
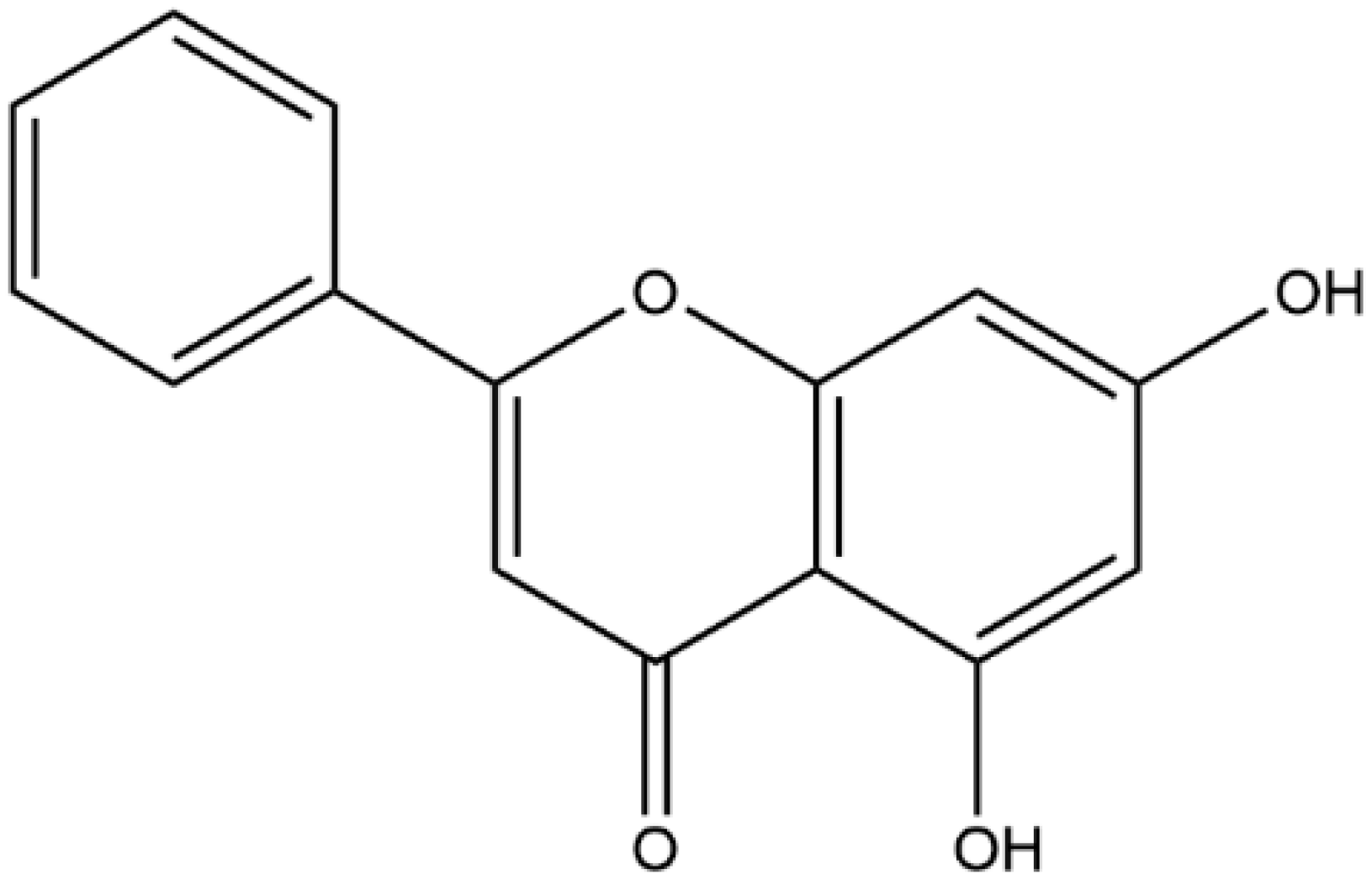
The Amorphous Solid Dispersion of Chrysin in Plasdone® S630 Demonstrates Improved Oral Bioavailability and Antihyperlipidemic Performance in Rats

Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Solubility Studies
2.3. Preparation of CHR-SD
2.4. Characterizations of CHR-SD
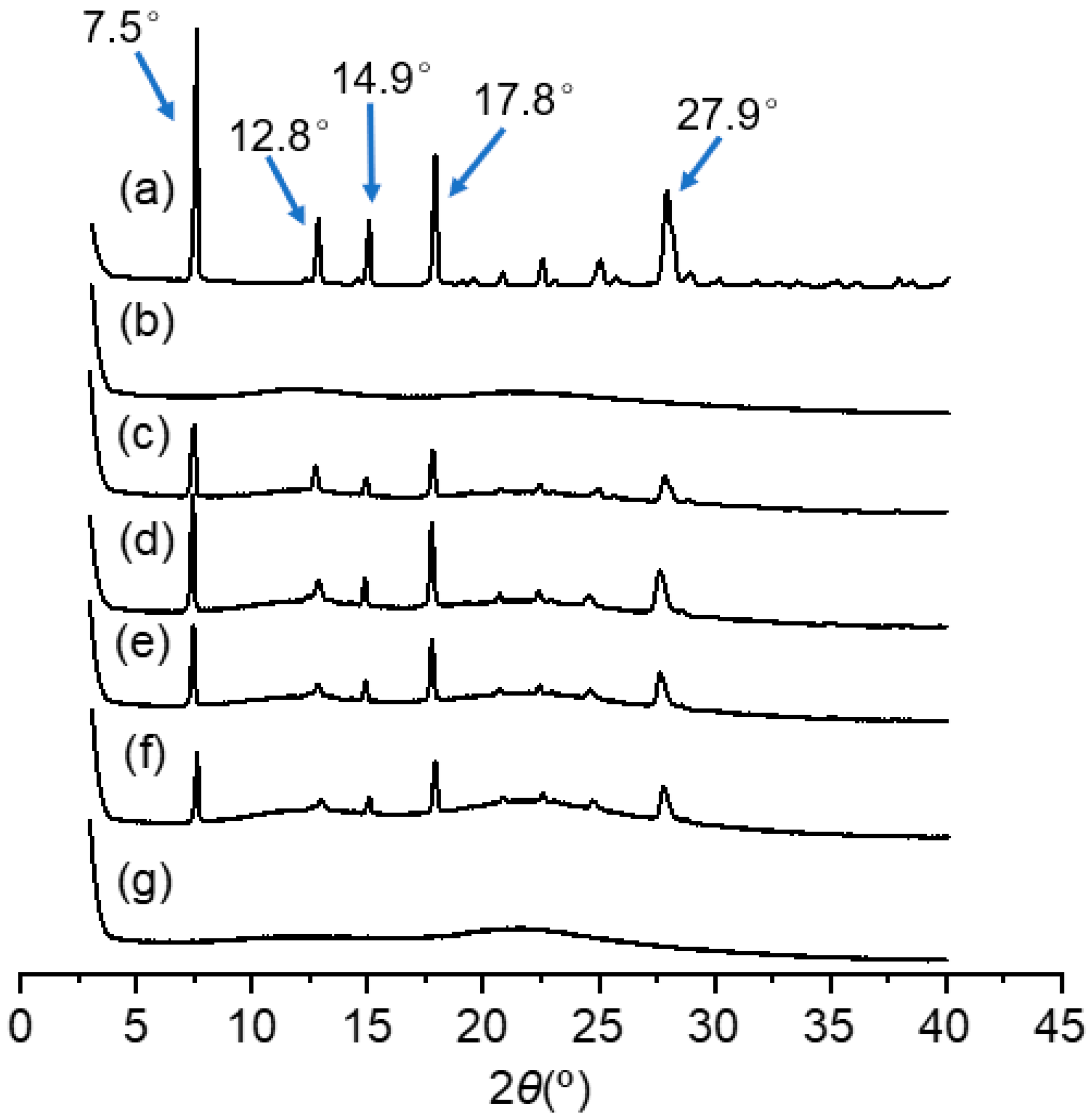
2.4.1. X-ray Diffraction (XRD)
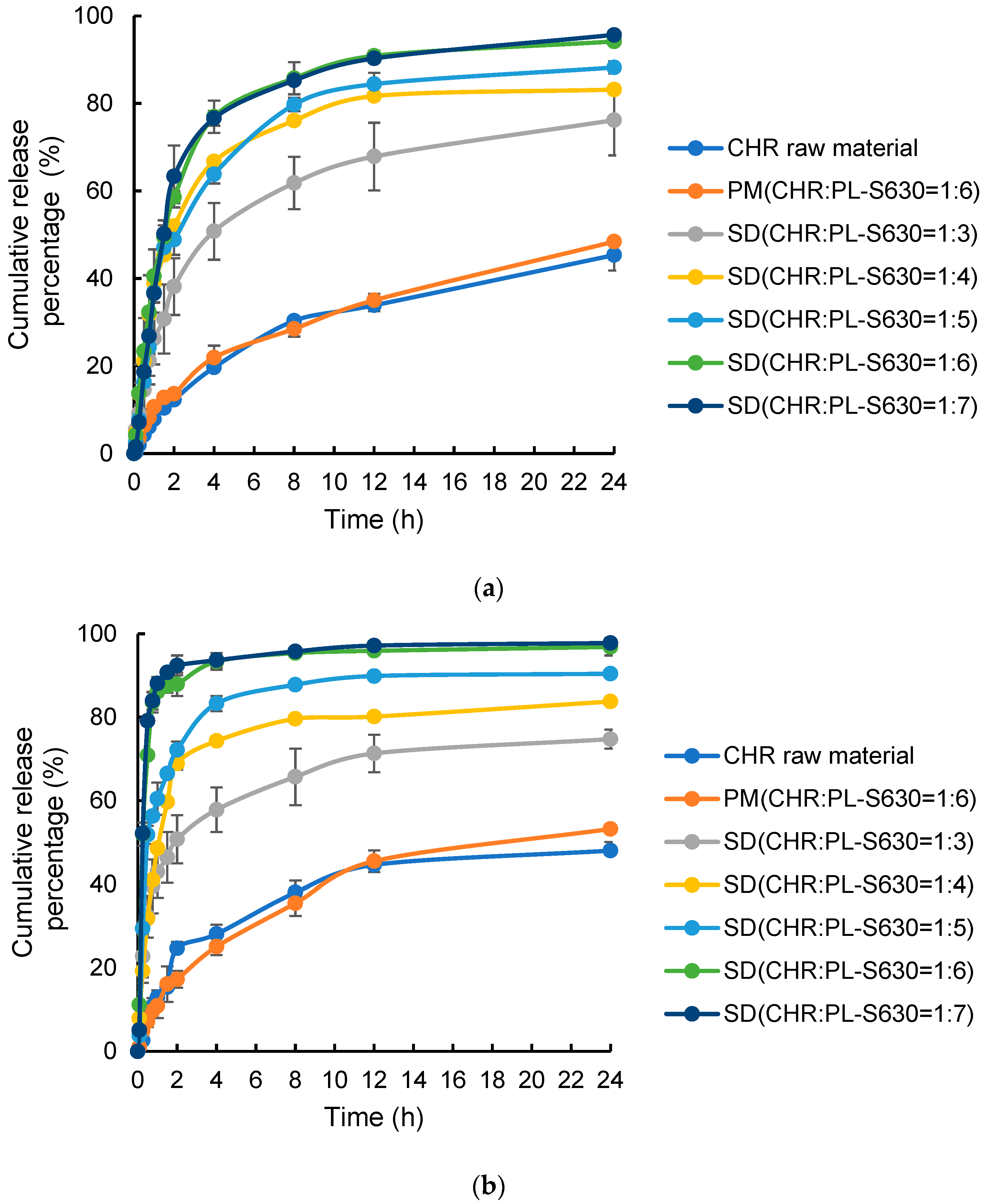
2.4.2. Dissolution Profiles
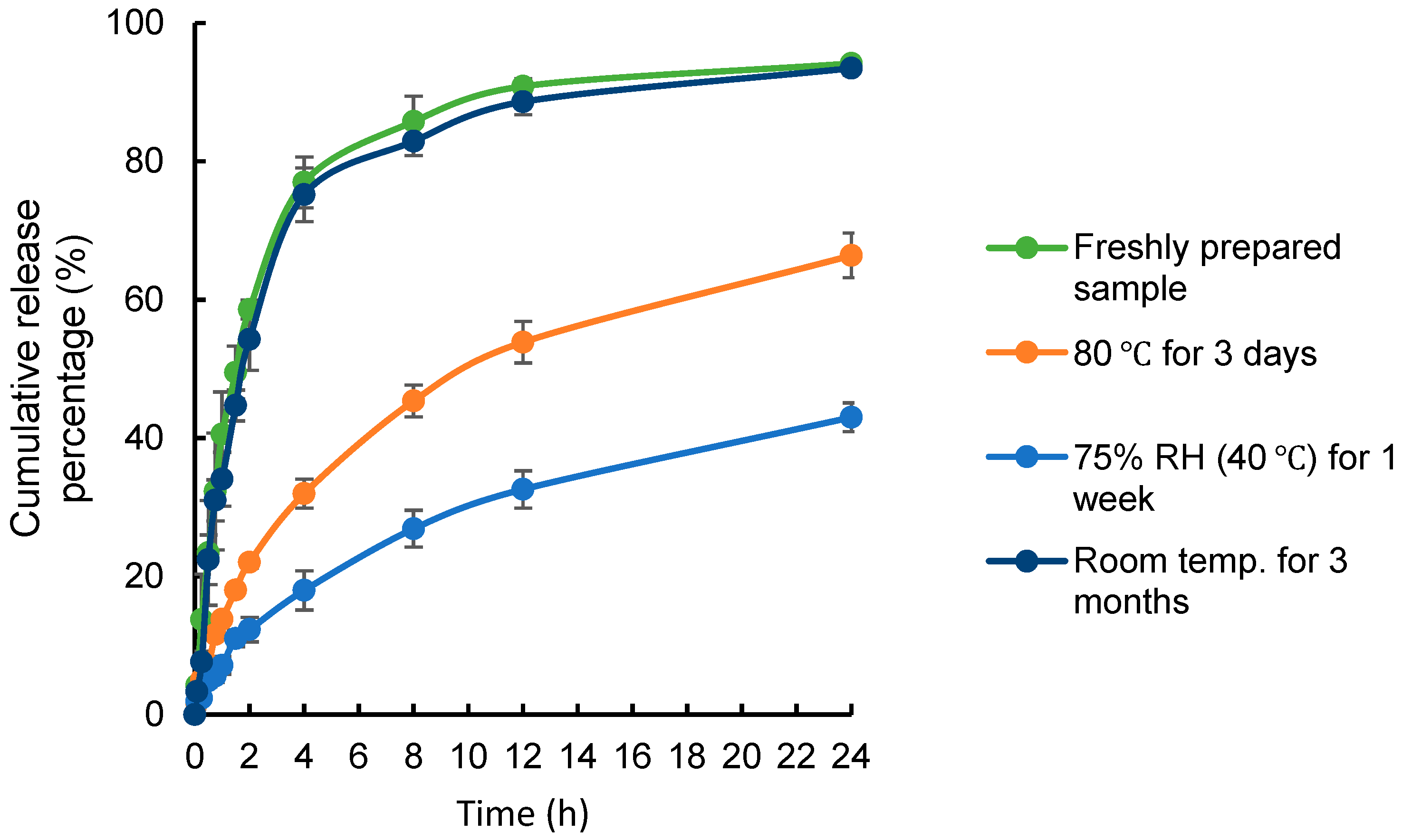
2.4.3. Stability Studies
2.5. In Situ Intestinal Perfusion Experiment
2.6. Plasma Pharmacokinetics
2.7. Antihyperlipidemic Performance
2.8. Statistical Analysis
3. Results and Discussion
3.1. Solubility Studies
3.2. Preparation of CHR-SD
3.3. Characterizations of CHR-SD
3.3.1. XRD
3.3.2. Dissolution
3.3.3. Stability
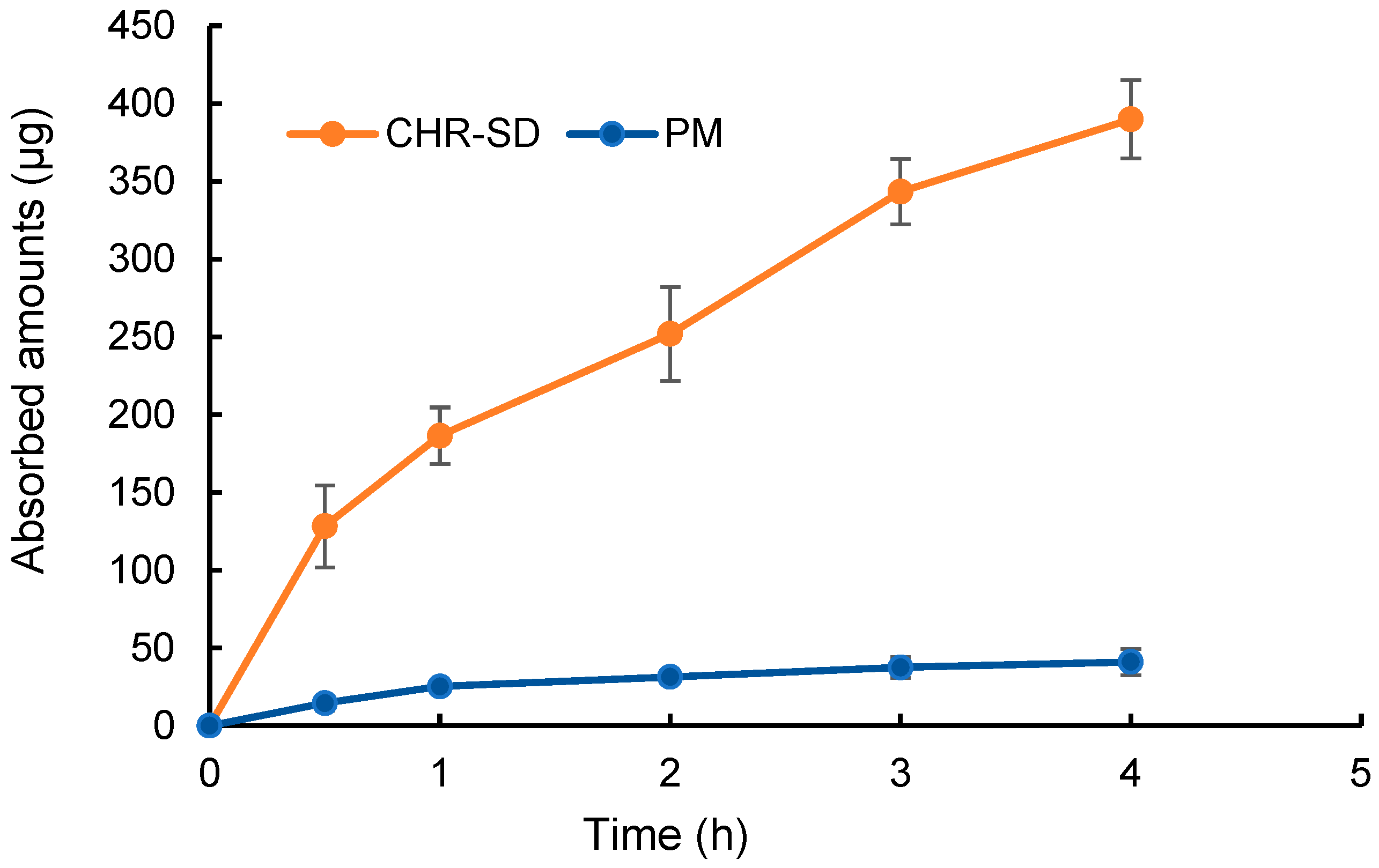
3.4. In-Situ Intestinal Perfusion Experiment
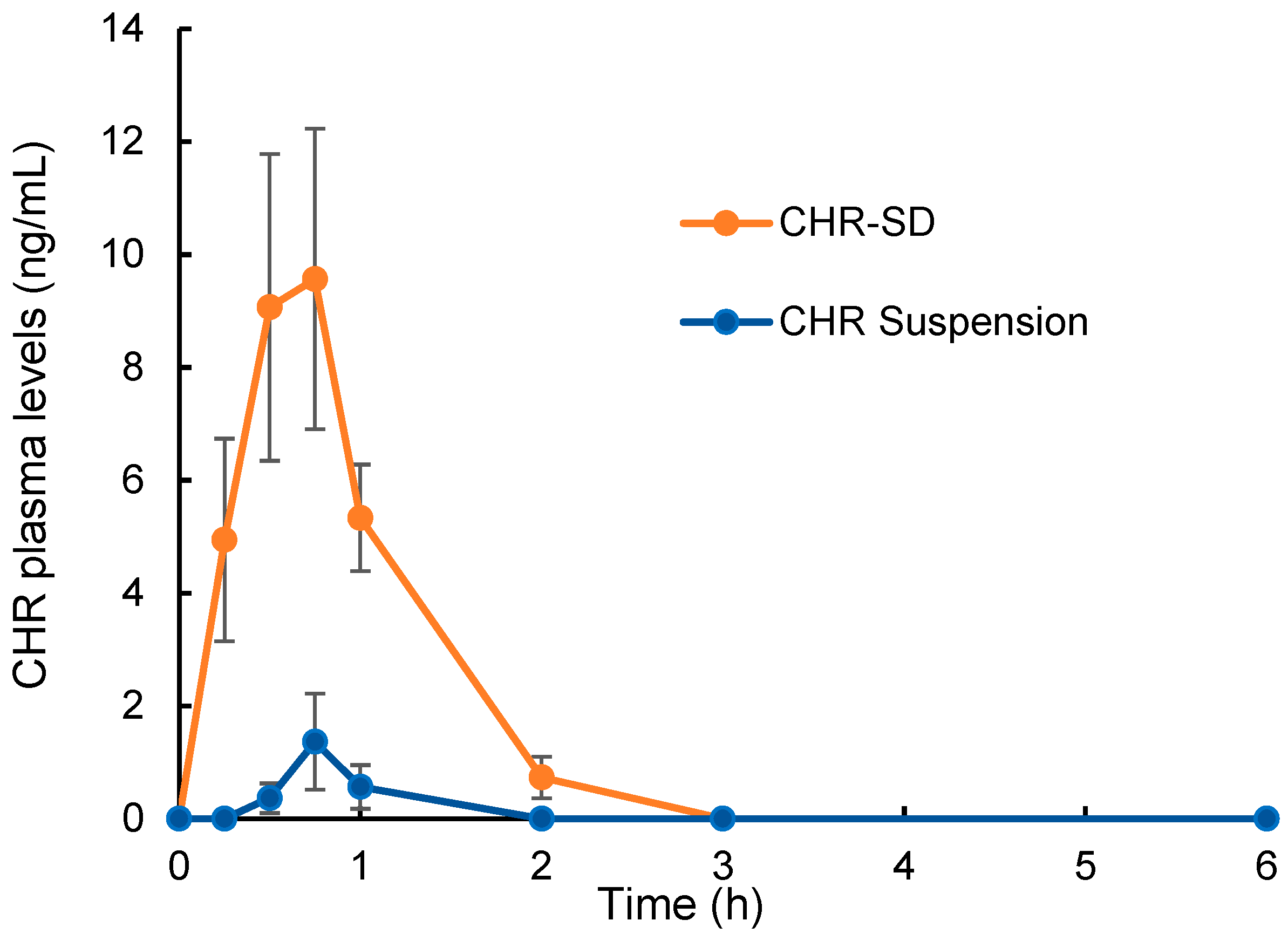
3.5. Bioavailability Study
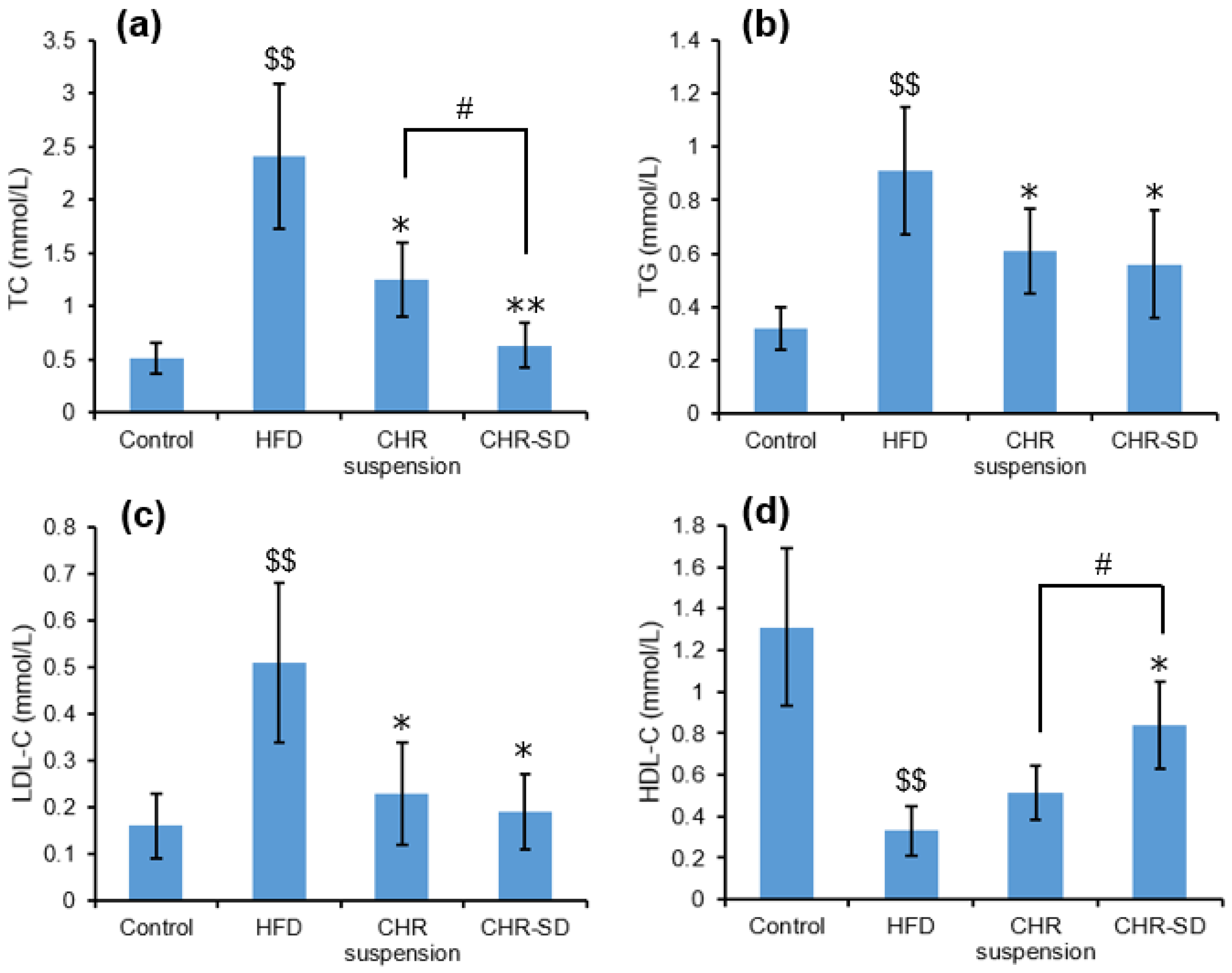
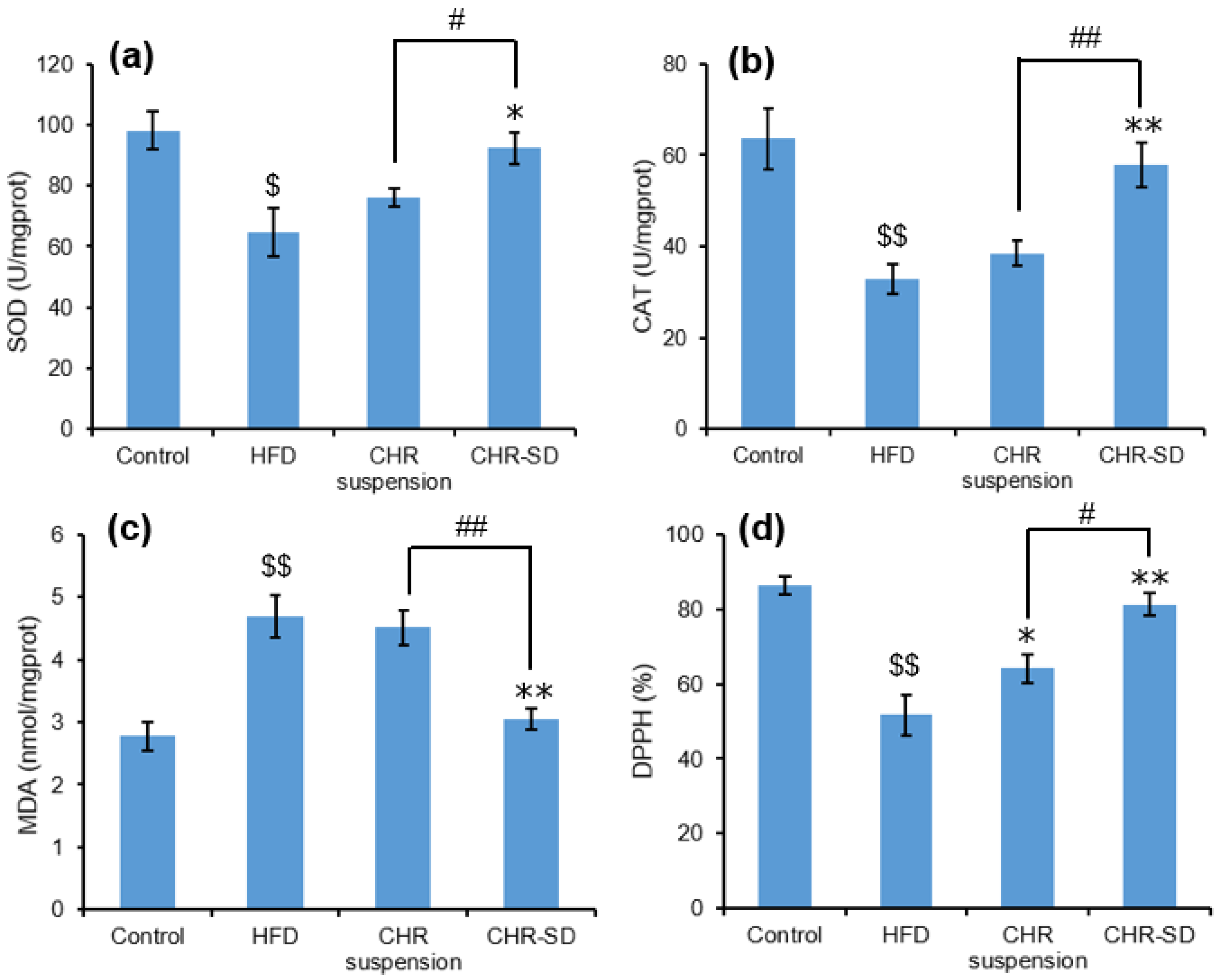
3.6. Antihyperlipidemic Performance
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- El-Tantawy, W.H.; Temraz, A. Natural Products for Controlling Hyperlipidemia: Review. Arch. Physiol. Biochem. 2019, 125, 128–135. [Google Scholar] [CrossRef]
- Wang, S.; Du, Q.; Meng, X.; Zhang, Y. Natural Polyphenols: A Potential Prevention and Treatment Strategy for Metabolic Syndrome. Food Funct. 2022, 13, 9734–9753. [Google Scholar] [CrossRef]
