
Cyclodextrins: Only Pharmaceutical Excipients or Full-Fledged Drug Candidates?
 ,
,  ,
,  ,
,  , and
, and
Abstract
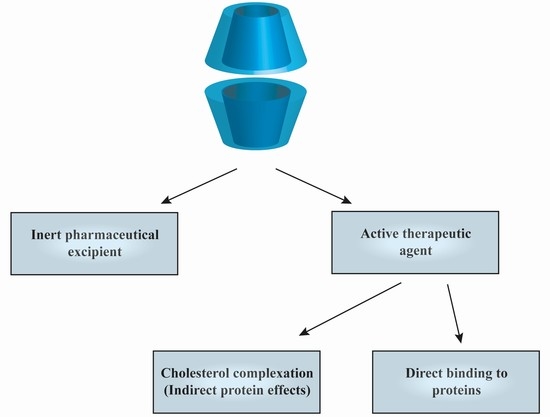
1. Introduction
2. Interactions between Cyclodextrins and Lipids
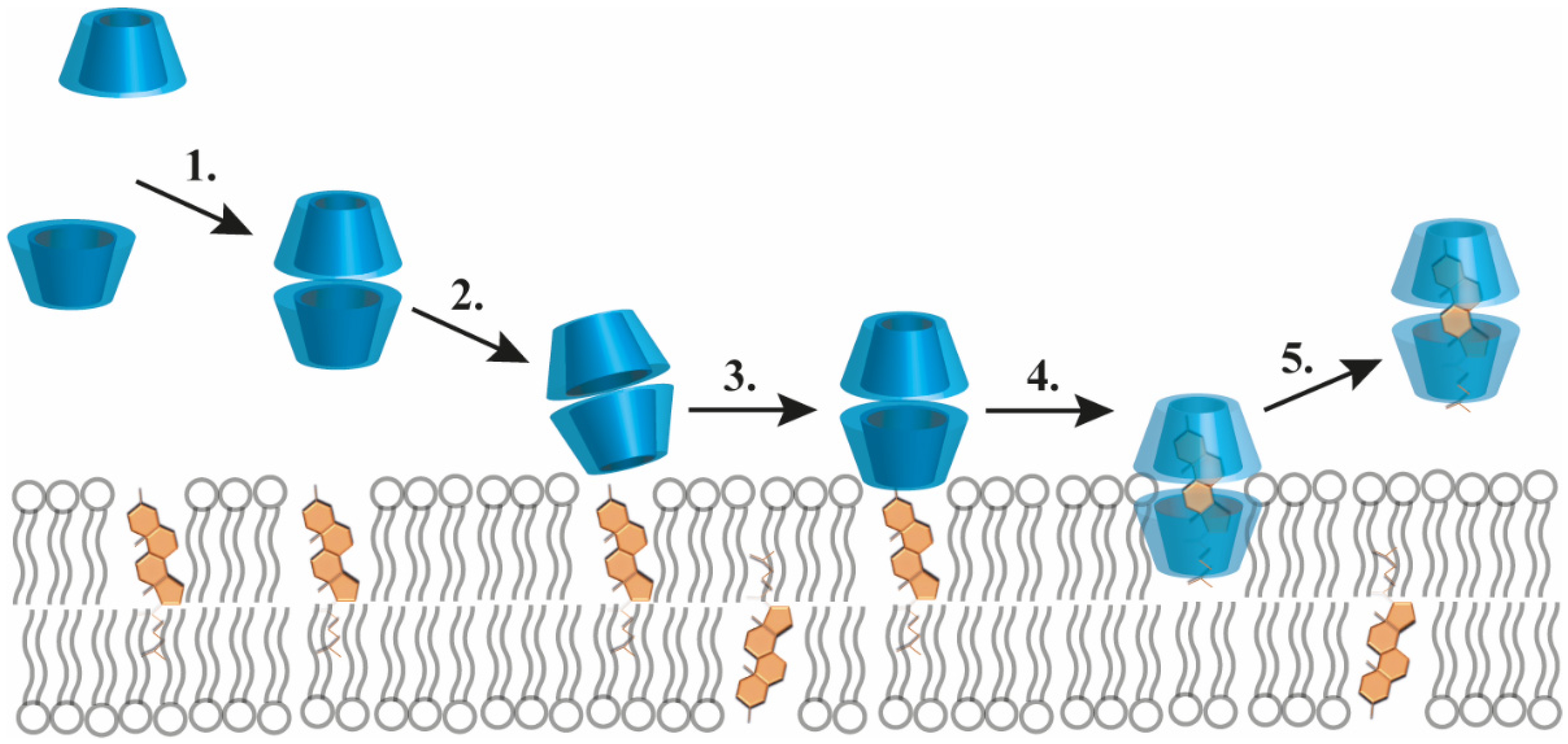
2.1. Cholesterol Complexation by Cyclodextrins
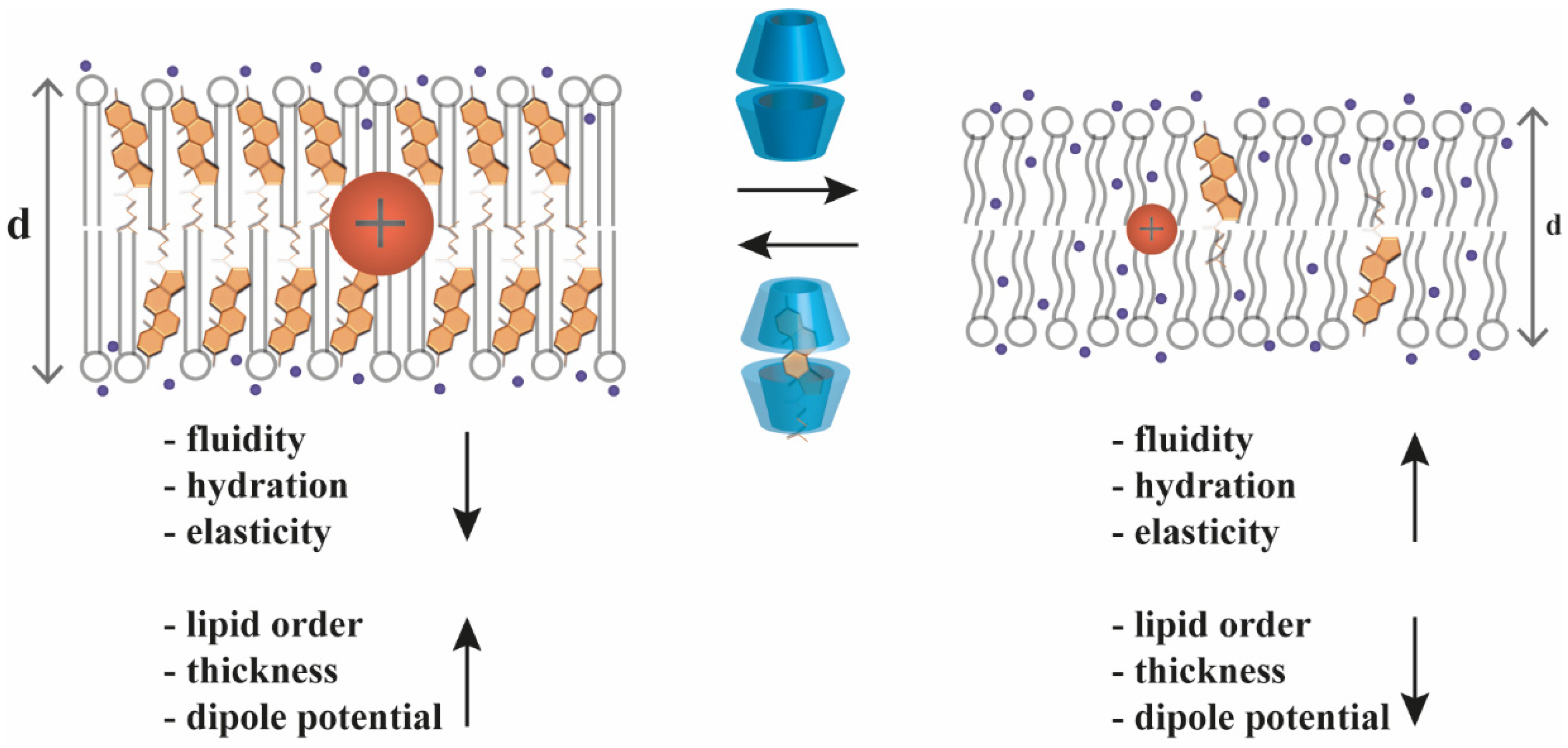
2.2. Cyclodextrin Effects on Biophysical Parameters of Cellular Membranes
2.3. Cyclodextrin-Induced Disruption of Lipid Raft Microdomains
2.4. Indirect Modulation of Protein Functions by Cyclodextrins via Alterations in Biophysical Parameters or Lateral Heterogeneity of Membranes
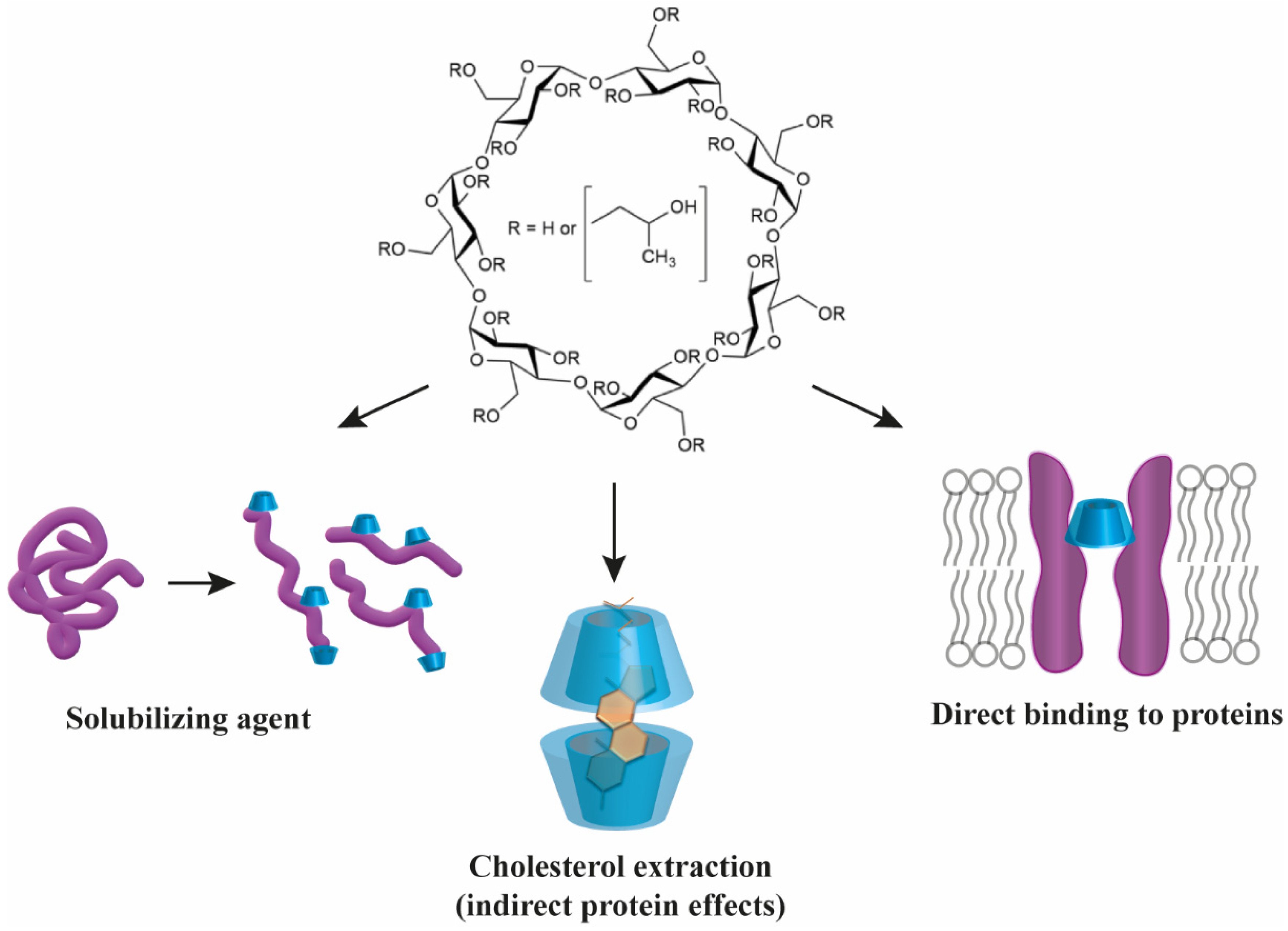
3. Direct Interactions between Cyclodextrins and Proteins
3.1. General Mechanisms of Direct Cyclodextrin–Protein Interactions
3.2. Bacterial, Fungal and Plant Carbohydrate-Binding Proteins
3.3. Aggregation-Prone Human Peptides and Proteins
3.3.1. Human Peptide Hormones
3.3.2. Amyloid-Forming Peptides and Proteins
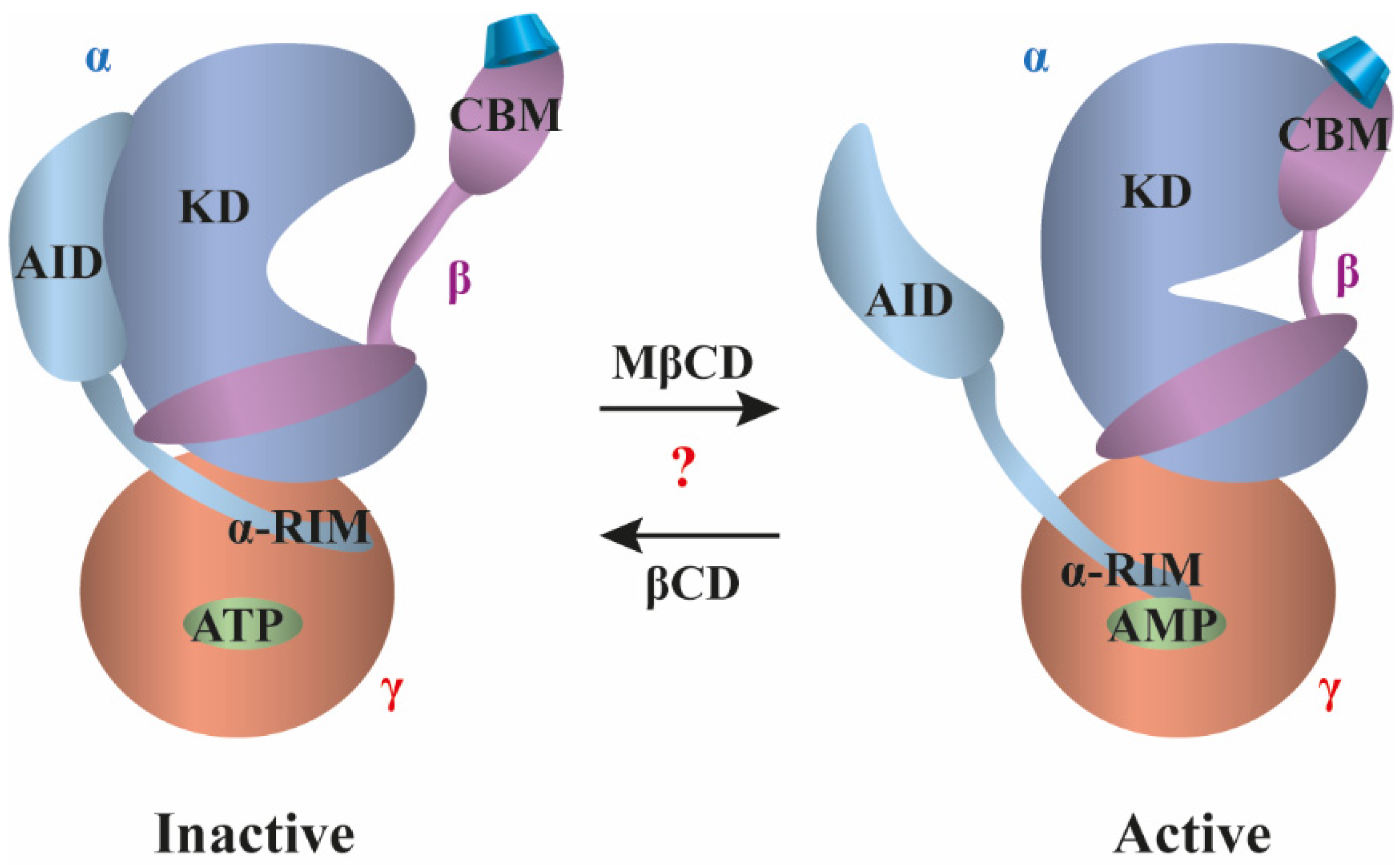
3.4. AMP-Activated Protein Kinase
3.5. Bacterial Pore-Forming Proteins
3.6. Human Pore-Forming Proteins
3.6.1. Connexins
3.6.2. GABAA Receptor
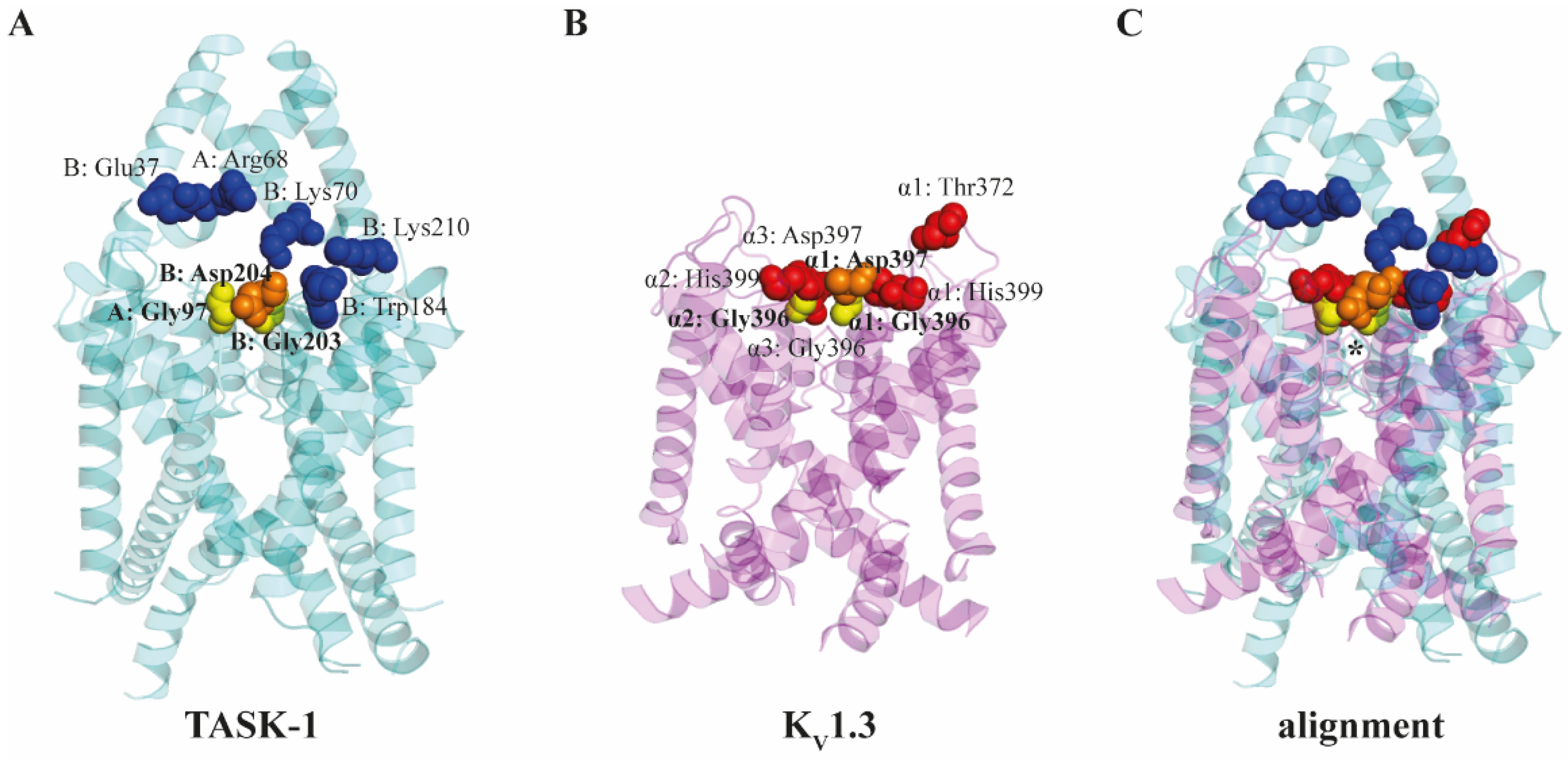
3.6.3. TASK Ion Channels
3.6.4. KV1.3 Ion Channel

