2.1. Polyesters
Several polyester-based copolymers with “allyl” functionality have been reported in the literature via ROP and post-functionalized by either click, epoxidation, or bromination reactions. For instance, Kost et al. [
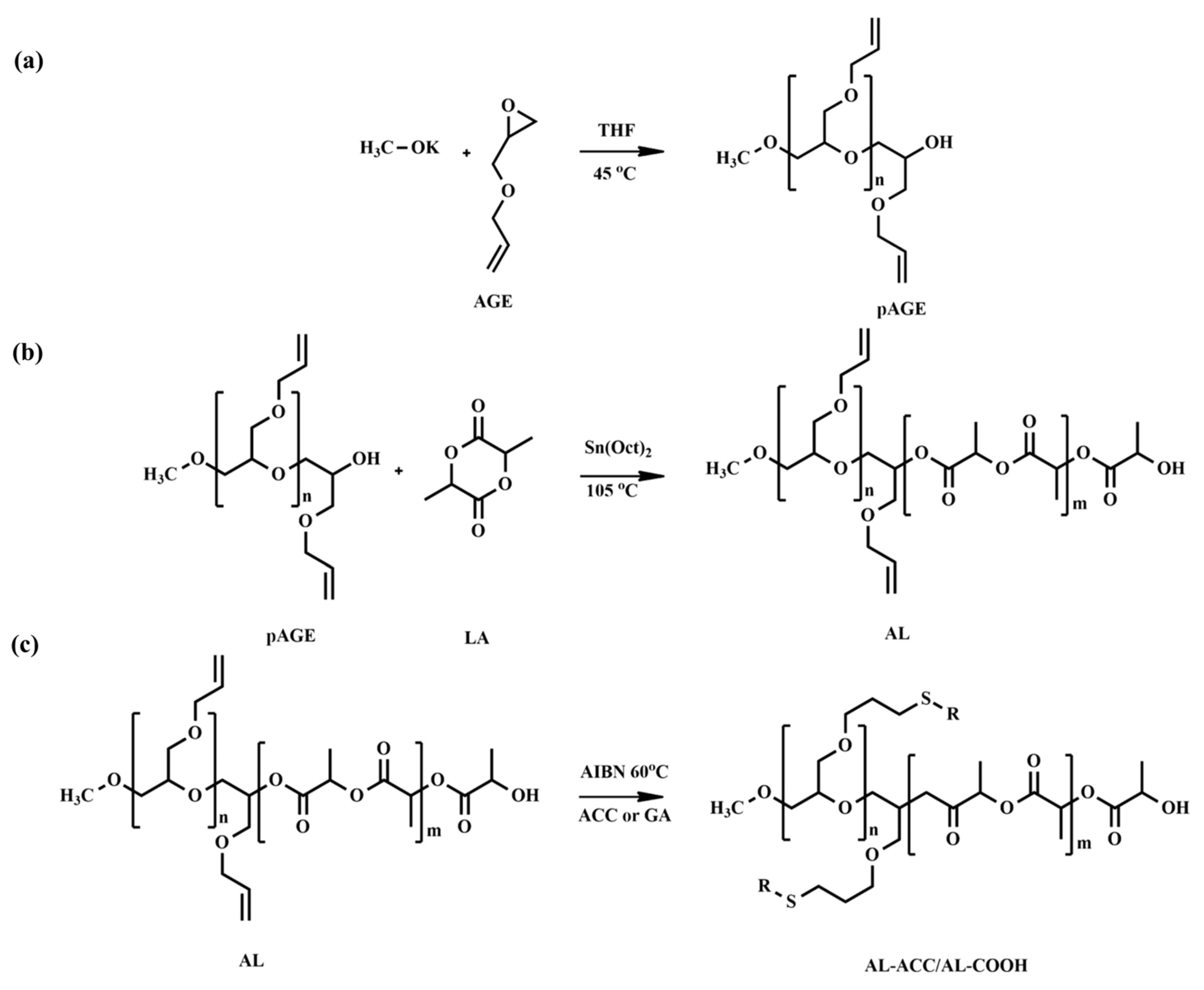
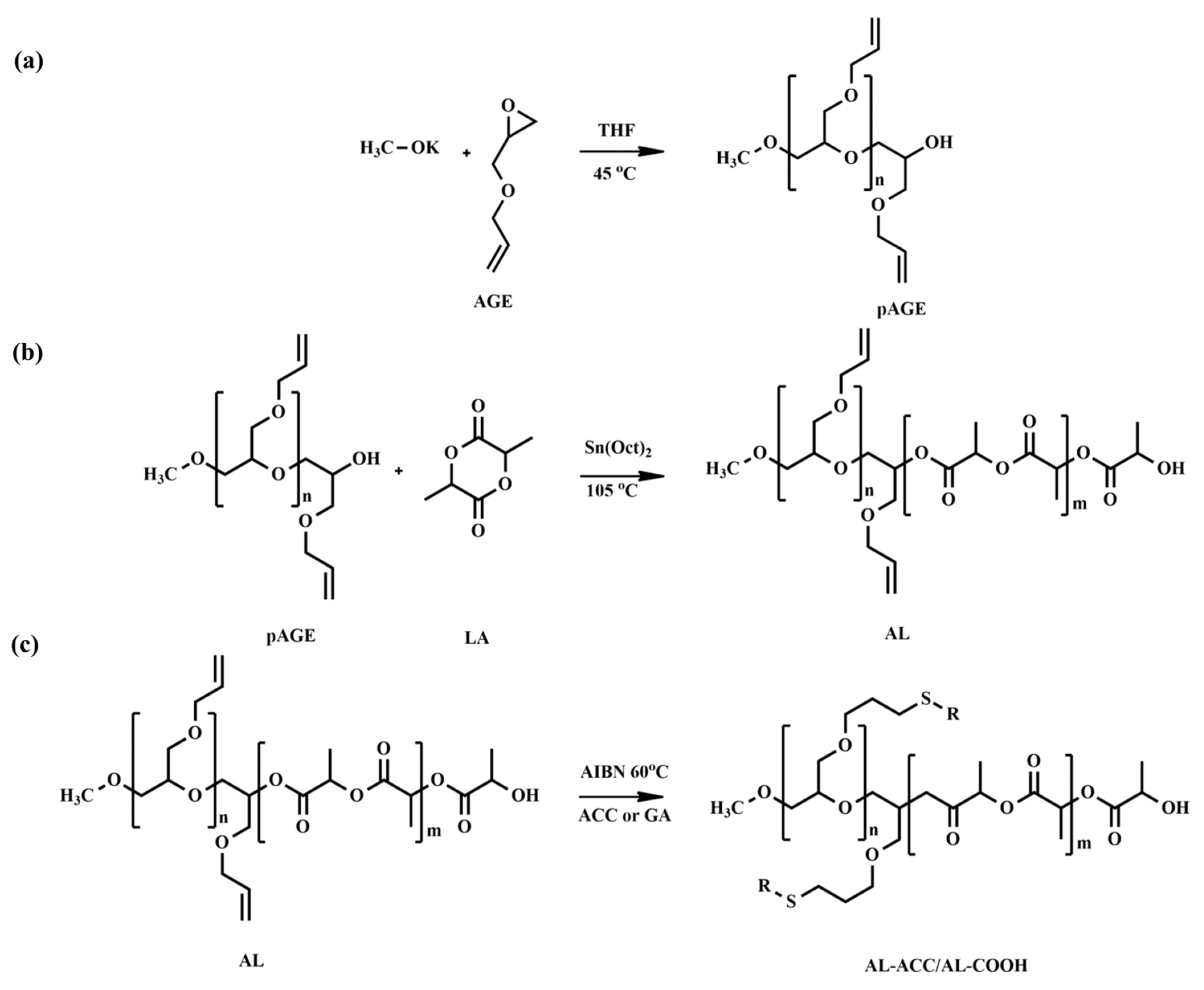
58] developed novel amorphous pH-tunable copolymers of lactide (LA) and allyl glycidyl ether (AGE) with functionalities for the efficient delivery of anti-cancer drugs. Poly(allyl glycidyl ether) (pAGE) was synthesized via anionic ROP and used as a macroinitiator for the ROP of LA to generate a copolymer, poly(allyl glycidyl ether-
co-lactide) (AL), with free allyl functional groups in the backbone. To synthesize pAGE, the authors utilized anionic ROP instead of cationic ROP, as a later method produced a low molecular weight polymer. The synthesis process of AL copolymers was performed at a relatively low temperature (105 °C) to prevent the isomerization of unsaturated bonds on pAGE. The proton nuclear magnetic resonance (
1H NMR) and size exclusion chromatography (SEC) analyses exhibited no significant isomerization of the double bond in the AL chains. The AL copolymers were not pH-responsive. Later, post-functionalization of the AL copolymers was performed by thiol-ene click reaction using thioglycolic acid (tGA) and N-acetyl-L-cysteine (ACC) to introduce pH-sensitive functional groups to the main chain pendants (
Figure 1). The resulting post-functionalized copolymers with either ACC (AL ACC) or tGA (AL COOH) were found to be sensitive to pH changes as these compounds serve carboxyl or amino functionalities.
The main focus of Kost et al.’s study [
58] was to design biocompatible pH-responsive nanocarriers, which will not only deliver the anti-cancer drugs effectively to the desired targets, but also preserve the balance between the responsiveness of the nanocarriers and their stability in physiological conditions. With the aims of obtaining high drug encapsulation efficiency (EE), narrow size dispersity, and relatively small-size nanoparticles (NPs), the authors conducted a nanoprecipitation method to prepare blank and doxorubicin (DOX)-loaded NPs using neat AL copolymer, post-functionalized (AL ACC) and (AL COOH) copolymers as the matrix and poly(ethylene glycol) methyl ether-b-poly(D,L-lactide) (PEGME-
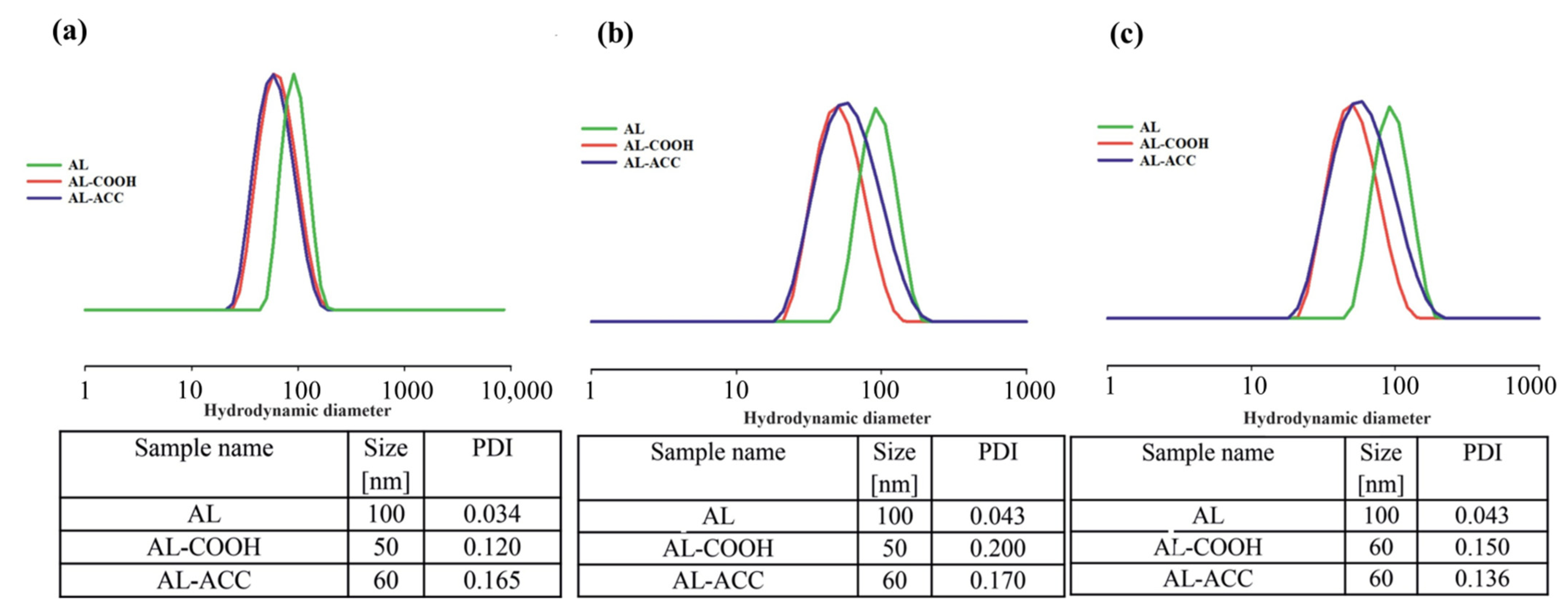
b-PDLLA) as the non-ionic surfactant. The size and dispersity of all prepared NPs in water, PBS (pH = 7.0), and acetic acid buffer (pH = 5.0) are presented in
Figure 2. The authors obtained larger NPs from non-functionalized AL copolymers (size 100 nm in all three media); this has been attributed to the rigidity of the pAGE chain due to the presence of unsaturated double bonds (which are known to prevent free rotation of molecules). On the other hand, functionalized copolymers produced NPs with a hydrodynamic size which was half that of their non-functionalized counterparts due to the increase in flexibility of the copolymer, with decrease in the number of allyl groups. Partial and full functionalization of allyl groups could be the factor in getting slightly smaller size and lower dispersity NPs from AL COOH, and slightly bigger and higher dispersity NPs from AL ACC copolymers, respectively. The authors assessed the change in size of NPs with respect to pH change by incubating NPs at 37 °C within 24 h and noticed no significant change in size of NPs in physiological and acidic conditions (pH = 7.0 and 5.0 respectively) with respect to the sizes in water. The stability of NPs with tGA and ACC in acidic pH 5.0 was accounted for by the formation of stable carboxylate moieties, negatively charged ions for both NPs with tGA and ACC, or the development of PEG corona on the surface of NPs with ACC. The authors’ explanation of the size of NPs in three different media was in agreement with studies in the literature [
69,
70,
71,
72,
73]. However, the authors did not discuss the dispersity of the prepared NPs.
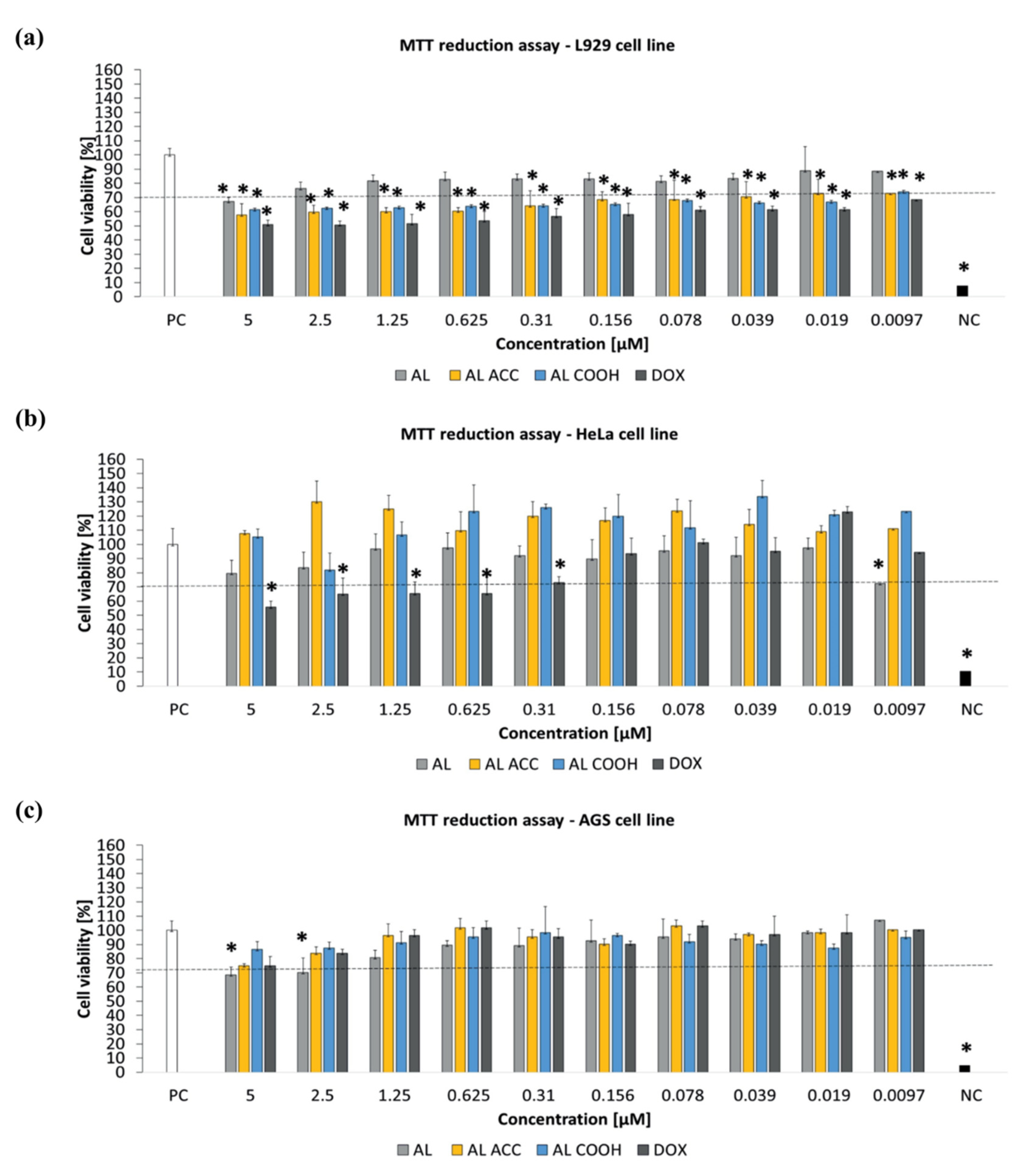
The observed EE for DOX was ~36% and the release study suggested reduction in burst release due to the supramolecular interactions between polymeric pedant groups and DOX. Moreover, a slower release rate was observed at pH = 5.0 due to the strongest interaction between the AC ACC or AC COOH functionalities with DOX. In addition, ACC is well recognized for its high antioxidant properties; therefore, ACC containing nanocarriers can contribute to enhancing cancer treatment during cancer therapy. A microculture tetrazolium assay (MTT) assay was performed using murine fibroblast cell line (L929), human breast cancer cell line (HeLa), and human gastric adenocarcinoma cancer cell line (AGS) to evaluate the toxicity of blank and DOX-loaded NPs. A complete culture medium and 0.03% H
2O
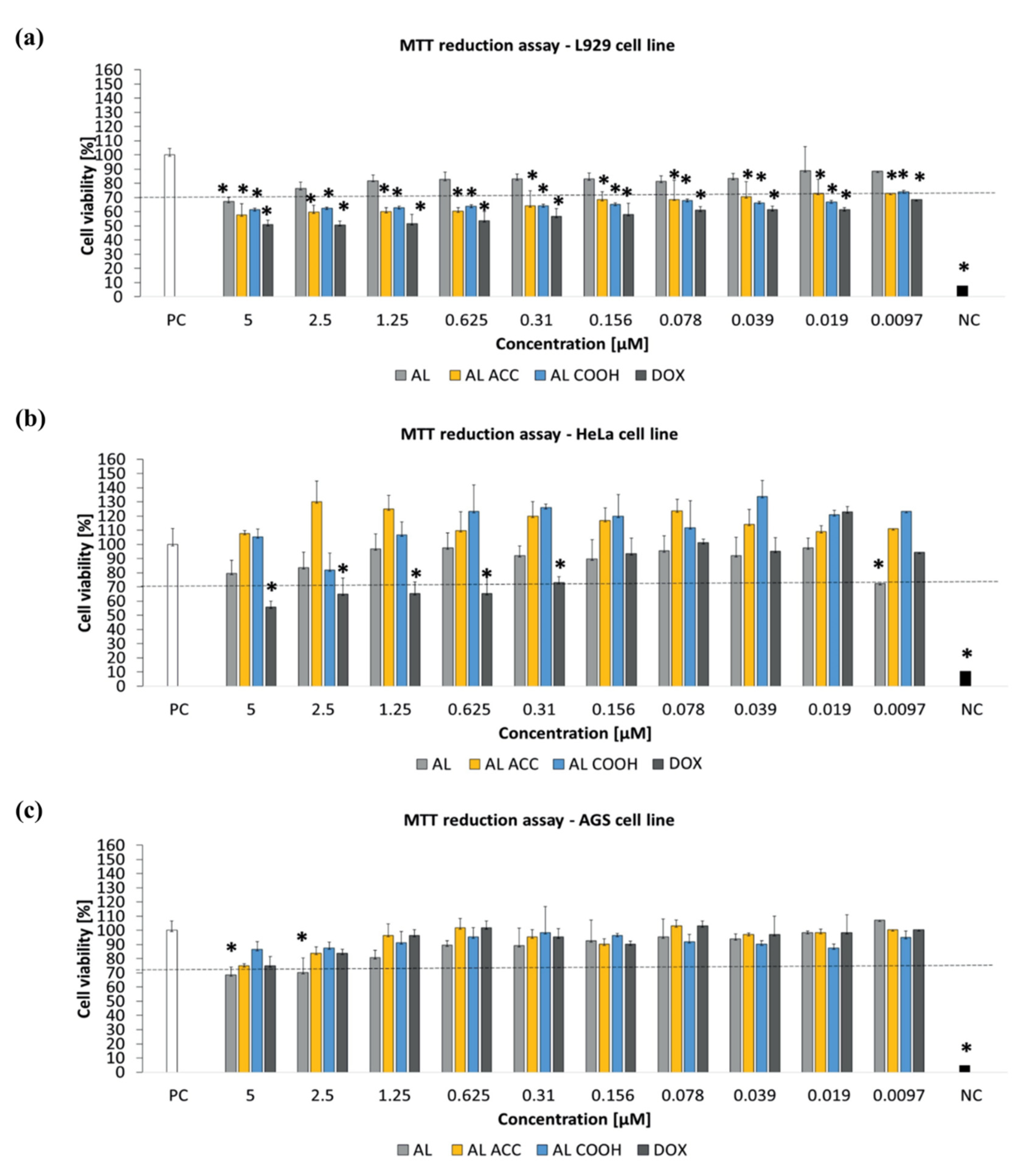
2 were considered as a positive control of cell viability (100% viable cells) and a negative control of cell viability (100% dead inactive cells), correspondingly. The MTT assay revealed the non-toxicity of all blank NPs against all three tested cell lines after 24 h incubation at 37 °C (
Figure 3). The authors stated that some tested compounds, e.g., DAPI (40, 6-diamino-2-phenylindole), might be subjected to Collagen type I secretion by endothelial or epithelial cells due to proregenerative activity, which eventually caused cell viability to pass over 100% in the case of the HeLa cell line. The concentration-dependent insignificant cytotoxicity of AL COOH and AL ACC NPs against the murine L929 fibroblasts observed in this study accounted for the strong interaction between tGA and ACC moieties of NPs and the cell membrane. This effect is supported by a study conducted by He [
1].