- Rufino, A.T.; Costa, V.M.; Carvalho, F.; Fernandes, E. Flavonoids as Antiobesity Agents: A Review. Med. Res. Rev. 2021, 41, 556–585. [Google Scholar] [CrossRef]
- Ross, J.A.; Kasum, C.M. Dietary Flavonoids: Bioavailability, Metabolic Effects, and Safety. Annu. Rev. Nutr. 2002, 22, 19–34. [Google Scholar] [CrossRef]
- Gao, S.; Siddiqui, N.; Etim, I.; Du, T.; Zhang, Y.; Liang, D. Developing Nutritional Component Chrysin as a Therapeutic Agent: Bioavailability and Pharmacokinetics Consideration, and ADME Mechanisms. Biomed. Pharmacother. 2021, 142, 112080. [Google Scholar] [CrossRef]
- El-Bassossy, H.M.; Abo-Warda, S.M.; Fahmy, A. Chrysin and Luteolin Alleviate Vascular Complications Associated with Insulin Resistance Mainly through PPAR-γ Activation. Am. J. Chin. Med. 2014, 42, 1153–1167. [Google Scholar] [CrossRef]
- Ramírez-Espinosa, J.; Saldaña-Ríos, J.; García-Jiménez, S.; Villalobos-Molina, R.; Ávila-Villarreal, G.; Rodríguez-Ocampo, A.; Bernal-Fernández, G.; Estrada-Soto, S. Chrysin Induces Antidiabetic, Antidyslipidemic and Anti-Inflammatory Effects in Athymic Nude Diabetic Mice. Molecules 2017, 23, 67. [Google Scholar] [CrossRef]
- Fenyvesi, F.; Nguyen, T.L.P.; Haimhoffer, Á.; Rusznyák, Á.; Vasvári, G.; Bácskay, I.; Vecsernyés, M.; Ignat, S.-R.; Dinescu, S.; Costache, M.; et al. Cyclodextrin Complexation Improves the Solubility and Caco-2 Permeability of Chrysin. Materials 2020, 13, 3618. [Google Scholar] [CrossRef]
