
Conceptual Framework of the Design of Pleiotropic Drugs against Alzheimer’s Disease


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. The Concept of Pleiotropic Active Drugs
2. The Application of the Concept of Pleiotropic Active Compounds to the Therapeutic Management of Alzheimer’s Disease
2.1. Conjugates with Immolable Chemical Bond
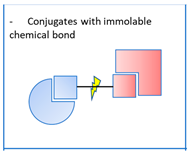 . Also known as mutual prodrugs or codrugs [7,8,9], these molecules have two structural units, each of which may have a different therapeutic target. The link between them is immolable and cleaved by various mechanisms: exposure to hydrogen peroxide, cleavage by a cysteine-dependent mechanism, hydrolysis of amide groups, etc. These entities do not have all the advantages of pleiotropic agents, since they release two active ingredients, each of which may be responsible for undesirable effects or interact with each other. Nevertheless, they make it possible to reduce the number of pharmaceutical doses and, above all, their characteristics as prodrugs confers on them an improved pharmacokinetic behaviour, such as the crossing of the blood–brain barrier, compared to that of the individual active substances, or avoid a toxicity which would otherwise be expressed. Examples include boronates releasing a derivative of tacrine, an acetylcholinesterase inhibitor, and ibuprofen, an anti-inflammatory drug (Figure 3) [10]. Ibuprofen was also associated with lipoic acid through diamino chemical linkers [9]. The strategy followed in these examples is to improve the Blood–brain barrier (BBB) permeability of the codrugs and the brain delivery of the two drugs by temporarily masking their acidic groups under ester, carbamate or amide ones. Another example concerns thio-isocyanates releasing memantine, an antagonist of N-methyl-D-aspartate (NMDA) receptors, inducing autophagy and neuroprotective effects, and hydrogen sulphide (H2S), with anti-inflammatory and anti-apoptotic properties [11]. On the other hand, benzamides were conceived to be cleaved into tacrine and probenecid, a neuroprotective agent (Figure 3) [12]. In this case, one of the objectives is to try to reduce the peripheral hepatotoxicity of tacrine which led to its withdrawing from the market.
. Also known as mutual prodrugs or codrugs [7,8,9], these molecules have two structural units, each of which may have a different therapeutic target. The link between them is immolable and cleaved by various mechanisms: exposure to hydrogen peroxide, cleavage by a cysteine-dependent mechanism, hydrolysis of amide groups, etc. These entities do not have all the advantages of pleiotropic agents, since they release two active ingredients, each of which may be responsible