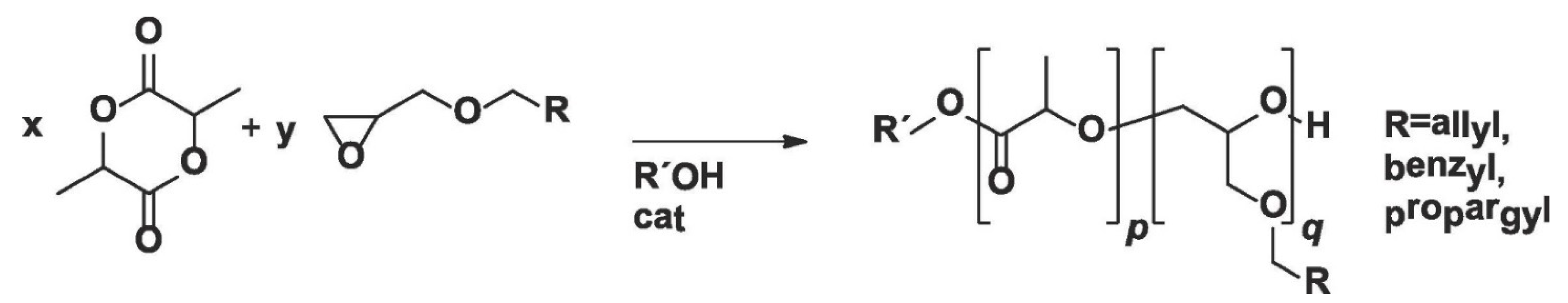
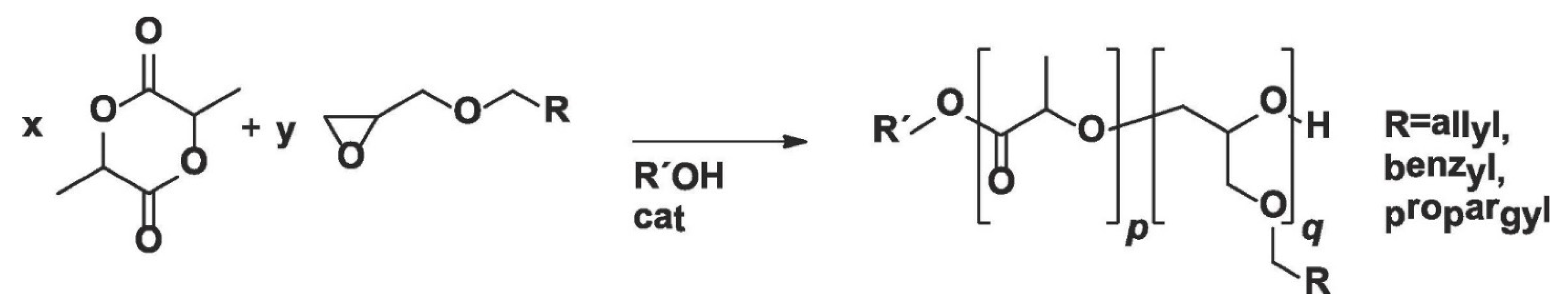
In a similar study, Pound-Lana et al. [
36] synthesized LA copolymer with three glycidyl ethers (allyl, benzyl, and propargyl glycidyl ethers) containing either allyl or propargyl groups for further modifications via ROP using tin(II)octoate or 1,5,7-triazabicyclo [4.4.0]dec-5-ene (TBD) as catalyst (
Figure 4). The authors observed that poly(lactide-co-allyl glycidyl ether) copolymer with 4.6 allyl groups per chain can be obtained by tin(II)octoate-catalyzed copolymerization of LA with AGE. However, the number of allyl groups per chain was reduced to less than one, and mostly PLA homopolymer was formed when TBD was used as the catalyst. The authors also found that a higher number of reactive groups per chain (up to 8.7 mole% of allyl functional groups) can be incorporated into the polymer backbone by increasing the feed ratio of AGE, but this comes with the expense of a decrease in molar mass of the copolymer. It was suggested that the decrease in molar mass was not due to systematic chain-termination but a chain transfer, as polydispersity values remained less than two. The authors proposed that, upon AGE incorporation, a chain is terminated to form a new polymer chain, which could be the main cause for the decrease in number average molecular weight (M
n) with increasing AGE content in the feed. Nadeau et al. [
41] reported similar observations, where a decrease in molar mass from 19,700 to 3300 g mol
−1 was observed when they polymerized LA in the presence of AGE with feed content varying from 2 to 30 mol%, using tetraphenyltin as a catalyst.
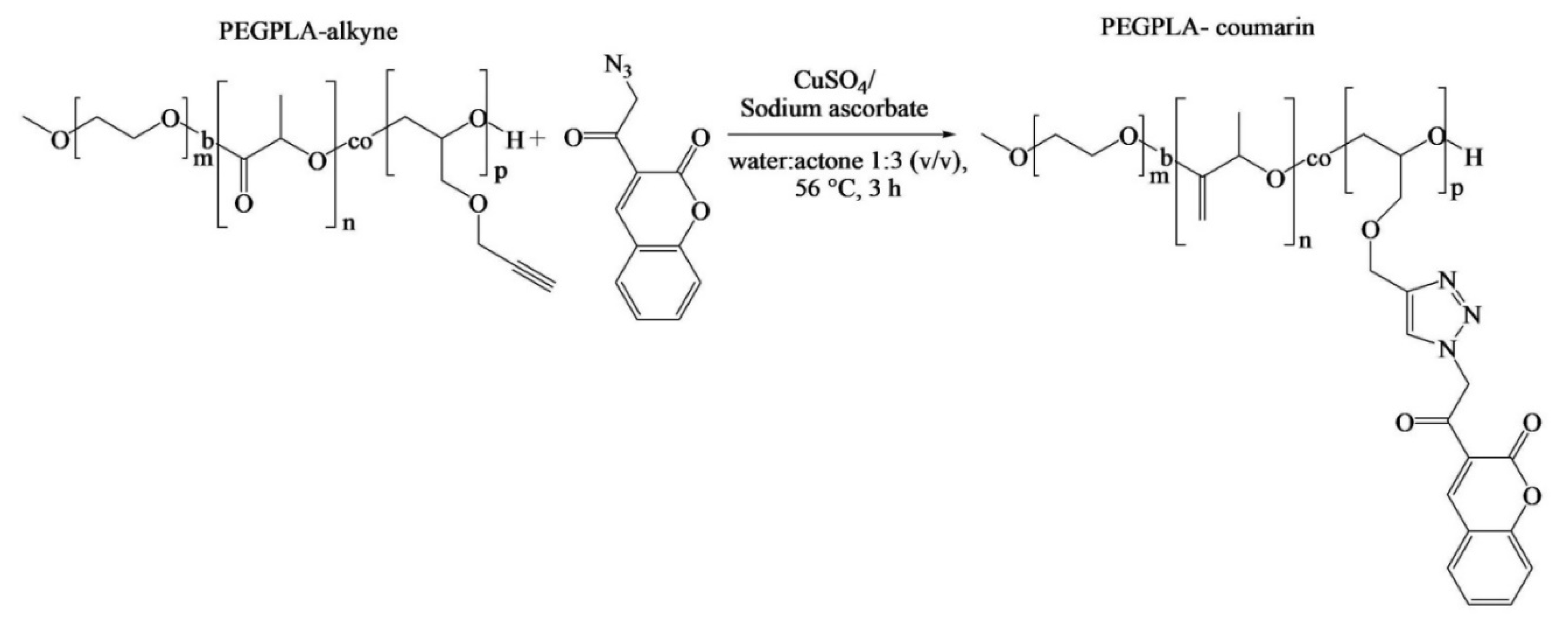
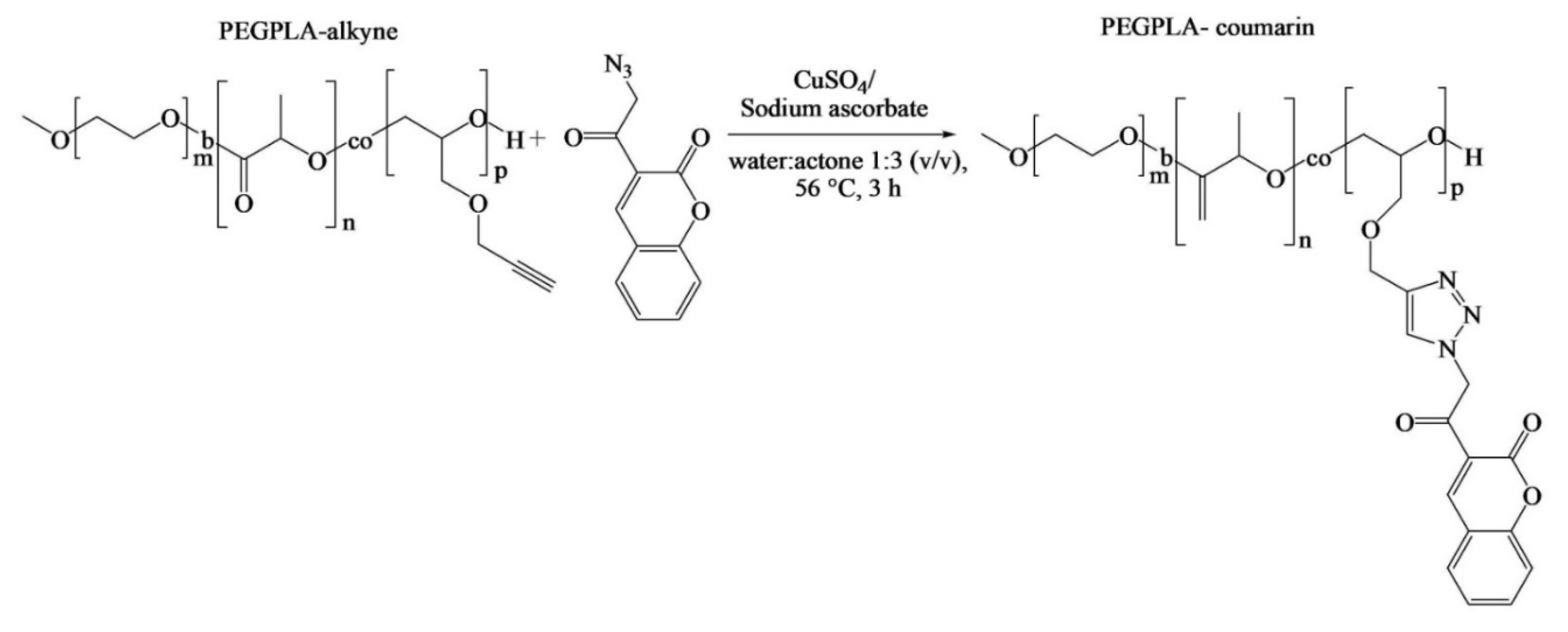
Although Pound-Lana [
36] synthesized block copolymers containing allyl groups, only alkyne-functional PEG-
b-P(LA-
co-PGE) copolymer was used for the investigation of its application. A fluorescent azidocoumarin, 3-(α-azidoacetyl)coumarin, was conjugated to the polymer via copper-catalyzed Huisgen-1,3-dipolar cycloaddition (
Figure 5). The authors proposed that the fluorescent labeled nanospheres could potentially act as drug carriers and imaging agents. However, the allyl-functional PEG-
b-P(LA-
co-AGE) copolymer synthesized in this study was not applied in fluorescent polymer nanosphere preparation; although, allyl groups of AGE unit in the copolymer could be tuned as per the desired application, or allow direct modification under “thiol-ene click” chemistry. Moreover, the facile modifications of AGE for grafting the part and inert character of the allyl group (in general alkene) under the polymerization conditions could be the principal advantage of using AGE.
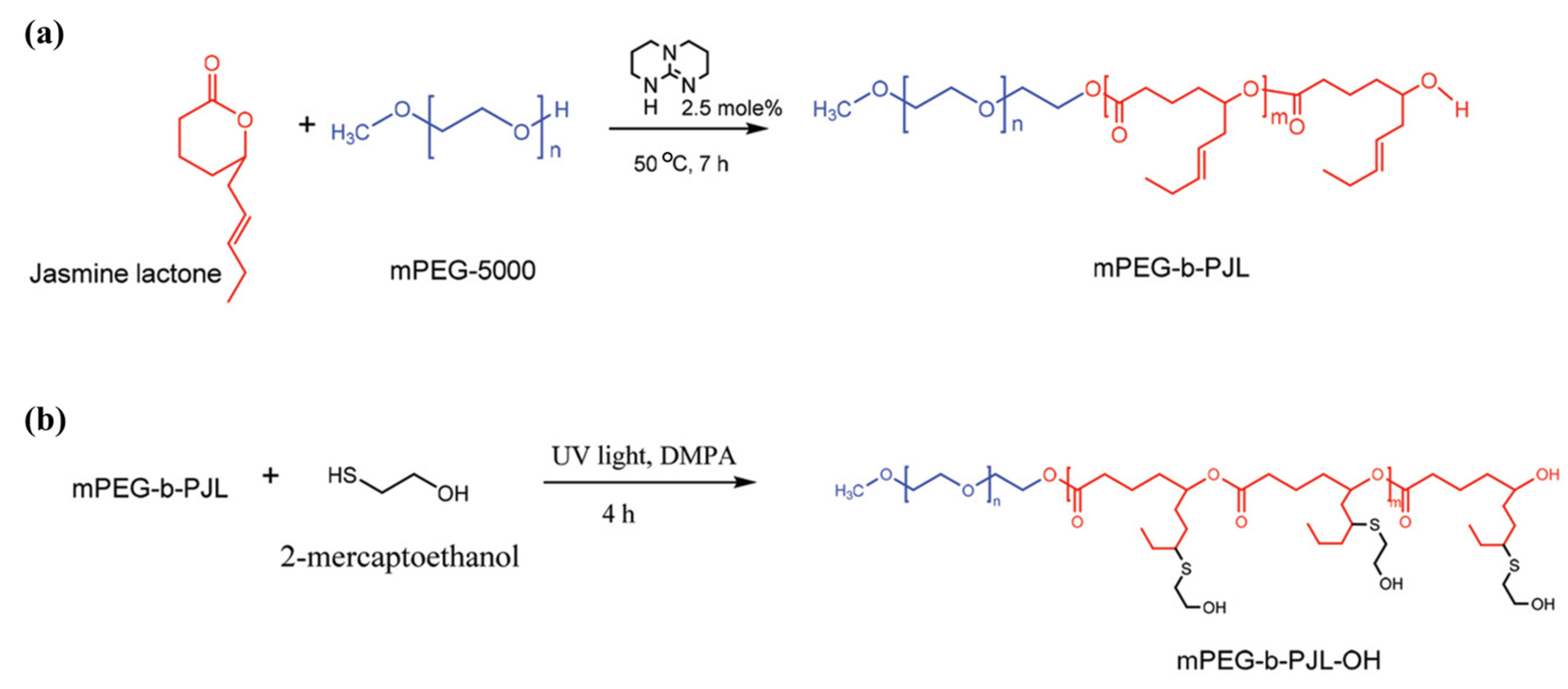
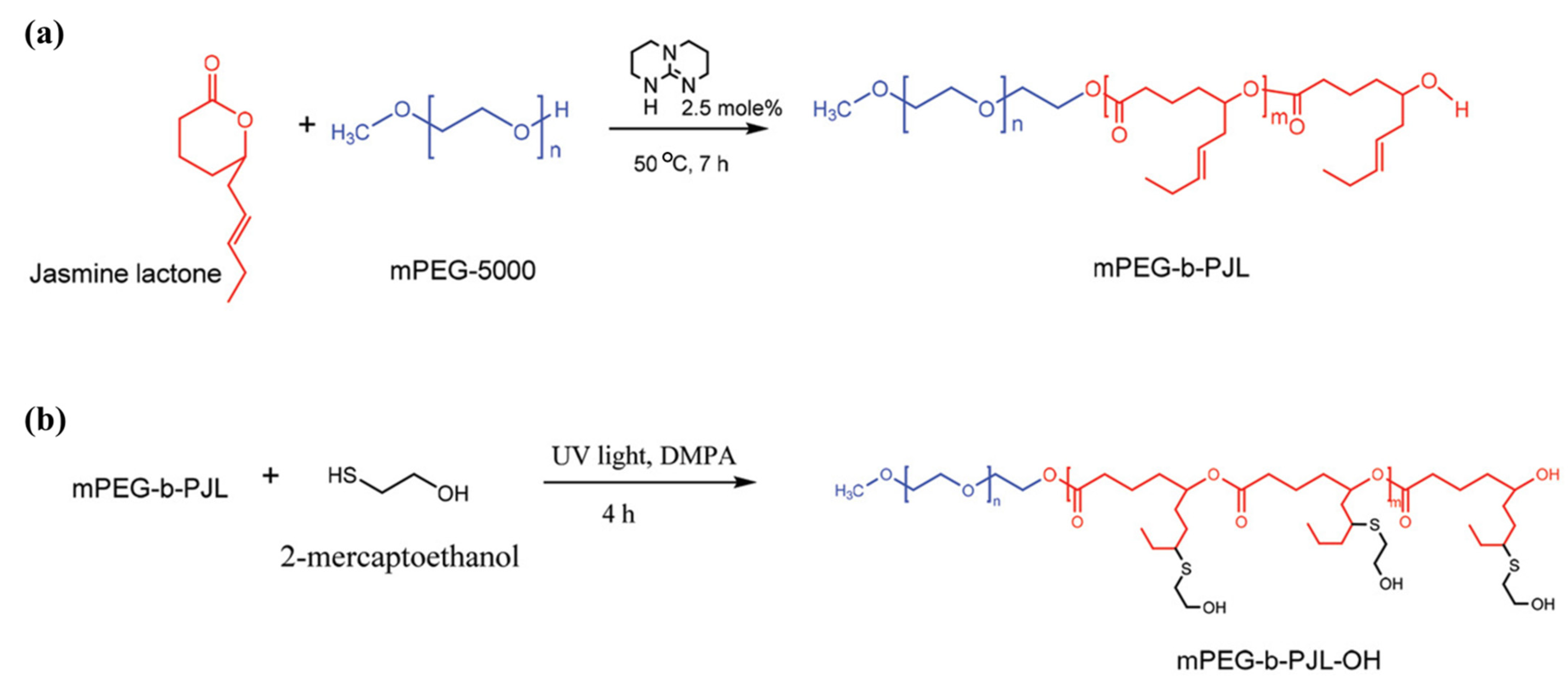
Recently, Bansal et al. [
59] took a smart approach and chose a commercially available starting lactone monomer that already contains free allyl groups on its chemical structure. The authors developed novel functional polymers from renewable feedstock jasmine lactone, using methoxy(polyethylene glycol) (mPEG) as initiator and TBD as catalyst. The post-functionalization of copolymer mPEG-
block-poly(jasmine lactone), (mPEG-
b-PJL) was then successfully demonstrated via UV assisted thiol-ene click chemistry to introduce hydroxyl, carboxyl, or amine functionality (
Figure 6). Although the authors reported that the introduction of hydroxyl and carboxyl groups to the polymer using respective thiol was found to be very efficient, insertion of amine functionality did not yield 100% conversion.
Bansal et al. [
59] utilized hydroxyl-terminated polymer (mPEG-
b-PJL-OH) for the preparation of polymer-drug conjugates (PDCs) using DOX as a model drug via redox-responsive disulfide linkage, i.e., mPEG-
b-PJL-S-S-DOX (PJL-DOX). The conjugation reaction followed a one-pot scheme; first, DOX reacted with DTPA, then, activation of the DTPA acid end group was followed by coupling of mPEG-
b-PJL-OH. The resultant product, PJL-DOX, self-assembled into micelles with an average hydrodynamic size of
~150 nm and demonstrated reduction-responsive DOX release. The authors observed that the PJL-DOX has the ability to stimulate the release of DOX due to the cleavage of disulfide linkage facilitated by glutathione (GSH). It was suggested that these stimuli-sensitive micelles could release the drug exclusively in cancerous cells in high amounts due to the presence of excessive GSH, and thus can mitigate the toxicity of cancerous drugs towards normal cells.
Le Devedec et al. [
60] prepared a series of well-defined poly(valerolactone)-
co-poly(allyl-δ-valerolactone) (PVL-
co-PAVL) copolymers for use in drug delivery. The authors synthesized PVL-
co-PAVL copolymers via metal-free ring-opening copolymerization of δ-Valerolactone (VL) and allyl δ-valerolactone (AVL) using TBD as a catalyst. In the synthesis process, the TBD catalyst was first placed in a flame-dried round two-neck Schlenk flask and dried under vacuum, and then anhydrous toluene and benzyl alcohol solvents were incorporated in the TBD-containing flask under argon and stirred for half an hour. Both VL and AVL monomers were distilled and transferred by cannulation into the reaction flask under positive pressure of argon. The polymerization reaction was conducted at room temperature for 6 h under argon atmosphere. After the polymerization was completed, the resulting slurry solution of copolymers was precipitated into cold methanol, and then re-dissolved in tetrahydrofuran (THF) and re-precipitated to the mixture of hexane/ethyl ether (3:7). The characteristics of copolymers are given in
Table 1.
