1. Introduction
Chronic liver disease (CLD) is responsible for more than 2 million annual deaths worldwide [
1]. Progressive CLD is caused by the continuous production and deposition of extracellular matrix components resulting in significant hepatic fibrosis and nodular distribution of the liver parenchyma, leading to organic dysfunction and liver failure [
2,
3]. Portal hypertension is an important complication of advanced CLD and is the consequence of a marked increase in intrahepatic vascular resistance (IHVR) together with an increased hepatic vascular tone [
4]. This increased vascular tone is strongly related to alterations in liver sinusoidal endothelial cells (LSECs) that, upon chronic injury, exhibit an imbalance of vasoactive molecules favoring vasoconstriction and increasing IHVR.
LSECs play a major role as sensors and drivers of CLD [
5]. Under physiological conditions, LSECs are responsible for maintaining hepatic homeostasis, metabolite transport and vascular tone, enhancing hepatocytes exposure to macromolecules from the portal circulation. Furthermore, LSECs possess anti-inflammatory and anti-fibrogenic properties by preventing the activation of Kupffer cells and hepatic stellate cells (HSCs) [
6]. However, under pathological conditions, LSECs dedifferentiate, becoming a common vascular endothelial phenotype and losing specialized functions [
7,
8]. Therefore, it is crucial to maintain the LSEC specialized phenotype to preserve hepatic function; this might be achieved by means of the direct delivery of vasoprotective drugs.
Statins, originally designed as inhibitors of the enzyme 3-hydroxy-3-methylglutaryl-co-enzyme A reductase (HMG-CoA reductase) of the cholesterol biosynthesis pathway [
9,
10], have been widely used for lipid-lowering purposes with a good safety profile in cardiovascular diseases. However, although the beneficial effects of statins in the cirrhotic liver have been demonstrated, their use in CLD has not yet been consolidated. Furthermore, different animal models of cirrhosis have shown that the use of statins (e.g., simvastatin, atorvastatin) exerts vasoprotective and antifibrotic effects, improving LSEC dysfunction, reducing HSC activation and achieving hepatic fibrosis regression. These remarkable beneficial effects were mainly dependent on the up-regulation of Krüppel-like factor 2 (KLF2) in hepatic cells after statins treatment [
11,
12] by enhancing the expression of their target genes, such as eNOS (endothelial nitric oxide synthase), or inhibiting the vasoconstrictive RhoA/ROCK (Ras homolog family member A/Rho-associated protein kinase) axis, among other pathways [
13,
14]. However, statins are not free of adverse effects, mainly including muscular but also hepatic toxicity [
15,
16]. As an example, in a previous study in a rat model with bile elimination impairment (bile duct ligation model, BDL) mimicking severely impaired liver function, the adverse events of simvastatin were magnified, reaching significant levels of mortality when using high doses [
13]. Therefore, the dose-dependent side effects of statins limit their efficacy, especially in populations with a higher risk of toxicity, such as patients with advanced CLD [
17].
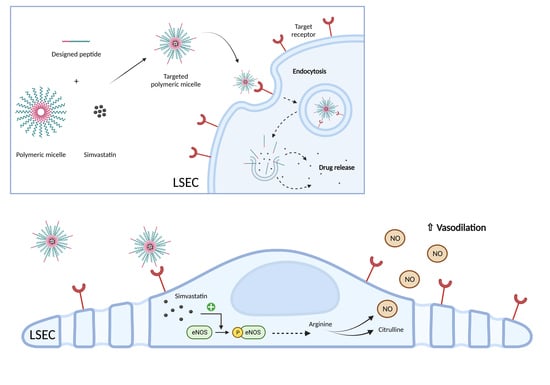
To overcome this limitation, we produced Pluronic-based polymeric micelles (PMs), loaded with chemically activated simvastatin [
18]. Briefly, the nanoparticle biodistribution was mainly hepatic and no signs of toxicity were detected. After one week of administration in the BDL model, these nanoparticles demonstrated superior effects compared to free simvastatin in reducing portal pressure, with a significant nanoparticle accumulation in LSECs, although they were also found in Kupffer cells, suggesting a possible loss of efficacy due to their clearance by macrophages. Therefore, it was proposed that an improvement to the formulation by means of functionalization with specific ligands was needed to increase the effect of nanoparticles in LSECs in a more specific manner [
19].
One of the outstanding properties of Pluronic PMs is their ability to be functionalized by adding peptide or protein molecules for active targeting to their hydrophilic surface to enhance drug delivery to specific sites in the body or specific receptors on cells. By doing this, the therapeutic efficacy of the cargo can be maximized while reducing systemic toxicity compared with untargeted micelles [
20]. When the ligands conjugated to PMs bind to their specific receptors on the cell membrane, endocytic internalization of those nanoparticles is promoted. Interestingly, LSECs have one of the highest endocytic capacities in the human body, providing an advantage for specific targeting [
21].
This study explores the development of nanoparticles specifically targeting LSECs for the delivery of simvastatin. We tested different peptides recognizing the binding sites of (i) the specific LSEC Fcγ-receptor IIb, expressed mainly in physiological conditions, (ii) the transmembrane glycoprotein thrombospondin receptor, unaltered during LSEC capillarization and (iii) the integrin αVβ3 receptor, highly expressed in dysfunctional LSECs.
2. Material and Methods
2.1. Reagents
Fluorenylmethyloxycarbonyl protecting group (Fmoc)-L-amino acids were obtained from CSBio (Menlo Park, CA, USA) or Matrix Innovation Inc. (Quebec, QC, Canada). O-(1H-6-chlorobenzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HCTU) was obtained from Luxembourg Bio Technologies Ltd. (Ness Ziona, Israel). Solvents for peptide synthesis such as piperidine, N,N-dimethylformamide (DMF), N,N-diisopropylethylamine (DIPEA), dichloromethane (DCM), trifluoroacetic acid (TFA), trisopropylsilane and acetonitrile were purchased from Bio-Lab Ltd. (Jerusalem, Israel). Ultrapure guanidinium chloride (GdmCl; Apollo Scientific Ltd., Stockport, UK) used in the buffer for dissolving the peptides. Pluronic F127 was provided by BASF (Ludwigshafen, Germany). Pure simvastatin, fluorescein 5-isothiocyanate (FITC), 5-([4,6-dichlorotriazin-2-yl])aminofluorescein (5-DTAF), maleic anhydride, 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC), 4′,6-diamidino-2-phenylindole (DAPI) and propidium iodide solution were obtained from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany). Other reagents such as the culture media, phosphate-buffered saline (PBS), trypsin, fetal bovine serum (FBS) and bovine serum albumin (BSA), were provided by Biowest (Nuaillé, France). All TaqMan probes were obtained from Applied Biosystems (Thermo Fisher Scientific, Waltham, MA, USA).