for undesirable effects or interact with each other. Nevertheless, they make it possible to reduce the number of pharmaceutical doses and, above all, their characteristics as prodrugs confers on them an improved pharmacokinetic behaviour, such as the crossing of the blood–brain barrier, compared to that of the individual active substances, or avoid a toxicity which would otherwise be expressed. Examples include boronates releasing a derivative of tacrine, an acetylcholinesterase inhibitor, and ibuprofen, an anti-inflammatory drug (Figure 3) [10]. Ibuprofen was also associated with lipoic acid through diamino chemical linkers [9]. The strategy followed in these examples is to improve the Blood–brain barrier (BBB) permeability of the codrugs and the brain delivery of the two drugs by temporarily masking their acidic groups under ester, carbamate or amide ones. Another example concerns thio-isocyanates releasing memantine, an antagonist of N-methyl-D-aspartate (NMDA) receptors, inducing autophagy and neuroprotective effects, and hydrogen sulphide (H2S), with anti-inflammatory and anti-apoptotic properties [11]. On the other hand, benzamides were conceived to be cleaved into tacrine and probenecid, a neuroprotective agent (Figure 3) [12]. In this case, one of the objectives is to try to reduce the peripheral hepatotoxicity of tacrine which led to its withdrawing from the market. 2.2. Non-Dissociable Conjugates
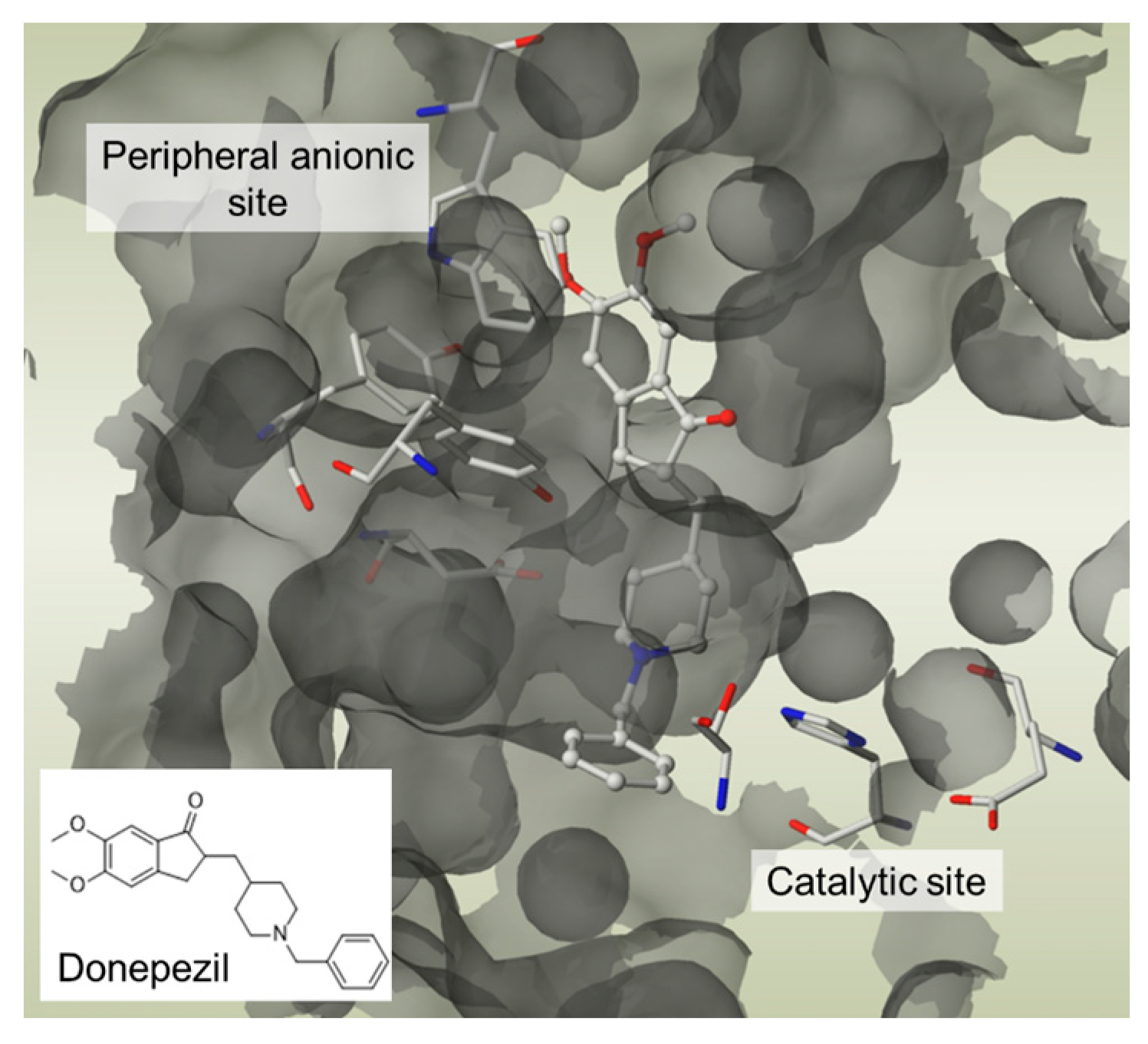
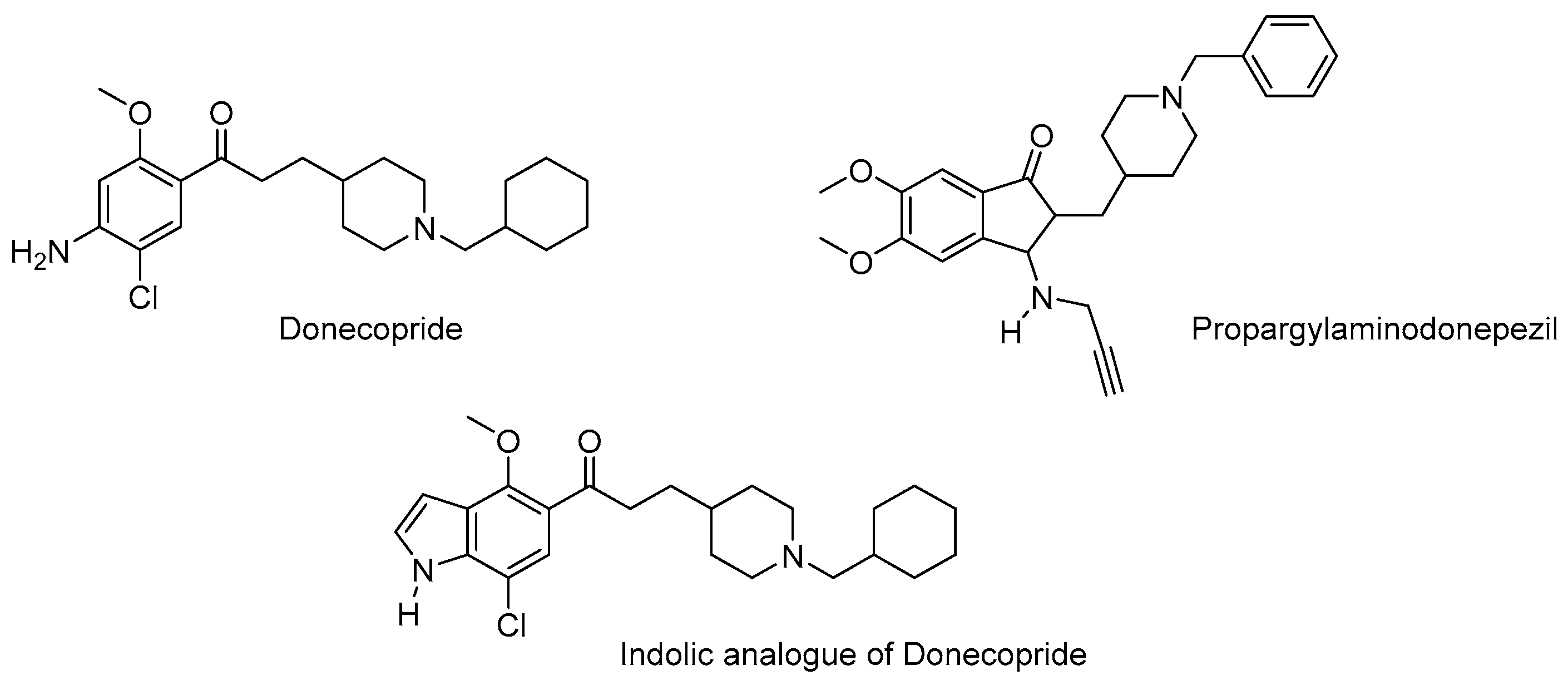
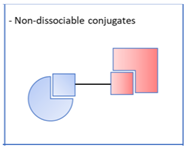 . Again, these entities are made of two structural moieties, each designed to recognise a different target [14,15,16,17,18,19,20]. However, this time, the chemical link between these elements is not cleavable. It is generally made up of aliphatic chains of variable length. These conjugates, often also called hybrids, should, in theory, be aimed at spatially close targets or at two distinct sites within the same protein. In this case, the length of the chemical bond should consider the distance separating the two therapeutic targets or the two targeted sites within a single target. This is the case, for example, with dual-site inhibitors of acetylcholinesterase (AChE), which interact with both the catalytic site of the enzyme and a peripheral site, which is described as anionic. These two sites are located on either side of an aromatic gorge within the enzyme, which is approximately 20 Å deep. For this reason, the number of atoms in the chemical bond in the structure of these dual interaction inhibitors is calculated to separate each of the two motifs by an adequate distance [21]. An example is donepezil, one of the anti-Alzheimer’s drugs currently on the market, which inhibits the catalytic site of AChE and thus temporarily alleviates the cholinergic deficiency that marks the disease, and also interacts with the peripheral anion site of the enzyme, preventing it from aggregating with the beta-amyloid peptide (Figure 4).