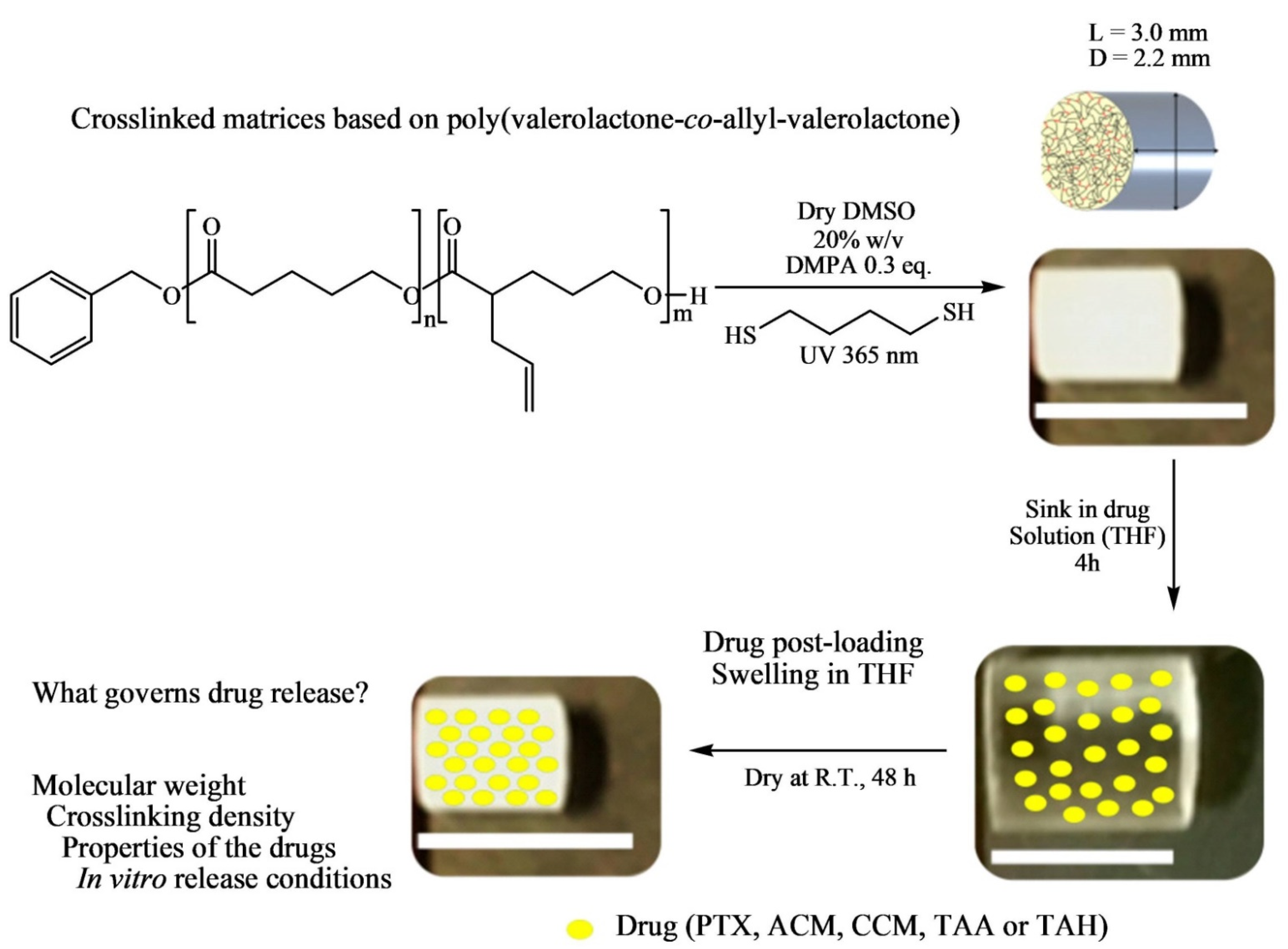
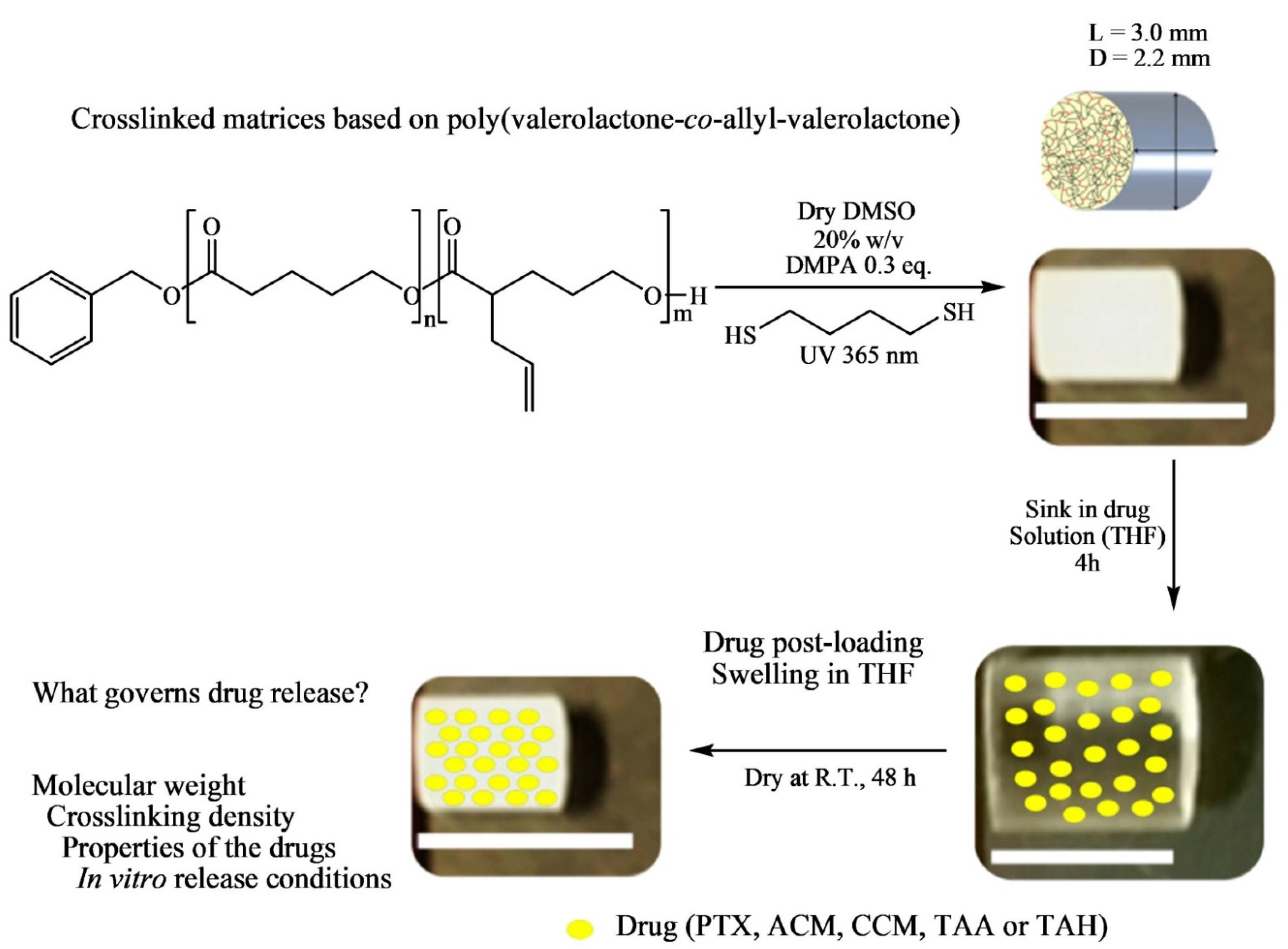
The post-synthesis modification was performed via UV-mediated crosslinking using 1,6-hexanedithiol (HDT) as crosslinker to generate solid cylindrical amorphous or semicrystalline polymeric matrices as potential implantable drug delivery systems (IDDS) (
Figure 7). In the post-functionalization, PVL-
co-PAVL copolymers (0.25 mol equivalents of 2,2-dimethoxy-2-phenylacetophenone (DMPA)), and HDT crosslinker (0.5 functional group molar equivalents) were fully dissolved in dry dimethyl sulfoxide (DMSO) solvent under heating. A 1 mL syringe (inner diameter, i.d. = 4.7 mm and d = 5 cm) was used as a scaffold to inject in the solution, and then the syringe was UV-irradiated (λ 365 nm) for 20 min placed in an upright position. The crosslinked polymer (CP) obtained by this thiol-ene click chemistry was syringed out and purified by solvent exchange in THF (washed extensively) and dried at room temperature. The maximum content of AVL reported in the copolymer was 28%.
The conventional way of drug-loaded polymeric microparticle (MP) preparation consists of co-dissolving polymer and drugs in the organic phase, followed by oil-in-water emulsification. However, as the crosslinking reagent HDT and catalyst DMPA, need to be removed after post-particle formation, the conventional way is considered as an inappropriate technique for the preparation of drug-loaded crosslinked copolymer MPs. To mitigate this concern, the authors adopted the “post-loading” swelling-equilibrium method to load all drugs investigated in this study (including paclitaxel, triamcinolone acetonide and hexacetonide, curcumin, and acetaminophen) within the crosslinked PVL-co-PAVL MPs. For this purpose, swelling/equilibration of dried CPs in saturated drug solutions was formulated by equilibrating around 15 mg of CPs in a 30 mg/mL drug containing 0.5 mL of THF for 4 h, and subsequently removing the surface-adsorbed drug through a brief rinse in fresh THF for 10 s. Finally, the drug-loaded CPs were dried at room temperature to evaporate the THF solvent.
The authors observed the swelling behavior of the four crosslinked copolymer matrices (CPs), i.e., CP15K (coral-like, heterogeneous surface morphology with densely packed folds), CP32K (smooth surface with uniform ridges) CP39K (smooth surface with uniform ridges), and CP7.5K (smooth surface with uniform ridges), in the CH2Cl2, THF, toluene, DMSO, and H2O solvents. The degree of swelling was predicted by using the group contribution method (GCM), i.e., CH2Cl2 > THF > toluene > DMSO > H2O. The same trend was witnessed for all four copolymer systems, from CP7.5K to CP39K. Though, CP39K and CP32K implants swelled well and kept structural integrity in CH2Cl2, owing to the high molecular weight of copolymers, CP7.5K and CP15K matrices broke into pieces after 2 h, accounted for by the low molecular weight of these copolymers. THF was chosen as the solvent for drug loading owing to the high degree of implant swelling without compromising structural integrity and drug solubility in THF. The four copolymer matrices swelled more in diameter than in length following 4 h of equilibration in THF.
From the same research group, Bao et al. [
74] prepared PVL-
co-PAVL MPs via an oil-in-water emulsification method, using PVA as stabilizer, followed by a thiol-ene click reaction, i.e., UV-assisted crosslinking of HDT reactive monomer. The objective was to introduce thiol functionality into the copolymer MPs and tailor the diameter of the MPs. In this formulation method, the authors prepared the oil phase by dissolving PVL-
co-PAVL copolymer, DMPA photoinitiator, and HDT crosslinker in dichloromethane (DCM). The water phase consisted of 5 mL of deionized (DI) water containing 5% PVA (
w/
v). The oil phase was then incorporated into the water phase and homogenized with the help of a Polytron™ 2500E Homogenizer to produce a MP suspension. Next, the thiol-ene click reaction was conducted by UV-irradiating the MP suspension to assist crosslinking of the copolymer. After evaporating the solvent, the sample was filtrated using a cell strainer (Fisherbrand, 40 mm mesh) to retain MPs above the size cut-off. The filtration residue was then suspended in DI water, followed by centrifugation to remove the surfactant. After that, MPs were washed with acetone extensively to get rid of unreacted HDT and DMPA. The scanning electron microscopy (SEM) analysis displayed that MPs had a smooth spherical morphology with an average diameter of 66 ± 13 µm and the differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA) analyses ensured that the crosslinking of the copolymer improved the integrity and thermal stability of the MPs.
In vitro evaluation of the Le Devedec study [
60] showed that the PVL-
co-PAVL MPs demonstrate sustained release of paclitaxel (PTX) for up to 19 days following first-order release kinetics where 60% cumulative fractional release was observed after six days, while 90% release was seen at the 19th day. In vitro degradation study indicated a slow but noticeable erosion, and thus suggested the applicability of these MPs for sustained drug release. The in vivo release of PTX from the MPs was found to be lower than predicted by the authors; based on the in vitro release studies, minimal tissue damage was observed at the administration site. The authors explained the lower release in the in vivo study to be due to the differences between the environment within the subcutaneous tissue (e.g., reduced aqueous volume) and in vitro release media.
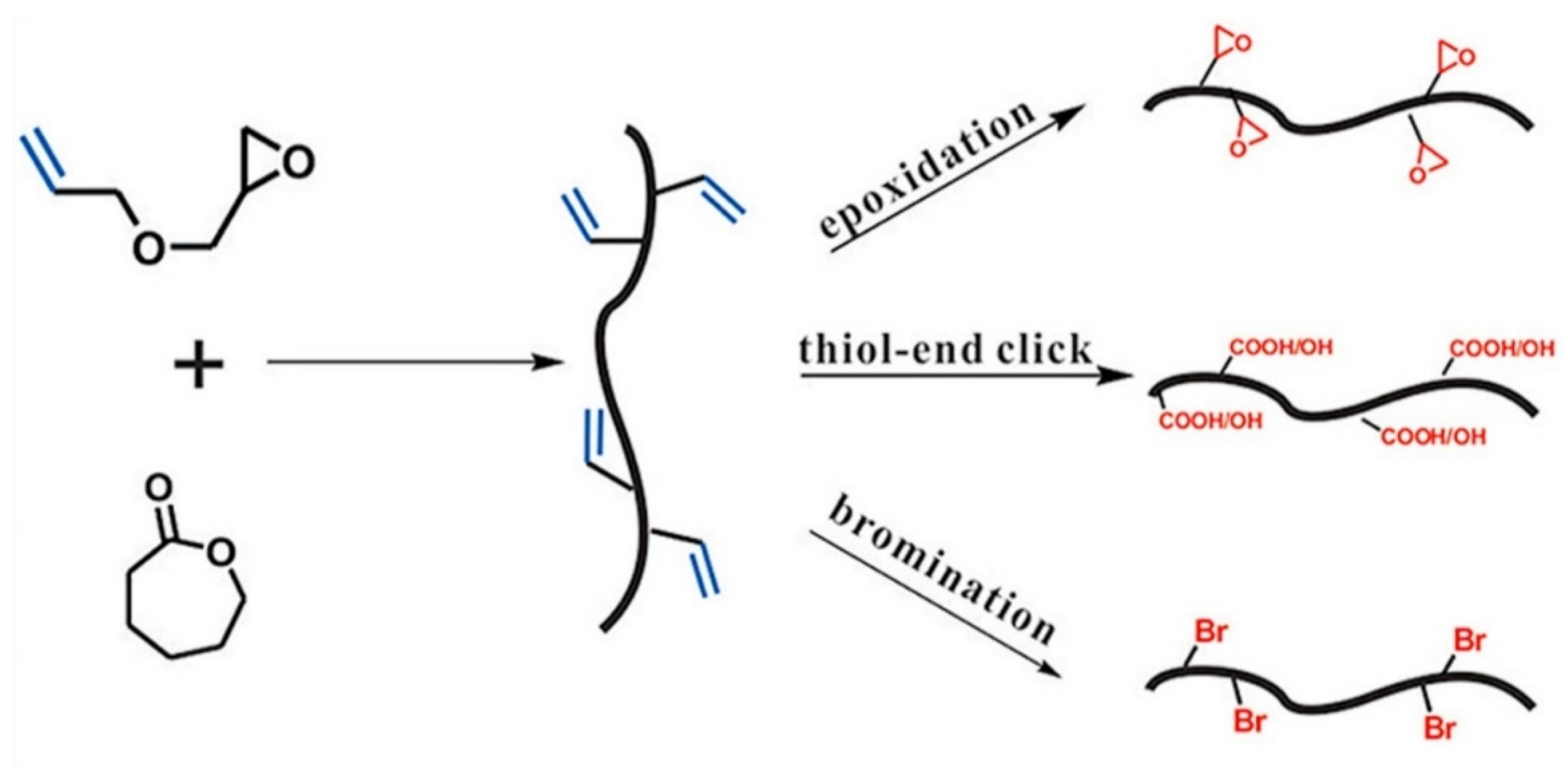
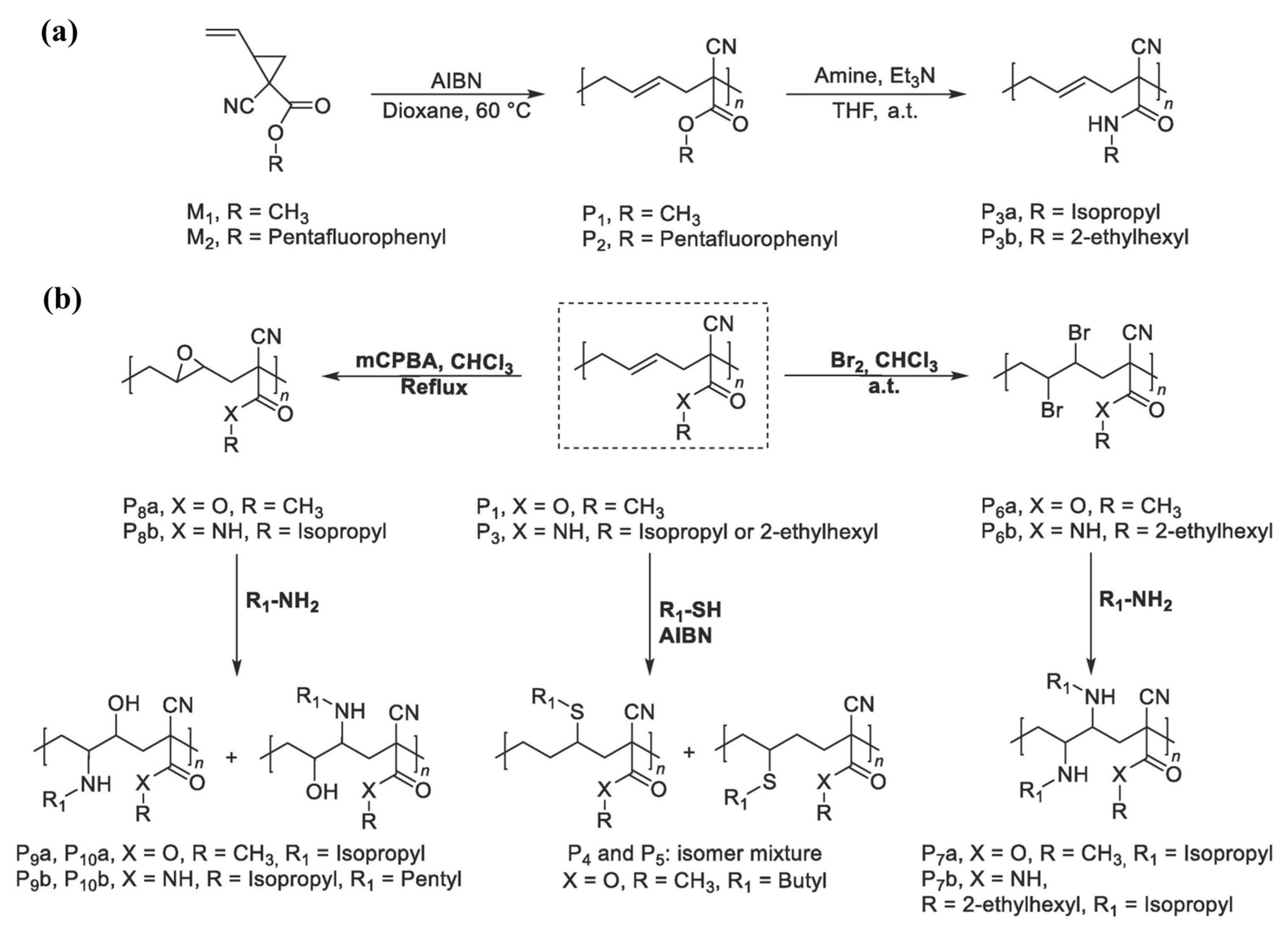
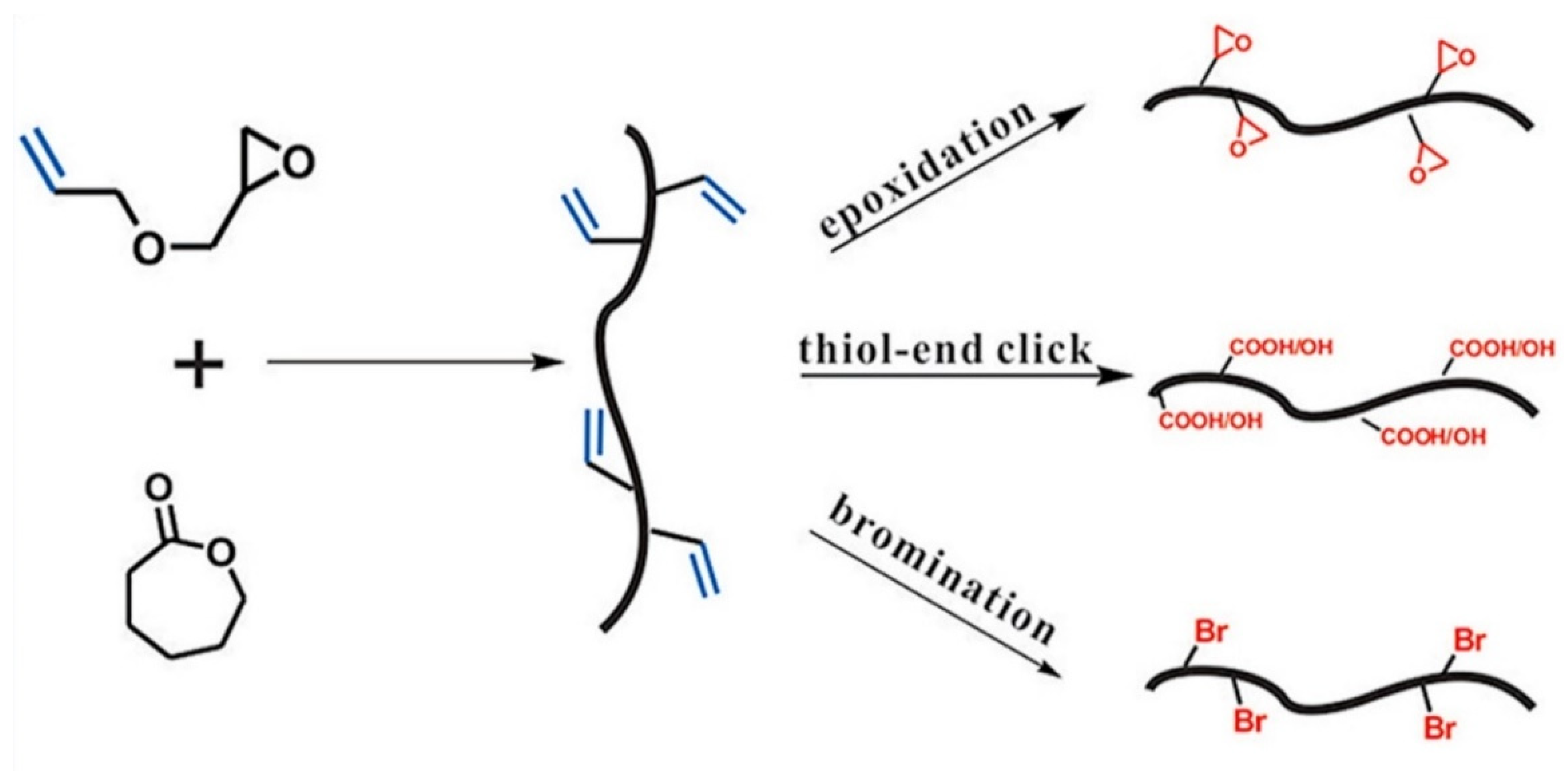
Yang et al. [
40] reported a scalable and facile strategy for the synthesis of an allyl functional aliphatic polyester via the ring-opening copolymerization of ε-caprolactone (CL) and AGE. This study also observed a similar trend report by Pound-Lana [
36], i.e., an increment of epoxide monomer in the polymerization feed ratio (with maximum incorporation of 16.7%), leading to a higher number of reactive allylic groups in the copolymer. This study also investigated the effects of temperature on polymerization and found that the amount of AGE in copolymer and AGE homopolymer synthesis increases with increased temperature. To prepare new functionalized polyesters, the pendant allyl groups of poly(CL-AGE) copolymer were post-functionalized by thiol-ene click, epoxidation, and bromination reactions. For this purpose, 2-mercaptoethanol, 3-chloroperoxybenzoic acid, and bromine were employed to insert functionalities separately (
Figure 8).