2.2. Animals
All animal procedures were conducted in accordance with European Union legislation on the protection of animals used for scientific purposes (Directive 2010/63/EU revising Directive 86/609/EEC), approved by the Animal Research Ethics Committee of the Vall d’Hebron Institut de Recerca (Barcelona, Spain).
Male Sprague Dawley CD rats (Charles River Laboratories, Saint-Germain-sur-l’Arbresle, France) were used to generate two models of CLD with portal hypertension and cirrhosis; intrahepatic portal hypertension was induced via secondary biliary cirrhosis, performing a common BDL for a period of 4 weeks on rats weighing 200–220 g, or via intraperitoneal injection of 250 mg/kg of thioacetamide (TAA) 2 days/week over a period of 8 weeks to rats weighing 125–150 g at baseline and weighed weekly to adjust the dose to the weight gain. Only male rats were used because female estrogen hormones modify hemodynamic parameters, increasing or decreasing their values randomly and unrelated to liver disease [
22]. All animals were housed under a constant temperature of 22 ± 2 °C and 50% humidity in a controlled 12/12 h light/dark cycle. They were fed ad libitum with a grain-based chow (SAFE 150; SAFE Complete Care Competence, Rosenberg, Germany) and had free access to water.
In addition, male BALB/cAnNRj mice (Janvier Labs, Le Genest-Saint-Isle, France) aged 5–6 weeks were used for the maximum tolerated dose assay performed at Cellvax facilities (Villejuif, France). Animal health status was specific and opportunistic pathogen-free (SOPF), and they were housed in polyethylene cages (<5 mice/cage) in a climate- and light-controlled environment in accordance with Cellvax’s approved standard operating procedures.
2.3. Simvastatin Activation
Prior to encapsulation or in vitro treatment, commercial pure simvastatin needs to be activated from its inactive lactone form to its β-hydroxyacid active form [
23]. For this purpose, 8 mg of simvastatin (0.019 mM) was dissolved in 0.2 mL of pure ethanol, with the subsequent addition of 0.3 mL of 0.1 N sodium hydroxide (NaOH). The solution was heated at 50 °C for 2 h and neutralized with hydrochloric acid (HCl) to pH 7.0. The resulting solution was brought to a final volume of 1 mL with distilled water, and aliquots were stored at −80 °C until use or lyophilized.
2.4. Synthesis of Peptide Ligands
All peptides were prepared via the solid-phase peptide synthesis (SPPS) technique [
24] using standard Fmoc-
L-amino acids on a CS136X peptide synthesizer (CSBio, Mountain View, CA, USA). Fmoc deprotection was performed twice using 20% piperidine in DMF for 5 min. The carboxylic acid functional group (-COOH) of each amino acid was activated with the coupling reagent HCTU in the presence of DIPEA for 5 min and added to the resin for coupling with constant shaking for 30 min at room temperature (RT). The resin was washed -three times with DMF and DCM, dried and cleaved with a cocktail of TFA/trisopropylsilane/water (ratio 94:3:3). After cleavage, the resin was removed by means of filtration, washed twice with neat TFA and bubbled with nitrogen for TFA removal. Afterwards, cold diethyl ether was added to precipitate the peptide, centrifuged (4000 rpm, 10 min) and ether was decanted. Cleaved peptide was dissolved in 0.1% TFA in GdmCl and lyophilized.
For the synthesis of fluorescently labelled peptides, Fmoc-L-Lys(Alloc)-OH was coupled on the C-terminus of the peptide chain to label it with FITC, allowing the free amine (NH2) of Lys to react with the fluorochrome.
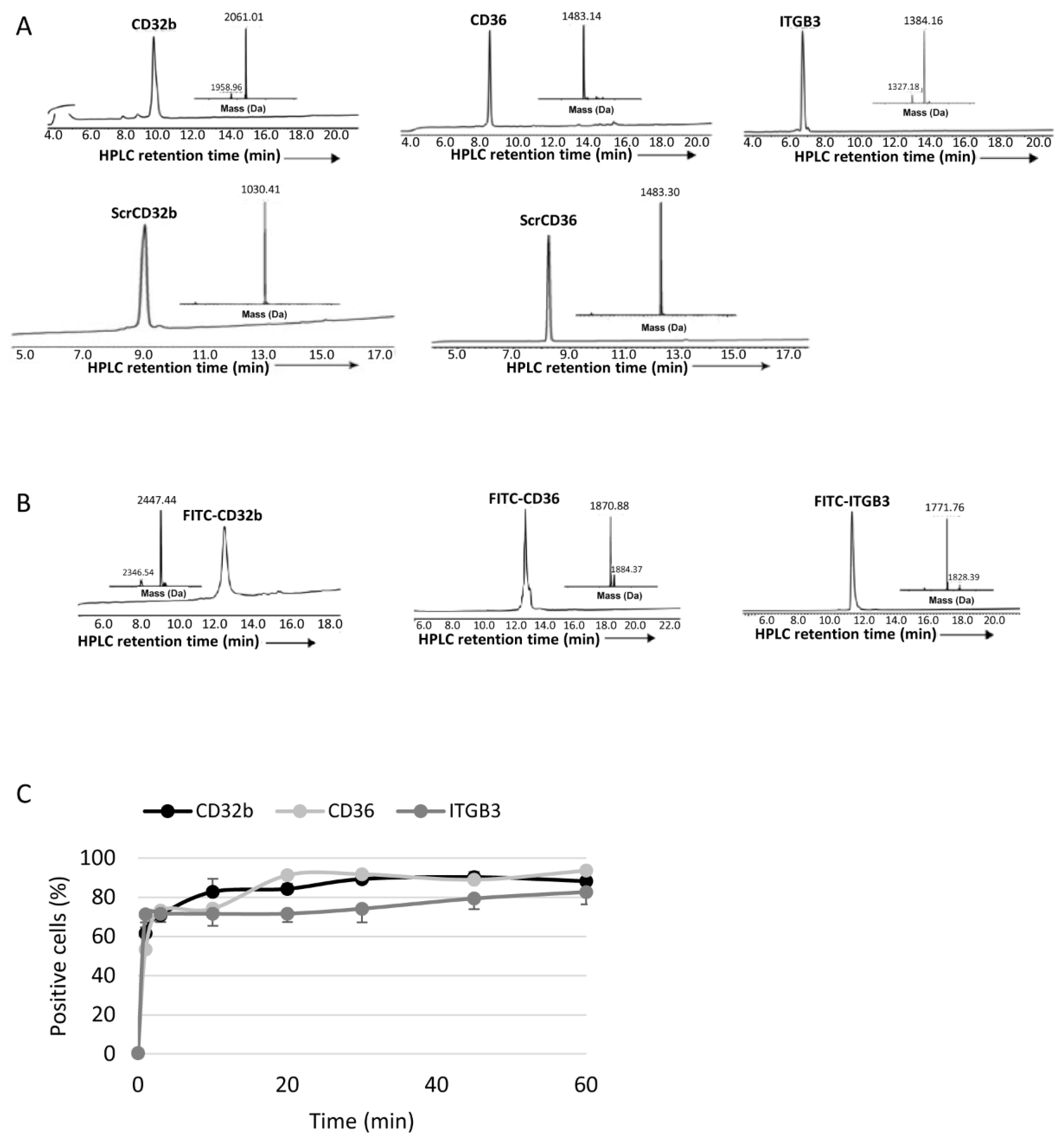
2.5. Purification and Characterization of Peptides
The analytical analyses RP-HPLC were performed on a reverse-phase Waters Alliance HPLC with UV detector (220 nm and 280 nm) using an X-Bridge C18 column (3.5 µm, 130 Å, 4.6 × 150 mm). Preparative RP-HPLC was performed on a Waters LCQ150 system (XSelect PPeptide CSHTM C18 column, 3.5 µm, 130 Å, 30 × 250 mm). Linear gradients of acetonitrile with 0.1% TFA (buffer B) and water with 0.1% TFA (buffer A) were used for all systems to elute peptides. The flow rates were 1 mL/min (analytical) and 10 mL/min (C4 preparative).