- Fang, Y.; Cao, W.; Xia, M.; Pan, S.; Xu, X. Study of Structure and Permeability Relationship of Flavonoids in Caco-2 Cells. Nutrients 2017, 9, 1301. [Google Scholar] [CrossRef]
- Baidya, D.; Kushwaha, J.; Mahadik, K.; Patil, S. Chrysin-Loaded Folate Conjugated PF127-F68 Mixed Micelles with Enhanced Oral Bioavailability and Anticancer Activity against Human Breast Cancer Cells. Drug Dev. Ind. Pharm. 2019, 45, 852–860. [Google Scholar] [CrossRef]
- Walle, T.; Otake, Y.; Brubaker, J.A.; Walle, U.K.; Halushka, P.V. Disposition and Metabolism of the Flavonoid Chrysin in Normal Volunteers. Br. J. Clin. Pharmacol. 2001, 51, 143–146. [Google Scholar] [PubMed]
- Kim, S.-M.; Jung, J.-I.; Chai, C.; Imm, J.-Y. Characteristics and Glucose Uptake Promoting Effect of Chrysin-Loaded Phytosomes Prepared with Different Phospholipid Matrices. Nutrients 2019, 11, 2549. [Google Scholar] [CrossRef]
- El-Hussien, D.; El-Zaafarany, G.M.; Nasr, M.; Sammour, O. Chrysin Nanocapsules with Dual Anti-Glycemic and Anti-Hyperlipidemic Effects: Chemometric Optimization, Physicochemical Characterization and Pharmacodynamic Assessment. Int. J. Pharm. 2021, 592, 120044. [Google Scholar] [CrossRef]