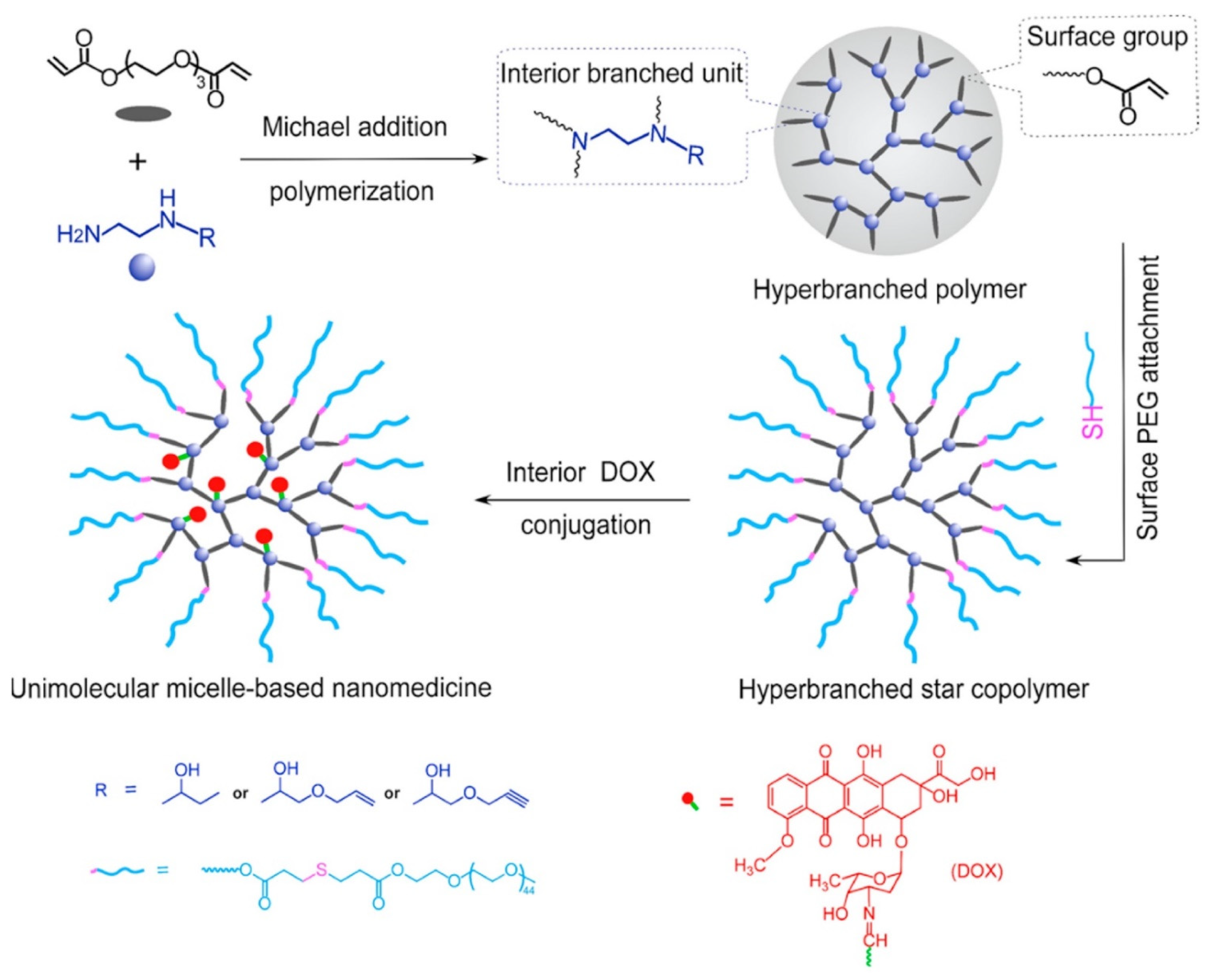
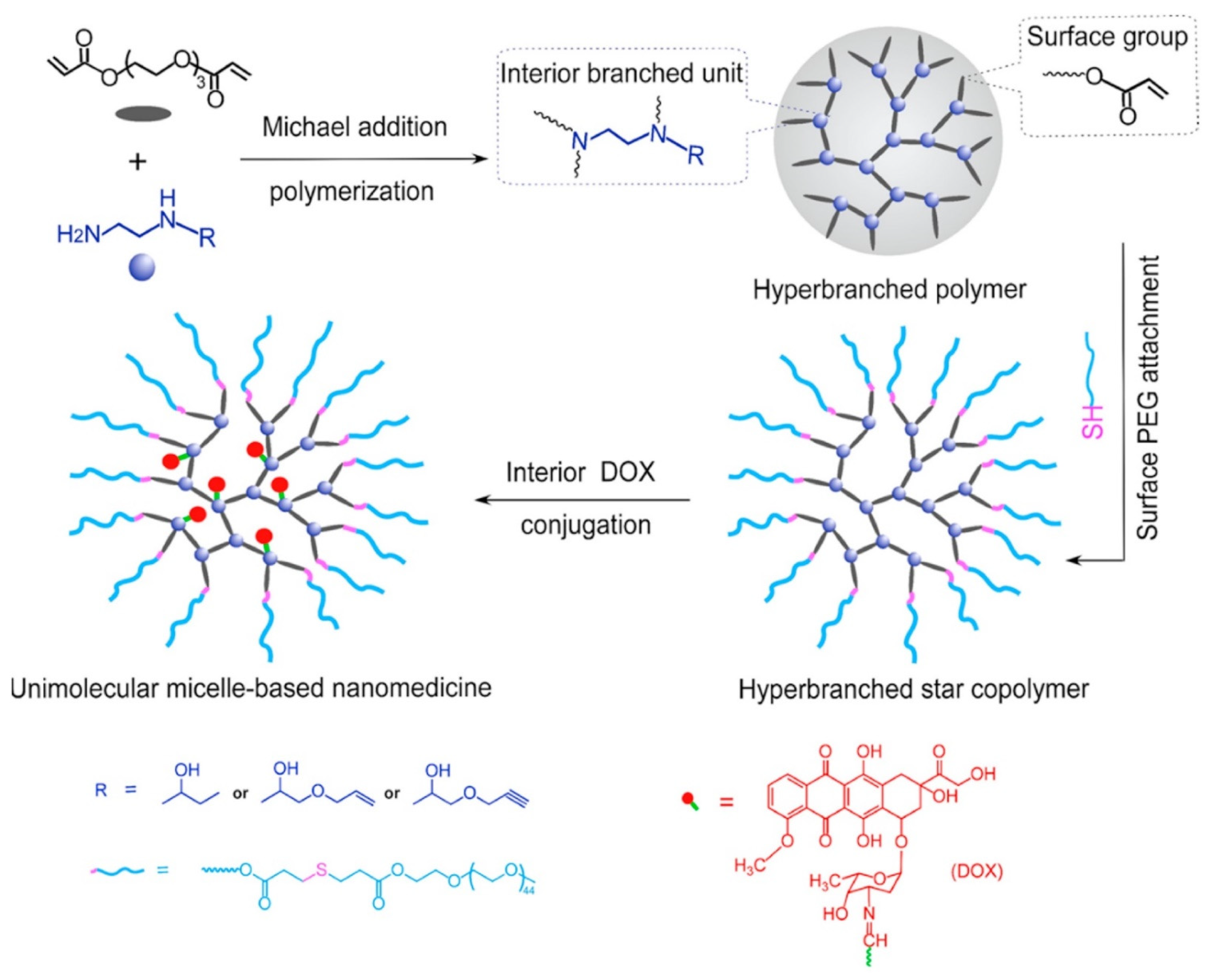
To avoid the thermodynamic stability issue related to traditional polymeric micelles, Liang et al. [
75] successfully designed and synthesized dense hyperbranched polymers with both terminal and internal “allyl” reactive groups. These reactive groups were subsequently utilized to attach hydrophilic chains at surface and interior drug conjugation (
Figure 9). Firstly, the authors synthesized a series of new B′B
2-R monomers, in which B stands for the hydrogen in primary amine, Bʹ stands for the hydrogen in secondary amine, while R is either the hydroxyl, alkene, or alkyne group that will not participate in the polymerization but will allow post-polymerization modification, by the ring-opening reaction of N-ethylethylenediamine with 1,2-epoxybutane, AGE, and propargyl glycidyl ether, called Hyperbranched poly(amino ester)s (HBPAE). Later, Poly(ethylene glycol) diacrylate (PEGDA), labelled as A
2 monomer, was allowed to react with BʹB
2-R monomers via Michael addition polymerization. The authors relied on the higher reactivity of primary amine in BʹB2-R to selectively react A
2 monomer acrylate group to synthesize AB
2 type intermediate with no gelation. Thereafter, hyperbranched star copolymers were synthesized via Michael addition reaction between PEG-SH, with terminal acrylate group of A
2 preserving the internal R group, thus, attaching PEG chains onto the surface of HBPAE-R. DOX was linked to the internal R groups via an acid-labile hydrazide bond to achieve a pH-responsive drug release. The DOX loading content for PEG-HBPAE-DOX was calculated to be ~32.0 wt% through the
1H NMR analysis. The size obtained via transmission electron microscopy (TEM) for PEG-HBPAE-OH and PEG-HBPAE-DOX micelles was 15.2 ± 2.1 and 14.2 ± 2.8 nm, respectively, whereas the hydrodynamic diameter measured by dynamic light scattering (DLS) was 32.4 and 30.4 nm, respectively. The in vitro release study for PEG-HBPAE-DOX was conducted at pHs of 7.4 and 6.0 to mimic normal extracellular pH and tumor microenvironment pH, respectively. The release rate observed at pH = 6.0 was higher (up to 95% after 48 h) compared to release at pH = 7.4, which reached a plateau after 6 h, indicating high stability under physiological conditions and pH-dependent drug release. The in vitro cytotoxicity of DOX, PEG-HBPAE-OH, and PEG-HBPAE-DOX was assessed on HeLa cells where the free nanocarrier exhibited almost no cytotoxicity even at high concentration (1.0 μg mL
−1), while the cell viability was only 16.0% in the PEG-HBPAE-DOX samples, with a drug concentration of 1.0 μg mL
−1.
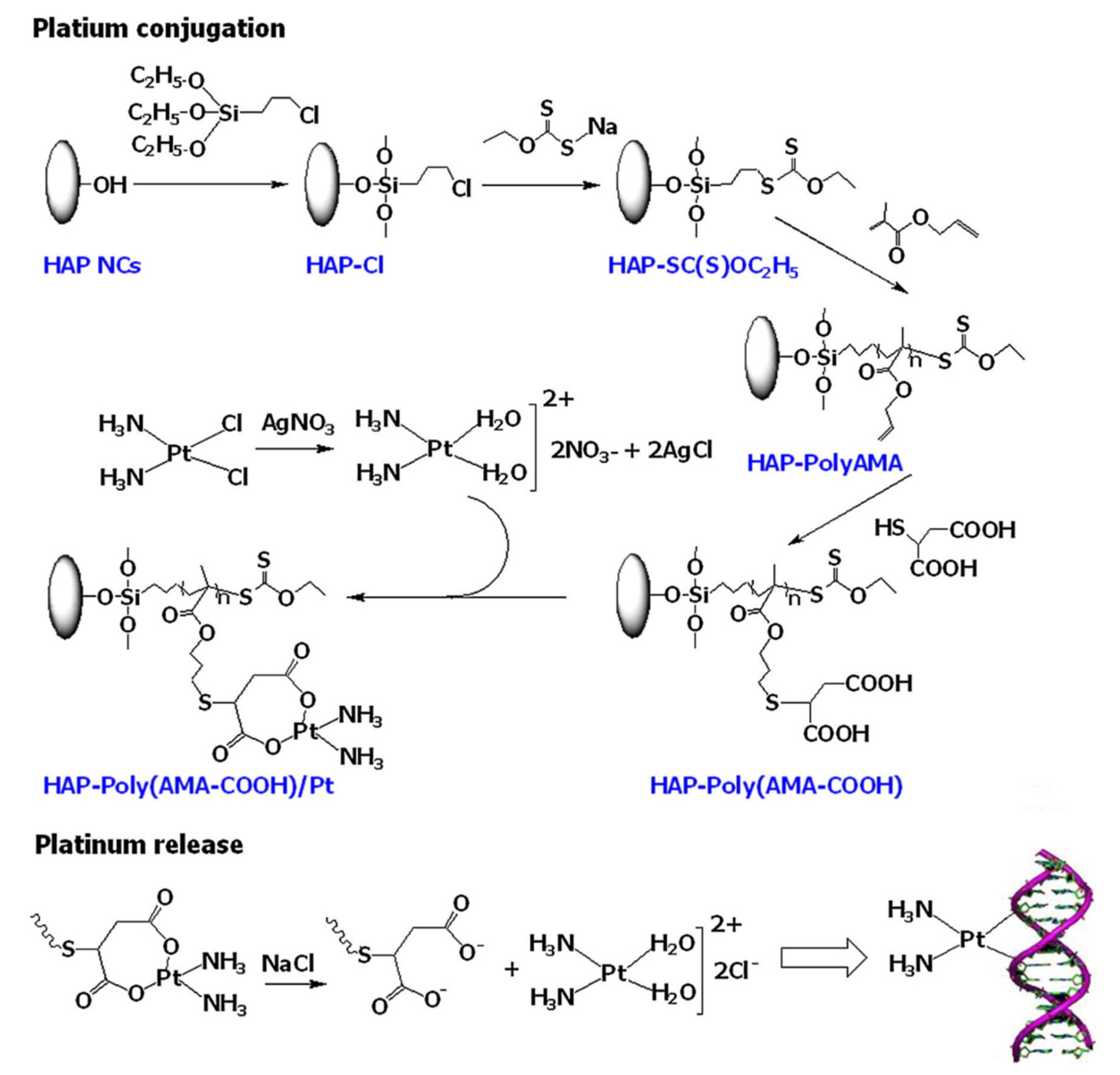
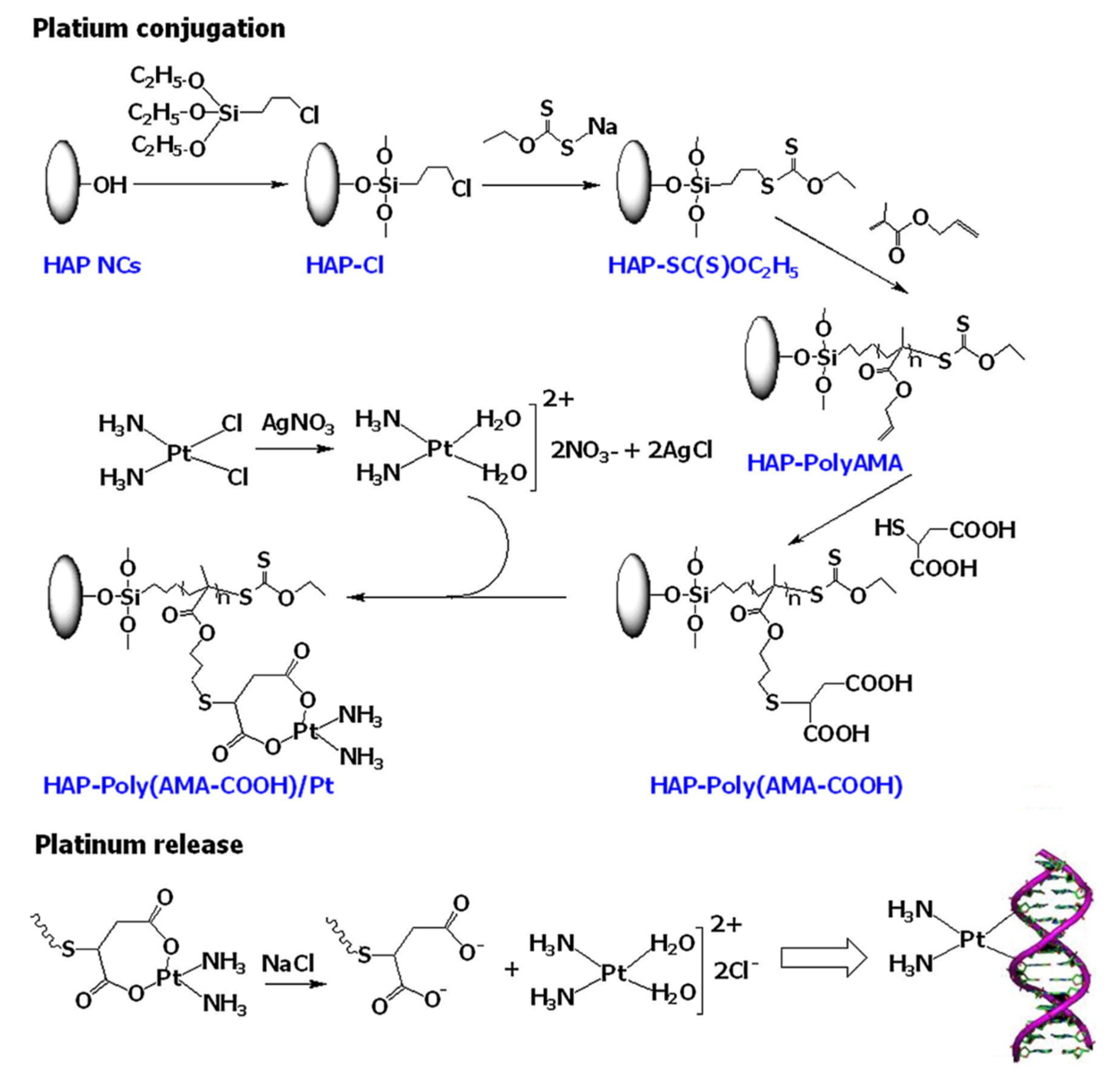
Bach et al. [
76] reported a facile method to synthesize a polymer grafted hydroxyapatite (HAP)-based drug delivery system for controlled and targeted drug release. Unfortunately, HAP is not degradable in the human body, and therefore introduction of polymer moieties to the backbone of HAP has been found to be a good strategy to improve the physicochemical properties of HAP. In this study, they utilized poly(allyl methacrylate) (Poly-AMA) to initially encapsulate HAP nanocrystals (NCs) through surface-initiated RAFT (SI-RAFT) polymerization. Thereafter, the pendant alkene in PolyAMA polymers was utilized to introduce carboxylic functionality via a thiol-ene reaction. Subsequently, PEG-HBPAE-DOX the HAP-Poly(AMA-COOH)/Pt complex was prepared by the interaction of HAP-PolyAMA nanohybrids with transiently generated cis-diamminediaqua platinum (II) species through the hydrolysis of cisplatin (
Figure 10). Field emission scanning electron microscopy (FE-SEM) revealed the morphology of HAP-Poly(AMA-COOH)/Pt complex, with Pt particles in the range of 10–50 nm decorating the surface of the HAP-Poly(AMA-COOH) nanohybrids. The release of the Pt species from the HAP-Poly(AMACOOH)/Pt complex in phosphate-buffered solution (pH = 7.4) and acetate buffer solution (pH = 5.0) was found to be pH-dependent. A faster release was observed in acidic pH, which is attributed to the hydrolysis of the HAP-Poly(AMA-COOH)/Pt complex.
The toxicity levels of HAP-Poly(AMA-COOH)/Pt complex and pure cisplatin were assessed against HeLa cells and A549 cells for 24, 48 h, and 72 h using MTT assay. Cisplatin and HAP-Poly(AMA-COOH)/Pt complex showed a dose-dependent cytotoxicity and enhanced cytotoxicity with longer incubation times. The cytotoxicity of pure cisplatin was higher compared to the HAP-Poly(AMA-COOH)/Pt complex against A549 cells at 24 h, and the cytotoxicity became more prominent when incubated for 48 h. Pure cisplatin also showed a higher cytotoxicity at 24 h on HeLa cells compared to the HAP-Poly(AMA-COOH)/Pt complex; however, the effect became nearly identical on prolonged exposure. Hence, HAP-Poly(AMA-COOH)/Pt complex nanohybrid is a promising nanocarrier for controlled drug delivery.