Lyophilized crude peptides were dissolved in acetonitrile in water with 0.1% TFA and purified by means of prep RP-HPLC, and peaks were collected and characterized by means of electrospray ionization mass spectrometry (ESI-MS) performed on a LCQ Fleet Ion Trap Mass Spectrometer (Thermo Scientific, Thermo Fisher Scientific, USA). Peptide masses were calculated from the experimental mass-to-charge ratios (m/z) from all the observed multiply charged species of peptides.
2.6. Synthesis of Carboxylated Polymer for Functionalization
For the synthesis of functionalized polymeric micelles (FPMs), Pluronic F127 was carboxylated (F127-COOH) by means of the maleic anhydride method [
25] to assist with the peptide ligand conjugation. F127 and maleic anhydride (ratio 1:11) were dissolved in distilled chloroform and allowed to react for 24 h under stirring at 70 °C in a condensation system to avoid any loss of solvent. The solution was concentrated and poured twice into an excess amount of iced cold diethyl ether to precipitate the reaction product. F127-COOH was dried via vacuum dehydration and collected as a white powder.
2.7. Production of Polymeric Micelles (PMs)
The synthesis of PMs was based on the thin-film hydration technique. Briefly, Pluronic F127 polymer was weighed and dissolved in an organic mixture of methanol/ethanol (1:1), which was removed under vacuum in a rotary evaporator and the formed thin film was left to dry overnight (O/N) at RT to eliminate any remaining solvent. The film was hydrated with PBS at RT and the aqueous solution was vortexed for 5 min, allowing polymers to self-assemble to form micelles. For PM functionalization, micelles were produced with a mixture of F127 and F127-COOH polymers (ratio 8:2), incubated with EDC dissolved in water (polymer/EDC ratio 1:1.5) and stirred for 30 min at RT to activate COOH groups. To conjugate the activated PMs with the peptide ligands, the native chemical ligation technique was utilized, where synthesized peptides, modified with a Cys residue at the N-terminus, were solubilized in PBS, added to the PM solution (peptide/PM ratio 1:100) and incubated under stirring for 2 h at RT. The obtained dispersion of PMs or FPMs was filtered through a 0.22 µm syringe filter for sterilization and the removal of eventual aggregates. All types of nanoparticles were lyophilized for long-term storage at RT until use.
For internalization studies, fluorescent micelles were synthesized using 10% 5-DTAF-labelled polymer obtained via the conjugation of Pluronic F127 polymer with 5-DTAF in an aqueous medium via nucleophilic aromatic substitution through an addition–elimination mechanism.
For the synthesis of drug-loaded micelles, activated simvastatin was dissolved at the desired concentration (20 or 40 mg/mL) in the organic methanol/ethanol mixture together with the polymer before the solvent evaporation step.
2.8. Physicochemical Characterization of PM
To confirm its correct functionalization, FPMs were analyzed by means of Fourier-transform infrared spectroscopy (FTIR) in the Preparation and Characterization of Soft-Materials Services at Institut de Ciència de Materials de Barcelona (Barcelona, Spain) using a Spectrum One FT-IR Spectrometer (PerkinElmer, Inc., Waltham, MA, USA), with an energy range of 450–4000 cm−1, equipped with the Universal Attenuated Total Reflectance (UATR) accessory.
Particles’ mean hydrodynamic diameter and polydispersity index were measured by means of dynamic light scattering (DLS), and zeta potential was measured via laser Doppler micro-electrophoresis using a Zetasizer Nano (Malvern Instruments, Malvern, UK) with an angle of 173° in a measurement range of 0.3 nm–10 μm and sensitivity of 0.1 mg/mL.
Particle shape and size were observed by means of transmission electron microscopy using the high-performance and high-contrast 120 kV JEM-1400Flash Electron Microscope (JEOL Ltd., Tokyo, Japan) from the Electron Microscopy Service at Univeristat Autònoma de Barcelona (Cerdanyola del Vallès, Spain). For their visualization, samples were placed on a copper carbon-coated grid and negatively stained with uranyl acetate for 1 min at RT. Gatan software V 2.32.888.0 (Gatan, Inc., Pleasanton, CA, USA) was used to process information and obtain measures from transmission electron microscopy images.
2.9. Encapsulation Efficiency
The encapsulation efficiency (EE) of simvastatin for each formulation was calculated according to Equation (1). The free drug present in the aqueous phase of the micelle synthesis was obtained via centrifugation with filtration (10,000 rpm, 10 min, 4 °C) using a Nanosep Centrifugal Device with Omega Membrane 10K (Pall Corporation, Port Washington, NY, USA) and analyzing the filtrate via UPLC-MS/MS (Xevo TQ Absolute; Waters Corporation, Los Angeles, CA, USA). All experiments were performed in triplicate at 20 °C.
2.10. In Vitro Drug Release Assay
The in vitro release profile of simvastatin from FPMs was assessed via the regular dialysis method, placing FPM-Sim (20 mg/mL) inside a Spectra/Por Float-A-Lyzer G2 dialysis device (MWCO 20 kDa; Spectrum Laboratories, Inc., Rancho Dominguez, CA, USA) immersed in a PBS pH 7.4 solution (1:100 dilution) and maintained at 37 °C under stirring. A 500 µL sample of released media was collected at predetermined time points (0.25, 0.5, 1, 3, 6, 9, 12, 24, 48 and 72 h) for simvastatin quantification via UPLC-MS/MS (Xevo TQ Absolute). This volume was replaced with fresh buffer at each time point. All formulations were analyzed in duplicate.
