023; 15(10):2511. https://doi.org/10.3390/pharmaceutics15102511
Chicago/Turabian StyleManoharadas, Salim, Basel F. Al-Rayes, Mohammed Abdulaziz M. Almuzaini, and Yasser Muteq Almohammadi. 2023. "Resensitisation of Methicillin-Resistant Staphylococcus aureus to Conventional Antibiotics in the Presence of an Engineered Enzybiotic" Pharmaceutics 15, no. 10: 2511. https://doi.org/10.3390/pharmaceutics15102511
APA StyleManoharadas, S., Al-Rayes, B. F., Almuzaini, M. A. M., & Almohammadi, Y. M. (2023). Resensitisation of Methicillin-Resistant Staphylococcus aureus to Conventional Antibiotics in the Presence of an Engineered Enzybiotic. Pharmaceutics, 15(10), 2511. https://doi.org/10.3390/pharmaceutics15102511