. Again, these entities are made of two structural moieties, each designed to recognise a different target [14,15,16,17,18,19,20]. However, this time, the chemical link between these elements is not cleavable. It is generally made up of aliphatic chains of variable length. These conjugates, often also called hybrids, should, in theory, be aimed at spatially close targets or at two distinct sites within the same protein. In this case, the length of the chemical bond should consider the distance separating the two therapeutic targets or the two targeted sites within a single target. This is the case, for example, with dual-site inhibitors of acetylcholinesterase (AChE), which interact with both the catalytic site of the enzyme and a peripheral site, which is described as anionic. These two sites are located on either side of an aromatic gorge within the enzyme, which is approximately 20 Å deep. For this reason, the number of atoms in the chemical bond in the structure of these dual interaction inhibitors is calculated to separate each of the two motifs by an adequate distance [21]. An example is donepezil, one of the anti-Alzheimer’s drugs currently on the market, which inhibits the catalytic site of AChE and thus temporarily alleviates the cholinergic deficiency that marks the disease, and also interacts with the peripheral anion site of the enzyme, preventing it from aggregating with the beta-amyloid peptide (Figure 4). 2.3. Pleiotropic Prodrugs
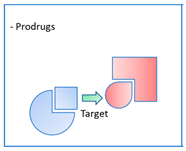 . Some multi-acting agents need to be administered in the form of prodrugs in order to overcome bioavailability problems or to mask a toxic profile. In this sense, they are not different from multi-conjugated agents, whether dissociable or not (see above), or from pleiotropic molecules making structural compromises (see below). These prodrugs are, in this case, activated by different mechanisms, such as the cleavage of sulphenamide or disulphide groups by thiol groups, such as those of glutathione [22], or the acid cleavage of Mannich bases [23] (Figure 6). These mechanisms do not involve proteins that could themselves be therapeutic targets. Pleiotropic prodrugs, on the other hand, are activated by a first therapeutic target that releases a second active molecule that is active towards a second target. This concept is relatively recent and has particular application in the treatment of neurodegenerative diseases such as Alzheimer’s disease.