2.11. Isolation and Culture of Primary Rat Liver Cells
All liver cells were isolated from healthy rats as follows.
2.11.1. LSECs and Kupffer cells
The liver was perfused through the portal vein for 10 min at a flow rate of 20 mL/min at 37 °C with Hanks’ Balanced Salt Solution (HBSS; Sigma-Aldrich, Merck KGaA, Darmstadt, Germany) without calcium and magnesium containing 12 mM HEPES (pH 7.4), 0.6 mM EGTA, 0.23 mM BSA and 1% heparin. Next, the liver was perfused with 0.15 mg/mL collagenase A (Roche, Merck KGaA, Darmstadt, Germany) in HBSS containing 12 mM HEPES (pH 7.4) and 4 mM calcium chloride dihydrate (CaCl
2·2H
2O) for 30 min at a flow rate of 5 mL/min at 37 °C, then excised and ex vivo digested with the same buffer for 10 min at 37 °C in constant agitation. Cells were passed through a 100 μm nylon filter, collected in cold Krebs’ buffer and centrifuged at 50×
g for 3 min at 4 °C to eliminate hepatocytes. The supernatant was then centrifuged at 800×
g for 10 min, and the pellet was resuspended in cold PBS to be centrifuged at 800×
g for 25 min through a two-step 25–50% Percoll gradient (Sigma-Aldrich, Merck KGaA, Darmstadt, Germany) at 4 °C. The interface of the gradient enriched in LSECs and Kupffer cells was collected, rinsed with PBS and centrifuged at 800×
g for 10 min. The pellet was resuspended in tempered correctly supplemented RPMI medium (10% FBS, 1%
L-glutamine, 1% penicillin-streptomycin, 1% amphotericin B, 0.1 mg/mL heparin and 0.05 mg/mL endothelial cell growth supplement), seeded into a culture dish and incubated for 30 min (37 °C, 5% CO
2). Kupffer cells were separated from LSECs via their selective adherence to the non-coated dish, being washed with PBS and maintained in RPMI at 37 °C (5% CO
2). Non-adherent LSECs were collected, seeded into a collagen-coated culture dish and incubated for 45 min (37 °C, 5% CO
2). Finally, they were washed with PBS and maintained in RPMI (37 °C, 5% CO
2) [
26].
2.11.2. Hepatocytes
After centrifuging the digested liver filtrate at 50×
g for 3 min at 4 °C, hepatocytes were found in the pellet, which were resuspended in cold PBS. This step was repeated 2–3 times to rinse the cells from cellular debris. Cells were then resuspended in tempered supplemented Dulbecco’s modified Eagle’s medium (DMEM) (10% FBS, 1%
L-glutamine, 1% penicillin-streptomycin, 1% amphotericin B, 1 μM dexamethasone and 0.002% insulin) and filtered with a 100 μm filter to eliminate residues of liver tissue. Hepatocytes were planted in a collagen-coated culture dish and incubated at 37 °C and 5% CO
2. After 4 h, cells were washed with PBS and cultured in maintenance DMEM medium (2% FBS, 1%
L-glutamine, 1% penicillin-streptomycin, 1% amphotericin B, 1 nM dexamethasone and 0.002% insulin) [
26].
2.11.3. Hepatic Stellate Cells
HSCs were isolated by perfusing the rat liver through the portal vein with Gey’s Balanced Salt Solution (GBSS; Sigma-Aldrich, Merck KGaA, Darmstadt, Germany) at a flow rate of 20 mL/min at 37 °C. The liver was then perfused with 1.5 mg/mL pronase E (Roche, Merck KGaA, Germany), 0.15 mg/mL collagenase A and 0.05 mg/mL DNase I (Roche, Merck KGaA, Germany) in GBSS solution for 30 min at a flow rate of 5 mL/min at 37 °C. The digested liver was excised and digested ex vivo, also with pronase E (0.4 mg/mL) and collagenase A and DNase I (0.1 mg/mL) for 10 min at 37 °C in agitation. The resulting suspension was filtered through a 100 μm nylon filter and centrifuged at 50×
g for 4 min at 21 °C. The supernatant was centrifuged at 800×
g for 5 min to eliminate the hepatocytes, and the obtained pellet was resuspended in GBSS and centrifuged in Optiprep Density Gradient Medium (11.5%) (Sigma-Aldrich, Merck KGaA, Darmstadt, Germany) at 1400×
g for 21 min. The fraction enriched in HSCs was collected, rinsed with GBBS and centrifuged at 800×
g for 5 min. The pellet was resuspended in tempered correctly supplemented Iscove’s modified Dulbecco’s medium (IMDM) (10% FBS, 1%
L-glutamine, 1% penicillin-streptomycin and 1% amphotericin B), seeded into a culture dish and incubated O/N at 37 °C (5% CO
2). The next day, HSCs were washed with PBS and maintained in IMDM (37 °C, 5% CO
2) [
27].
2.12. Cellular Uptake of Peptide Ligands and Micelles
To determine the in vitro uptake of peptide ligands and FPM, healthy LSECs were seeded into a 96-well plate and treated with 15 μl of FITC-peptide ligands (0.75 mg/mL) or 10 μL of 5-DTAF-labelled FPMs (100 mg/mL polymer) at different time points: from 1 min to 1 h (peptide ligands) or 4 h (FPMs). As negative control, some LSECs were not treated. Next, cells were washed with PBS and for 5–10 min at 37 °C. Once the cells were detached after incubation with 30 μL of trypsin 10×, 120 μL of PBS at 5% FBS with DAPI (1:1000 dilution) was added per well and the plate was read by means of flow cytometry on a BD LSRFortessa Cell Analyzer (BD, Franklin Lakes, NJ, USA), detecting the percentage of positive cells for the fluorescent staining of peptide ligands or FPMs. Each condition was analyzed in triplicate.
For the in vivo uptake of micelles, healthy rats received an intravenous dose of 5-DTAF-labelled PMs or FPMs (100 mg/kg of polymer). Untreated rats were used as negative controls. The following day, LSECs, Kupffer cells, HSCs and hepatocytes were isolated and cultured. Cells were then lifted from the plate with trypsin 10×, or 1× for hepatocytes, for 5–10 min at 37 °C, collected and centrifuged at 800× g for 5 min. The pellet was resuspended in PBS at 5% FBS with DAPI (1:1000 dilution), or propidium iodide (1:50 dilution) in the case of HSCs, and cells were analyzed by means of flow cytometry. Results were obtained from three or five animals for each of the study groups, with three determinations per sample.