4. Potential Clinical Applicability of Cyclodextrin Interactions with Cholesterol and Proteins
4.1. Cyclodextrins as Anti-Aggregative Excipients in Peptide and Protein Drug Formulations
4.2. Cyclodextrins as Active Anti-Aggregative and Cholesterol-Depleting Agents in Human Amyloid-Related Neurodegenerative Diseases
4.3. Cyclodextrins as Active Cholesterol-Extracting Agents in Atherosclerosis
4.4. Cyclodextrins as Active Cholesterol-Extracting Agents and Direct Modulators of Protein Function in Niemann–Pick Type C Disease
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Davis, M.E.; Brewster, M.E. Cyclodextrin-based pharmaceutics: Past, present and future. Nat. Rev. Drug Discov. 2004, 3, 1023–1035. [Google Scholar] [CrossRef]
- Serno, T.; Geidobler, R.; Winter, G. Protein stabilization by cyclodextrins in the liquid and dried state. Adv. Drug Deliv. Rev. 2011, 63, 1086–1106. [Google Scholar] [CrossRef]
- Szejtli, J. Introduction and General Overview of Cyclodextrin Chemistry. Chem. Rev. 1998, 98, 1743–1754. [Google Scholar] [CrossRef] [PubMed]
- Agency, E.M. Cyclodextrins Used as Excipients. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/questions-answers-cyclodextrins-used-excipients-medicinal-products-human-use_en.pdf (accessed on 13 November 2022).