. Some multi-acting agents need to be administered in the form of prodrugs in order to overcome bioavailability problems or to mask a toxic profile. In this sense, they are not different from multi-conjugated agents, whether dissociable or not (see above), or from pleiotropic molecules making structural compromises (see below). These prodrugs are, in this case, activated by different mechanisms, such as the cleavage of sulphenamide or disulphide groups by thiol groups, such as those of glutathione [22], or the acid cleavage of Mannich bases [23] (Figure 6). These mechanisms do not involve proteins that could themselves be therapeutic targets. Pleiotropic prodrugs, on the other hand, are activated by a first therapeutic target that releases a second active molecule that is active towards a second target. This concept is relatively recent and has particular application in the treatment of neurodegenerative diseases such as Alzheimer’s disease.2.4. Pleiotropic Structural Compromises
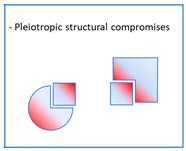 . A pleiotropic structural compromise is a molecule specifically designed to have structural elements that respond to several pharmacophores with distinct biological effects. As a result, this multi-acting entity must be able to bind indifferently to several targets and interact with each of them in such a way as to determine a synergistic therapeutic effect. The first condition for this approach to be successful is the necessary structural homology that must exist between the intended targets, so that the same drug can bind to both of them. This homology, illustrated for example by the phylogenetic tree of kinases, explains why it is possible to design pleiotropic agents that can “selectively” inhibit several kinases of therapeutic interest in oncology [34].
. A pleiotropic structural compromise is a molecule specifically designed to have structural elements that respond to several pharmacophores with distinct biological effects. As a result, this multi-acting entity must be able to bind indifferently to several targets and interact with each of them in such a way as to determine a synergistic therapeutic effect. The first condition for this approach to be successful is the necessary structural homology that must exist between the intended targets, so that the same drug can bind to both of them. This homology, illustrated for example by the phylogenetic tree of kinases, explains why it is possible to design pleiotropic agents that can “selectively” inhibit several kinases of therapeutic interest in oncology [34].3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ehrlich, P. Experimental Researches on Specific Therapy; Pergamon Press Ltd., Ed.; The collected papers of Paul Ehrlich; Himmelweit: London, UK, 1960; pp. 106–117. [Google Scholar]
- Handler, N.; Bushmann, H. Multiple ligands in neurodegenerative diseases. In Drug Selectivity: An Evolving Concept in Medicinal Chemistry; Handler, N., Bushmann, H., Eds.; Wiley-VCH Verlag GmbH & Co.: Hoboken, NJ, USA, 2017; ISBN 9783527335381. [Google Scholar]
- Cavalli, A.; Bolognesi, M.L.; Minarini, A.; Rosini, M.; Tumiatti, V.; Recanatini, M.; Melchiorre, C. Multi-Target Directed Ligands to combat neurodegenerative diseases. J. Med. Chem. 2008, 51, 347–372. [Google Scholar] [CrossRef] [PubMed]
- Parvin, N.; Jozwiak, K. Effects of linkers and substitutions on Multi-Target Directed Ligands for Alzheimer’s diseases: Emerging paradigms and strategies. Int. J. Mol. Sci. 2022, 23, 6085. [Google Scholar] [CrossRef] [PubMed]
- De Simone, A.; Tumiatti, V.; Andrisano, V.; Milelli, A. Glycogen Synthase Kinase 3β: A New Gold Rush in Anti-Alzheimer’s Disease Multitarget Drug Discovery? J. Med. Chem. 2021, 64, 26–41. [Google Scholar] [CrossRef]