2.13. Determination of Simvastatin in Muscle Tissue
A single dose of oral (inactive) or intravenous (active) simvastatin, and PM-Sim, FPM-CD32b-Sim, FPM-CD36-Sim or FPM-CD32b-CD36-Sim was administered at 20 mg/kg to healthy rats for simvastatin detection in muscle after 10 h of treatment (n = 3 animals/group). A sample of ground quadriceps femoris was dissolved in methanol/distilled water (1:1 volume, final concentration 0.2 g muscle/mL) and sonicated (3 × 15 s). The homogenate was centrifuged (13,000× g, 10 min, 4 °C) and the supernatant was processed for simvastatin extraction: 50 μL of muscle homogenate was mixed with 125 μL of acetonitrile and vortexed for 30 s. Then, 25 μL of 5 M ammonium formate (NH4HCO2) buffer (pH 4) was added and the mixture was vortexed again. The sample was centrifuged (13,000× g, 10 min, 4 °C) and the supernatant was analyzed via UPLC-MS/MS (Xevo TQ Absolute) for the quantification of active simvastatin.
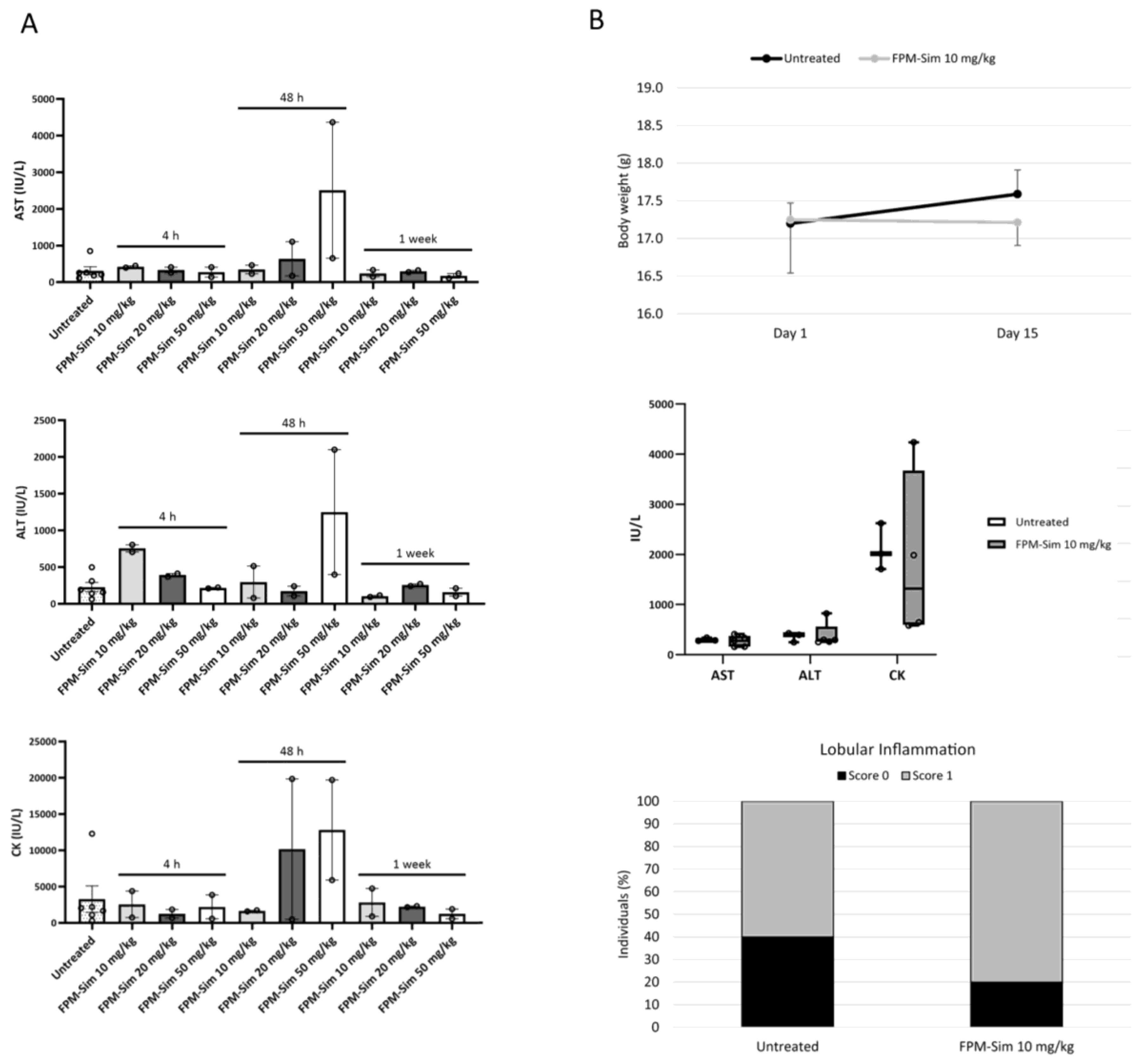
2.14. Maximum Tolerated Dose
To study the safety profile of simvastatin-loaded FPMs, a three-phase maximum tolerated dose assay was conducted, in which acute toxicity (phase 1 and phase 2) and subacute toxicity (phase 3) of treatment with the FPM-CD36-Sim formulation (referred to as FPM-Sim) were evaluated. After each phase, serum and liver tissue samples were obtained from the animals for biochemical and histological analysis, respectively. All phases were performed in healthy mice, and the weight and condition of the animals were monitored throughout the process.
2.14.1. Acute Toxicity: Phase 1 and Phase 2
In phase 1, different doses of encapsulated simvastatin (10, 20 and 50 mg/kg) were tested by administering a single intravenous dose and drawing samples at different times (4 h, 48 h and 1 week) post-treatment (n = 2 animals/group). Untreated animals were used as the control group. The dose that showed no toxicity or mortality (FPM-CD36-Sim 10 mg/kg) was further evaluated in phase 2, where it was given intravenously 3 days/week for 2 weeks to monitor the cumulative effect of the drug on the animals compared to the control condition (n = 5 animals/group).
2.14.2. Subacute Toxicity: Phase 3
In the subacute toxicity phase, FPM-CD36-Sim 10 mg/kg was administered intravenously 5 days/week for 3 weeks, simulating a longer treatment situation. For this phase, a vehicle group receiving intravenous saline injections was conducted as a control (n = 10 animals/group).
2.15. Animal Treatment
In order to assess the efficacy of FPM-Sim in BDL rats, animals were randomly assigned to each of the following study groups: control (untreated) (n = 9), oral simvastatin 10 mg/kg (n = 15), PM-Sim 5 mg/kg (n = 9), FPM-CD32b-Sim 5 mg/kg (n = 9), FPM-CD36-Sim 5 mg/kg (n = 14) and FPM-CD32b-CD36-Sim 5 mg/kg (n = 10). Rats were treated from day 22 of BDL and administered daily for 7 days.