- Matencio, A.; Hoti, G.; Monfared, Y.K.; Rezayat, A.; Pedrazzo, A.R.; Caldera, F.; Trotta, F. Cyclodextrin Monomers and Polymers for Drug Activity Enhancement. Polymers 2021, 13, 1684. [Google Scholar] [CrossRef] [PubMed]
- Lagiewka, J.; Girek, T.; Ciesielski, W. Cyclodextrins-Peptides/Proteins Conjugates: Synthesis, Properties and Applications. Polymers 2021, 13, 1759. [Google Scholar] [CrossRef]
- Gidwani, B.; Vyas, A. A Comprehensive Review on Cyclodextrin-Based Carriers for Delivery of Chemotherapeutic Cytotoxic Anticancer Drugs. Biomed. Res. Int. 2015, 2015, 198268. [Google Scholar] [CrossRef] [PubMed]
- Haimhoffer, Á.; Rusznyák, Á.; Réti-Nagy, K.; Vasvári, G.; Váradi, J.; Vecsernyés, M.; Bácskay, I.; Fehér, P.; Ujhelyi, Z.; Fenyvesi, F. Cyclodextrins in Drug Delivery Systems and Their Effects on Biological Barriers. Sci. Pharm. 2019, 87, 33. [Google Scholar] [CrossRef]
- Szente, L.; Fenyvesi, E. Cyclodextrin-Lipid Complexes: Cavity Size Matters. Struct. Chem. 2017, 28, 479–492. [Google Scholar] [CrossRef]
- Zidovetzki, R.; Levitan, I. Use of cyclodextrins to manipulate plasma membrane cholesterol content: Evidence, misconceptions and control strategies. Biochim. Biophys. Acta 2007, 1768, 1311–1324. [Google Scholar] [CrossRef] [PubMed]
- Szente, L.; Singhal, A.; Domokos, A.; Song, B. Cyclodextrins: Assessing the Impact of Cavity Size, Occupancy, and Substitutions on Cytotoxicity and Cholesterol Homeostasis. Molecules 2018, 23, 1228. [Google Scholar] [CrossRef]
- Kilsdonk, E.P.; Yancey, P.G.; Stoudt, G.W.; Bangerter, F.W.; Johnson, W.J.; Phillips, M.C.; Rothblat, G.H. Cellular cholesterol efflux mediated by cyclodextrins. J. Biol. Chem. 1995, 270, 17250–17256. [Google Scholar] [CrossRef] [PubMed]
- Yancey, P.G.; Rodrigueza, W.V.; Kilsdonk, E.P.; Stoudt, G.W.; Johnson, W.J.; Phillips, M.C.; Rothblat, G.H. Cellular cholesterol efflux mediated by cyclodextrins. Demonstration Of kinetic pools and mechanism of efflux. J. Biol. Chem. 1996, 271, 16026–16034. [Google Scholar] [CrossRef] [PubMed]
- Lopez, C.A.; de Vries, A.H.; Marrink, S.J. Molecular mechanism of cyclodextrin mediated cholesterol extraction. PLoS Comput. Biol. 2011, 7, e1002020. [Google Scholar] [CrossRef] [PubMed]
- Lopez, C.A.; de Vries, A.H.; Marrink, S.J. Computational microscopy of cyclodextrin mediated cholesterol extraction from lipid model membranes. Sci. Rep. 2013, 3, 2071. [Google Scholar] [CrossRef] [PubMed]
- Christian, A.E.; Haynes, M.P.; Phillips, M.C.; Rothblat, G.H. Use of cyclodextrins for manipulating cellular cholesterol content. J. Lipid Res. 1997, 38, 2264–2272. [Google Scholar] [CrossRef]
- Zakany, F.; Kovacs, T.; Panyi, G.; Varga, Z. Direct and indirect cholesterol effects on membrane proteins with special focus on potassium channels. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 1865, 158706. [Google Scholar] [CrossRef]
- Zakany, F.; Pap, P.; Papp, F.; Kovacs, T.; Nagy, P.; Peter, M.; Szente, L.; Panyi, G.; Varga, Z. Determining the target of membrane sterols on voltage-gated potassium channels. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 312–325. [Google Scholar] [CrossRef]
- Barbera, N.; Ayee, M.A.A.; Akpa, B.S.; Levitan, I. Differential Effects of Sterols on Ion Channels: Stereospecific Binding vs Stereospecific Response. Curr. Top. Membr. 2017, 80, 25–50. [Google Scholar] [CrossRef]
- Zakany, F.; Szabo, M.; Batta, G.; Karpati, L.; Mandity, I.M.; Fulop, P.; Varga, Z.; Panyi, G.; Nagy, P.; Kovacs, T. An omega-3, but Not an omega-6 Polyunsaturated Fatty Acid Decreases Membrane Dipole Potential and Stimulates Endo-Lysosomal Escape of Penetratin. Front. Cell Dev. Biol. 2021, 9, 647300. [Google Scholar] [CrossRef]
- Leeb, F.; Maibaum, L. Spatially Resolving the Condensing Effect of Cholesterol in Lipid Bilayers. Biophys. J. 2018, 115, 2179–2188. [Google Scholar] [CrossRef]
- Owen, D.M.; Williamson, D.J.; Magenau, A.; Gaus, K. Sub-resolution lipid domains exist in the plasma membrane and regulate protein diffusion and distribution. Nat. Commun. 2012, 3, 1256. [Google Scholar] [CrossRef] [PubMed]
- Sezgin, E.; Waithe, D.; Bernardino de la Serna, J.; Eggeling, C. Spectral imaging to measure heterogeneity in membrane lipid packing. Chemphyschem 2015, 16, 1387–1394. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Benda, A.; Kwiatek, J.; Owen, D.M.; Gaus, K. Time-Resolved Laurdan Fluorescence Reveals Insights into Membrane Viscosity and Hydration Levels. Biophys. J. 2018, 115, 1498–1508. [Google Scholar] [CrossRef] [PubMed]
- Oncul, S.; Klymchenko, A.S.; Kucherak, O.A.; Demchenko, A.P.; Martin, S.; Dontenwill, M.; Arntz, Y.; Didier, P.; Duportail, G.; Mely, Y. Liquid ordered phase in cell membranes evidenced by a hydration-sensitive probe: Effects of cholesterol depletion and apoptosis. Biochim. Biophys. Acta 2010, 1798, 1436–1443. [Google Scholar] [CrossRef]
- Parasassi, T.; Gratton, E.; Yu, W.M.; Wilson, P.; Levi, M. Two-photon fluorescence microscopy of laurdan generalized polarization domains in model and natural membranes. Biophys. J. 1997, 72, 2413–2429. [Google Scholar] [CrossRef]
- Bongiovanni, M.N.; Godet, J.; Horrocks, M.H.; Tosatto, L.; Carr, A.R.; Wirthensohn, D.C.; Ranasinghe, R.T.; Lee, J.E.; Ponjavic, A.; Fritz, J.V.; et al. Multi-dimensional super-resolution imaging enables surface hydrophobicity mapping. Nat. Commun. 2016, 7, 13544. [Google Scholar] [CrossRef]
- Hung, W.C.; Lee, M.T.; Chung, H.; Sun, Y.T.; Chen, H.; Charron, N.E.; Huang, H.W. Comparative Study of the Condensing Effects of Ergosterol and Cholesterol. Biophys. J. 2016, 110, 2026–2033. [Google Scholar] [CrossRef][Green Version]
- Henriksen, J.; Rowat, A.C.; Brief, E.; Hsueh, Y.W.; Thewalt, J.L.; Zuckermann, M.J.; Ipsen, J.H. Universal behavior of membranes with sterols. Biophys. J. 2006, 90, 1639–1649. [Google Scholar] [CrossRef] [PubMed]
- Niemela, P.S.; Ollila, S.; Hyvonen, M.T.; Karttunen, M.; Vattulainen, I. Assessing the nature of lipid raft membranes. PLoS Comput. Biol. 2007, 3, e34. [Google Scholar] [CrossRef]
- Pan, J.; Mills, T.T.; Tristram-Nagle, S.; Nagle, J.F. Cholesterol perturbs lipid bilayers nonuniversally. Phys. Rev. Lett. 2008, 100, 198103. [Google Scholar] [CrossRef]
- Samuli Ollila, O.H.; Rog, T.; Karttunen, M.; Vattulainen, I. Role of sterol type on lateral pressure profiles of lipid membranes affecting membrane protein functionality: Comparison between cholesterol, desmosterol, 7-dehydrocholesterol and ketosterol. J. Struct. Biol. 2007, 159, 311–323. [Google Scholar] [CrossRef]
- Chen, Z.; Rand, R.P. The influence of cholesterol on phospholipid membrane curvature and bending elasticity. Biophys. J. 1997, 73, 267–276. [Google Scholar] [CrossRef]
- Kollmitzer, B.; Heftberger, P.; Rappolt, M.; Pabst, G. Monolayer spontaneous curvature of raft-forming membrane lipids. Soft Matter 2013, 9, 10877–10884. [Google Scholar] [CrossRef]
- O’Shea, P. Intermolecular interactions with/within cell membranes and the trinity of membrane potentials: Kinetics and imaging. Biochem. Soc. Trans. 2003, 31, 990–996. [Google Scholar] [CrossRef] [PubMed]
- Wang, L. Measurements and implications of the membrane dipole potential. Annu. Rev. Biochem. 2012, 81, 615–635. [Google Scholar] [CrossRef] [PubMed]
- Haldar, S.; Kanaparthi, R.K.; Samanta, A.; Chattopadhyay, A. Differential effect of cholesterol and its biosynthetic precursors on membrane dipole potential. Biophys. J. 2012, 102, 1561–1569. [Google Scholar] [CrossRef]
- Shen, H.; Deng, M.; Wu, Z.; Zhang, J.; Zhang, Y.; Gao, C.; Cen, C. Effect of Cholesterol on Membrane Dipole Potential: Atomistic and Coarse-Grained Molecular Dynamics Simulations. J. Chem. Theory Comput. 2018, 14, 3780–3795. [Google Scholar] [CrossRef] [PubMed]
- Bandari, S.; Chakraborty, H.; Covey, D.F.; Chattopadhyay, A. Membrane dipole potential is sensitive to cholesterol stereospecificity: Implications for receptor function. Chem. Phys. Lipids 2014, 184, 25–29. [Google Scholar] [CrossRef][Green Version]
- Davis, S.; Davis, B.M.; Richens, J.L.; Vere, K.A.; Petrov, P.G.; Winlove, C.P.; O’Shea, P. alpha-Tocopherols modify the membrane dipole potential leading to modulation of ligand binding by P-glycoprotein. J. Lipid Res. 2015, 56, 1543–1550. [Google Scholar] [CrossRef] [PubMed]
- Jafurulla, M.; Rao, B.D.; Sreedevi, S.; Ruysschaert, J.M.; Covey, D.F.; Chattopadhyay, A. Stereospecific requirement of cholesterol in the function of the serotonin1A receptor. Biochim. Biophys. Acta 2014, 1838, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, T.; Sohajda, T.; Szente, L.; Nagy, P.; Panyi, G.; Varga, Z.; Zakany, F. Cyclodextrins Exert a Ligand-like Current Inhibitory Effect on the KV1.3 Ion Channel Independent of Membrane Cholesterol Extraction. Front. Mol. Biosci. 2021, 8, 735357. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, K.; Liu, P.; Lagerholm, B.C. The Lateral Organization and Mobility of Plasma Membrane Components. Cell 2019, 177, 806–819. [Google Scholar] [CrossRef] [PubMed]
- Lingwood, D.; Simons, K. Lipid rafts as a membrane-organizing principle. Science 2010, 327, 46–50. [Google Scholar] [CrossRef] [PubMed]
- Dart, C. Lipid microdomains and the regulation of ion channel function. J. Physiol. 2010, 588, 3169–3178. [Google Scholar] [CrossRef] [PubMed]
- Egawa, J.; Pearn, M.L.; Lemkuil, B.P.; Patel, P.M.; Head, B.P. Membrane lipid rafts and neurobiology: Age-related changes in membrane lipids and loss of neuronal function. J. Physiol. 2016, 594, 4565–4579. [Google Scholar] [CrossRef]