- Kumar, N.; Kumar, V.; Anand, P.; Kumar, V.; Dwivedi, A.R.; Kumar, V. Advancements in the development of multi-target directed ligands for the treatment of Alzheimer’s disease. Bioorg. Med. Chem. 2022, 61, 116742. [Google Scholar] [CrossRef] [PubMed]
- Das, N.; Dhanawat, M.; Dash, B.; Nagarwal, R.C.; Shrivastava, S.K. Codrug: An efficient approach for drug optimization. Eur. J. Pharm. Sci. 2010, 41, 571–588. [Google Scholar] [CrossRef]
- Vu, C.B.; Bemis, J.E.; Benson, E.; Bista, P.; Carney, D.; Fahrner, R.; Lee, D.; Liu, F.; Lonkar, P.; Milne, J.C.; et al. Synthesis and characterization of fatty acid conjugates of niacin and salicylic acid. J. Med. Chem. 2016, 59, 1217–1231. [Google Scholar] [CrossRef]
- Albertini, C.; Salerno, A.; de Sena Murteira Pinheiro, P.; Bolognesi, M.L. From combinations to multitarget-directed ligands: A continuum in Alzheimer’s disease polypharmacology. Med. Res. Rev. 2021, 41, 2606–2633. [Google Scholar] [CrossRef]
- Liu, Z.; Zhang, B.; Xia, S.; Fang, L.; Gou, S. ROS-responsive and multifunctional anti-alzheimer prodrugs: Tacrine-ibuprofen hybrids via a phenylboronate linker. Eur. J. Med. Chem. 2021, 212, 112997. [Google Scholar] [CrossRef]
- Sestito, S.; Daniele, S.; Pietrobono, D.; Citi, V.; Bellusci, L.; Chiellini, G.; Calderone, V.; Martini, C.; Rapposelli, S. Memantine prodrug as a new agent for Alzheimer’s disease. Sci. Rep. 2019, 9, 4612. [Google Scholar] [CrossRef]
- Gong, T.; Huang, Y.; Zhang, Z.-R.; Li, L.L. Synthesis and characterization of 9-[P-(N,N-dipropylsulfamide)] benzoylamino-1,2,3,4-4H-acridine- A potential prodrug for the CNS delivery of tacrine. J. Drug Target. 2004, 12, 177–182. [Google Scholar] [CrossRef]
- Liargkova, T.; Hadjipavlou-Litina, D.J.; Koukoulitsa, C.; Voulgari, E.; Avgoustakis, C. Simple chalcones and bis-chalcones ethers as possible pleiotropic agents. J. Enzyme Inhib. Med. Chem. 2016, 31, 302–313. [Google Scholar] [CrossRef] [PubMed]
- Gody, J.; Zareba, P.; Lazewska, D.; Stary, D.; Reiner-Link, D.; Frank, A.; Latacz, G.; Mogilski, S.; Kaleta, M.; Doroz-Płonka, A.; et al. Cyanobiphenyls: Novel H3 receptor ligands with cholinesterase and MAO B inhibitory activity as multitarget compounds for potential treatment of Alzheimer’s disease. Bioorg. Chem. 2021, 114, 105129. [Google Scholar] [CrossRef] [PubMed]
- George, N.; Akhtar, M.J.; Al Balushi, K.A.; Khan, S.A. Rational drug design strategies for the development of promising multi-target directed indole hybrids as Anti-Alzheimer agents. Bioorg. Chem. 2022, 127, 105941. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.; Freschi, M.; de Camargo Nascente, L.; Salerno, A.; de Melo Viana Teixeira, S.; Nachon, F.; Chantegreil, F.; Soukup, O.; Prchal, L.; Malaguti, M.; et al. Sustainable drug discovery of multi-target-directed ligands for Alzheimer’s disease. J. Med. Chem. 2021, 64, 4972–4990. [Google Scholar] [CrossRef]
- Ismaili, L.; Monnin, J.; Etievant, A.; Arribas, R.L.; Viejo, L.; Refouvelet, B.; Soukup, O.; Janockova, J.; Hepnarova, V.; Korabecny, J.; et al. (±)-BIGI-3h: Pentatarget-directed ligand combining cholinesterase, monoamine oxidase, and glycogen synthase kinase 3β inhibition with calcium channel antagonism and antiaggregating properties for Alzheimer’s disease. ACS Chem. Neurosci. 2021, 12, 1328–1342. [Google Scholar] [CrossRef]
- Javed, M.A.; Ashraf, N.; Jan, M.S.; Mahnashi, M.H.; Alqahtani, Y.S.; Alyami, B.A.; Alqarni, A.O.; Asiri, Y.I.; Ikram, M.; Sadiq, A.; et al. Structural modification, in vitro, in vivo, ex vivo, and in silico exploration of pyrimidine and pyrrolidine cores for targeting enzymes associated with neuroinflammation and cholinergic deficit in Alzheimer’s disease. ACS Chem. Neurosci. 2021, 12, 4123–4143. [Google Scholar] [CrossRef]