In the case of the TAA model, animals were randomly distributed in each study group as follows: control (untreated) (n = 9), oral simvastatin 10 mg/kg (n = 10), PM-Sim 5 mg/kg (n = 10), FPM-CD32b-Sim 5 mg/kg (n = 10), FPM-CD36-Sim 5 mg/kg (n = 10) and FPM-CD32b-CD36-Sim 5 mg/kg (n = 10). Rats were treated during the last 2 weeks of the 8-week model generation, receiving 5 doses/week (10 total doses).
2.16. Hemodynamic Measurements
The measurement of hemodynamic parameters was performed in fasted conditions (O/n) 90 min after the last treatment administration. Mean arterial pressure (MAP; mmHg) was measured by means of catheterization of the femoral artery and portal pressure (PP; mmHg) via ileocolic vein catheterization using highly sensitive pressure transducers from a PowerLab data acquisition device associated with the physiological data analysis software LabChart 5.0 (ADInstruments, Dunedin, New Zealand). Superior mesenteric artery (SMA) blood flow (SMABF; (mL/min)·100 g) and portal blood flow (PBF; (mL/min)·100 g) were evaluated with a 1.0 mm-diameter ultrasonic perivascular flowprobe connected to a TS420 Perivascular Flow Module (Transonic Systems Inc., Ithaca, NY, USA). SMA resistance (SMAR; (mmHg·min)/(mL·100 g)) and IHVR ((mmHg·min)/(mL·100 g)) were calculated as ((MAP-PP)/SMABF) and (PP/PBF), respectively. Once the hemodynamic study was completed, blood was collected for biochemistry, and liver tissue samples were collected for histological and molecular analysis.
2.17. Biochemical Analysis
Fasting blood and serum samples were analyzed to determine creatinine, total bilirubin, alanine aminotransferase (AST), aspartate aminotransferase (ALT), alkaline phosphatase (ALP), creatine kinase (CK), total cholesterol, triglycerides and albumin values. Samples were measured in the automatized CORE laboratory from Hospital Vall d’Hebron, by means of the analytical platform ATELLICA Solution, using Standard CE Mark-approved IVDR diagnostic kits provided by Siemens Healthineers (Erlangen, Germany).
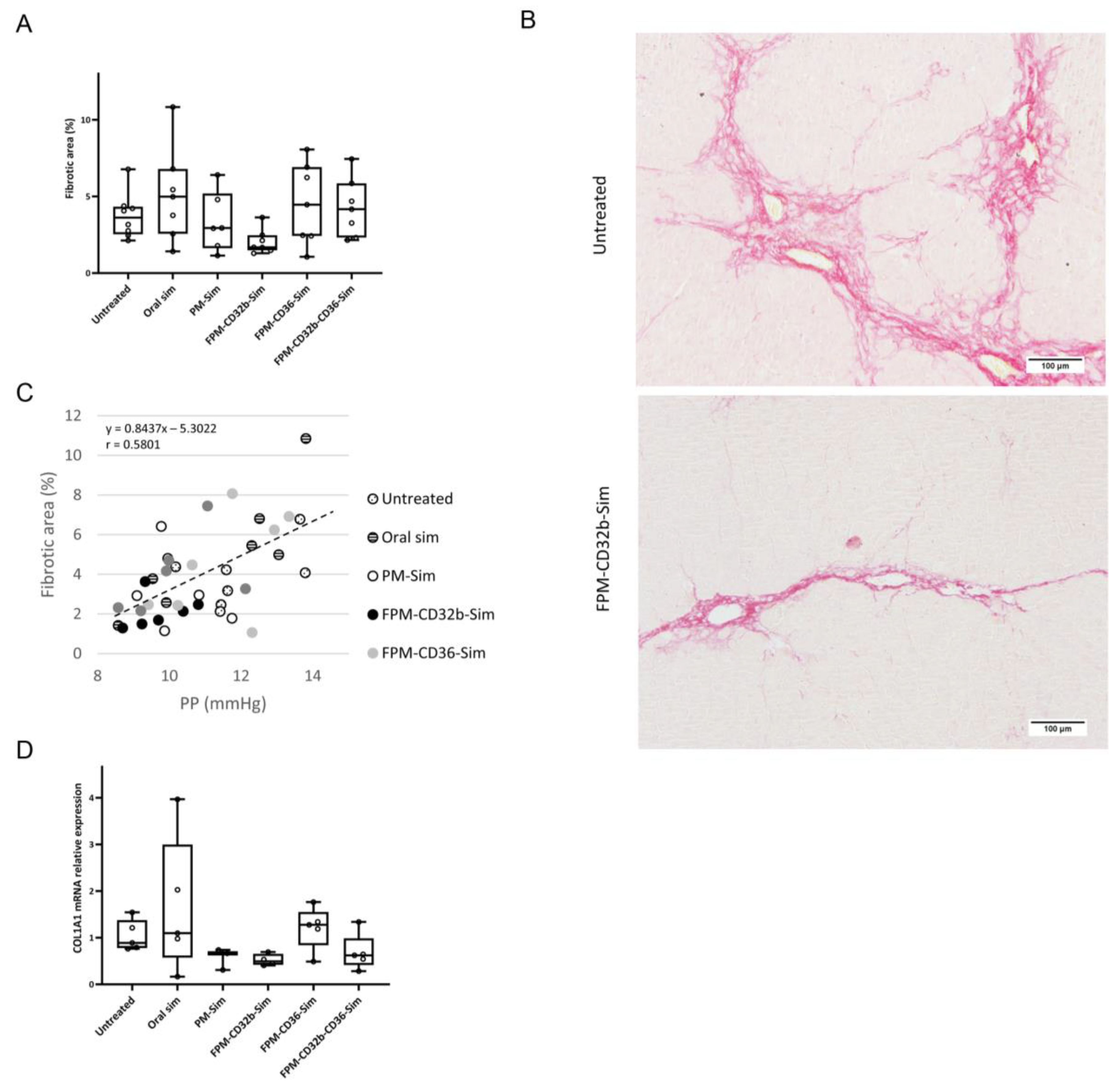
2.18. Sirius Red Staining
Liver samples were fixed in 4% paraformaldehyde, embedded in liquid paraffin at 65 °C, and sectioned in 4 μm-thick slices. Once hydrated, samples were dried and stained with 0.1% Picro-Sirius red for 1 h at RT under gentle agitation. Samples were then mounted with DPX rapid mounting medium (Panreac Química SLU, Castellar del Vallès, Spain).