- Newman, A.; Knipp, G.; Zografi, G. Assessing the Performance of Amorphous Solid Dispersions. J. Pharm. Sci. 2012, 101, 1355–1377. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Chen, J.; Zhang, Z.; Wang, F.; Wang, L.; Lin, Y.; Zhang, X.; Liu, J. Solubilization of Luteolin in PVP40 Solid Dispersion Improves Inflammation-Induced Insulin Resistance in Mice. Eur. J. Pharm. Sci. 2022, 174, 106188. [Google Scholar] [CrossRef] [PubMed]
- Ishimoto, K.; Shimada, Y.; Ohno, A.; Otani, S.; Ago, Y.; Maeda, S.; Lin, B.; Nunomura, K.; Hino, N.; Suzuki, M.; et al. Physicochemical and Biochemical Evaluation of Amorphous Solid Dispersion of Naringenin Prepared Using Hot-Melt Extrusion. Front. Nutr. 2022, 9, 850103. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Lee, Y.; Song, J.G.; Han, H.-K. Improved In vivo Effect of Chrysin as an Absorption Enhancer Via the Preparation of Ternary Solid Dispersion with Brij®L4 and Aminoclay. Curr. Drug Deliv. 2018, 16, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Castro, G.T.; Ferretti, F.H.; Blanco, S.E. Determination of the Overlapping PKa Values of Chrysin Using UV–Vis Spectroscopy and ab Initio Methods. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2005, 62, 657–665. [Google Scholar] [CrossRef]
- Van Eerdenbrugh, B.; Baird, J.A.; Taylor, L.S. Crystallization Tendency of Active Pharmaceutical Ingredients Following Rapid Solvent Evaporation—Classification and Comparison with Crystallization Tendency from Undercooled Melts. J. Pharm. Sci. 2010, 99, 3826–3838. [Google Scholar] [CrossRef]