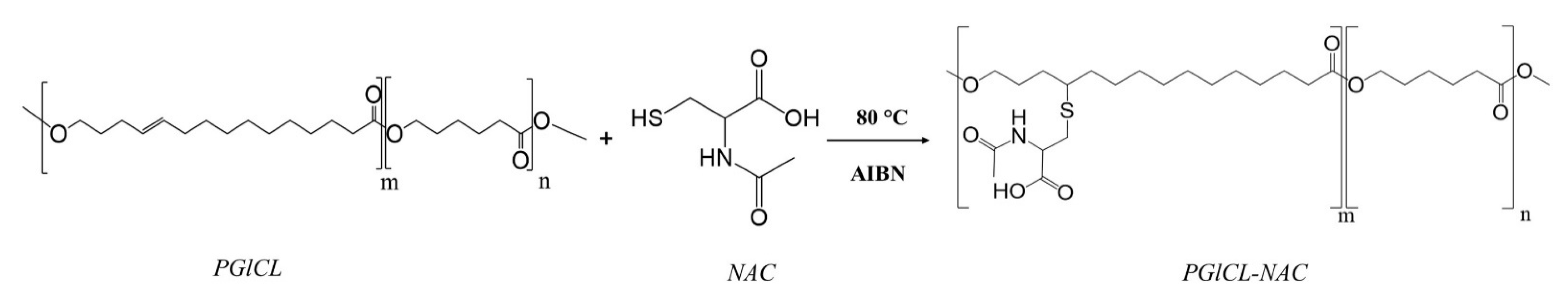
Polyglobalide (PGl) is a biocompatible and non-toxic polyester that is obtained through ROP of globalide and retains the double bond on its main skeleton after polymerization. Thus, the design of copolymers of CL and globalide (Gl) would add wide versatility to the polymer’s biomedical applications as it opens doors for post-polymerization modification via thiol-ene coupling reaction. Therefore, Guindani et al., [
77] incorporated the antimucolytic and antioxidant, N-acetylcysteine (NAC), to poly(globalide-
co-ε-caprolactone) (PGlCL) copolymer. NAC is a hydrophilic molecule and bears a thiol group that allows conjugation by thiol-ene reaction; moreover, the formed thio-ether linkages could enhance the affinity of the polymer for water and reduce its degree of crystallinity. Herein, PGlCl was synthesized by ROP using supercritical carbon dioxide (scCO
2) as solvent in a fixed mass ratio of 1:2 (CO
2:monomers) with variation of the Gl/CL mass ratios as follows: 10/90, 25/75, 50/50, 75/25, and 90/10. Thereafter, PGlCL with varying Gl/CL ratios were reacted with NAC via thiol-ene reaction resulting in the functionalized copolymer, PGlCL-NAC (
Figure 11). The functionalization of PGlCL containing a 10/90 Gl/CL resulted in lower Tm PGlCL-NAC samples; however, the degree of crystallinity (χ
c) did not practically change after functionalization. It was suggested that this result could be ascribed to weaker intermolecular forces in the crystalline arrangement of the material due to the addition of NAC. The free volume increase due to branching, which allows for an easier chain movement of the chains, reduces the energy needed to overcome the secondary intermolecular forces between the chains of the crystalline phase. Generally, amorphous polymers exhibit higher degradation rates than semi-crystalline polymers, and hence they are promising candidates for biomedical applications. Polymer samples with amorphous characteristics were then assessed for their water affinity through contact angle assay. PGlCL with different Gl/CL ratios presented contact angle values around 88°, and accordingly may be considered hydrophobic materials. However, PGlCL-NAC demonstrated lower contact angle values, varying from ~60° to ~47°. The contact angle decrease as a result of the increased hydrophilicity via the attachment of the hydrophilic NAC yields a polymer more suitable for cell attachment and drug delivery, and allows for a better degradation rate. NAC is a renowned antioxidant and, consequently, after functionalization, PGlCL-NAC also presented antioxidant activity. PGlCL-NAC with a Gl/CL ratio of 50/50 was assessed for its antioxidant potential through 1,1-diphenyl-2-picrylhydrazil (DPPH) and 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulphonic acid) (ABTS) assays. The non-functionalized PGlCL showed no antioxidant activity in both assays while free form of NAC showed a strong antioxidant activity in DPPH (EC50 = 4.31 ± 0.03 μg mL
−1) and ABTS (EC50 = 137 ± 3 μg mL
−1). PGlCL-NAC presented EC50 = 4065 ± 157 μg mL
−1 and EC50 = 1553 ± 22 μg mL
−1 in DPPH and ABTS assays, respectively.
2.2. Polyethers
Insertion of allyl functional (poly)glycidyl ether as building blocks in polyether- leads to a wide range of promising applications for these materials. In this section, we have discussed the synthesis of such polymers and its post-functionalization, if any.
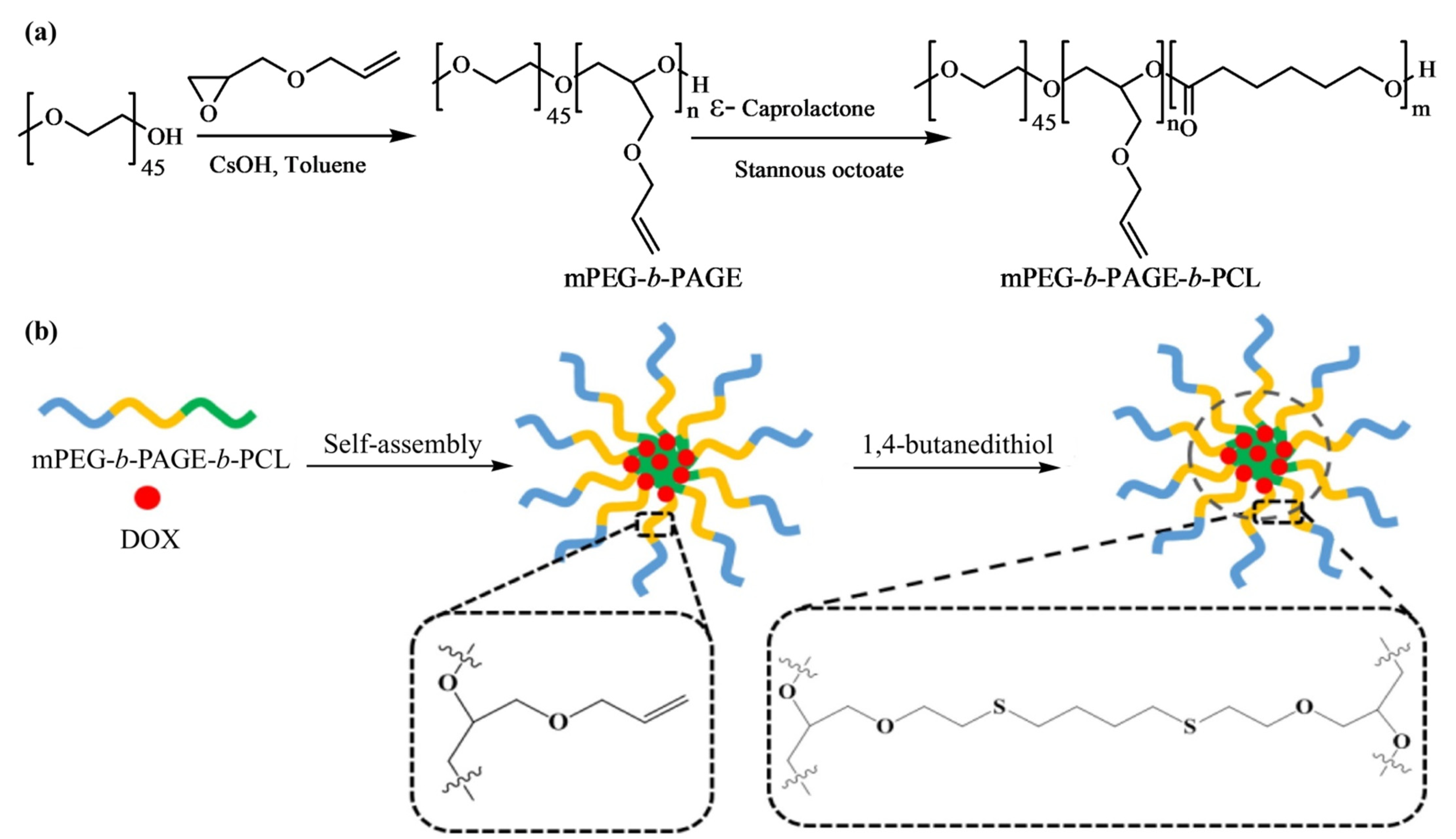
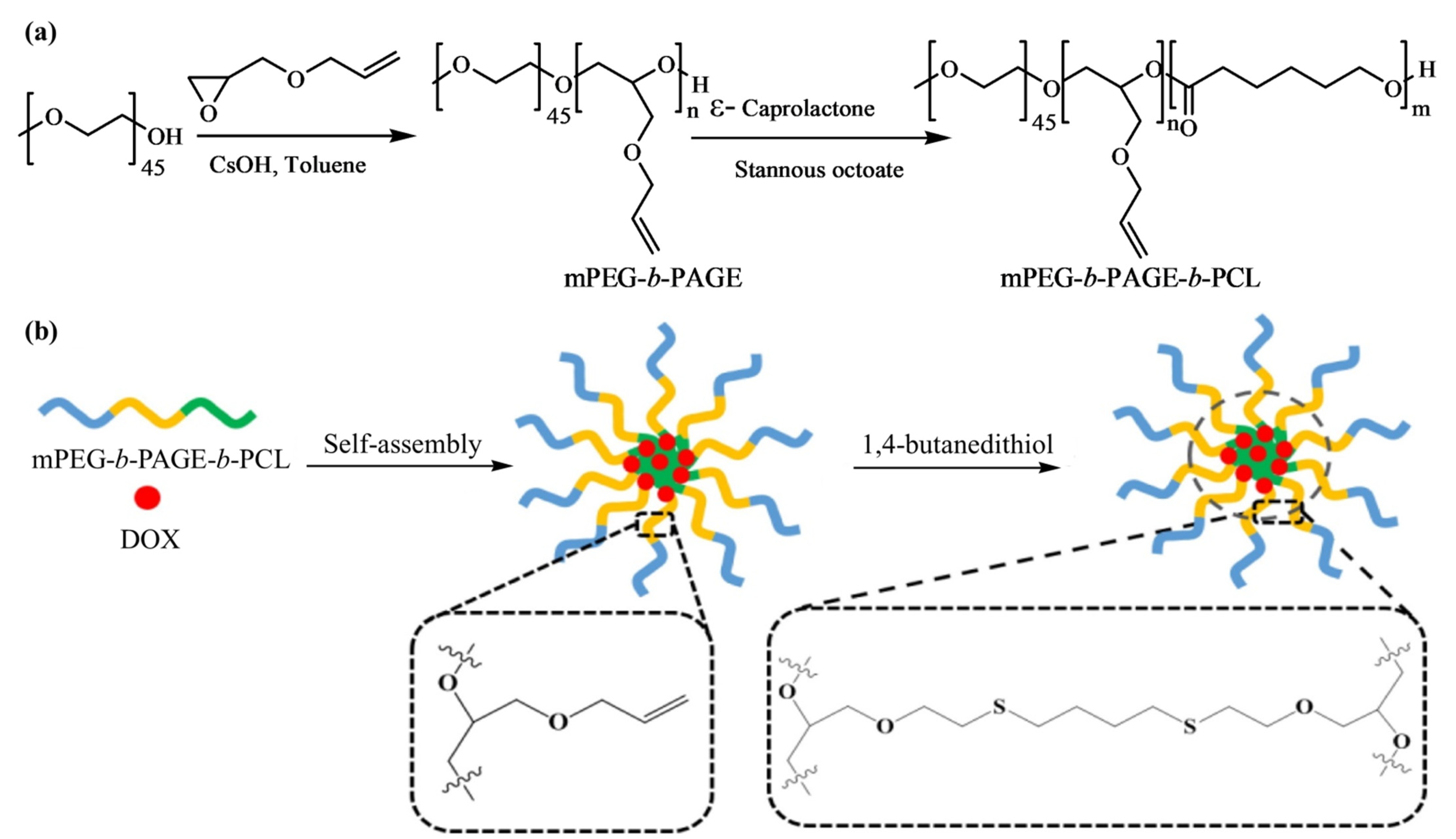
Lu et al. [
51] designed and synthesized amphiphilic triblock copolymer, methoxy poly(ethylene glycol)-
b-poly(allyl glycidyl ether)-
b-poly(ε-caprolactone), (mPEG-
b-PAGE-
b-PCL) with different hydrophobic lengths using two successive ROPs and post-functionalized by a thiol-ene click reaction, displayed in
Figure 12. The macroinitiator mPEG−OH was employed for the deprotonation of the terminal -OH group and then subjected to the anionic ROP of AGE to obtain mPEG-
b-PAGE diblock copolymers. Later, different amounts of CL were incorporated into the system to synthesize the triblock copolymer mPEG-
b-PAGE-
b-PCL with distinct hydrophobic lengths.
Blank micelles were prepared by dissolving mPEG-b-PAGE-b-PCL triblock copolymers in THF, followed by adding copolymer solution into pure water drop-wise at room temperature with continuous stirring. The blank micelles were obtained by evaporating THF solvent through rotary evaporation. To prepare crosslinked mPEG-b-PAGE-b-PCL triblock copolymers micelles, 1,4-butanedithiol crosslinker was added into the micelle dispersion solution and irradiated the dispersion by a UV lamp (λ 365 nm). The centrifugal ultrafiltration technique was applied to remove residual crosslinker and cut off low-molecular weight fraction (M.W. 3500).
The triblock copolymers could self-assemble into highly stable polymeric spherical micelles in an aqueous solution. The micelles formation depends on the hydrophobic interactions between the drug and the hydrophobic segments of the amphiphilic polymers. The micelles encapsulate DOX and subsequently undergo interface crosslinking by a thio-ene reaction with 1,4-butanedithiol. The covalently crosslinked network ultimately enhances the stability of the micelles but decreases its size to some extent, compared to that of the non-crosslinked micelles. The authors attributed this slight decrease in size to the shrinkage of the inner core of the micelles. The DLS results indicate that the diameter of the DOX-loaded micelles was dependent on the hydrophobic PCL block length in the copolymer composition. The maximum drug loading content and entrap efficiency were calculated as 8.62% and 47.16%, respectively, for the mPEG-b-PAGE-b-PCL50/DOX micelles with a size of 99.28 nm. The in vitro cytotoxicity was determined by MTT assay with human oral epidermoid carcinoma (KB) and human gastric carcinoma (SGC) cell lines. The results showed that the DOX-loaded micelles could be effectively endocytosed by cancer cells and possessed good antitumor efficacy. In addition, pH-responsive DOX release profile was observed for both DOX-loaded noncrosslinked micelles (NCLMs) and crosslinked micelles (CLMs). As DOX is known for its enhanced protonation affinity and higher solubility in acidic environments, micelles achieved a rapid drug release in a weakly acidic intracellular environment. However, the cumulative release rate of DOX-loaded CLMs was lower than that of the DOX-loaded NCLMs at both pH 7.4 and 5.0 because the crosslinking layer hindered the release of DOX from the core of the micelles. The authors also investigated the tumor targeting efficiency of CLMs and NCLMs using in vivo fluorescence imaging in 4T1 tumor-bearing BALB/c mice; the dye used was the commercially available lipophilic dye DiR. They observed that the CLMs rapidly accumulated in tumors and had a better passive targeting ability than NCLMs due to their good stability, as the fluorescence intensity of DiR-loaded CLMs in the tumor (13.3%) was 1.67-fold higher than that of the NCLMs (9.3%). The authors summarized that the interface crosslinking strategy from the triblock copolymer mPEG-b-PAGE-b-PCL could improve the stability of the micelles in vivo and holds great promising applications in future cancer therapy.
Clamor et al. [
78] reported a well-controlled ROP of ε-allyl caprolactone using Mg(BHT)
2(THF)
2 to yield an allyl-functionalized lactone, ε-allyl-ε-caprolactone (AεPCL), suitable for post-polymerization modification with a 95% monomer conversion (
Figure 13). The synthesized PCL was subsequently post-functionalized by photo-initiated thiol-ene addition on the pendant allyl-functionality using various alkyl thiols to produce lipophilic polyesters with tuned lipophilicity and crystallinity. An increasing solubility in n-dodecane was observed with increased alkyl chain length on the PCL backbone. Furthermore, crystallinity increased with alkyl chain length; a change in crystallinity from amorphous to semicrystalline was observed when the alkyl length reached 10 carbon atoms in length, likely due to effective chain interaction among the alkyl pendant groups. The authors demonstrated that the physical and thermal properties of PCL can be altered by varying the alkyl functionality. The tailoring of the polymer microstructure and solubility could be advantageous in exploring PCL in new nanostructures and (bio)material applications.