- Nozal, V.; Garcia-Rubia, A.; Cuevas, E.P.; Pérez, C.; Tosat-Bitrián, C.; Bartolomé, F.; Carro, E.; Ramírez, D.; Palomo, V.; Martínez, A. From kinase inhibitors to multitarget ligands as powerful drug leads for Alzheimer’s disease using protein-templated synthesis. Angew. Chem. Int. Ed. 2021, 60, 19344–19354. [Google Scholar] [CrossRef]
- Mazej, T.; Knez, D.; Meden, A.; Gobec, S.; Sova, M. 4-Phenethyl-1-propargylpiperidine-derived dual inhibitors of butyrylcholinesterase and monoamine oxidase B. Molecules 2021, 26, 4118. [Google Scholar] [CrossRef]
- Zhang, P.; Wang, Z.; Mou, C.; Zou, J.; Xie, Y.; Liu, Z.; Naman, C.B.; Mao, Y.; Wei, J.; Huang, X.; et al. Design and synthesis of novel tacrine-dipicolylamine dimers that are multiple-target-directed ligands with potential to treat Alzheimer’s disease. Bioorg. Chem. 2021, 116, 105387. [Google Scholar] [CrossRef]
- Markowicz-Piasecka, M.; Sikora, J.; Mateusiak, L.; Mikiciuk-Olasik, E.; Huttunen, K.M. Metformin and its sulfenamide prodrugs inhibit human cholinesterase activity. Oxid. Med. Cell. Longev. 2017, 2017, 7303096. [Google Scholar] [CrossRef]
- Purgatorio, R.; Candia, M.; Catto, M.; Rullo, M.; Pisani, L.; Denora, N.; Carrieri, A.; Nevskaya, A.A.; Voskressensky, L.G.; Altomare, C.D. Evaluation of water-soluble Mannich base prodrugs of 2,3,4,5-tetrahydroazepino [4,3-b]indol-1(6H)-one as multitarget-directed agents for Alzheimer’s disease. ChemMedChem 2021, 16, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Weinstock, M.; Goren, T.; Youdim, M.B.H. Development of a novel neuroprotective drug (TV3326) for the treatment of Alzheimer’s disease, with cholinesterase and monoamine oxidase inhibitory activities. Drug Dev. Res. 2000, 50, 216–222. [Google Scholar] [CrossRef]
- Youdim, M.B.; Weinstock, M. Molecular basis of neuroprotective activities of rasagiline and the anti-Alzheimer drug TV3326 [(N-propargyl-(3R)aminoindan-5-yl)-ethyl methyl carbamate]. Cell. Mol. Neurobiol. 2001, 21, 555–573. [Google Scholar] [CrossRef] [PubMed]
- Zohar, K.; Giladi, E.; Eliyahu, T.; Linial, M. Oxidative stress and its modulation by ladostigil alter the expression of abundant long non-coding RNAs in SH-SY5Y cells. Non-Coding RNA 2022, 8, 72. [Google Scholar] [CrossRef]
- Zohar, K.; Lezmi, E.; Eliyahu, T.; Linial, M. Ladostigil Attenuates Induced Oxidative Stress in Human Neuroblast-like SH-SY5Y Cells. Biomedicines 2021, 9, 1251. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Youdim, M.B.H.; Fridkin, M. Site-activated multifunctional chelator with acetylcholinesterase and neuroprotective–neurorestorative moieties for Alzheimer’s therapy. J. Med. Chem. 2009, 52, 4095–4098. [Google Scholar] [CrossRef]
- Huang, W.; Liang, M.; Li, Q.; Zheng, X.; Zhang, C.; Wang, Q.; Tang, L.; Zhang, Z.; Wang, B.; Shen, Z. Development of the “Hidden” multifunctional agents for Alzheimer’s disease. Eur. J. Med. Chem. 2019, 177, 247–258. [Google Scholar] [CrossRef]
- Toublet, F.-X.; Lalut, J.; Hatat, B.; Lecoutey, C.; Davis, A.; Since, M.; Corvaisier, S.; Freret, T.; Sopková-de Oliveira Santos, J.; Claeysen, S.; et al. Pleiotropic prodrugs: Design of a dual butyrylcholinesterase inhibitor and 5-HT6 receptor antagonist with therapeutic interest in Alzheimer’s disease. Eur. J. Med. Chem. 2021, 210, 113059. [Google Scholar] [CrossRef]
- Zheng, H.; Youdim, M.B.H.; Fridkin, M. Site-activated chelators targeting acetylcholinesterase and monoamine oxidase for Alzheimer’s therapy. ACS Chem. Biol. 2010, 5, 603–610. [Google Scholar] [CrossRef]