- Serajuddin, A.T.M. Solid Dispersion of Poorly Water-soluble Drugs: Early Promises, Subsequent Problems, and Recent Breakthroughs. J. Pharm. Sci. 1999, 88, 1058–1066. [Google Scholar] [CrossRef] [PubMed]
- Zecevic, D.E.; Evans, R.C.; Paulsen, K.; Wagner, K.G. From Benchtop to Pilot Scale–Experimental Study and Computational Assessment of a Hot-Melt Extrusion Scale-up of a Solid Dispersion of Dipyridamole and Copovidone. Int. J. Pharm. 2018, 537, 132–139. [Google Scholar] [CrossRef]
- Choi, J.-S.; Lee, S.-E.; Jang, W.S.; Byeon, J.C.; Park, J.-S. Tadalafil Solid Dispersion Formulations Based on PVP/VA S-630: Improving Oral Bioavailability in Rats. Eur. J. Pharm. Sci. 2017, 106, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Hurley, D.; Carter, D.; Foong Ng, L.Y.; Davis, M.; Walker, G.M.; Lyons, J.G.; Higginbotham, C.L. An Investigation of the Inter-Molecular Interaction, Solid-State Properties and Dissolution Properties of Mixed Copovidone Hot-Melt Extruded Solid Dispersions. J. Drug Deliv. Sci. Technol. 2019, 53, 101132. [Google Scholar] [CrossRef]
- Khougaz, K.; Clas, S. Crystallization Inhibition in Solid Dispersions of MK-0591 and Poly(Vinylpyrrolidone) Polymers. J. Pharm. Sci. 2000, 89, 1325–1334. [Google Scholar] [CrossRef]