- Pollet, H.; Conrard, L.; Cloos, A.S.; Tyteca, D. Plasma Membrane Lipid Domains as Platforms for Vesicle Biogenesis and Shedding? Biomolecules 2018, 8, 94. [Google Scholar] [CrossRef]
- Sezgin, E.; Levental, I.; Mayor, S.; Eggeling, C. The mystery of membrane organization: Composition, regulation and roles of lipid rafts. Nat. Rev. Mol. Cell Biol. 2017, 18, 361–374. [Google Scholar] [CrossRef]
- Simons, K.; Ehehalt, R. Cholesterol, lipid rafts, and disease. J. Clin. Investig. 2002, 110, 597–603. [Google Scholar] [CrossRef]
- Gaus, K.; Kritharides, L.; Schmitz, G.; Boettcher, A.; Drobnik, W.; Langmann, T.; Quinn, C.M.; Death, A.; Dean, R.T.; Jessup, W. Apolipoprotein A-1 interaction with plasma membrane lipid rafts controls cholesterol export from macrophages. FASEB J. 2004, 18, 574–576. [Google Scholar] [CrossRef] [PubMed]
- Gaus, K.; Rodriguez, M.; Ruberu, K.R.; Gelissen, I.; Sloane, T.M.; Kritharides, L.; Jessup, W. Domain-specific lipid distribution in macrophage plasma membranes. J. Lipid Res. 2005, 46, 1526–1538. [Google Scholar] [CrossRef] [PubMed]
- Rouquette-Jazdanian, A.K.; Pelassy, C.; Breittmayer, J.P.; Aussel, C. Revaluation of the role of cholesterol in stabilizing rafts implicated in T cell receptor signaling. Cell. Signal. 2006, 18, 105–122. [Google Scholar] [CrossRef]
- Frankel, D.J.; Pfeiffer, J.R.; Surviladze, Z.; Johnson, A.E.; Oliver, J.M.; Wilson, B.S.; Burns, A.R. Revealing the topography of cellular membrane domains by combined atomic force microscopy/fluorescence imaging. Biophys. J. 2006, 90, 2404–2413. [Google Scholar] [CrossRef]
- Sanchez, S.A.; Gunther, G.; Tricerri, M.A.; Gratton, E. Methyl-beta-cyclodextrins preferentially remove cholesterol from the liquid disordered phase in giant unilamellar vesicles. J. Membr. Biol. 2011, 241, 1–10. [Google Scholar] [CrossRef] [PubMed]
- de Meyer, F.J.; Rodgers, J.M.; Willems, T.F.; Smit, B. Molecular simulation of the effect of cholesterol on lipid-mediated protein-protein interactions. Biophys. J. 2010, 99, 3629–3638. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.G. How lipids affect the activities of integral membrane proteins. Biochim. Biophys. Acta 2004, 1666, 62–87. [Google Scholar] [CrossRef] [PubMed]
- Saka, S.K.; Honigmann, A.; Eggeling, C.; Hell, S.W.; Lang, T.; Rizzoli, S.O. Multi-protein assemblies underlie the mesoscale organization of the plasma membrane. Nat. Commun. 2014, 5, 4509. [Google Scholar] [CrossRef] [PubMed]
- Levental, I.; Lyman, E. Regulation of membrane protein structure and function by their lipid nano-environment. Nat. Rev. Mol. Cell Biol. 2022. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.F. Soft Matter in Lipid-Protein Interactions. Annu. Rev. Biophys. 2017, 46, 379–410. [Google Scholar] [CrossRef] [PubMed]
- Yuan, C.; O’Connell, R.J.; Feinberg-Zadek, P.L.; Johnston, L.J.; Treistman, S.N. Bilayer thickness modulates the conductance of the BK channel in model membranes. Biophys. J. 2004, 86, 3620–3633. [Google Scholar] [CrossRef] [PubMed]
- daCosta, C.J.; Dey, L.; Therien, J.P.; Baenziger, J.E. A distinct mechanism for activating uncoupled nicotinic acetylcholine receptors. Nat. Chem. Biol. 2013, 9, 701–707. [Google Scholar] [CrossRef] [PubMed]
- Cornelius, F. Modulation of Na,K-ATPase and Na-ATPase activity by phospholipids and cholesterol. I. Steady-state kinetics. Biochemistry 2001, 40, 8842–8851. [Google Scholar] [CrossRef]
- Botelho, A.V.; Huber, T.; Sakmar, T.P.; Brown, M.F. Curvature and hydrophobic forces drive oligomerization and modulate activity of rhodopsin in membranes. Biophys. J. 2006, 91, 4464–4477. [Google Scholar] [CrossRef]
- Soubias, O.; Teague, W.E., Jr.; Hines, K.G.; Gawrisch, K. Rhodopsin/lipid hydrophobic matching-rhodopsin oligomerization and function. Biophys. J. 2015, 108, 1125–1132. [Google Scholar] [CrossRef] [PubMed]
- Teague, W.E., Jr.; Soubias, O.; Petrache, H.; Fuller, N.; Hines, K.G.; Rand, R.P.; Gawrisch, K. Elastic properties of polyunsaturated phosphatidylethanolamines influence rhodopsin function. Faraday Discuss. 2013, 161, 383–395; discussion 419–459. [Google Scholar] [CrossRef] [PubMed]
- Bruno, A.; Costantino, G.; de Fabritiis, G.; Pastor, M.; Selent, J. Membrane-sensitive conformational states of helix 8 in the metabotropic Glu2 receptor, a class C GPCR. PLoS ONE 2012, 7, e42023. [Google Scholar] [CrossRef]
- Mondal, S.; Johnston, J.M.; Wang, H.; Khelashvili, G.; Filizola, M.; Weinstein, H. Membrane driven spatial organization of GPCRs. Sci. Rep. 2013, 3, 2909. [Google Scholar] [CrossRef]
- Gutierrez, M.G.; Mansfield, K.S.; Malmstadt, N. The Functional Activity of the Human Serotonin 5-HT1A Receptor Is Controlled by Lipid Bilayer Composition. Biophys. J. 2016, 110, 2486–2495. [Google Scholar] [CrossRef] [PubMed]
- Prasanna, X.; Sengupta, D.; Chattopadhyay, A. Cholesterol-dependent Conformational Plasticity in GPCR Dimers. Sci. Rep. 2016, 6, 31858. [Google Scholar] [CrossRef] [PubMed]
- Needham, S.R.; Roberts, S.K.; Arkhipov, A.; Mysore, V.P.; Tynan, C.J.; Zanetti-Domingues, L.C.; Kim, E.T.; Losasso, V.; Korovesis, D.; Hirsch, M.; et al. EGFR oligomerization organizes kinase-active dimers into competent signalling platforms. Nat. Commun. 2016, 7, 13307. [Google Scholar] [CrossRef]
- Haselwandter, C.A.; MacKinnon, R. Piezo’s membrane footprint and its contribution to mechanosensitivity. eLife 2018, 7, e41968. [Google Scholar] [CrossRef]
- Lundbaek, J.A.; Birn, P.; Hansen, A.J.; Sogaard, R.; Nielsen, C.; Girshman, J.; Bruno, M.J.; Tape, S.E.; Egebjerg, J.; Greathouse, D.V.; et al. Regulation of sodium channel function by bilayer elasticity: The importance of hydrophobic coupling. Effects of Micelle-forming amphiphiles and cholesterol. J. Gen. Physiol. 2004, 123, 599–621. [Google Scholar] [CrossRef]
- Eckford, P.D.; Sharom, F.J. Interaction of the P-glycoprotein multidrug efflux pump with cholesterol: Effects on ATPase activity, drug binding and transport. Biochemistry 2008, 47, 13686–13698. [Google Scholar] [CrossRef]
- Gimpl, G.; Burger, K.; Fahrenholz, F. Cholesterol as modulator of receptor function. Biochemistry 1997, 36, 10959–10974. [Google Scholar] [CrossRef]
- Niu, S.L.; Mitchell, D.C.; Litman, B.J. Manipulation of cholesterol levels in rod disk membranes by methyl-beta-cyclodextrin: Effects on receptor activation. J. Biol. Chem. 2002, 277, 20139–20145. [Google Scholar] [CrossRef] [PubMed]
- Cox, C.D.; Zhang, Y.; Zhou, Z.; Walz, T.; Martinac, B. Cyclodextrins increase membrane tension and are universal activators of mechanosensitive channels. Proc. Natl. Acad. Sci. USA 2021, 118, e2104820118. [Google Scholar] [CrossRef]
- Richens, J.L.; Lane, J.S.; Bramble, J.P.; O’Shea, P. The electrical interplay between proteins and lipids in membranes. Biochim. Biophys. Acta 2015, 1848, 1828–1836. [Google Scholar] [CrossRef]
- Ostroumova, O.S.; Efimova, S.S.; Schagina, L.V. Probing amphotericin B single channel activity by membrane dipole modifiers. PLoS ONE 2012, 7, e30261. [Google Scholar] [CrossRef]
- Pearlstein, R.A.; Dickson, C.J.; Hornak, V. Contributions of the membrane dipole potential to the function of voltage-gated cation channels and modulation by small molecule potentiators. Biochim. Biophys. Acta Biomembr. 2017, 1859, 177–194. [Google Scholar] [CrossRef]
- Clarke, R.J. Dipole-Potential-Mediated Effects on Ion Pump Kinetics. Biophys. J. 2015, 109, 1513–1520. [Google Scholar] [CrossRef]
- Kovacs, T.; Batta, G.; Hajdu, T.; Szabo, A.; Varadi, T.; Zakany, F.; Csomos, I.; Szollosi, J.; Nagy, P. The Dipole Potential Modifies the Clustering and Ligand Binding Affinity of ErbB Proteins and Their Signaling Efficiency. Sci. Rep. 2016, 6, 35850. [Google Scholar] [CrossRef]
- Batta, G.; Karpati, L.; Henrique, G.F.; Toth, G.; Tarapcsak, S.; Kovacs, T.; Zakany, F.; Mandity, I.M.; Nagy, P. Statin-boosted cellular uptake and endosomal escape of penetratin due to reduced membrane dipole potential. Br. J. Pharmacol. 2021, 178, 3667–3681. [Google Scholar] [CrossRef] [PubMed]
- Batta, G.; Soltesz, L.; Kovacs, T.; Bozo, T.; Meszar, Z.; Kellermayer, M.; Szollosi, J.; Nagy, P. Alterations in the properties of the cell membrane due to glycosphingolipid accumulation in a model of Gaucher disease. Sci. Rep. 2018, 8, 157. [Google Scholar] [CrossRef]
- Kovacs, T.; Batta, G.; Zakany, F.; Szollosi, J.; Nagy, P. The dipole potential correlates with lipid raft markers in the plasma membrane of living cells. J. Lipid Res. 2017, 58, 1681–1691. [Google Scholar] [CrossRef] [PubMed]
- Hajdu, P.; Varga, Z.; Pieri, C.; Panyi, G.; Gaspar, R., Jr. Cholesterol modifies the gating of Kv1.3 in human T lymphocytes. Pflug. Arch. 2003, 445, 674–682. [Google Scholar] [CrossRef]
- Wong, W.; Schlichter, L.C. Differential recruitment of Kv1.4 and Kv4.2 to lipid rafts by PSD-95. J. Biol. Chem. 2004, 279, 444–452. [Google Scholar] [CrossRef]
- Martens, J.R.; Sakamoto, N.; Sullivan, S.A.; Grobaski, T.D.; Tamkun, M.M. Isoform-specific localization of voltage-gated K+ channels to distinct lipid raft populations. Targeting of Kv1.5 to caveolae. J. Biol. Chem. 2001, 276, 8409–8414. [Google Scholar] [CrossRef]
- Martens, J.R.; Navarro-Polanco, R.; Coppock, E.A.; Nishiyama, A.; Parshley, L.; Grobaski, T.D.; Tamkun, M.M. Differential targeting of Shaker-like potassium channels to lipid rafts. J. Biol. Chem. 2000, 275, 7443–7446. [Google Scholar] [CrossRef]
- Rudakova, E.; Wagner, M.; Frank, M.; Volk, T. Localization of Kv4.2 and KChIP2 in lipid rafts and modulation of outward K+ currents by membrane cholesterol content in rat left ventricular myocytes. Pflug. Arch. 2015, 467, 299–309. [Google Scholar] [CrossRef]