2.19. Immunohistochemistry
Immunohistochemistry detection of CD32b receptor was carried out on frozen OCT-embedded 8 µm sections of rat liver. Sections were incubated with primary antibody against SE-1 (Cd32) at 1/100 dilution (NB110–68095, Novus Biologicals, Centennial, CO, USA), followed by EnVision + Dual Link System-HRP (Dako, Glostrup, Denmark) and visualized with the VIP substrate kit (Vector, Burlingame, CA, USA). Samples were counterstained with hematoxylin. Quantitative analysis of stained areas was carried out with Image J software version 1.54d (
http://imagej.org (accessed on 13 September 2023)) [
28] on ten fields per sample, randomly captured at 10× magnification with an optical microscope Olympus BX61 (Olympus, Hamburg, Germany).
2.20. Gene Expression Analysis
Healthy LSECs were treated O/N (37 °C, 5% CO2) with free activated simvastatin or PM/FPM-Sim at a drug concentration of 2.5 μM. Control wells were treated with empty (Ø) micelles. Liver samples from treated rats were collected in RNAlater stabilization Solution (Invitrogen, Thermo Fisher Scientific, USA) and kept for 1 week at 4 °C. Total RNA was extracted from liver cells or tissue and converted to cDNA. From each sample, 20 ng of cDNA was amplified with specific TaqMan probes for COL1A1 (collagen type I alpha 1 chain; Rn 01463848_m1) and KLF2 (Rn 01420496_gH). The relative gene expression was normalized to GAPDH (glyceraldehyde-3-phosphate dehydrogenase; Rn 99999916_s1). Each sample was analyzed in triplicate.
2.21. Western Blot
Rat liver samples containing 60 µg of protein were run on a NuPAGE Bis-Tris 4–12% SDS-PAGE (Invitrogen, Thermo Fisher Scientific, USA) under denaturing conditions, blotted onto a polyvinylidene difluoride (PVDF) blotting membrane and incubated with the appropriate dilution of the primary antibody for eNOS (1:500 in TTBS 1X; BD, USA), p-eNOS (1:250 in 5% BSA; Cell Signaling Technology, Danvers, MA, USA) and KLF2 (1:200 in 5% BSA; Santa Cruz Biotechnology, Santa Cruz, CA, USA). GAPDH antibody (1:5000 in TTBS 1X, Ambion, Austin, TX, USA) was used as the loading control. The electrophoretic bands of Western blots were detected through digital chemiluminescence development using an Odyssey Fc Imaging System (LI-COR Biosciences, Lincoln, NE, USA). Protein quantification was performed with Image Studio Lite software version 5.2 (LI-COR Biosciences, USA).
2.22. Statistical Analysis
IBM SPSS Statistics 20 (IBM, Armonk, NY, USA) was used for statistical analysis. Quantitative results were expressed as mean ± standard error of the mean (SEM) and compared with analysis of variance followed by unpaired Student’s t test (between two groups) or one-way analysis of variance (ANOVA) with Tukey’s HSD post hoc correction (among three or more groups). When data were not normally distributed, nonparametric tests were applied, using the Mann–Whitney U test to compare two groups and the Kruskal–Wallis test for multiple comparisons. Pearson’s correlation coefficient was calculated to show the correlation of two parameters. A p-value ≤ 0.05 was considered statistically significant.
4. Discussion
In an attempt to provide a solution for improving the therapeutic window of statins for the treatment of advanced CLD, this work addresses the optimization of a simvastatin delivery system enhancing the targeting of LSECs. The optimization included the design and functionalization with peptide ligands of Pluronic-based PMs loaded with simvastatin. We studied the in vitro characteristics in primary cultures of liver cells, and the in vivo effect in animal models of CLD, demonstrating the increased therapeutic potential of the loaded drug by reducing portal hypertension and liver fibrosis in a non-decompensated CLD animal model.
To increase the accumulation of nanoparticles in LSECs, peptide ligands recognizing three LSEC receptors were designed and synthesized according to various expression criteria (either present in functional, dysfunctional or in both LSEC differentiation stages) and their ability to enter into primary LSECs.
The coupling of these peptides on the surface of PMs generated three FPM formulations with different features, but with homogeneous particle shape and size, and a zeta potential close to neutrality. This was due to the use of polyethylene glycol (PEG) at the hydrophilic end of the polymer, which prevents aggregation between the uncharged nanoparticles [
30]. The effect of an encapsulated drug can be maximized and the side-effects of the drug minimized only if a micelle is stable enough to retain most of the drug until the target is reached. Our result indicated that FPMs were thermodynamically stable, since no simvastatin was released until at least 72 h under neutral pH conditions, simulating the blood circulation [
31,
32].
In vitro internalization in primary cultures of LSECs of the three FPM formulations with the specific ligands for CD32b, CD36 and integrin αVβ3 receptors demonstrated that specific ligand–receptor interactions plus passive targeting promoted uptake by a greater number of healthy LSECs than the micelles conjugated with their scrambled variants.
Simvastatin has been shown to reduce portal hypertension through the putative reduction in IHVR by means of several mechanisms, including the induction of KLF2 expression, related to the stimulation of a vasoprotective phenotype in LSECs [
11,
12,
33]. This effect was confirmed by the expected overexpression of KLF2 in healthy isolated LSECs treated with free or encapsulated simvastatin. We also ruled out any possible effect of the empty functionalized nanodevices acting only as an inert vehicle for the delivery of the loaded drug.
Intravenous treatment of healthy rats with the different fluorescent formulations confirms that, in all cases, functionalization confers greater efficiency in entering the main liver cells than passive targeting; the capture of FPMs was clearly superior to that achieved by non-functionalized PMs, and LSECs were the liver cells with the highest uptake. Furthermore, in endothelial cells, all three types of functionalization were equally effective. In vivo cell internalization experiments also confirmed that the receptors selected as LSEC targets for our nanoparticles were also expressed in healthy Kupffer cells and hepatocytes, allowing functionalized nanoparticles to enter both cell types quite efficiently. This was expected for CD36 and integrin α
Vβ
3, as their expression in the liver is elevated not only in LSECs, but also in other hepatic cell types [
34,
35]. However, CD32b, an FcγR conferring LSECs the highest endocytic capacity of any cell in the human body, is known to be the most specific marker of these cells in the liver. Yet, recent studies have shown that CD32b is not only expressed in LSECs but also in Kupffer cells, with a liver level expression of 90% and 10%, respectively [
36].
The differential accumulation of simvastatin in muscle depending on the different formulations used was a key factor in this study. It is well known that nanoparticles tend to accumulate passively in the liver because this organ is part of the reticuloendothelial system [
37,
38]. Accordingly, the use of nanoparticle encapsulation decreased the presence of active drug detected in the muscle compared to free simvastatin administered either orally or intravenously. This effect is due to the lower uptake of nanoparticles by skeletal muscle compared with the higher natural retention of nanoparticles in the liver, regardless of their functionalization, since both PMs and FPMs showed an equal reduction in the amount of active simvastatin detected in the muscle.