- Scheiner, M.; Hoffmann, M.; He, F.; Poeta, E.; Chatonnet, A.; Monti, B.; Maurice, T.; Decker, M. Selective pseudo-irreversible butyrylcholinesterase inhibitors transferring antioxidant moieties to the enzyme show pronounced neuroprotective efficacy in vitro and in vivo in an Alzheimer’s disease mouse model. J. Med. Chem. 2021, 64, 9302–9320. [Google Scholar] [CrossRef]
- Verheijen, J.C.; Wiig, K.A.; Du, S.; Connors, S.L.; Martin, A.N.; Ferreira, J.P.; Slepnev, V.I.; Kochendörfer, U. Novel carbamate cholinesterase inhibitors that release biologically active amines following enzyme inhibition. Bioorg. Med. Chem. Lett. 2009, 19, 3243–3246. [Google Scholar] [CrossRef] [PubMed]
- Iancu, G.; Serban, D.; Badiu, C.D.; Tanasescu, C.; Tudosie, M.S.; Tudor, C.; Costea, D.O.; Zgura, A.; Iancu, R.; Vasile, D. Tyrosine kinase inhibitors in breast cancer. Exp. Ther. Med. 2022, 23, 114. [Google Scholar] [CrossRef] [PubMed]
- Lecoutey, C.; Hedou, D.; Freret, T.; Giannoni, P.; Gaven, F.; Since, M.; Bouet, V.; Ballandonne, C.; Corvaisier, S.; Malzert Fréon, A.; et al. Design of Donecopride, a Dual Serotonin Subtype 4 Receptor Agonist/Acetylcholinesterase Inhibitor with Potential Interest for Alzheimer’s Disease Treatment. Proc. Natl. Acad. Sci. USA 2014, 111, E3825–E3830. [Google Scholar] [CrossRef] [PubMed]
- Giannoni, P.; Gaven, F.; de Bundel, D.; Baranger, K.; Marchetti-Gauthier, E.; Roman, F.S.; Valjent, E.; Marin, P.; Bockaert, J.; Rivera, S.; et al. Early administration of RS 67333, a specific 5-HT4 receptor agonist, prevents amyloidogenesis and behavioral deficits in the 5XFAD mouse model of Alzheimer’s disease. Front. Aging Neurosci. 2013, 5, 96. [Google Scholar] [CrossRef]
- Baranger, K.; Giannoni, P.; Girard, S.D.; Girot, S.; Gaven, F.; Stephan, D.; Migliorati, M.; Khrestchatisky, M.; Bockaert, J.; Marchetti-Gauthier, E.; et al. Chronic treatments with a 5-HT4 receptor agonist decrease amyloid pathology in the entorhinal cortex and learning and memory deficits in the 5xFAD mouse model of Alzheimer’s disease. Neuropharmacology 2017, 126, 128–141. [Google Scholar] [CrossRef]
- Hughes, R.E.; Nikolic, K.; Ramsay, R.R. One for All? Hitting Multiple Alzheimer’s Disease Targets with One Drug. Front. Neurosci. 2016, 10, 177. [Google Scholar] [CrossRef]
- Guieu, B.; Lecoutey, C.; Legay, R.; Davis, A.; Sopkova de Oliveira Santos, J.; Altomare, C.D.; Catto, M.; Rochais, C.; Dallemagne, P. First synthesis of racemic trans propargylamino-donepezil, a pleiotrope agent able to both inhibit AChE and MAO-B, with potential interest against Alzheimer’s disease. Molecules 2021, 26, 80. [Google Scholar] [CrossRef]
- Lalut, J.; Santoni, G.; Karila, D.; Lecoutey, C.; Davis, A.; Nachon, F.; Silman, I.; Sussman, J.; Weik, M.; Maurice, T.; et al. Novel Multitarget-Directed Ligands targeting acetylcholinesterase and σ1 receptors as lead compounds for treatment of Alzheimer’s disease: Synthesis, evaluation, and structural characterization of their complexes with acetylcholinesterase. Eur. J. Med. Chem. 2018, 162, 234–248. [Google Scholar] [CrossRef]
- Galimberti, D.; Scarpini, E. Idalopirdine as a treatment for Alzheimer’s disease. Expert Opin. Investig. Drugs 2015, 24, 981–987. [Google Scholar] [CrossRef]
- Lecoutey, C.; Legay, R.; Davis, A.; Sopková-de Oliveira Santos, J.; Dallemagne, P.; Rochais, C. Development of novel potential pleiotropic compounds of interest in Alzheimer ’s disease treatment through rigidification strategy. Molecules 2021, 26, 2536. [Google Scholar] [CrossRef]