- Jimenez-Garduno, A.M.; Mitkovski, M.; Alexopoulos, I.K.; Sanchez, A.; Stuhmer, W.; Pardo, L.A.; Ortega, A. KV10.1 K+-channel plasma membrane discrete domain partitioning and its functional correlation in neurons. Biochim. Biophys. Acta 2014, 1838, 921–931. [Google Scholar] [CrossRef]
- Balijepalli, R.C.; Delisle, B.P.; Balijepalli, S.Y.; Foell, J.D.; Slind, J.K.; Kamp, T.J.; January, C.T. Kv11.1 (ERG1) K+ channels localize in cholesterol and sphingolipid enriched membranes and are modulated by membrane cholesterol. Channels 2007, 1, 263–272. [Google Scholar] [CrossRef]
- O’Connell, K.M.; Loftus, R.; Tamkun, M.M. Localization-dependent activity of the Kv2.1 delayed-rectifier K+ channel. Proc. Natl. Acad. Sci. USA 2010, 107, 12351–12356. [Google Scholar] [CrossRef]
- Babiychuk, E.B.; Smith, R.D.; Burdyga, T.; Babiychuk, V.S.; Wray, S.; Draeger, A. Membrane cholesterol regulates smooth muscle phasic contraction. J. Membr. Biol. 2004, 198, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Lam, R.S.; Shaw, A.R.; Duszyk, M. Membrane cholesterol content modulates activation of BK channels in colonic epithelia. Biochim. Biophys. Acta 2004, 1667, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kim, Y.J.; Choi, L.M.; Lee, K.; Park, H.K.; Choi, S.Y. Muscarinic Receptors and BK Channels Are Affected by Lipid Raft Disruption of Salivary Gland Cells. Int. J. Mol. Sci. 2021, 22, 4780. [Google Scholar] [CrossRef] [PubMed]
- Tajima, N.; Itokazu, Y.; Korpi, E.R.; Somerharju, P.; Kakela, R. Activity of BK(Ca) channel is modulated by membrane cholesterol content and association with Na+/K+-ATPase in human melanoma IGR39 cells. J. Biol. Chem. 2011, 286, 5624–5638. [Google Scholar] [CrossRef] [PubMed]
- Saeki, T.; Suzuki, Y.; Yamamura, H.; Takeshima, H.; Imaizumi, Y. A junctophilin-caveolin interaction enables efficient coupling between ryanodine receptors and BKCa channels in the Ca2+ microdomain of vascular smooth muscle. J. Biol. Chem. 2019, 294, 13093–13105. [Google Scholar] [CrossRef]
- Pristera, A.; Baker, M.D.; Okuse, K. Association between tetrodotoxin resistant channels and lipid rafts regulates sensory neuron excitability. PLoS ONE 2012, 7, e40079. [Google Scholar] [CrossRef]
- Wang, H.; Haas, M.; Liang, M.; Cai, T.; Tian, J.; Li, S.; Xie, Z. Ouabain assembles signaling cascades through the caveolar Na+/K+-ATPase. J. Biol. Chem. 2004, 279, 17250–17259. [Google Scholar] [CrossRef]
- Bergdahl, A.; Gomez, M.F.; Dreja, K.; Xu, S.Z.; Adner, M.; Beech, D.J.; Broman, J.; Hellstrand, P.; Sward, K. Cholesterol depletion impairs vascular reactivity to endothelin-1 by reducing store-operated Ca2+ entry dependent on TRPC1. Circ. Res. 2003, 93, 839–847. [Google Scholar] [CrossRef]
- Startek, J.B.; Talavera, K. Lipid Raft Destabilization Impairs Mouse TRPA1 Responses to Cold and Bacterial Lipopolysaccharides. Int. J. Mol. Sci. 2020, 21, 3826. [Google Scholar] [CrossRef]
- Qi, Y.; Andolfi, L.; Frattini, F.; Mayer, F.; Lazzarino, M.; Hu, J. Membrane stiffening by STOML3 facilitates mechanosensation in sensory neurons. Nat. Commun. 2015, 6, 8512. [Google Scholar] [CrossRef]
- Ridone, P.; Pandzic, E.; Vassalli, M.; Cox, C.D.; Macmillan, A.; Gottlieb, P.A.; Martinac, B. Disruption of membrane cholesterol organization impairs the activity of PIEZO1 channel clusters. J. Gen. Physiol. 2020, 152, e201912515. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, T.; Zakany, F.; Nagy, P. It Takes More than Two to Tango: Complex, Hierarchal, and Membrane-Modulated Interactions in the Regulation of Receptor Tyrosine Kinases. Cancers 2022, 14, 944. [Google Scholar] [CrossRef] [PubMed]
- Pike, L.J.; Casey, L. Cholesterol levels modulate EGF receptor-mediated signaling by altering receptor function and trafficking. Biochemistry 2002, 41, 10315–10322. [Google Scholar] [CrossRef]
- Ringerike, T.; Blystad, F.D.; Levy, F.O.; Madshus, I.H.; Stang, E. Cholesterol is important in control of EGF receptor kinase activity but EGF receptors are not concentrated in caveolae. J. Cell Sci. 2002, 115, 1331–1340. [Google Scholar] [CrossRef]
- Saffarian, S.; Li, Y.; Elson, E.L.; Pike, L.J. Oligomerization of the EGF receptor investigated by live cell fluorescence intensity distribution analysis. Biophys. J. 2007, 93, 1021–1031. [Google Scholar] [CrossRef]
- Yavas, S.; Machan, R.; Wohland, T. The Epidermal Growth Factor Receptor Forms Location-Dependent Complexes in Resting Cells. Biophys. J. 2016, 111, 2241–2254. [Google Scholar] [CrossRef]
- Westover, E.J.; Covey, D.F.; Brockman, H.L.; Brown, R.E.; Pike, L.J. Cholesterol depletion results in site-specific increases in epidermal growth factor receptor phosphorylation due to membrane level effects. Studies with cholesterol enantiomers. J. Biol. Chem. 2003, 278, 51125–51133. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, L.; Du, J.; Li, Y.; Yang, H.; Li, C.; Li, H.; Hu, H. Lipid raft localization of epidermal growth factor receptor alters matrix metalloproteinase-1 expression in SiHa cells via the MAPK/ERK signaling pathway. Oncol. Lett. 2016, 12, 4991–4998. [Google Scholar] [CrossRef]
- Manes, S.; del Real, G.; Lacalle, R.A.; Lucas, P.; Gomez-Mouton, C.; Sanchez-Palomino, S.; Delgado, R.; Alcami, J.; Mira, E.; Martinez, A.C. Membrane raft microdomains mediate lateral assemblies required for HIV-1 infection. EMBO Rep. 2000, 1, 190–196. [Google Scholar] [CrossRef]
- Eroglu, C.; Brugger, B.; Wieland, F.; Sinning, I. Glutamate-binding affinity of Drosophila metabotropic glutamate receptor is modulated by association with lipid rafts. Proc. Natl. Acad. Sci. USA 2003, 100, 10219–10224. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Xu, W.; Yoon, S.I.; Chen, C.; Chong, P.L.; Liu-Chen, L.Y. Cholesterol reduction by methyl-beta-cyclodextrin attenuates the delta opioid receptor-mediated signaling in neuronal cells but enhances it in non-neuronal cells. Biochem. Pharmacol. 2007, 73, 534–549. [Google Scholar] [CrossRef] [PubMed]
- Kumari, R.; Castillo, C.; Francesconi, A. Agonist-dependent signaling by group I metabotropic glutamate receptors is regulated by association with lipid domains. J. Biol. Chem. 2013, 288, 32004–32019. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Chu, J.; Qiu, Y.; Loh, H.H.; Law, P.Y. Agonist-selective signaling is determined by the receptor location within the membrane domains. Proc. Natl. Acad. Sci. USA 2008, 105, 9421–9426. [Google Scholar] [CrossRef] [PubMed]
- Ohtani, Y.; Irie, T.; Uekama, K.; Fukunaga, K.; Pitha, J. Differential effects of alpha-, beta- and gamma-cyclodextrins on human erythrocytes. Eur. J. Biochem. 1989, 186, 17–22. [Google Scholar] [CrossRef]
- Horský, J.; Pitha, J. Inclusion complexes of proteins: Interaction of cyclodextrins with peptides containing aromatic amino acids studied by competitive spectrophotometry. J. Incl. Phenom. Mol. Recognit. Chem. 1994, 18, 291–300. [Google Scholar] [CrossRef]
- Ramanathan, R.; Prokai, L. Electrospray ionization mass spectrometric study of encapsulation of amino acids by cyclodextrins. J. Am. Soc. Mass Spectrom. 1995, 6, 866–871. [Google Scholar] [CrossRef]
- Kahle, C.; Holzgrabe, U. Determination of binding constants of cyclodextrin inclusion complexes with amino acids and dipeptides by potentiometric titration. Chirality 2004, 16, 509–515. [Google Scholar] [CrossRef]
- Caso, J.V.; Russo, L.; Palmieri, M.; Malgieri, G.; Galdiero, S.; Falanga, A.; Isernia, C.; Iacovino, R. Investigating the inclusion properties of aromatic amino acids complexing beta-cyclodextrins in model peptides. Amino Acids 2015, 47, 2215–2227. [Google Scholar] [CrossRef]
- Arsiccio, A.; Rospiccio, M.; Shea, J.E.; Pisano, R. Force Field Parameterization for the Description of the Interactions between Hydroxypropyl-beta-Cyclodextrin and Proteins. J. Phys. Chem. B 2021, 125, 7397–7405. [Google Scholar] [CrossRef]
- Koushik, K.N.; Bandi, N.; Kompella, U.B. Interaction of [D-Trp6, Des-Gly10] LHRH ethylamide and hydroxy propyl beta-cyclodextrin (HPbetaCD): Thermodynamics of interaction and protection from degradation by alpha-chymotrypsin. Pharm. Dev. Technol. 2001, 6, 595–606. [Google Scholar] [CrossRef] [PubMed]
- Khajehpour, M.; Troxler, T.; Nanda, V.; Vanderkooi, J.M. Melittin as model system for probing interactions between proteins and cyclodextrins. Proteins 2004, 55, 275–287. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Geib, T.; Volmer, D.A. Determining the Binding Sites of beta-Cyclodextrin and Peptides by Electron-Capture Dissociation High Resolution Tandem Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2015, 26, 1143–1149. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Schmidt, A.K.; Cottaz, S.; Driguez, H.; Schulz, G.E. Structure of cyclodextrin glycosyltransferase complexed with a derivative of its main product beta-cyclodextrin. Biochemistry 1998, 37, 5909–5915. [Google Scholar] [CrossRef] [PubMed]
- Yokota, T.; Tonozuka, T.; Shimura, Y.; Ichikawa, K.; Kamitori, S.; Sakano, Y. Structures of Thermoactinomyces vulgaris R-47 alpha-amylase II complexed with substrate analogues. Biosci. Biotechnol. Biochem. 2001, 65, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S.; Kim, M.S.; Cho, H.S.; Kim, J.I.; Kim, T.J.; Choi, J.H.; Park, C.; Lee, H.S.; Oh, B.H.; Park, K.H. Cyclomaltodextrinase, neopullulanase, and maltogenic amylase are nearly indistinguishable from each other. J. Biol. Chem. 2002, 277, 21891–21897. [Google Scholar] [CrossRef] [PubMed]
- Adachi, M.; Mikami, B.; Katsube, T.; Utsumi, S. Crystal structure of recombinant soybean beta-amylase complexed with beta-cyclodextrin. J. Biol. Chem. 1998, 273, 19859–19865. [Google Scholar] [CrossRef]