- Tang, X.C.; Pikal, M.J.; Taylor, L.S. A Spectroscopic Investigation of Hydrogen Bond Patterns in Crystalline and Amorphous Phases in Dihydropyridine Calcium Channel Blockers. Pharm. Res. 2002, 19, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Wegiel, L.A.; Mauer, L.J.; Edgar, K.J.; Taylor, L.S. Crystallization of Amorphous Solid Dispersions of Resveratrol during Preparation and Storage—Impact of Different Polymers. J. Pharm. Sci. 2013, 102, 171–184. [Google Scholar] [CrossRef]
- Kaparekar, P.S.; Poddar, N.; Anandasadagopan, S.K. Fabrication and Characterization of Chrysin—A Plant Polyphenol Loaded Alginate -Chitosan Composite for Wound Healing Application. Colloids Surf. B Biointerfaces 2021, 206, 111922. [Google Scholar] [CrossRef] [PubMed]
- Butreddy, A.; Sarabu, S.; Bandari, S.; Batra, A.; Lawal, K.; Chen, N.N.; Bi, V.; Durig, T.; Repka, M.A. Influence of PlasdoneTM S630 Ultra—An Improved Copovidone on the Processability and Oxidative Degradation of Quetiapine Fumarate Amorphous Solid Dispersions Prepared via Hot-Melt Extrusion Technique. AAPS PharmSciTech 2021, 22, 196. [Google Scholar] [CrossRef]
- Wilson, V.; Lou, X.; Osterling, D.J.; Stolarik, D.F.; Jenkins, G.; Gao, W.; Zhang, G.G.Z.; Taylor, L.S. Relationship between Amorphous Solid Dispersion in Vivo Absorption and in Vitro Dissolution: Phase Behavior during Dissolution, Speciation, and Membrane Mass Transport. J. Control Release 2018, 292, 172–182. [Google Scholar] [CrossRef]