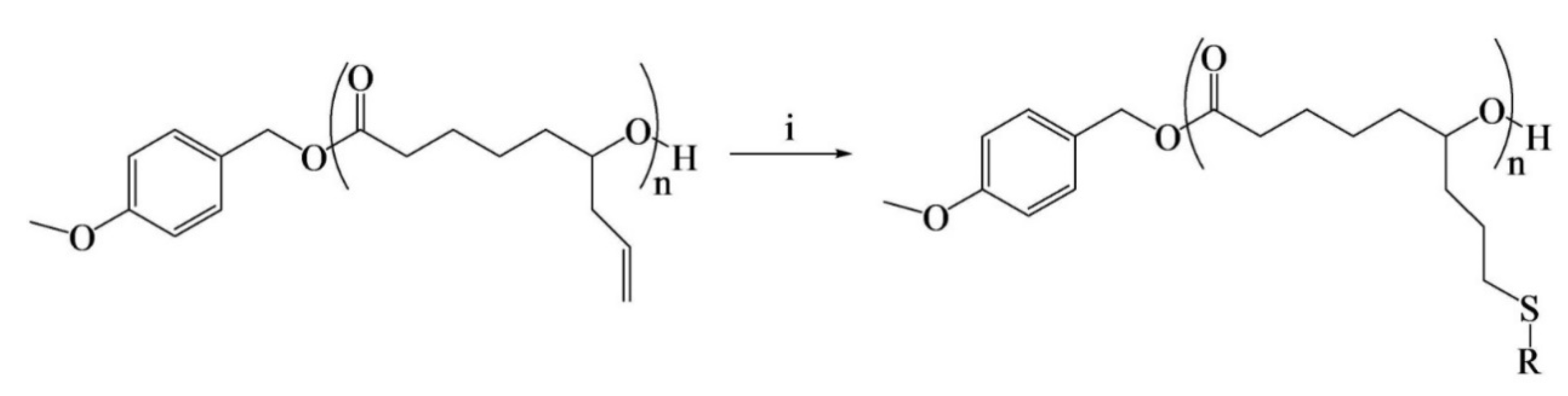
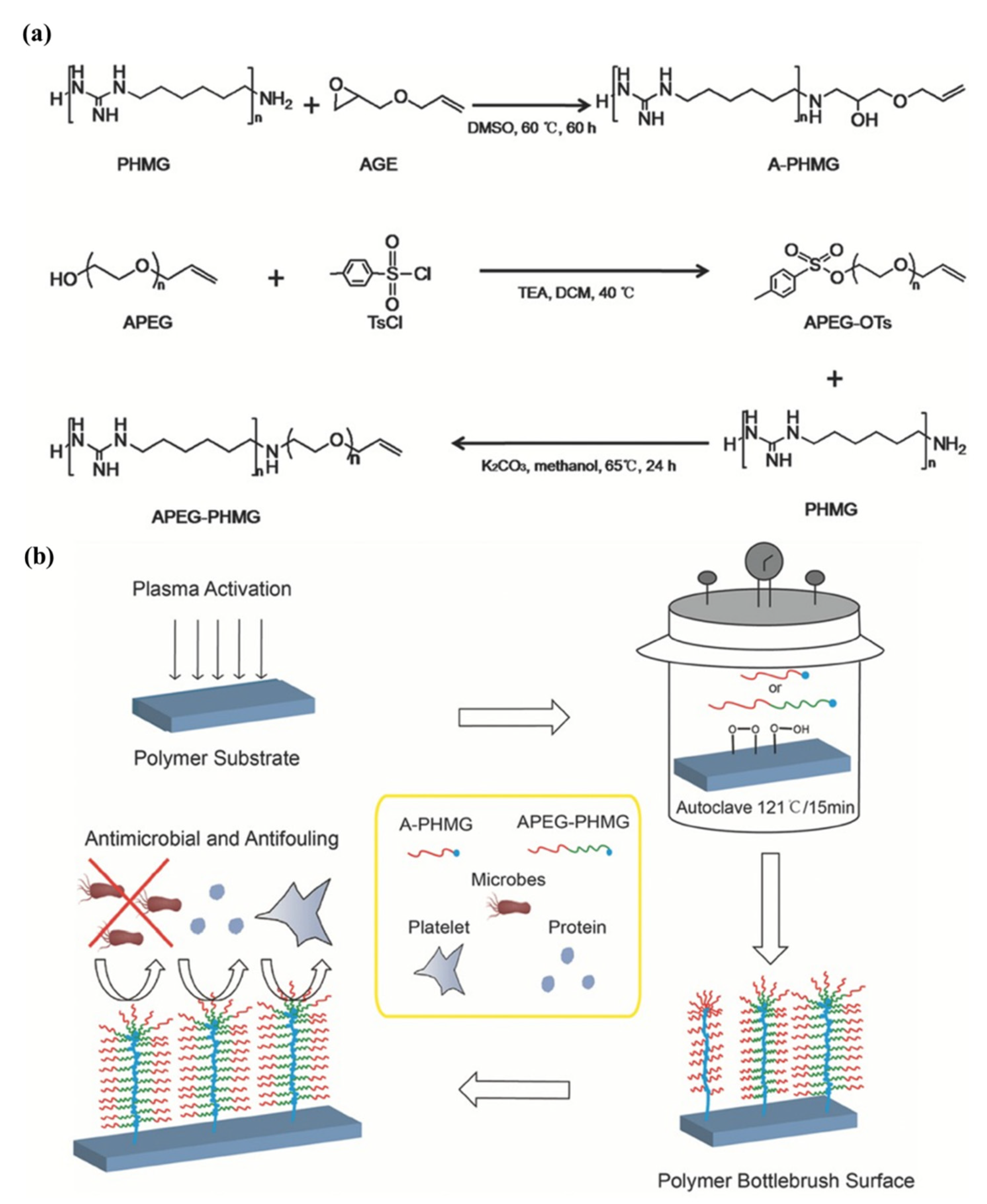
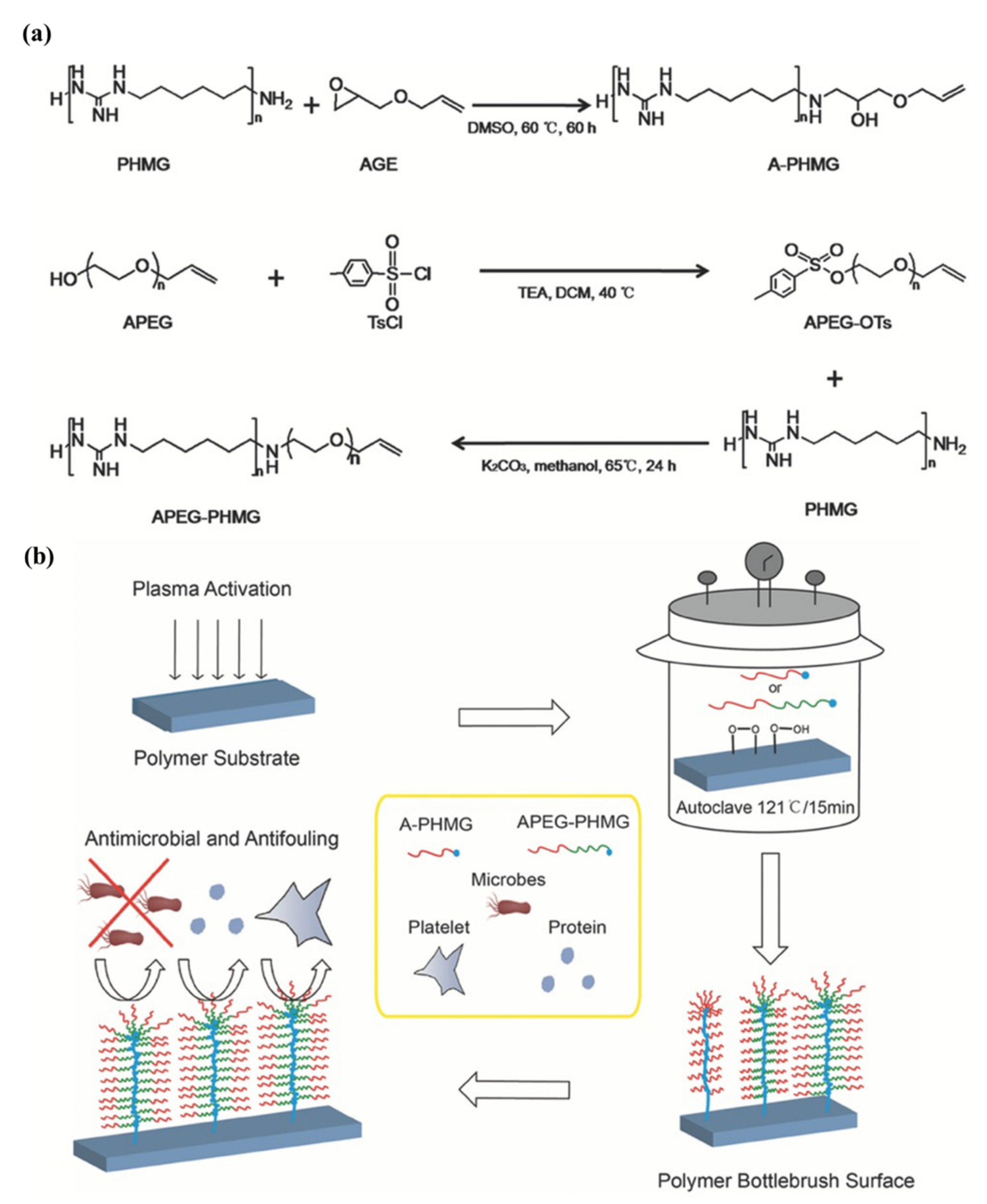
Su et al. [
37] successfully designed and synthesized allyl-terminated polyethylene glycol-polyhexamethylene guanidine block copolymer, APEG-PHMG, consisting of antimicrobial, antifouling, and surface-tethering segments in a facile, up-scalable, and inexpensive way. Later, APEG-PHMG oligomers were grafted on polydimethylsiloxane (PDMS) silicone rubber substrates by a facile plasma/autoclave-induced surface grafting polymerization method to form a permanently bonded bottlebrush-like surface coating. The authors basically modified the –NH
2 end-group of polyhexamethylene guanidine (PHMG) with AGE and, ultimately, obtained allyl-functionalized oligomers. The authors first prepared hetero-bifunctional PEG with allyl and tosyl groups (APEG-OTs) using PEGs with 1200 and 2400 Da molecular weights, where end hydroxyl group of APEG was reacted with 4-toluene sulfonyl chloride (TsCl) to produce APEG-OTs. After that, thermally and chemically stable broad-spectrum antimicrobial agent polyhexamethylene guanidine (PHMG) was conjugated with APEG-OTs to produce the block copolymer (APEG-PHMG). Separately, an allyl-terminated PHMG was synthesized by reaction with AGE. The synthesis process of oligomers and schematic of plasma/autoclave-assisted grafting is depicted in
Figure 14. Argon plasma (13.56 MHz, 40 W, and 25 sccm) was applied on the PDMS surface for 5 min to generate reactive free radicals and, subsequently, plasma-treated PDMS was exposed to air for 15 min to produce relatively stable peroxide or hydroperoxide groups on the treated surface via reaction between reactive free radicals on the PDMS surface, and oxygen and water in the air. The activated PDMS surfaces were then immersed in 5 wt% of A-PHMG or APEG-PHMG oligomer solution in vials, separately. After that, sample vials were autoclaved at 121 °C for 15 min. In this autoclaving process, reactive free radicals were generated again on the PDMS surface through decomposition of peroxide/hydroperoxide groups on the silicone surface under high autoclaving temperature. These newly generated free radicals initiated the polymerization of the allyl groups. The main function of allyl functionality of A-PHMG/APEG-PHMG oligomers was the formation of a permanent covalent bond between oligomers and the plasma-activated silicone surfaces, using allyl group as this group is very reactive to peroxide/hydroperoxide groups generated on silicone substrate via plasma activation and the reaction is very fast.
The antimicrobial activity of AGE and APEG modified PHMG oligomers were examined both in solution and coating form against gram-negative, gram-positive bacteria, and fungus. In this study, Pseudomonas aeruginosa (ATCC 27853), Staphylococcus aureus (ATCC 2921), and
Fusarium solani (ATCC 36031) were considered as gram-negative, gram-positive bacteria, and fungus, correspondingly. In solution form, a broth microdilution minimum inhibitory concentration (MIC) assay was performed to determine the antimicrobial activity; results are shown in
Table 2. The authors observed that both A-PHMG and APEG1
200/
2400-PHMG coatings displayed potent broad-spectrum and reusable antimicrobial activity against gram-positive bacterium (
S. aureus), gram-negative bacterium (
P. aeruginosa), and fungus (
F. solani), whereas APEG
1200/
2400-PHMG coatings exhibited superior antifouling activity and long-term reusability to A-PHMG coating. Contrarily, APEG
2400-PHMG coating demonstrates the most effective in vitro anti-biofilm and protein/platelet-resistant properties, as well as excellent hemo/biocompatibility. In addition, APEG
2400-PHMG showed high efficacy towards the inhabitation of the bacteria growth, significantly, and prevented implant-associated infection caused by
P. aeruginosa in a rodent subcutaneous infection model.
The outcomes of the study indicated that antimicrobial and antifouling APEG-PHMG dual-functional diblock copolymer coating has great potential to prevent bacterial colonization and biofilm formation on biomedical implants, and subsequently in combating biomedical devices/implant-associated infections.
2.3. Poly(Ester-Anhydride)s
Poly(ester-anhydride) copolymers are frequently synthesized to combine the individual properties of polyester and polyanhydride polymers in a single polymer, with various applications. Polyesters display mostly bulk erosion mechanisms and polyanhydrides exhibit surface erosion mechanisms during hydrolytic degradation, whereas poly(ester-anhydride)s demonstrate a two-stage degradation mechanism. In poly(ester-anhydride)s, first, a rapid degradation in molecular weight of the polymer occurred through polymer chain breaks at the hydrolytically labile anhydride linkage, and then, a slower degradation of remaining oligomers took place. The rate of the second stage, the degradation of the remaining oligomers, is determined by the composition of polyester prepolymers [
79]. Numerous studies have been conducted to prepare functional poly(ester-anhydride)s bearing allyl pendant groups to combine the individual properties of polyesters and polyanhydrides, and to control the degradation rate and mode of degradation by manipulating the polymer composition.
Jaszcz et al. published three separate studies discussing the synthesis and post-functionalization of functional poly(ester-anhydride)s, based on succinic acid [
48], on oligosuccinate, and aliphatic diacids. Furthermore, they studied the hydrolytic degradation behavior [
49] and epoxidation of pendant allyl groups in poly(ester-anhydride)s, proposed for application in drug delivery [
50]. The purpose of introducing allyl groups to poly(ester-anhydride)s was that the allyl groups could be converted into epoxide groups through epoxidation reaction, and the epoxide functionality in poly(ester-anhydride)s could create an interesting perspective for chemical coupling of drugs to poly(ester-anhydride)s carrier, e.g., via microspheres.
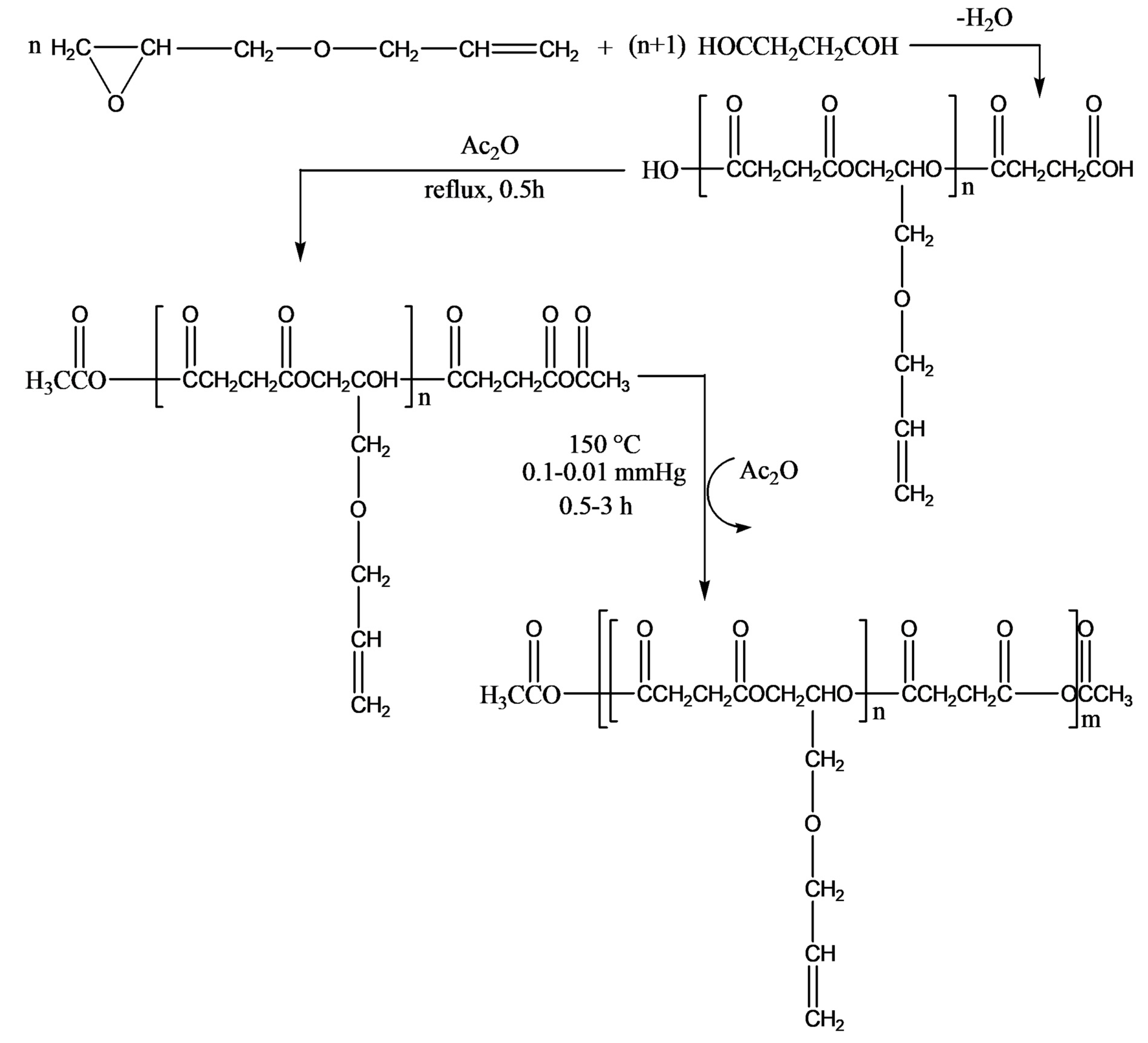
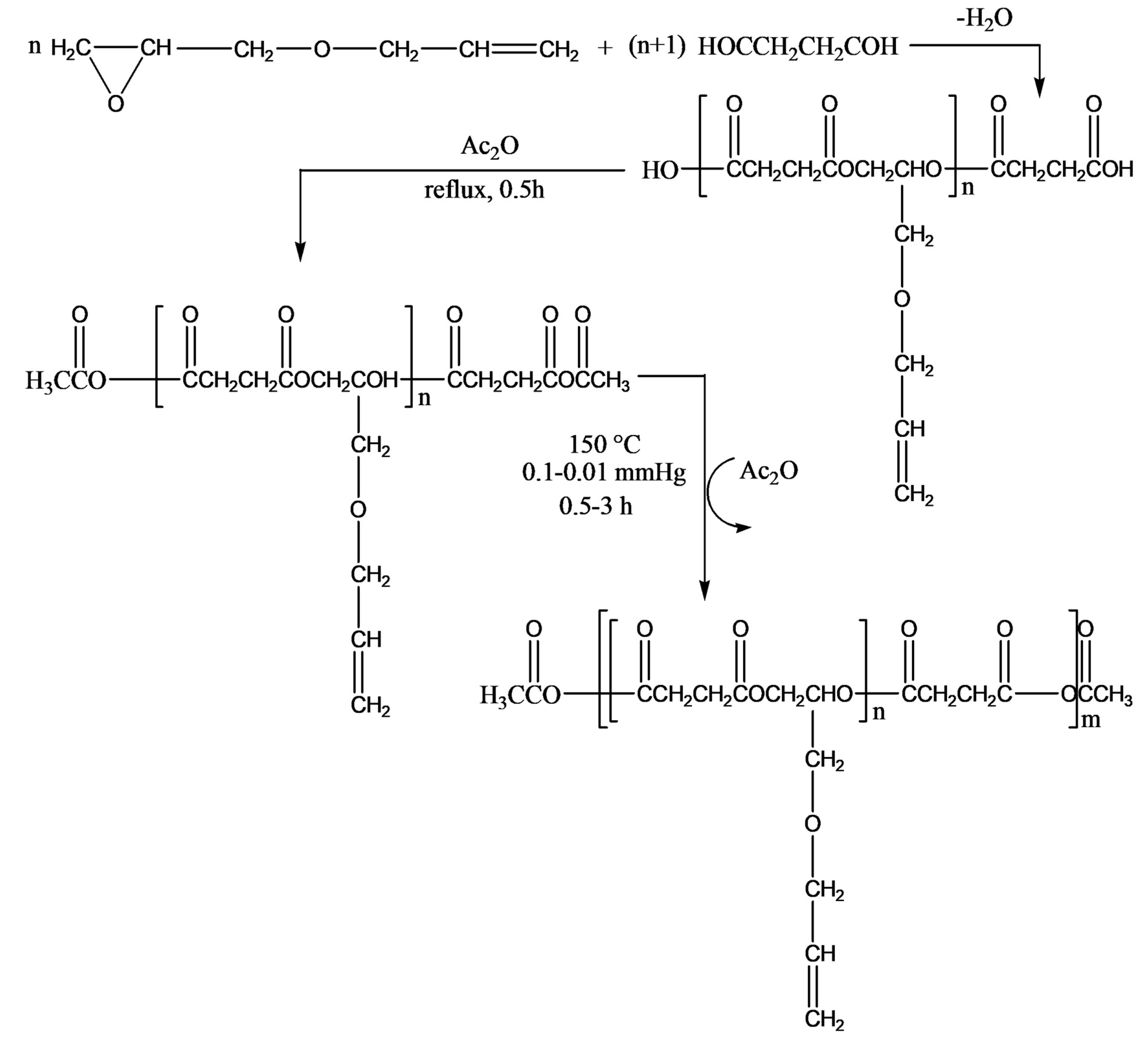
The authors described a three-step synthesis of succinic acid-based functional poly(ester-anhydride)s bearing allyl groups in the side chains, as shown in
Figure 15. The steps include (1) preparation of carboxyl-terminated functional oligoesters (OSAGE) by melt condensation of AGE with an excess of diacid (DA), succinic acid, or carbonic acid; (2) conversion of carboxyl end groups of the macromer to mixed anhydride groups by refluxing in acetic anhydride; and (3) preparation of poly(ester-anhydride)s from ester-anhydride prepolymers by melt polycondensation.
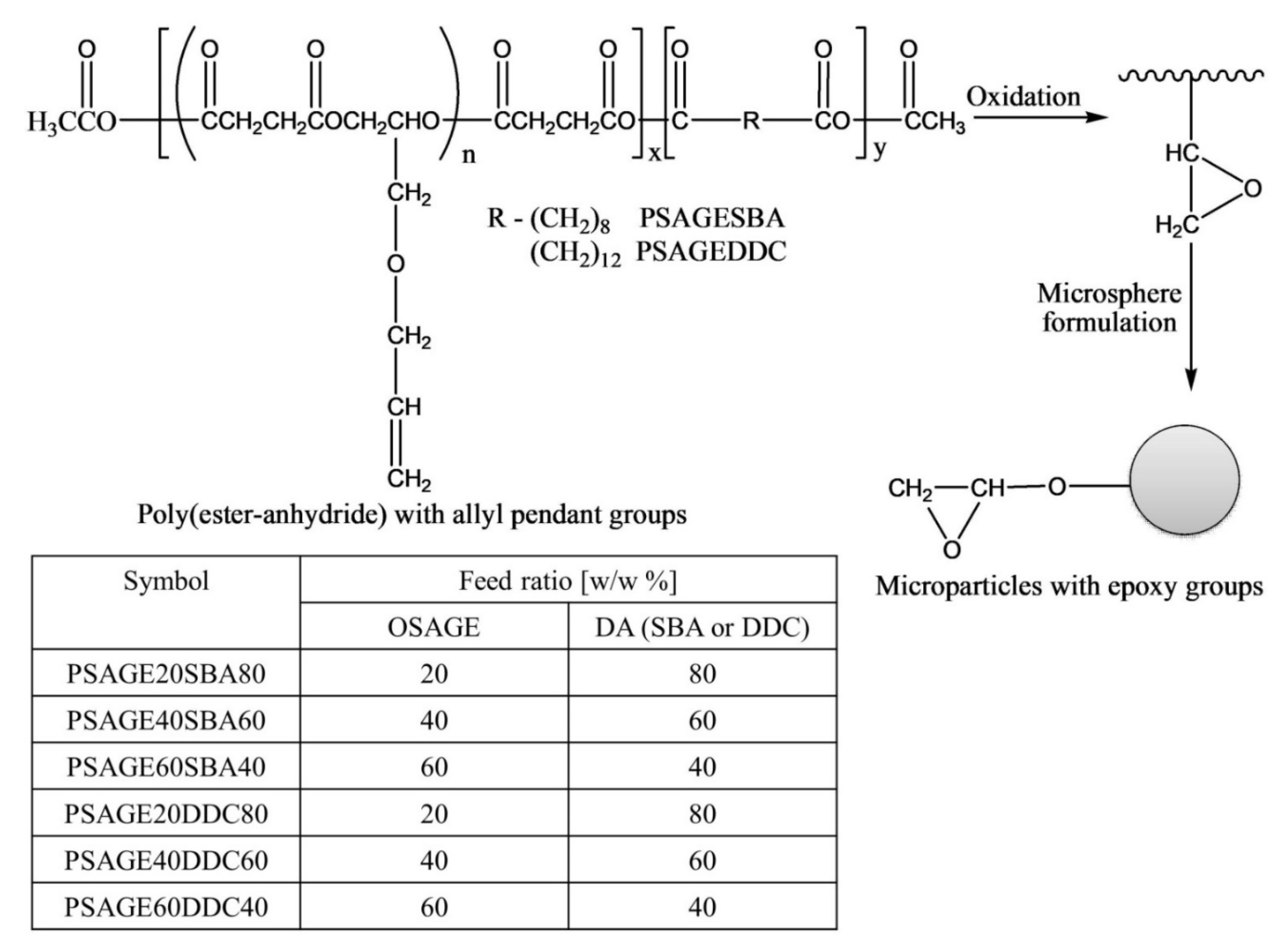
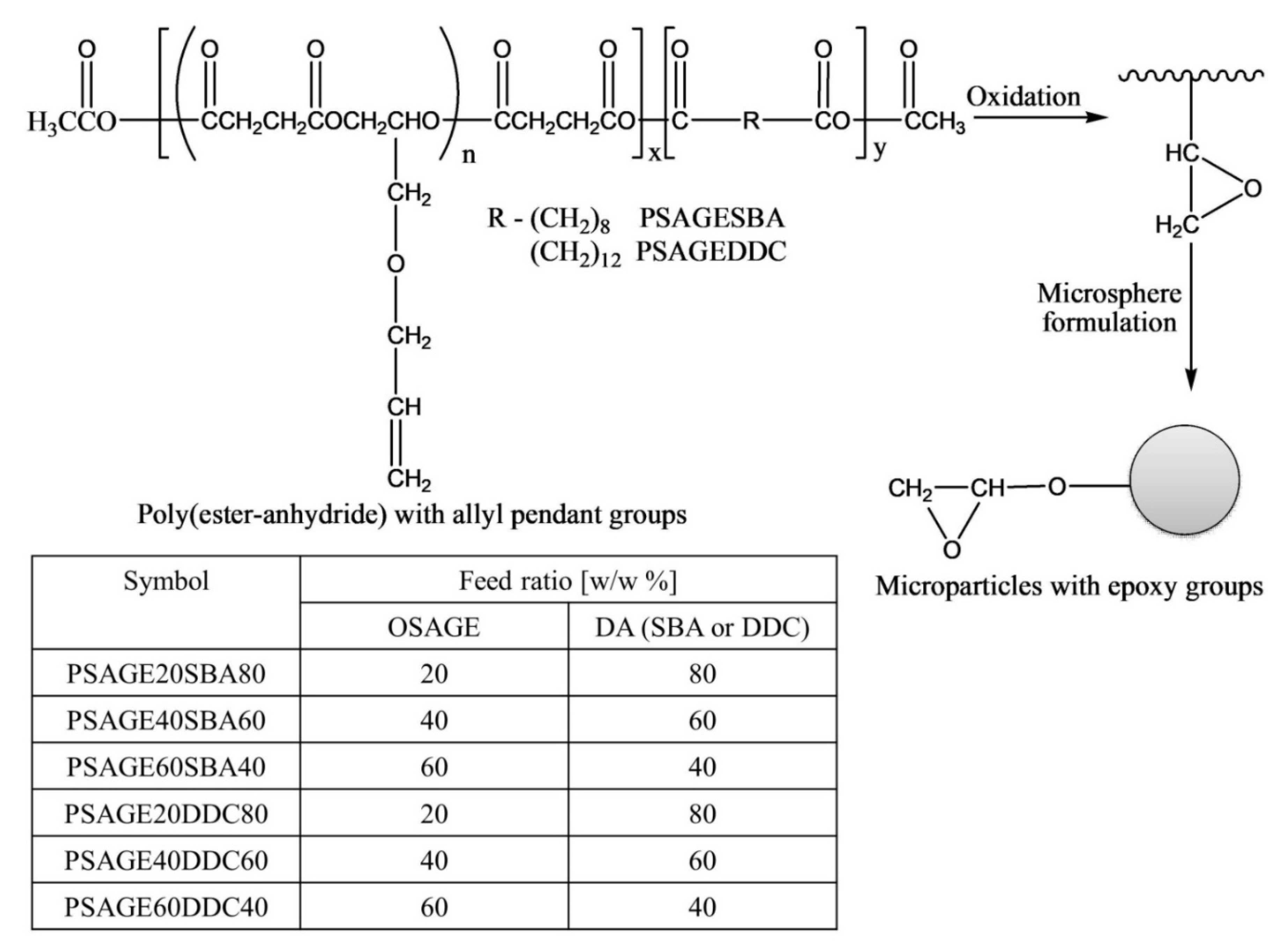
The authors post-functionalized poly(ester-anhydride) with allyl groups to epoxy groups via oxidation using m-chloroperbenzoic acid (MCPBA) with 100% conversion (
Figure 16) [
50]. However, it was observed that the conversion of allyl groups into glycidyl was greatly dependent on the content of pendant allyl groups in poly(ester-anhydride) (OSAGE to DA ratio), the concentration of a polymer in the reaction solution, the amount of MCPBA, and the reaction time.