- Sharff, A.J.; Rodseth, L.E.; Quiocho, F.A. Refined 1.8-A structure reveals the mode of binding of beta-cyclodextrin to the maltodextrin binding protein. Biochemistry 1993, 32, 10553–10559. [Google Scholar] [CrossRef]
- Matsumoto, N.; Yamada, M.; Kurakata, Y.; Yoshida, H.; Kamitori, S.; Nishikawa, A.; Tonozuka, T. Crystal structures of open and closed forms of cyclo/maltodextrin-binding protein. FEBS J. 2009, 276, 3008–3019. [Google Scholar] [CrossRef]
- Sorimachi, K.; Le Gal-Coeffet, M.F.; Williamson, G.; Archer, D.B.; Williamson, M.P. Solution structure of the granular starch binding domain of Aspergillus niger glucoamylase bound to beta-cyclodextrin. Structure 1997, 5, 647–661. [Google Scholar] [CrossRef]
- Vester-Christensen, M.B.; Abou Hachem, M.; Svensson, B.; Henriksen, A. Crystal structure of an essential enzyme in seed starch degradation: Barley limit dextrinase in complex with cyclodextrins. J. Mol. Biol. 2010, 403, 739–750. [Google Scholar] [CrossRef]
- Li, X.; Bai, Y.; Ji, H.; Jin, Z. The binding mechanism between cyclodextrins and pullulanase: A molecular docking, isothermal titration calorimetry, circular dichroism and fluorescence study. Food Chem. 2020, 321, 126750. [Google Scholar] [CrossRef] [PubMed]
- Saka, N.; Iwamoto, H.; Malle, D.; Takahashi, N.; Mizutani, K.; Mikami, B. Elucidation of the mechanism of interaction between Klebsiella pneumoniae pullulanase and cyclodextrin. Acta Crystallogr. D Struct. Biol. 2018, 74, 1115–1123. [Google Scholar] [CrossRef] [PubMed]
- Aachmann, F.L.; Otzen, D.E.; Larsen, K.L.; Wimmer, R. Structural background of cyclodextrin-protein interactions. Protein Eng. 2003, 16, 905–912. [Google Scholar] [CrossRef]
- Tavornvipas, S.; Hirayama, F.; Takeda, S.; Arima, H.; Uekama, K. Effects of cyclodextrins on chemically and thermally induced unfolding and aggregation of lysozyme and basic fibroblast growth factor. J. Pharm. Sci. 2006, 95, 2722–2729. [Google Scholar] [CrossRef] [PubMed]
- Rospiccio, M.; Arsiccio, A.; Winter, G.; Pisano, R. The Role of Cyclodextrins against Interface-Induced Denaturation in Pharmaceutical Formulations: A Molecular Dynamics Approach. Mol. Pharm. 2021, 18, 2322–2333. [Google Scholar] [CrossRef]
- Otzen, D.E.; Knudsen, B.R.; Aachmann, F.; Larsen, K.L.; Wimmer, R. Structural basis for cyclodextrins’ suppression of human growth hormone aggregation. Protein Sci. 2002, 11, 1779–1787. [Google Scholar] [CrossRef]
- Dotsikas, Y.; Loukas, Y.L. Kinetic degradation study of insulin complexed with methyl-beta cyclodextrin. Confirmation of complexation with electrospray mass spectrometry and (1)H NMR. J. Pharm. Biomed. Anal. 2002, 29, 487–494. [Google Scholar] [CrossRef]
- Zhang, L.; Zhu, W.; Song, L.; Wang, Y.; Jiang, H.; Xian, S.; Ren, Y. Effects of hydroxylpropyl-beta-cyclodextrin on in vitro insulin stability. Int. J. Mol. Sci. 2009, 10, 2031–2040. [Google Scholar] [CrossRef] [PubMed]
- Bucur, P.; Fulop, I.; Sipos, E. Insulin Complexation with Cyclodextrins-A Molecular Modeling Approach. Molecules 2022, 27, 465. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, D.; Jucker, M. The amyloid state of proteins in human diseases. Cell 2012, 148, 1188–1203. [Google Scholar] [CrossRef]
- Camilleri, P.; Haskins, N.J.; Howlett, D.R. beta-Cyclodextrin interacts with the Alzheimer amyloid beta-A4 peptide. FEBS Lett. 1994, 341, 256–258. [Google Scholar] [CrossRef]
- Qin, X.R.; Abe, H.; Nakanishi, H. NMR and CD studies on the interaction of Alzheimer beta-amyloid peptide (12–28) with beta-cyclodextrin. Biochem. Biophys. Res. Commun. 2002, 297, 1011–1015. [Google Scholar] [CrossRef]
- Danielsson, J.; Jarvet, J.; Damberg, P.; Graslund, A. Two-site binding of beta-cyclodextrin to the Alzheimer Abeta(1-40) peptide measured with combined PFG-NMR diffusion and induced chemical shifts. Biochemistry 2004, 43, 6261–6269. [Google Scholar] [CrossRef] [PubMed]
- Wahlstrom, A.; Cukalevski, R.; Danielsson, J.; Jarvet, J.; Onagi, H.; Rebek, J., Jr.; Linse, S.; Graslund, A. Specific binding of a beta-cyclodextrin dimer to the amyloid beta peptide modulates the peptide aggregation process. Biochemistry 2012, 51, 4280–4289. [Google Scholar] [CrossRef] [PubMed]
- Ren, B.; Jiang, B.; Hu, R.; Zhang, M.; Chen, H.; Ma, J.; Sun, Y.; Jia, L.; Zheng, J. HP-beta-cyclodextrin as an inhibitor of amyloid-beta aggregation and toxicity. Phys. Chem. Chem. Phys. 2016, 18, 20476–20485. [Google Scholar] [CrossRef] [PubMed]
- Prior, M.; Lehmann, S.; Sy, M.S.; Molloy, B.; McMahon, H.E. Cyclodextrins inhibit replication of scrapie prion protein in cell culture. J. Virol. 2007, 81, 11195–11207. [Google Scholar] [CrossRef] [PubMed]
- Gautam, S.; Karmakar, S.; Bose, A.; Chowdhury, P.K. beta-cyclodextrin and curcumin, a potent cocktail for disaggregating and/or inhibiting amyloids: A case study with alpha-synuclein. Biochemistry 2014, 53, 4081–4083. [Google Scholar] [CrossRef]
- Gautam, S.; Karmakar, S.; Batra, R.; Sharma, P.; Pradhan, P.; Singh, J.; Kundu, B.; Chowdhury, P.K. Polyphenols in combination with beta-cyclodextrin can inhibit and disaggregate alpha-synuclein amyloids under cell mimicking conditions: A promising therapeutic alternative. Biochim. Biophys. Acta Proteins Proteom. 2017, 1865, 589–603. [Google Scholar] [CrossRef]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, L.; Zhou, X.E.; Ke, J.; de Waal, P.W.; Gu, X.; Tan, M.H.; Wang, D.; Wu, D.; Xu, H.E.; et al. Structural basis of AMPK regulation by adenine nucleotides and glycogen. Cell Res. 2015, 25, 50–66. [Google Scholar] [CrossRef] [PubMed]
- Polekhina, G.; Gupta, A.; van Denderen, B.J.; Feil, S.C.; Kemp, B.E.; Stapleton, D.; Parker, M.W. Structural basis for glycogen recognition by AMP-activated protein kinase. Structure 2005, 13, 1453–1462. [Google Scholar] [CrossRef] [PubMed]
- McBride, A.; Ghilagaber, S.; Nikolaev, A.; Hardie, D.G. The glycogen-binding domain on the AMPK beta subunit allows the kinase to act as a glycogen sensor. Cell Metab. 2009, 9, 23–34. [Google Scholar] [CrossRef]
- Dai, S.; Dulcey, A.E.; Hu, X.; Wassif, C.A.; Porter, F.D.; Austin, C.P.; Ory, D.S.; Marugan, J.; Zheng, W. Methyl-beta-cyclodextrin restores impaired autophagy flux in Niemann-Pick C1-deficient cells through activation of AMPK. Autophagy 2017, 13, 1435–1451. [Google Scholar] [CrossRef]
- Gu, L.Q.; Braha, O.; Conlan, S.; Cheley, S.; Bayley, H. Stochastic sensing of organic analytes by a pore-forming protein containing a molecular adapter. Nature 1999, 398, 686–690. [Google Scholar] [CrossRef]
- Gu, L.Q.; Cheley, S.; Bayley, H. Prolonged residence time of a noncovalent molecular adapter, beta-cyclodextrin, within the lumen of mutant alpha-hemolysin pores. J. Gen. Physiol. 2001, 118, 481–494. [Google Scholar] [CrossRef]
- Banerjee, A.; Mikhailova, E.; Cheley, S.; Gu, L.Q.; Montoya, M.; Nagaoka, Y.; Gouaux, E.; Bayley, H. Molecular bases of cyclodextrin adapter interactions with engineered protein nanopores. Proc. Natl. Acad. Sci. USA 2010, 107, 8165–8170. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.Q.; Dalla Serra, M.; Vincent, J.B.; Vigh, G.; Cheley, S.; Braha, O.; Bayley, H. Reversal of charge selectivity in transmembrane protein pores by using noncovalent molecular adapters. Proc. Natl. Acad. Sci. USA 2000, 97, 3959–3964. [Google Scholar] [CrossRef] [PubMed]
- Egwolf, B.; Luo, Y.; Walters, D.E.; Roux, B. Ion selectivity of alpha-hemolysin with beta-cyclodextrin adapter. II. Multi-ion effects studied with grand canonical Monte Carlo/Brownian dynamics simulations. J. Phys. Chem. B 2010, 114, 2901–2909. [Google Scholar] [CrossRef]
- Orlik, F.; Andersen, C.; Danelon, C.; Winterhalter, M.; Pajatsch, M.; Bock, A.; Benz, R. CymA of Klebsiella oxytoca outer membrane: Binding of cyclodextrins and study of the current noise of the open channel. Biophys. J. 2003, 85, 876–885. [Google Scholar] [CrossRef]
- Locke, D.; Koreen, I.V.; Liu, J.Y.; Harris, A.L. Reversible pore block of connexin channels by cyclodextrins. J. Biol. Chem. 2004, 279, 22883–22892. [Google Scholar] [CrossRef]
- Pytel, M.; Mercik, K.; Mozrzymas, J.W. Interaction between cyclodextrin and neuronal membrane results in modulation of GABA(A) receptor conformational transitions. Br. J. Pharmacol. 2006, 148, 413–422. [Google Scholar] [CrossRef]
- Zuniga, R.; Mancilla, D.; Rojas, T.; Vergara, F.; Gonzalez, W.; Catalan, M.A.; Zuniga, L. A Direct Interaction between Cyclodextrins and TASK Channels Decreases the Leak Current in Cerebellar Granule Neurons. Biology 2022, 11, 1097. [Google Scholar] [CrossRef] [PubMed]
- Rodstrom, K.E.J.; Kiper, A.K.; Zhang, W.; Rinne, S.; Pike, A.C.W.; Goldstein, M.; Conrad, L.J.; Delbeck, M.; Hahn, M.G.; Meier, H.; et al. A lower X-gate in TASK channels traps inhibitors within the vestibule. Nature 2020, 582, 443–447. [Google Scholar] [CrossRef]
- Feske, S.; Wulff, H.; Skolnik, E.Y. Ion channels in innate and adaptive immunity. Annu. Rev. Immunol. 2015, 33, 291–353. [Google Scholar] [CrossRef]
- Kavanaugh, M.P.; Varnum, M.D.; Osborne, P.B.; Christie, M.J.; Busch, A.E.; Adelman, J.P.; North, R.A. Interaction between tetraethylammonium and amino acid residues in the pore of cloned voltage-dependent potassium channels. J. Biol. Chem. 1991, 266, 7583–7587. [Google Scholar] [CrossRef]
- Gilquin, B.; Braud, S.; Eriksson, M.A.; Roux, B.; Bailey, T.D.; Priest, B.T.; Garcia, M.L.; Menez, A.; Gasparini, S. A variable residue in the pore of Kv1 channels is critical for the high affinity of blockers from sea anemones and scorpions. J. Biol. Chem. 2005, 280, 27093–27102. [Google Scholar] [CrossRef] [PubMed]