To determine the dose and schedule of FPM-Sim for the efficacy studies in experimental models of liver diseases, the safety profile was assessed in healthy mice by establishing the maximum tolerated dose in a phased assay, studying the use of different simvastatin doses, as well as different administration patterns. The use of FPM-Sim at high doses of 20 and 50 mg/kg resulted in transient elevations in liver transaminases and CK in some individuals, and these doses were discarded to avoid further toxicity in animal models. In contrast, the 10 mg/kg dose showed no evidence of toxicity in any of the established phases and was scaled in subsequent efficacy studies in rat experimental models of liver disease.
The first efficacy evaluation in an advanced CLD model such as BDL was aimed at improving the PP-lowering effect achieved by PM-encapsulated simvastatin in a previous study [
18] using the new functionalized formulations while maintaining the lower levels of toxicity compared with free simvastatin. Encapsulated simvastatin resulted in significantly less body weight reduction at the end of treatment in cirrhotic animals compared to free simvastatin, and lower levels of hepatic and muscular toxicity markers. On the other hand, the use of nanoparticles triggered a significant increase in serum triglycerides and, in a more moderate way, in total cholesterol. This may be explained by the fact that the PM is made from Pluronic F127 (or polaxamer P407), which has been shown to induce a transient hyperlipidemic effect caused by a temporary reduction in the number of fenestrae in LSECs in a dose-dependent manner [
39]. Consequently, given that BDL animals at baseline have an advanced capillarized endothelium and a marked reduction in fenestrae due to the endothelial dysfunction induced by bile accumulation [
40], the addition of an extra element that closes the fenestrae, even temporarily, may lead to an impaired transendothelial transfer of lipoproteins from sinusoidal blood to the extracellular space of Disse.
In terms of efficacy, oral simvastatin, although causing greater toxicity, was the most effective treatment in significantly reducing PP compared to the untreated control group, while the functionalized nanoparticles FPM-CD32b and mixed-FPM only caused a discrete reduction in the effect of simvastatin on PP compared to the non-functionalized PMs. It is worth mentioning that the BDL model used in the present study, mimicking an advanced CLD, resulted in severely affected animals with unusually high levels of PP in the control group and increased liver transaminase values.
We then performed a second in vivo efficacy study in a model of cirrhosis induced by TAA, a widely used model for its high reproducibility and homogeneity of results, its low mortality, and a lower systemic toxicity [
41]. Moreover, the model was developed only for 8 weeks, yielding cirrhotic animals but in a non-decompensated stage of the disease. Simvastatin was administered in different formulations during the last 2 weeks of model generation. In this non-decompensated model, simvastatin induced lower toxicity in all aspects: the body weight of animals was not reduced, and the slight decrease in body weight observed in the groups that received intravenous treatment might be associated with the stress caused by the administration route rather than the drug itself. Furthermore, transaminases and CK values were also diminished in all TAA animals compared to BDL. In addition, the increase in total cholesterol and triglycerides observed in BDL rats was of lower magnitude, which could be explained by the lesser degree of capillarization expected from a more moderate CLD model represented by the 8-week TAA animals.
The reduced toxicity observed in this model was accompanied by a lower PP in the untreated control group compared with the previous BDL model. Treatment with encapsulated simvastatin further reduced PP, with a significant difference in the FPM-CD32b and mixed-FPM formulations. Moreover, the PP-lowering effect achieved by FPM-CD32b-Sim was also significant versus FPM-CD36-Sim and oral free simvastatin. To elucidate this reduction effect, we evaluated the correlation between PP and fibrosis, confirming that the greater the area of liver fibrosis, the greater the PP increase, and vice versa [
3,
42]. Collagen expression followed the same trend, and in the case of FPM-CD32b-Sim treatment, it was reduced to half compared to the untreated TAA group. Quantification of proteins involved in signaling pathways related to vasoprotection induced by statins administration [
14,
43,
44] showed unaltered expression of KLF2 between treated and untreated animals, suggesting that the dose used in in vivo treatments is not enough to increase the values of this transcription factor. Nonetheless, in line with the improvement in PP reduction, the p-eNOS/eNOS ratio was significantly elevated by FPM-CD32b-Sim and mixed-FPM-Sim, suggesting an amelioration of endothelial dysfunction by stimulating nitric oxide production through the targeted release of simvastatin into LSECs of TAA-induced cirrhotic rats.
We interpret the greater efficacy demonstrated by the FPM-CD32b-Sim nanoparticles in the TAA model as a consequence of enhanced targeting by the specific peptide ligand due to higher levels of CD32b expression in LSECs from this model compared with the BDL model. In this regard, immunohistochemical staining to identify the CD32b antigen in liver sections of rats from both experimental models was significantly higher in TAA than in BDL rats. This is also supported by a recent study conducted in our laboratory [
45], in which the percentage of CD32b
+ LSECs was estimated by means of cell sorting, establishing that the loss of CD32b during the capillarization process might display different features depending on the etiology, disease stage and mechanisms causing liver injury.
One limitation of this work was performing the efficacy studies only in male animals. In our study, we wanted to evaluate as a proof of concept the FPM efficiency by means of the ability to reduce portal pressure in cirrhotic individuals. The vast majority of published studies on the hemodynamic disturbances occurring in liver disease use male rats or mice, because female estrogen hormones modify hemodynamic parameters, increasing or decreasing their values randomly and unrelated to liver disease [
22]. However, we acknowledge that to have a complete picture of the efficacy demonstrated by the FPM-CD32b-Sim nanoparticles, further studies are needed using both sexes.
In summary, there are three important observations in this study. First, the adequate functionalization of nanoparticles may provide a noticeable positive effect for passive targeting. Second, it seems clear that, even though the specific targeting of LSECs can be lost gradually in pathological situations, it is more effective to choose specificity over other alternatives where the target may be abundant but ubiquitously expressed. Third, the stage of the disease is a key factor in the use of PM nanoparticles in liver diseases. Our results indicate that in advanced decompensated models, nanoparticles are able to decrease the toxicity caused by simvastatin but, on the other hand, their effectiveness is limited. By contrast, in a less severe stage of the disease, simvastatin encapsulation increases the nanoparticles’ hepatic beneficial impact and minimizes the possible secondary effect of the poloxamer device.
In conclusion, active targeting of a Pluronic-based nanodevice by means of functionalization with peptide ligands for the specific administration of simvastatin towards LSECs reduces muscle and liver toxicity, but without a clear portal pressure reduction, in a decompensated model of cirrhosis. However, in a non-decompensated model of CLD, functionalization with CD32b ligand enhances the efficacy of simvastatin, reducing PP values as well reducing liver fibrosis. This conclusion favors the use of CD32b-functionalized PMs as potential nanotransporters for vasoprotective drugs targeting the sinusoidal endothelial cells in the liver.


 ,
, 














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}