The microspheres were fabricated by emulsion solvent evaporation method using parent and oxidized polymers. The microparticles obtained in this study were stable and spherical in shape (with diameter 4.7–9.2 μm in size distribution D
v/D
n = 1.2–1.4) and no holes or pores were observed on their surfaces. Microspheres obtained from allyl functional polymers were smoother, bigger, and had a broader size distribution in comparison with microspheres prepared with epoxy-functional polymers (
Figure 17). Authors also suggested that, owing to the higher polarity of epoxide groups with respect to allyl groups, the epoxy-functionalized microspheres did not show agglomerations and coalescence in an aqueous medium. Moreover, the epoxide groups can be covalently bonded with the functionalities of drugs, such as amines, and can be potentially utilized as polymeric drug carriers [
50]. In the preliminary trials, the authors conducted several reactions between amine-containing model compounds, such as isopropylamine, a-amino-x-methoxy-poly(ethylene glycol)s, spermine, and spermidine, and the epoxy-functionalized poly(ester-anhydride) microspheres. It was observed that the model amine compounds can covalently bond to the surfaces of the microspheres, which was confirmed by the
1H NMR or ATR-IR spectra as epoxy groups peaks disappeared. However, the consumption of anhydride bonds was also observed simultaneously. The two factors which determined the ratio of epoxide groups to anhydride bonds consumption were the basic character of amine compounds and the reaction conditions.
Both initial and oxidized poly(ester-anhydride)s microspheres were subjected to hydrolytic degradation in phosphate buffer (pH = 7.4) at 37 °C for 10 days, followed by weight loss determination due to hydrolytic degradation as a function of immersion time in buffer solution by gel permeation chromatography (GPC). Two general trends were identified from GPC results: (1) among the microspheres prepared from unoxidized and oxidized poly(ester-anhydride)s, the unoxidized microspheres degraded somewhat faster than the respective oxidized ones, and (2) microspheres obtained from poly(ester-anhydride) containing sebacic acid (SBA) degraded faster compared to poly(ester-anhydride) containing dodecanedicarboxylic acid (DDC) microspheres. After 7 days of hydrolytic degradation, the weight loss was calculated. For the unoxidized and oxidized PSAGE20SBA80 microspheres, approximately 90% and 60% weight loss was observed, respectively. Whereas a weight loss of 54% and 45% was determined for unoxidized and oxidized PSAGE20DDC80 microspheres, respectively. Both oxidized SAGE20DDC80 and PSAGE20SBA80 microspheres produced water soluble products by hydrolytic degradation. However, oxidized SAGE20DDC80 microspheres took longer to degrade entirely (more than a month) in comparison to oxidized SAGE20DDC80 microspheres (over three weeks).
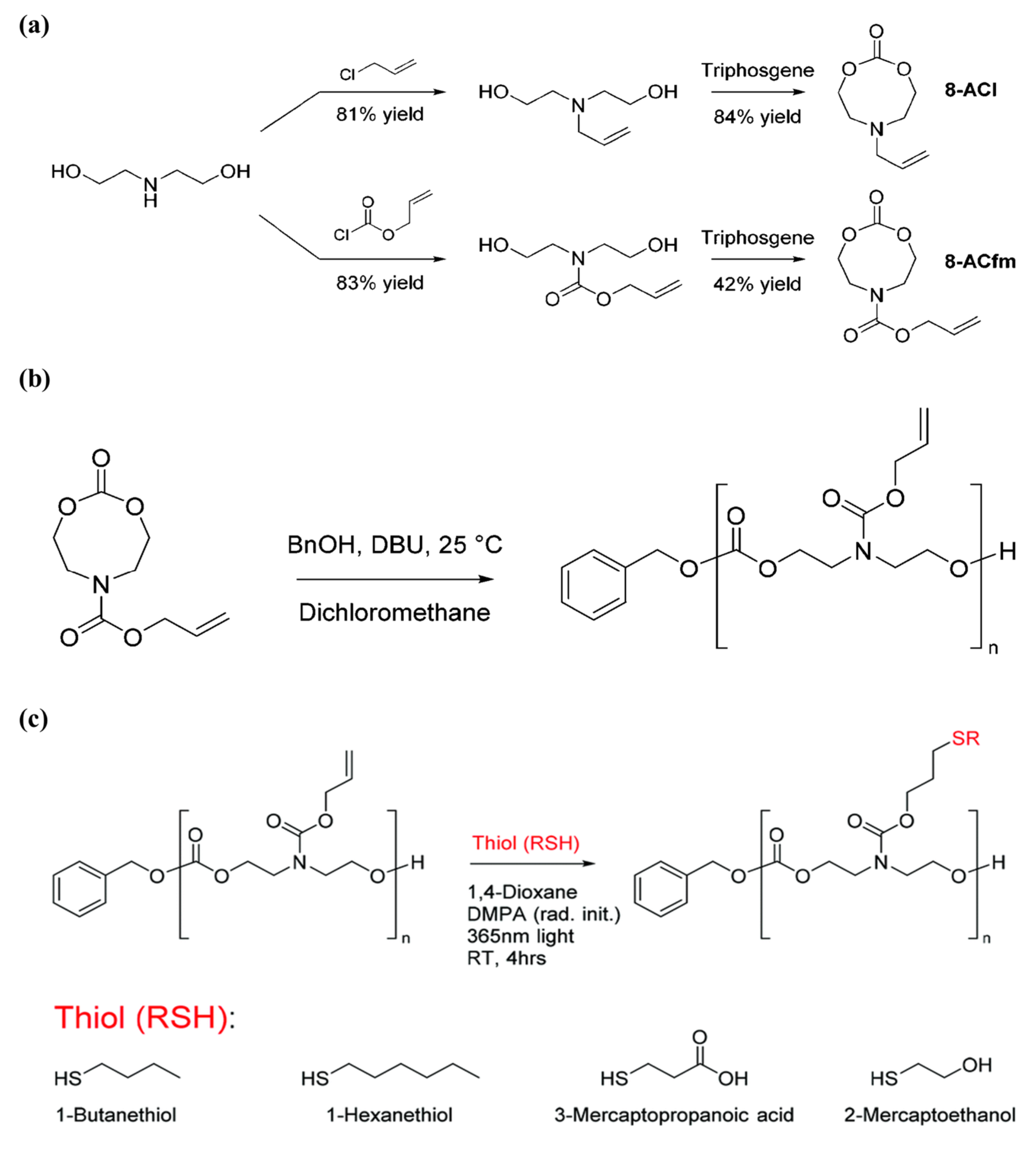
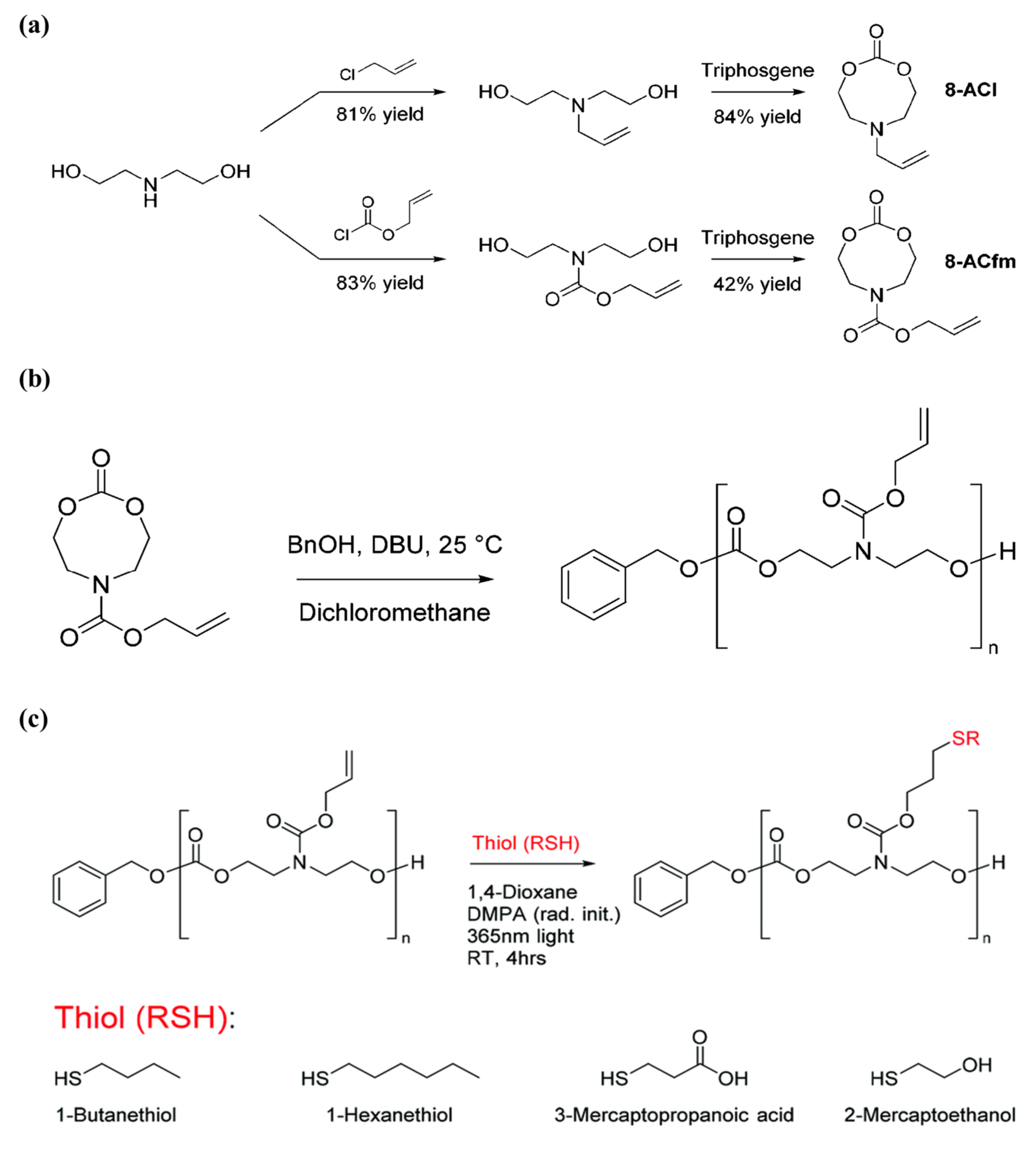
Aliphatic polycarbonates are degradable materials with low toxicity and high compatibility, and thus are an important class of polymers in biomedical applications. Designing and polymerizing cyclic carbonates carrying allyl pendants that could be easily modified with thiol–ene reaction is a dominant approach in post-polymerization functionalization for polymers based on cyclic carbonates. The method is selective, facile, and yields high conversions and high reaction rates; however, the long reaction time needed for the allyl functional monomer to yield high conversions while maintaining its end-group fidelity remains a challenge. Therefore, Yuen et al. [
80] incorporated allyl functionalities into N-substituted eight-membered cyclic carbonates, renowned for being more reactive than six-membered cyclic carbonates, to improve the reactivity of cyclic carbonate monomers that contain allyl functionality. The authors further utilized the allyl groups in the polymer by enabling modification of the polymer and, hence, widening the extent of the versatility of the carbonate-based polymers. Firstly, the authors synthesized two different allyl-bearing N-substituted eight-membered cyclic carbonates, 6-allyl-1,3,6-dioxazocan-2-one (8-ACl) and allyl 2-oxo-1,3,6-dioxazocane-6-carboxylate (8-ACfm), in yields of 84% and 42%, respectively, as shown in
Figure 18A. Thereafter, homopolymerization of the monomers 8-ACl and 8-ACfm was explored using the organocatalyst 1,8-di- azabicyclo[5 .4.0]undec-7-ene (DBU) (
Figure 18B). They used 10% DBU for catalysis and monitored the polymerization and conversion through
1H NMR, in which 8-ACfm was able to achieve 97% monomer conversion in 10 min; on the other hand, 8-ACl reached only 14% monomer conversion after 1 h. The observed variability in reactivity between 8-ACl and 8-ACfm was attributed to the different reactivity of the N-substituent pendant group of the cyclic carbonate between the two monomers. Later, the allyl bearing polymer was then modified with four thiols, 1-butanethiol, 1-hexanethiol, 3-mercaptopropionic acid, and 2-mercaptoethanol, as presented in
Figure 18C. The efficiency of the modifications was very high with ≥90% conversion for all cases, as calculated by
1H NMR spectroscopy. Moreover, the authors managed to successfully form a gel through reacting allyl-bearing polycarbonates, with HDT as a crosslinker. Importantly, the superior polymerization kinetics of 8-ACfm allows its copolymerization with other cyclic monomers in a reasonable amount of time. The N-substituted monomer, 8-ACfm, was copolymerized with the commercially available trimethylene carbonate (TMC) with ≥97% within 1 h. The thiol–ene radical additions with 1-butanethiol were performed successfully for the prepared co-polymer, which suggested that N-substituted eight-membered cyclic carbonates chemistry is an attractive approach for producing functional biodegradable aliphatic polycarbonates.
2.4. Polysaccharides
Polysaccharides derived from natural resources are renewable, inexpensive, often biodegradable, biocompatible, generally nontoxic, and demonstrate excellent properties, including aqueous solubility, stability, and excellent swelling ability. Owing to these outstanding characteristics, polysaccharides are being utilized as suitable biomaterials in many biomedical applications, such as the delivery of drugs and therapeutics, protein encapsulation, wound healing, tissue regeneration, and bioimaging. Among many widely-known polysaccharides, chitosan (CS), derived from chitin via de-acetylation, primarily composed of D-glucosamine and N-acetyl glucosamine units with a β-(1–4)-linkage, is the second most abundant natural polysaccharide, after cellulose. Its biodegradable, biocompatible, non-toxic, and non-allergenic properties, as well as its antimicrobial, antioxidant, anti-tumor, and anti-inflammatory activities, make chitosan one of the most studied and frequently investigated biomaterials for a wide range of biomedical applications. Owing to its important biological properties, chitosan is considered an immunoadjuvant, anti-thrombogenic, and anti-cholesteric agent [
81,
82,
83,
84]. It is a very popular excipient in the pharmaceutical industry. Several research innovations have been made on chitosan as a polymer matrix for drug delivery [
81,
85], tissue engineering, and regenerative medicines. On account of its high versatility, chitosan can be processed into many physical forms, such as micro or nano-sized particles, fibers, gels, beads, films, sponges, scaffolds etc., for oral and parenteral drug delivery, and tissue engineering [
86,
87]. End group functionalization of chitosan by inserting allyl (ene) functionality opens new doors for promising applications of these materials. In this section, we have discussed the synthesis of such allyl-functionalized chitosan-based polymers.