- Irie, T.; Uekama, K. Cyclodextrins in peptide and protein delivery. Adv. Drug Deliv. Rev. 1999, 36, 101–123. [Google Scholar] [CrossRef]
- Lovatt, M.; Cooper, A.; Camilleri, P. Energetics of cyclodextrin-induced dissociation of insulin. Eur. Biophys. J. 1996, 24, 354–357. [Google Scholar] [CrossRef] [PubMed]
- Tavornvipas, S.; Tajiri, S.; Hirayama, F.; Arima, H.; Uekama, K. Effects of hydrophilic cyclodextrins on aggregation of recombinant human growth hormone. Pharm. Res. 2004, 21, 2369–2376. [Google Scholar] [CrossRef]
- Karran, E.; Mercken, M.; De Strooper, B. The amyloid cascade hypothesis for Alzheimer’s disease: An appraisal for the development of therapeutics. Nat. Rev. Drug Discov. 2011, 10, 698–712. [Google Scholar] [CrossRef]
- Prince, M.; Bryce, R.; Albanese, E.; Wimo, A.; Ribeiro, W.; Ferri, C.P. The global prevalence of dementia: A systematic review and metaanalysis. Alzheimer’s Dement. 2013, 9, 63–75.e62. [Google Scholar] [CrossRef] [PubMed]
- Zakany, F.; Kovacs, T.; Szente, L.; Varga, Z. Chapter 34—Cyclodextrins as promising therapeutics against cholesterol overload. In Cholesterol; Bukiya, A.N., Dopico, A.M., Eds.; Academic Press: Cambridge, MA, USA, 2022; pp. 927–967. [Google Scholar]
- Haass, C.; Kaether, C.; Thinakaran, G.; Sisodia, S. Trafficking and proteolytic processing of APP. Cold Spring Harb. Perspect. Med. 2012, 2, a006270. [Google Scholar] [CrossRef] [PubMed]
- Long, J.M.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef] [PubMed]
- Cole, S.L.; Grudzien, A.; Manhart, I.O.; Kelly, B.L.; Oakley, H.; Vassar, R. Statins cause intracellular accumulation of amyloid precursor protein, beta-secretase-cleaved fragments, and amyloid beta-peptide via an isoprenoid-dependent mechanism. J. Biol. Chem. 2005, 280, 18755–18770. [Google Scholar] [CrossRef]
- Czech, C.; Burns, M.P.; Vardanian, L.; Augustin, A.; Jacobsen, H.; Baumann, K.; Rebeck, G.W. Cholesterol independent effect of LXR agonist TO-901317 on gamma-secretase. J. Neurochem. 2007, 101, 929–936. [Google Scholar] [CrossRef]
- Ehehalt, R.; Keller, P.; Haass, C.; Thiele, C.; Simons, K. Amyloidogenic processing of the Alzheimer beta-amyloid precursor protein depends on lipid rafts. J. Cell Biol. 2003, 160, 113–123. [Google Scholar] [CrossRef]
- Guardia-Laguarta, C.; Coma, M.; Pera, M.; Clarimon, J.; Sereno, L.; Agullo, J.M.; Molina-Porcel, L.; Gallardo, E.; Deng, A.; Berezovska, O.; et al. Mild cholesterol depletion reduces amyloid-beta production by impairing APP trafficking to the cell surface. J. Neurochem. 2009, 110, 220–230. [Google Scholar] [CrossRef]
- Kojro, E.; Gimpl, G.; Lammich, S.; Marz, W.; Fahrenholz, F. Low cholesterol stimulates the nonamyloidogenic pathway by its effect on the alpha -secretase ADAM 10. Proc. Natl. Acad. Sci. USA 2001, 98, 5815–5820. [Google Scholar] [CrossRef]
- von Arnim, C.A.; von Einem, B.; Weber, P.; Wagner, M.; Schwanzar, D.; Spoelgen, R.; Strauss, W.L.; Schneckenburger, H. Impact of cholesterol level upon APP and BACE proximity and APP cleavage. Biochem. Biophys. Res. Commun. 2008, 370, 207–212. [Google Scholar] [CrossRef]
- Xiong, H.; Callaghan, D.; Jones, A.; Walker, D.G.; Lue, L.F.; Beach, T.G.; Sue, L.I.; Woulfe, J.; Xu, H.; Stanimirovic, D.B.; et al. Cholesterol retention in Alzheimer’s brain is responsible for high beta- and gamma-secretase activities and Abeta production. Neurobiol. Dis. 2008, 29, 422–437. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.Y.; Kwon, O.H.; Park, M.K.; Kim, T.W.; Chung, S. Elevated cellular cholesterol in Familial Alzheimer’s presenilin 1 mutation is associated with lipid raft localization of beta-amyloid precursor protein. PLoS ONE 2019, 14, e0210535. [Google Scholar] [CrossRef]
- Cho, Y.Y.; Kwon, O.H.; Chung, S. Preferred Endocytosis of Amyloid Precursor Protein from Cholesterol-Enriched Lipid Raft Microdomains. Molecules 2020, 25, 5490. [Google Scholar] [CrossRef] [PubMed]
- Marquer, C.; Devauges, V.; Cossec, J.C.; Liot, G.; Lecart, S.; Saudou, F.; Duyckaerts, C.; Leveque-Fort, S.; Potier, M.C. Local cholesterol increase triggers amyloid precursor protein-Bace1 clustering in lipid rafts and rapid endocytosis. FASEB J. 2011, 25, 1295–1305. [Google Scholar] [CrossRef] [PubMed]
- Barbero-Camps, E.; Roca-Agujetas, V.; Bartolessis, I.; de Dios, C.; Fernandez-Checa, J.C.; Mari, M.; Morales, A.; Hartmann, T.; Colell, A. Cholesterol impairs autophagy-mediated clearance of amyloid beta while promoting its secretion. Autophagy 2018, 14, 1129–1154. [Google Scholar] [CrossRef]
- Yalcin, A.; Soddu, E.; Turunc Bayrakdar, E.; Uyanikgil, Y.; Kanit, L.; Armagan, G.; Rassu, G.; Gavini, E.; Giunchedi, P. Neuroprotective Effects of Engineered Polymeric Nasal Microspheres Containing Hydroxypropyl-beta-cyclodextrin on beta-Amyloid (1–42)-Induced Toxicity. J. Pharm. Sci. 2016, 105, 2372–2380. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.S.; Stavrides, P.; Kumar, A.; Jiang, Y.; Mohan, P.S.; Ohno, M.; Dobrenis, K.; Davidson, C.D.; Saito, M.; Pawlik, M.; et al. Cyclodextrin has conflicting actions on autophagy flux in vivo in brains of normal and Alzheimer model mice. Hum. Mol. Genet. 2017, 26, 843–859. [Google Scholar] [CrossRef]
- Yao, J.; Ho, D.; Calingasan, N.Y.; Pipalia, N.H.; Lin, M.T.; Beal, M.F. Neuroprotection by cyclodextrin in cell and mouse models of Alzheimer disease. J. Exp. Med. 2012, 209, 2501–2513. [Google Scholar] [CrossRef]
- Bendor, J.T.; Logan, T.P.; Edwards, R.H. The function of alpha-synuclein. Neuron 2013, 79, 1044–1066. [Google Scholar] [CrossRef]
- Du, X.Y.; Xie, X.X.; Liu, R.T. The Role of α-Synuclein Oligomers in Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 8645. [Google Scholar] [CrossRef]
- Goedert, M. Alpha-synuclein and neurodegenerative diseases. Nat. Rev. Neurosci. 2001, 2, 492–501. [Google Scholar] [CrossRef]
- Bar-On, P.; Rockenstein, E.; Adame, A.; Ho, G.; Hashimoto, M.; Masliah, E. Effects of the cholesterol-lowering compound methyl-beta-cyclodextrin in models of alpha-synucleinopathy. J. Neurochem. 2006, 98, 1032–1045. [Google Scholar] [CrossRef] [PubMed]
- Kilpatrick, K.; Zeng, Y.; Hancock, T.; Segatori, L. Genetic and chemical activation of TFEB mediates clearance of aggregated alpha-synuclein. PLoS ONE 2015, 10, e0120819. [Google Scholar] [CrossRef]
- Atger, V.M.; de la Llera Moya, M.; Stoudt, G.W.; Rodrigueza, W.V.; Phillips, M.C.; Rothblat, G.H. Cyclodextrins as catalysts for the removal of cholesterol from macrophage foam cells. J. Clin. Investig. 1997, 99, 773–780. [Google Scholar] [CrossRef]
- Coisne, C.; Hallier-Vanuxeem, D.; Boucau, M.C.; Hachani, J.; Tilloy, S.; Bricout, H.; Monflier, E.; Wils, D.; Serpelloni, M.; Parissaux, X.; et al. beta-Cyclodextrins Decrease Cholesterol Release and ABC-Associated Transporter Expression in Smooth Muscle Cells and Aortic Endothelial Cells. Front. Physiol. 2016, 7, 185. [Google Scholar] [CrossRef]
- Liu, S.M.; Cogny, A.; Kockx, M.; Dean, R.T.; Gaus, K.; Jessup, W.; Kritharides, L. Cyclodextrins differentially mobilize free and esterified cholesterol from primary human foam cell macrophages. J. Lipid Res. 2003, 44, 1156–1166. [Google Scholar] [CrossRef]
- Kritharides, L.; Kus, M.; Brown, A.J.; Jessup, W.; Dean, R.T. Hydroxypropyl-beta-cyclodextrin-mediated efflux of 7-ketocholesterol from macrophage foam cells. J. Biol. Chem. 1996, 271, 27450–27455. [Google Scholar] [CrossRef] [PubMed]
- Ao, M.; Wu, L.; Zhou, X.; Chen, Y. Methyl-beta-Cyclodextrin Impairs the Monocyte-Adhering Ability of Endothelial Cells by Down-Regulating Adhesion Molecules and Caveolae and Reorganizing the Actin Cytoskeleton. Biol. Pharm. Bull. 2016, 39, 1029–1034. [Google Scholar] [CrossRef] [PubMed]
- Montecucco, F.; Lenglet, S.; Carbone, F.; Boero, S.; Pelli, G.; Burger, F.; Roth, A.; Bertolotto, M.; Nencioni, A.; Cea, M.; et al. Treatment with KLEPTOSE(R) CRYSMEB reduces mouse atherogenesis by impacting on lipid profile and Th1 lymphocyte response. Vascul. Pharmacol. 2015, 72, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Bakke, S.S.; Aune, M.H.; Niyonzima, N.; Pilely, K.; Ryan, L.; Skjelland, M.; Garred, P.; Aukrust, P.; Halvorsen, B.; Latz, E.; et al. Cyclodextrin Reduces Cholesterol Crystal-Induced Inflammation by Modulating Complement Activation. J. Immunol. 2017, 199, 2910–2920. [Google Scholar] [CrossRef]
- He, J.; Yang, Y.; Zhou, X.; Zhang, W.; Liu, J. Shuttle/sink model composed of beta-cyclodextrin and simvastatin-loaded discoidal reconstituted high-density lipoprotein for enhanced cholesterol efflux and drug uptake in macrophage/foam cells. J. Mater. Chem. B 2020, 8, 1496–1506. [Google Scholar] [CrossRef]
- Singh, R.K.; Lund, F.W.; Haka, A.S.; Maxfield, F.R. High-density lipoprotein or cyclodextrin extraction of cholesterol from aggregated LDL reduces foam cell formation. J. Cell Sci. 2019, 132, jcs237271. [Google Scholar] [CrossRef]
- Irie, T.; Fukunaga, K.; Garwood, M.K.; Carpenter, T.O.; Pitha, J.; Pitha, J. Hydroxypropylcyclodextrins in parenteral use. II: Effects on transport and disposition of lipids in rabbit and humans. J. Pharm. Sci. 1992, 81, 524–528. [Google Scholar] [CrossRef]
- Walenbergh, S.M.; Houben, T.; Hendrikx, T.; Jeurissen, M.L.; van Gorp, P.J.; Vaes, N.; Olde Damink, S.W.; Verheyen, F.; Koek, G.H.; Lutjohann, D.; et al. Weekly Treatment of 2-Hydroxypropyl-beta-cyclodextrin Improves Intracellular Cholesterol Levels in LDL Receptor Knockout Mice. Int. J. Mol. Sci. 2015, 16, 21056–21069. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, X.; Yu, B.; Peng, X.; Liu, Y.; Wang, A.; Zhao, D.; Pang, D.; OuYang, H.; Tang, X. Cyclodextrin Ameliorates the Progression of Atherosclerosis via Increasing High-Density Lipoprotein Cholesterol Plasma Levels and Anti-inflammatory Effects in Rabbits. J. Cardiovasc. Pharmacol. 2019, 73, 334–342. [Google Scholar] [CrossRef]