- Dong, D.; Quan, E.; Yuan, X.; Xie, Q.; Li, Z.; Wu, B. Sodium Oleate-Based Nanoemulsion Enhances Oral Absorption of Chrysin through Inhibition of UGT-Mediated Metabolism. Mol. Pharm. 2017, 14, 2864–2874. [Google Scholar] [CrossRef]
- Ahmed, T.A.; Elimam, H.; Alrifai, A.O.; Nadhrah, H.M.; Masoudi, L.Y.; Sairafi, W.O.; El-Say, K.M. Rosuvastatin Lyophilized Tablets Loaded with Flexible Chitosomes for Improved Drug Bioavailability, Anti-Hyperlipidemic and Anti-Oxidant Activity. Int. J. Pharm. 2020, 588, 119791. [Google Scholar] [CrossRef] [PubMed]
- Rani, N.; Bharti, S.; Bhatia, J.; Tomar, A.; Nag, T.C.; Ray, R.; Arya, D.S. Inhibition of TGF-β by a Novel PPAR-γ Agonist, Chrysin, Salvages β-receptor Stimulated Myocardial Injury in Rats through MAPKs-dependent Mechanism. Nutr. Metab. 2015, 12, 11. [Google Scholar] [CrossRef] [PubMed]
- Meshram, S.; Verma, V.K.; Mutneja, E.; Sahu, A.K.; Malik, S.; Mishra, P.; Bhatia, J.; Arya, D.S. Evidence-based Mechanistic Role of Chrysin towards Protection of Cardiac Hypertrophy and Fibrosis in Rats. Br. J. Nutr. 2023, 129, 1105–1118. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | CHR-SD | CHR Suspension |
|---|---|---|
| Tmax (h) | 0.62 ± 0.12 | 0.71 ± 0.09 |
| Cmax (ng/mL) | 9.57 ± 2.66 ** | 1.37 ± 0.85 |
| AUC0–6 h (h·ng/mL) | 13.81 ± 2.70 ** | 0.33 ± 0.18 |
| Groups | Hepatic Indexes (%) | WAT Indexes (%) |
|---|---|---|
| Control | 2.86 ± 0.29 | 3.89 ± 0.51 |
| HFD | 4.23 ± 0.38 | 8.46 ± 1.58 |