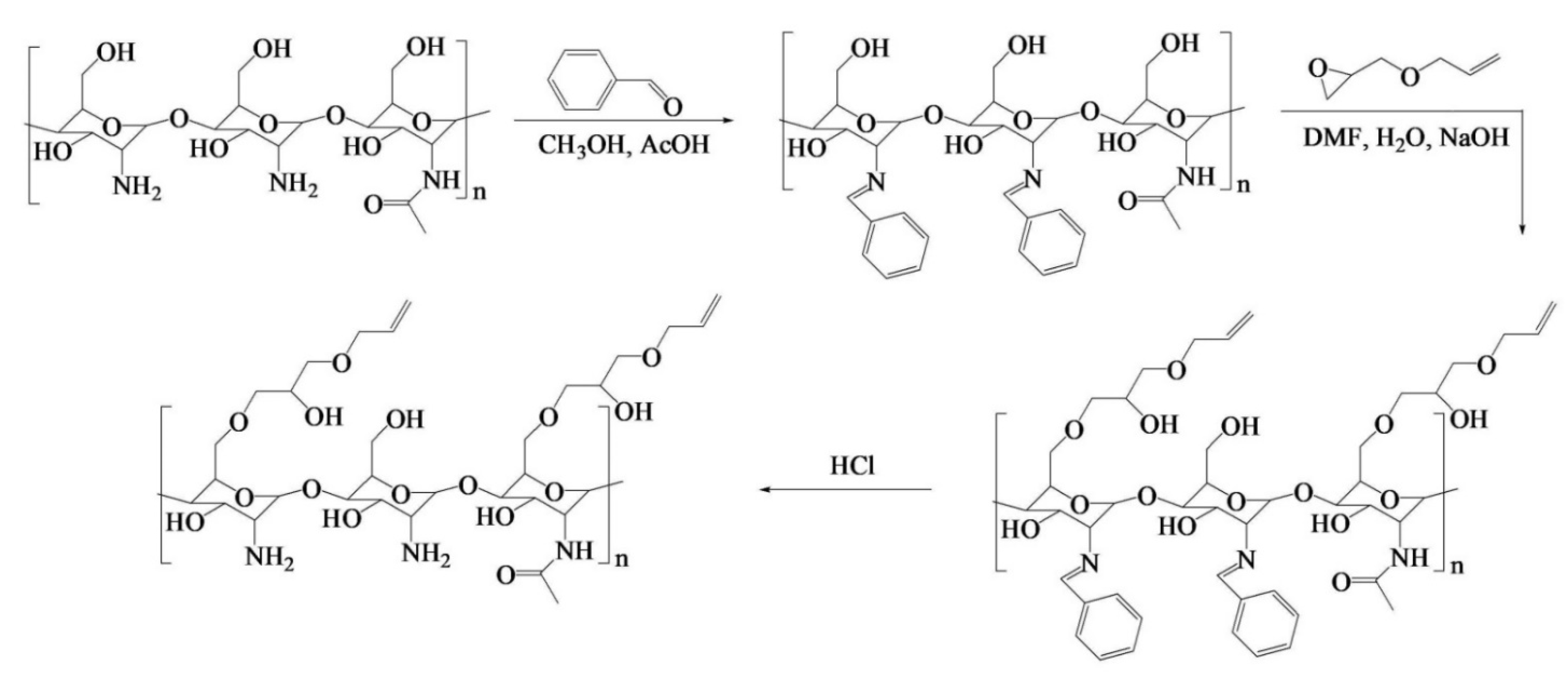
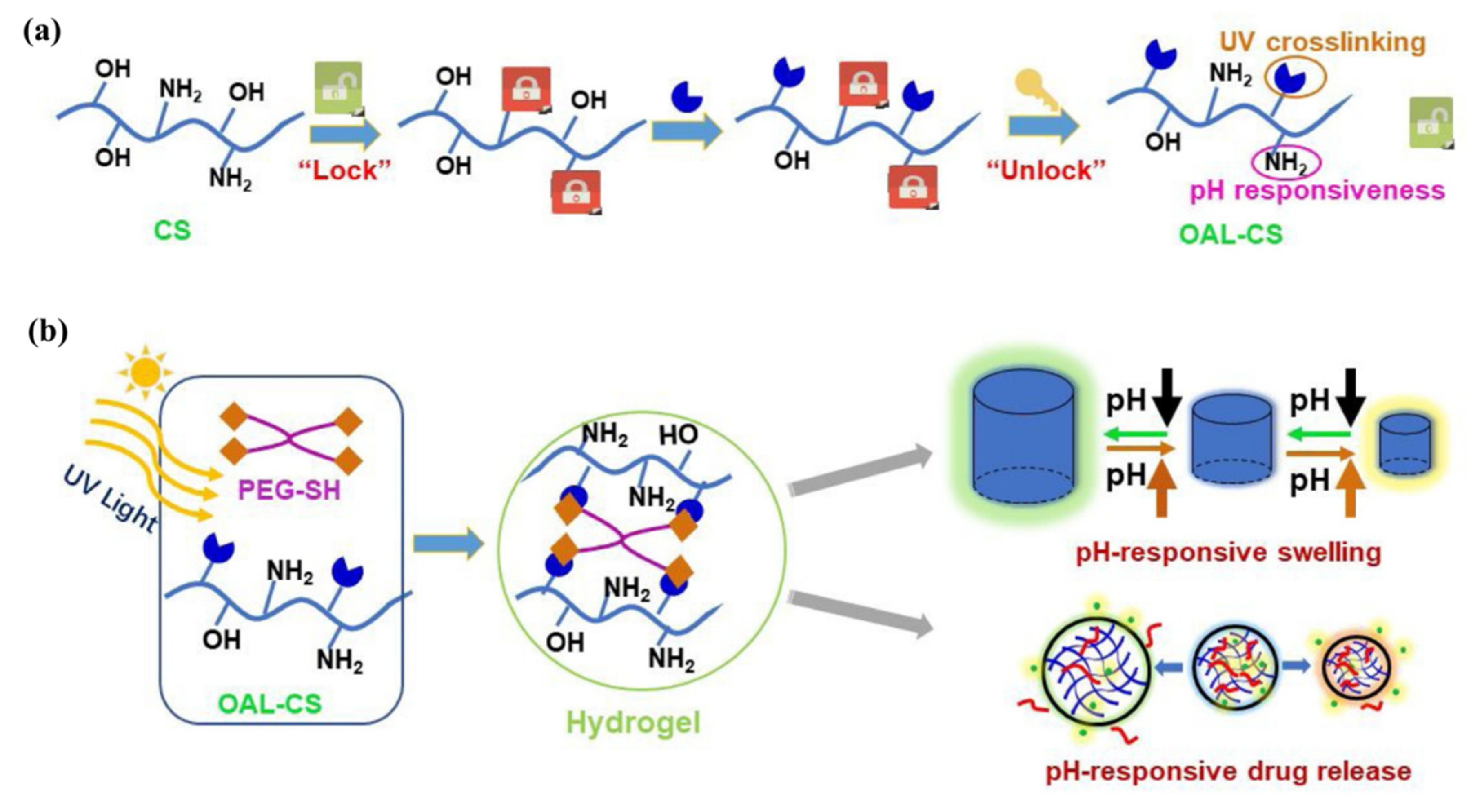
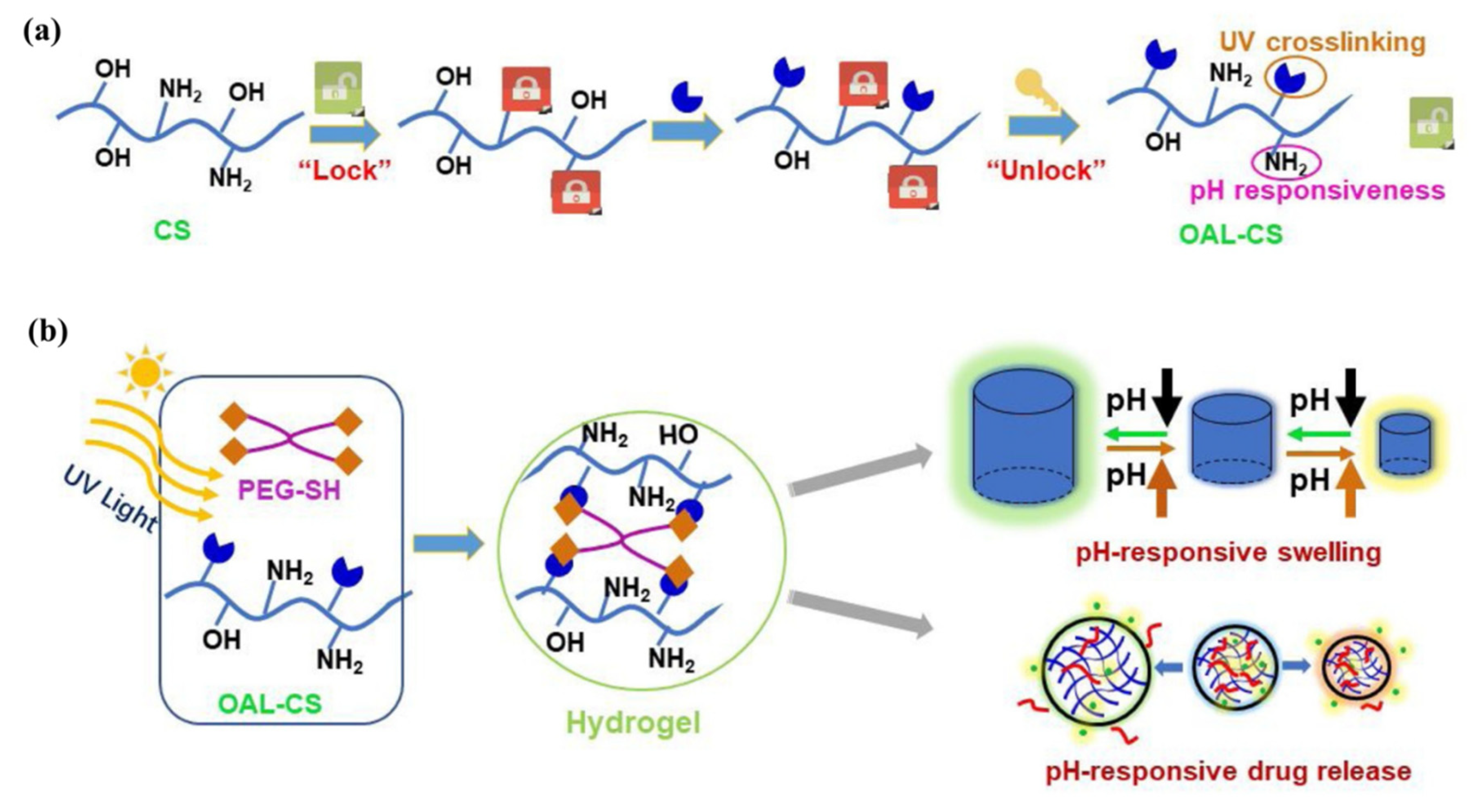
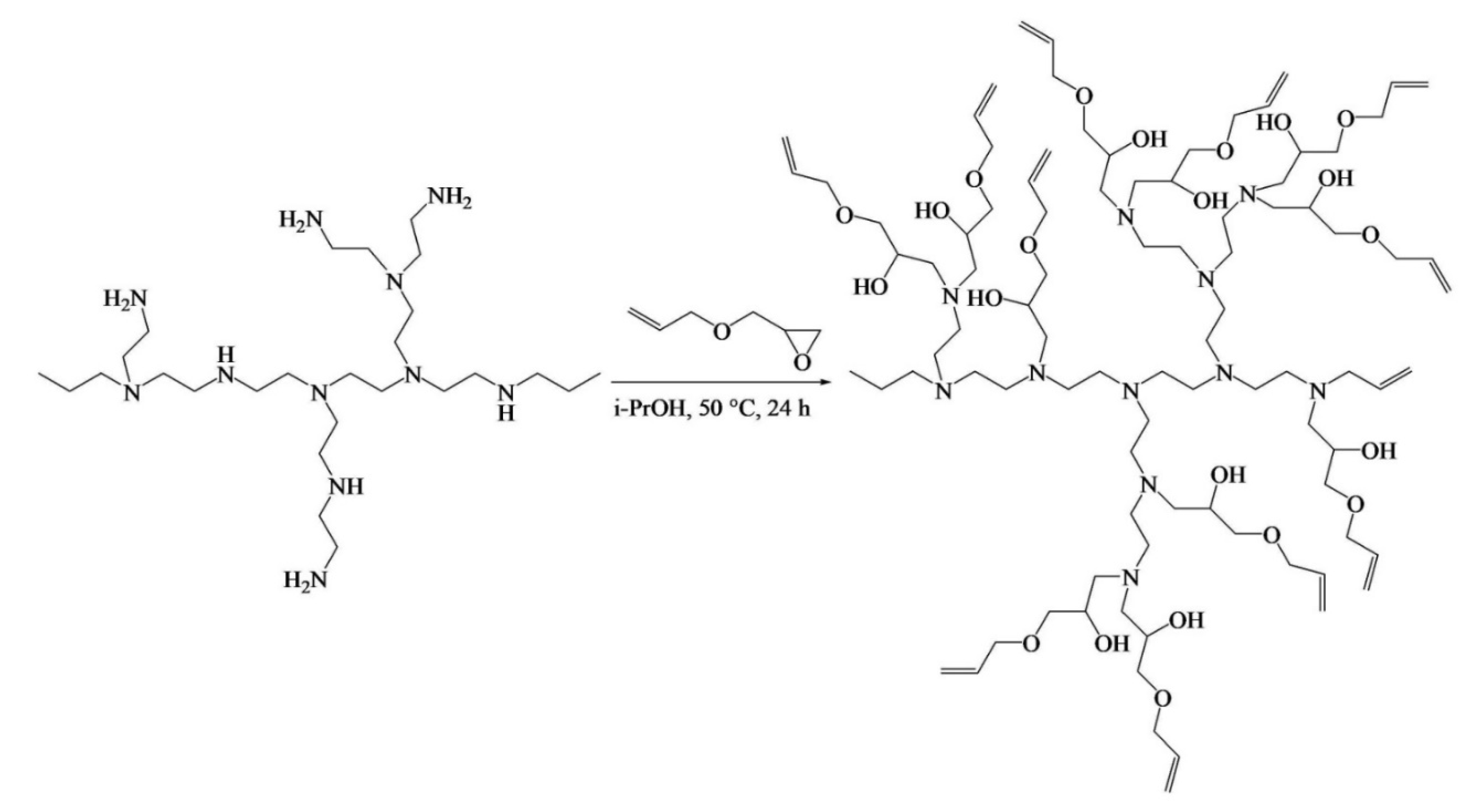
Ding et al. [
88] engineered a pH-responsive UV-crosslinkable C
6 O-allyl chitosan (OAL-CS) polymer hydrogel. The authors synthesized OAL-CS via a three-step reaction: (1) a Schiff’s base reaction of C
2-NH
2 with benzaldehyde to suppress the activity of the amino group, (2) a ring-opening reaction of epoxy group of AGE with C
6-OH of CS to graft UV-crosslinkable allyl groups on CS, and (3) removal of protective groups in dilute hydrochloric acid, as depicted in
Figure 19. The authors adopted the protection of amino groups to preserve the pH responsiveness of the native CS and to ensure that the epoxy groups reacted solely with the hydroxyl groups (C
6-OH) (
Figure 20a). The authors post-functionalized OAL-CS via UV-induced “thiol-ene” click chemistry to demonstrate the drug delivery capability of the polymer (
Figure 20b).
Hydrogel of OAL-CS was prepared by dissolving the polymer in PBS containing four-arm PEG-SH as crosslinker. The final mixture solution was turned into gel within 30 s under low-dose UV irradiation. Later, the pH-responsive swelling and shrinkage for modulating the small molecular drug, DOX, and macromolecular drug, bovine serum albumin (BSA) release, were investigated in PBS buffer solutions at pH = 5.0, 6.8, and 8.0. The authors found that the release behavior of the two drugs was different, and significantly dependent on the pH of the solution, while the release of the drugs occurred mainly by their free diffusion from the hydrogel. In the case of DOX, about 96% of the DOX load was released within 4 h in basic pH (pH = 8.0 PBS) and this value was 53% more than the DOX released within the same duration in neutral pH (pH = 6.8 PBS). On the other hand, after three days, the cumulative release of BSA at pH = 8.0 was only 27%, while 53% and 62% releases were observed at pH = 6.8 and 5.0, respectively. The pH-responsive shrinkage behavior of hydrogel delayed the release of BSA by 49%, whereas the free diffusion, together with extrusion of hydrogel, surprisingly promoted the release of DOX by 81%. It is noted that, due to the degradation of hydrogel in acidic pH = 5.0, a quick release of BSA was observed after 6 days. The product developed in this study has the properties of being worked as a patterned microgel and rapid transdermal curing hydrogel in vivo, with potential for pH-responsive drug delivery and other biomedical applications.
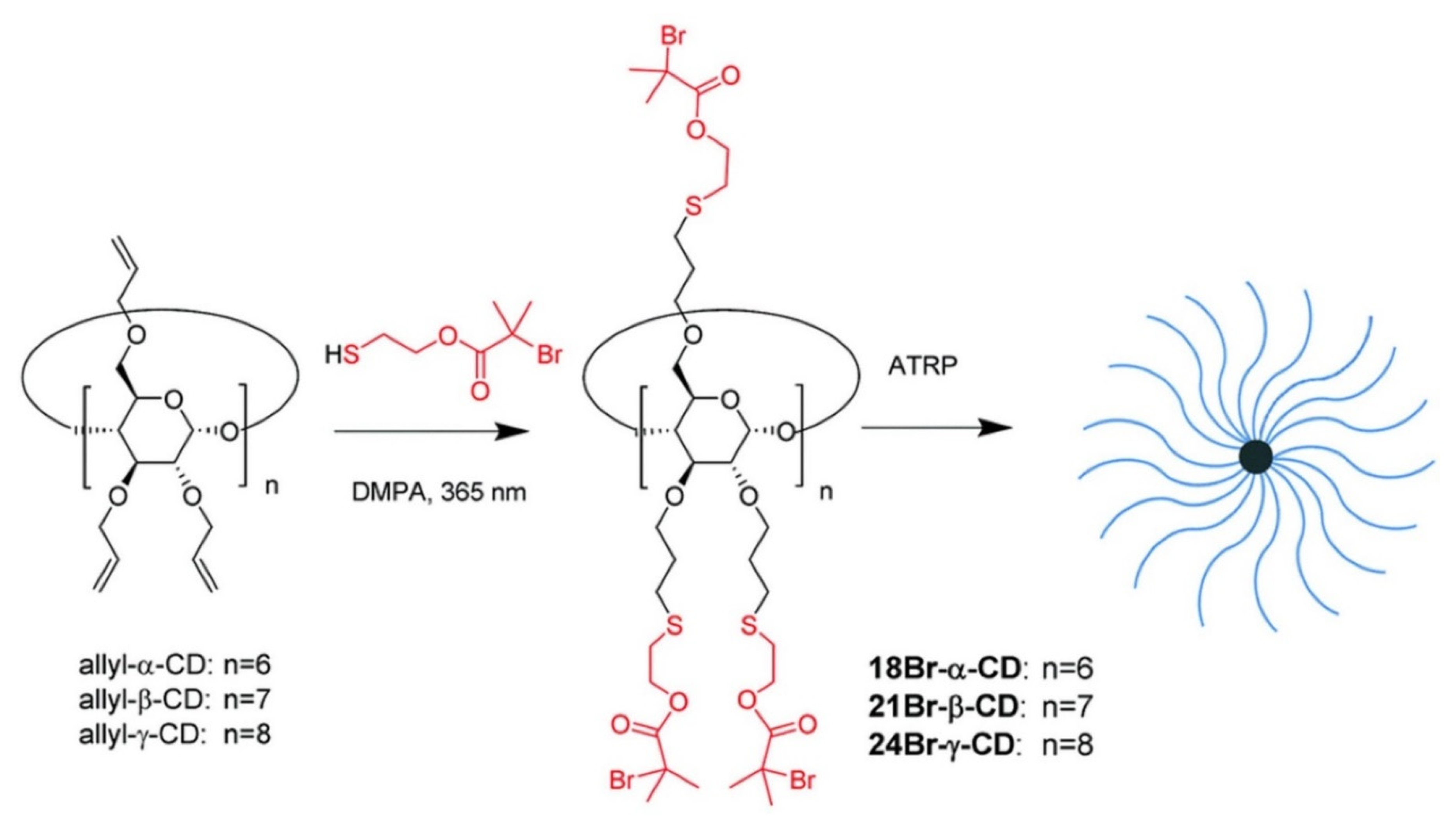
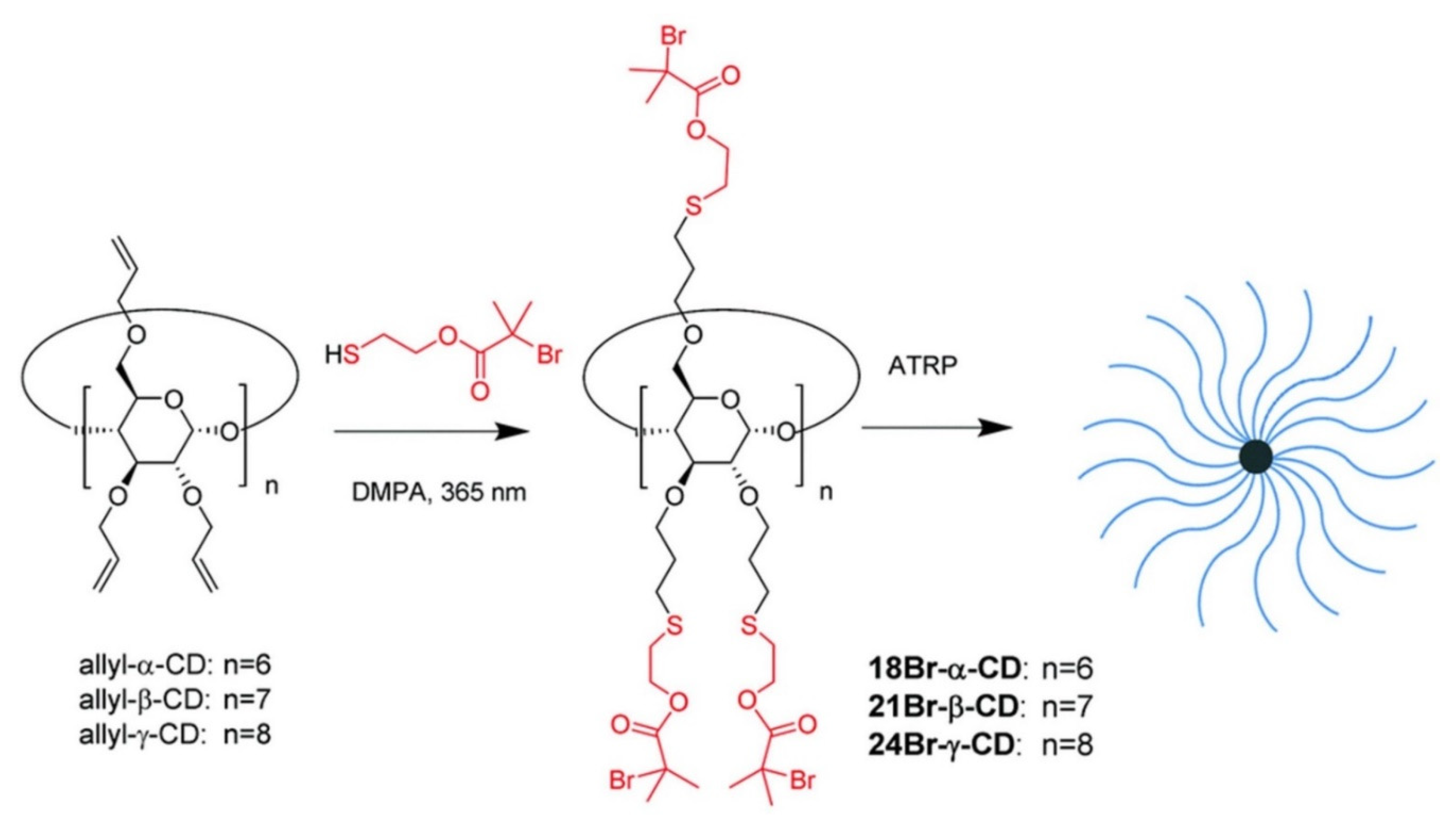
Yi [
89] utilized a thiol-ene photoclick strategy to efficiently synthesize multifunctional initiators based on cyclodextrine (CD) cores (
Figure 21). The synthesized α-, β-, and γ-CD cores were successfully employed in a “core-first” approach to produce well-defined multiarm star polymers via atom transfer radical polymerization (ATRP), which has been demonstrated as one of the most versatile polymerization techniques for building architecturally complex polymers. Multiarm star polymers are a unique class of branched polymers with a large number of linear arms jointly connected to a central core. Their unique solid and solution properties, due to compact globular architecture and high arm density, make them attractive for a wide range of applications. The author produced the multifunctional core initiator 21BR-S-β-CD by a general procedure of thiol-ene photoclick chemistry, dissolving perallylated β-CD (allyl-β-CD) and DMPA in thiol 2-mercaptoethyl-2-bromo-2-methylpropanoate (HS-EBiB) with a yield of above 90%. In a similar manner, 18Br-S-α-CD and 24Br-S-γ-CD core initiators were synthesized from allyl-α-CD and allyl-γ-CD, respectively, in high yields. Yi also synthesized a new functional thiol 2-mercaptoethyl-2-chloropropanoate (HS-ECP), and in a photoclick reaction with allyl-β-CD, 21Cl-S-β-CD was successfully synthesized. Using 21Br-S-β-CD as the multifunctional core initiator, 21-arm star polymers based on poly(tert-butyl acrylate) (PtBA), polystyrene (PS) and PMMA were successfully prepared. 21-arm poly(N-isopropylacrylamide) (PNIPAM) stars were made from 21Cl-S-β-CD. 18-Arm and 24-arm PtBA stars were produced from 18Br-S-β-CD and 24Br-S-β-CD, respectively. The author expects that the approach could be applied in growing patterned polymer brushes on planar substrates, such as silicon chips and glass slides surfaces.


 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}