- Bautista-Aguilera, Ó.; Hagenow, S.; Palomino-Antolin, A.; Farré-Alins, V.; Ismaili, L.; Joffrin, P.L.; Jimeno, M.L.; Soukup, O.; Janočková, J.; Kalinowsky, L.; et al. Multitarget-directed ligands combining cholinesterase and monoamine oxidase inhibition with histamine H. Angew. Chem. Int. Ed. Engl. 2017, 56, 12765–12769. [Google Scholar] [CrossRef] [PubMed]
- Bautista-Aguilera, Ó.; Budni, J.; Mina, F.; Medeiros, E.B.; Deuther-Conrad, W.; Entrena, J.M.; Moraleda, I.; Iriepa, I.; Lopez-Munoz, F.; Marco-Contelles, J. Contilisant, a tetratarget small molecule for Alzheimer’s disease therapy combining cholinesterase, monoamine oxidase inhibition, and H3R antagonism with S1R agonism profile. J. Med. Chem. 2018, 61, 6937–6943. [Google Scholar] [CrossRef] [PubMed]
- Jourdan, J.-P.; Since, M.; El Kihel, L.; Lecoutey, C.; Corvaisier, S.; Legay, R.; Sopková-de Oliveira Santos, J.; Cresteil, T.; Malzert-Fréon, A.; Rochais, C.; et al. Benzylphenylpyrrolizinones with anti-amyloid and radical scavenging effects, potentially useful in Alzheimer’s disease treatment. ChemMedChem 2017, 12, 913–916. [Google Scholar] [CrossRef] [PubMed]
- Villard, V.; Espallergues, J.; Keller, E.; Vamvakides, A.; Maurice, T. Anti-amnesic and neuroprotective potentials of the mixed muscarinic receptor/sigma 1 (σ1) ligand ANAVEX2-73, a novel aminotetrahydrofuran derivative. J. Psychopharmacol. 2011, 25, 1101–1117. [Google Scholar] [CrossRef]
- Lahmy, V.; Long, R.; Morin, D.; Villard, V.; Maurice, T. Mitochondrial protection by the mixed muscarinic/σ1 ligand ANAVEX2-73, a tetrahydrofuran derivative, in Aβ25-35 peptide-injected mice, a nontransgenic Alzheimer’s disease model. Front. Cell. Neurosci. 2014, 8, 463. [Google Scholar]
- Today AsN. Available online: https://alzheimersnewstoday.com/anavex-2-73 (accessed on 28 July 2023).
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03790709 (accessed on 28 July 2023).
- Freret, T.; Bouet, V.; Quiedeville, A.; Nee, G.; Dallemagne, P.; Rochais, C.; Boulouard, M. Synergistic effect of acetylcholinesterase inhibition (Donepezil) and 5-HT4 receptor activation (RS67333) on object recognition in mice. Behav. Brain Res. 2012, 230, 304–308. [Google Scholar] [CrossRef]
- Sopkova-de Oliveira Santos, J.; Lesnard, A.; Agondanou, J.H.; Dupont, N.; Godard, A.M.; Stiebing, S.; Rochais, C.; Fabis, F.; Dallemagne, P.; Bureau, R.; et al. Virtual screening discovery of new acetylcholinesterase inhibitors issued from CERMN chemical library. J. Chem. Inf. Model. 2010, 50, 422–428. [Google Scholar] [CrossRef]
- Abatematteo, F.S.; Niso, M.; Contino, M.; Leopoldo, M.; Abate, C. Multi-Target Directed Ligands (MTDLs) Binding the σ1 Receptor as Promising Therapeutics: State of the Art and Perspectives. Int. J. Mol. Sci. 2021, 22, 6359. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guiselin, T.; Lecoutey, C.; Rochais, C.; Dallemagne, P. Conceptual Framework of the Design of Pleiotropic Drugs against Alzheimer’s Disease. Pharmaceutics 2023, 15, 2382. https://doi.org/10.3390/pharmaceutics15102382
Guiselin T, Lecoutey C, Rochais C, Dallemagne P. Conceptual Framework of the Design of Pleiotropic Drugs against Alzheimer’s Disease. Pharmaceutics. 2023; 15(10):2382. https://doi.org/10.3390/pharmaceutics15102382
Chicago/Turabian StyleGuiselin, Thomas, Cédric Lecoutey, Christophe Rochais, and Patrick Dallemagne. 2023. "Conceptual Framework of the Design of Pleiotropic Drugs against Alzheimer’s Disease" Pharmaceutics 15, no. 10: 2382. https://doi.org/10.3390/pharmaceutics15102382
APA StyleGuiselin, T., Lecoutey, C., Rochais, C., & Dallemagne, P. (2023). Conceptual Framework of the Design of Pleiotropic Drugs against Alzheimer’s Disease. Pharmaceutics, 15(10), 2382. https://doi.org/10.3390/pharmaceutics15102382