| CHR suspension | 3.57 ± 0.34 * | 6.22 ± 1.21 * |
| CHR-SD | 3.11 ± 0.21 * | 4.47 ± 0.79 ** |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, C.; Liu, X.; Zhao, R.; Yang, M.; Liu, W.; Dai, Q.; Bao, X.; Chen, Y.; Ma, J. The Amorphous Solid Dispersion of Chrysin in Plasdone® S630 Demonstrates Improved Oral Bioavailability and Antihyperlipidemic Performance in Rats. Pharmaceutics 2023, 15, 2378. https://doi.org/10.3390/pharmaceutics15102378
Wang C, Liu X, Zhao R, Yang M, Liu W, Dai Q, Bao X, Chen Y, Ma J. The Amorphous Solid Dispersion of Chrysin in Plasdone® S630 Demonstrates Improved Oral Bioavailability and Antihyperlipidemic Performance in Rats. Pharmaceutics. 2023; 15(10):2378. https://doi.org/10.3390/pharmaceutics15102378
Chicago/Turabian StyleWang, Chenhui, Xiaowei Liu, Ruihan Zhao, Meiqing Yang, Wenqian Liu, Qiuyang Dai, Xiaofeng Bao, Yong Chen, and Jun Ma. 2023. "The Amorphous Solid Dispersion of Chrysin in Plasdone® S630 Demonstrates Improved Oral Bioavailability and Antihyperlipidemic Performance in Rats" Pharmaceutics 15, no. 10: 2378. https://doi.org/10.3390/pharmaceutics15102378
APA StyleWang, C., Liu, X., Zhao, R., Yang, M., Liu, W., Dai, Q., Bao, X., Chen, Y., & Ma, J. (2023). The Amorphous Solid Dispersion of Chrysin in Plasdone® S630 Demonstrates Improved Oral Bioavailability and Antihyperlipidemic Performance in Rats. Pharmaceutics, 15(10), 2378. https://doi.org/10.3390/pharmaceutics15102378