- Zimmer, S.; Grebe, A.; Bakke, S.S.; Bode, N.; Halvorsen, B.; Ulas, T.; Skjelland, M.; De Nardo, D.; Labzin, L.I.; Kerksiek, A.; et al. Cyclodextrin promotes atherosclerosis regression via macrophage reprogramming. Sci. Transl. Med. 2016, 8, 333ra50. [Google Scholar] [CrossRef]
- Guo, J.; Li, D.; Tao, H.; Li, G.; Liu, R.; Dou, Y.; Jin, T.; Li, L.; Huang, J.; Hu, H.; et al. Cyclodextrin-Derived Intrinsically Bioactive Nanoparticles for Treatment of Acute and Chronic Inflammatory Diseases. Adv. Mater. 2019, 31, e1904607. [Google Scholar] [CrossRef]
- Kim, H.; Han, J.; Park, J.H. Cyclodextrin polymer improves atherosclerosis therapy and reduces ototoxicity. J. Control. Release 2020, 319, 77–86. [Google Scholar] [CrossRef]
- Kim, H.; Kumar, S.; Kang, D.W.; Jo, H.; Park, J.H. Affinity-Driven Design of Cargo-Switching Nanoparticles to Leverage a Cholesterol-Rich Microenvironment for Atherosclerosis Therapy. ACS Nano 2020, 14, 6519–6531. [Google Scholar] [CrossRef]
- Hammond, N.; Munkacsi, A.B.; Sturley, S.L. The complexity of a monogenic neurodegenerative disease: More than two decades of therapeutic driven research into Niemann-Pick type C disease. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 1109–1123. [Google Scholar] [CrossRef]
- Vanier, M.T. Niemann-Pick disease type C. Orphanet J. Rare Dis. 2010, 5, 16. [Google Scholar] [CrossRef]
- Walkley, S.U.; Suzuki, K. Consequences of NPC1 and NPC2 loss of function in mammalian neurons. Biochim. Biophys. Acta 2004, 1685, 48–62. [Google Scholar] [CrossRef]
- Sarkar, S.; Carroll, B.; Buganim, Y.; Maetzel, D.; Ng, A.H.; Cassady, J.P.; Cohen, M.A.; Chakraborty, S.; Wang, H.; Spooner, E.; et al. Impaired autophagy in the lipid-storage disorder Niemann-Pick type C1 disease. Cell Rep. 2013, 5, 1302–1315. [Google Scholar] [CrossRef]
- Rosenbaum, A.I.; Zhang, G.; Warren, J.D.; Maxfield, F.R. Endocytosis of beta-cyclodextrins is responsible for cholesterol reduction in Niemann-Pick type C mutant cells. Proc. Natl. Acad. Sci. USA 2010, 107, 5477–5482. [Google Scholar] [CrossRef]
- Peake, K.B.; Vance, J.E. Normalization of cholesterol homeostasis by 2-hydroxypropyl-beta-cyclodextrin in neurons and glia from Niemann-Pick C1 (NPC1)-deficient mice. J. Biol. Chem. 2012, 287, 9290–9298. [Google Scholar] [CrossRef]
- Yu, D.; Swaroop, M.; Wang, M.; Baxa, U.; Yang, R.; Yan, Y.; Coksaygan, T.; DeTolla, L.; Marugan, J.J.; Austin, C.P.; et al. Niemann-Pick Disease Type C: Induced Pluripotent Stem Cell-Derived Neuronal Cells for Modeling Neural Disease and Evaluating Drug Efficacy. J. Biomol. Screen 2014, 19, 1164–1173. [Google Scholar] [CrossRef]
- Maetzel, D.; Sarkar, S.; Wang, H.; Abi-Mosleh, L.; Xu, P.; Cheng, A.W.; Gao, Q.; Mitalipova, M.; Jaenisch, R. Genetic and chemical correction of cholesterol accumulation and impaired autophagy in hepatic and neural cells derived from Niemann-Pick Type C patient-specific iPS cells. Stem Cell Rep. 2014, 2, 866–880. [Google Scholar] [CrossRef]
- Abi-Mosleh, L.; Infante, R.E.; Radhakrishnan, A.; Goldstein, J.L.; Brown, M.S. Cyclodextrin overcomes deficient lysosome-to-endoplasmic reticulum transport of cholesterol in Niemann-Pick type C cells. Proc. Natl. Acad. Sci. USA 2009, 106, 19316–19321. [Google Scholar] [CrossRef]
- Chen, F.W.; Li, C.; Ioannou, Y.A. Cyclodextrin induces calcium-dependent lysosomal exocytosis. PLoS ONE 2010, 5, e15054. [Google Scholar] [CrossRef]
- Vacca, F.; Vossio, S.; Mercier, V.; Moreau, D.; Johnson, S.; Scott, C.C.; Montoya, J.P.; Moniatte, M.; Gruenberg, J. Cyclodextrin triggers MCOLN1-dependent endo-lysosome secretion in Niemann-Pick type C cells. J. Lipid Res. 2019, 60, 832–843. [Google Scholar] [CrossRef]
- Feltes, M.; Gale, S.E.; Moores, S.; Ory, D.S.; Schaffer, J.E. Monitoring the itinerary of lysosomal cholesterol in Niemann-Pick Type C1-deficient cells after cyclodextrin treatment. J. Lipid Res. 2020, 61, 403–412. [Google Scholar] [CrossRef] [PubMed]
- Singhal, A.; Szente, L.; Hildreth, J.E.K.; Song, B. Hydroxypropyl-beta and -gamma cyclodextrins rescue cholesterol accumulation in Niemann-Pick C1 mutant cell via lysosome-associated membrane protein 1. Cell Death Dis. 2018, 9, 1019. [Google Scholar] [CrossRef] [PubMed]
- Hoque, S.; Kondo, Y.; Sakata, N.; Yamada, Y.; Fukaura, M.; Higashi, T.; Motoyama, K.; Arima, H.; Higaki, K.; Hayashi, A.; et al. Differential Effects of 2-Hydroxypropyl-Cyclodextrins on Lipid Accumulation in Npc1-Null Cells. Int. J. Mol. Sci. 2020, 21, 898. [Google Scholar] [CrossRef] [PubMed]
- Soga, M.; Ishitsuka, Y.; Hamasaki, M.; Yoneda, K.; Furuya, H.; Matsuo, M.; Ihn, H.; Fusaki, N.; Nakamura, K.; Nakagata, N.; et al. HPGCD outperforms HPBCD as a potential treatment for Niemann-Pick disease type C during disease modeling with iPS cells. Stem Cells 2015, 33, 1075–1088. [Google Scholar] [CrossRef] [PubMed]
- Singhal, A.; Krystofiak, E.S.; Jerome, W.G.; Song, B. 2-Hydroxypropyl-gamma-cyclodextrin overcomes NPC1 deficiency by enhancing lysosome-ER association and autophagy. Sci. Rep. 2020, 10, 8663. [Google Scholar] [CrossRef]
- Liu, B.; Li, H.; Repa, J.J.; Turley, S.D.; Dietschy, J.M. Genetic variations and treatments that affect the lifespan of the NPC1 mouse. J. Lipid Res. 2008, 49, 663–669. [Google Scholar] [CrossRef]
- Liu, B.; Turley, S.D.; Burns, D.K.; Miller, A.M.; Repa, J.J.; Dietschy, J.M. Reversal of defective lysosomal transport in NPC disease ameliorates liver dysfunction and neurodegeneration in the npc1-/- mouse. Proc. Natl. Acad. Sci. USA 2009, 106, 2377–2382. [Google Scholar] [CrossRef]
- Davidson, C.D.; Ali, N.F.; Micsenyi, M.C.; Stephney, G.; Renault, S.; Dobrenis, K.; Ory, D.S.; Vanier, M.T.; Walkley, S.U. Chronic cyclodextrin treatment of murine Niemann-Pick C disease ameliorates neuronal cholesterol and glycosphingolipid storage and disease progression. PLoS ONE 2009, 4, e6951. [Google Scholar] [CrossRef]
- Davidson, C.D.; Fishman, Y.I.; Puskas, I.; Szeman, J.; Sohajda, T.; McCauliff, L.A.; Sikora, J.; Storch, J.; Vanier, M.T.; Szente, L.; et al. Efficacy and ototoxicity of different cyclodextrins in Niemann-Pick C disease. Ann. Clin. Transl. Neurol. 2016, 3, 366–380. [Google Scholar] [CrossRef]
- Ramirez, C.M.; Liu, B.; Taylor, A.M.; Repa, J.J.; Burns, D.K.; Weinberg, A.G.; Turley, S.D.; Dietschy, J.M. Weekly cyclodextrin administration normalizes cholesterol metabolism in nearly every organ of the Niemann-Pick type C1 mouse and markedly prolongs life. Pediatr. Res. 2010, 68, 309–315. [Google Scholar] [CrossRef]
- Camargo, F.; Erickson, R.P.; Garver, W.S.; Hossain, G.S.; Carbone, P.N.; Heidenreich, R.A.; Blanchard, J. Cyclodextrins in the treatment of a mouse model of Niemann-Pick C disease. Life Sci. 2001, 70, 131–142. [Google Scholar] [CrossRef]
- Aqul, A.; Liu, B.; Ramirez, C.M.; Pieper, A.A.; Estill, S.J.; Burns, D.K.; Liu, B.; Repa, J.J.; Turley, S.D.; Dietschy, J.M. Unesterified cholesterol accumulation in late endosomes/lysosomes causes neurodegeneration and is prevented by driving cholesterol export from this compartment. J. Neurosci. 2011, 31, 9404–9413. [Google Scholar] [CrossRef] [PubMed]
- Vite, C.H.; Bagel, J.H.; Swain, G.P.; Prociuk, M.; Sikora, T.U.; Stein, V.M.; O’Donnell, P.; Ruane, T.; Ward, S.; Crooks, A.; et al. Intracisternal cyclodextrin prevents cerebellar dysfunction and Purkinje cell death in feline Niemann-Pick type C1 disease. Sci. Transl. Med. 2015, 7, 276ra26. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, M.; Shraishi, K.; Wada, K.; Ishitsuka, Y.; Doi, H.; Maeda, M.; Mizoguchi, T.; Eto, J.; Mochinaga, S.; Arima, H.; et al. Effects of intracerebroventricular administration of 2-hydroxypropyl-beta-cyclodextrin in a patient with Niemann-Pick Type C disease. Mol. Genet. Metab. Rep. 2014, 1, 391–400. [Google Scholar] [CrossRef]
- Matsuo, M.; Togawa, M.; Hirabaru, K.; Mochinaga, S.; Narita, A.; Adachi, M.; Egashira, M.; Irie, T.; Ohno, K. Effects of cyclodextrin in two patients with Niemann-Pick Type C disease. Mol. Genet. Metab. 2013, 108, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Megias-Vericat, J.E.; Company-Albir, M.J.; Garcia-Robles, A.A.; Poveda, J.L. Use of 2-Hydroxypropyl-Beta-Cyclodextrin for Niemann-Pick Type C Disease. In Cyclodextrin—A Versatile Ingredient; Arora, P., Dhingra, N., Eds.; IntechOpen: London, UK, 2017. [Google Scholar]
- Garcia-Robles, A.A.; Company-Albir, M.J.; Megias-Vericat, J.E.; Fernandez-Megia, M.J.; Perez-Miralles, F.C.; Lopez-Briz, E.; Alcala-Vicente, C.; Galeano, I.; Casanova, B.; Poveda, J.L. Use of 2 hydroxypropyl-beta-cyclodextrin therapy in two adult Niemann Pick Type C patients. J. Neurol. Sci. 2016, 366, 65–67. [Google Scholar] [CrossRef] [PubMed]
- Maarup, T.J.; Chen, A.H.; Porter, F.D.; Farhat, N.Y.; Ory, D.S.; Sidhu, R.; Jiang, X.; Dickson, P.I. Intrathecal 2-hydroxypropyl-beta-cyclodextrin in a single patient with Niemann-Pick C1. Mol. Genet. Metab. 2015, 116, 75–79. [Google Scholar] [CrossRef]
- Crumling, M.A.; King, K.A.; Duncan, R.K. Cyclodextrins and Iatrogenic Hearing Loss: New Drugs with Significant Risk. Front. Cell Neurosci. 2017, 11, 355. [Google Scholar] [CrossRef]
- Ory, D.S.; Ottinger, E.A.; Farhat, N.Y.; King, K.A.; Jiang, X.; Weissfeld, L.; Berry-Kravis, E.; Davidson, C.D.; Bianconi, S.; Keener, L.A.; et al. Intrathecal 2-hydroxypropyl-beta-cyclodextrin decreases neurological disease progression in Niemann-Pick disease, type C1: A non-randomised, open-label, phase 1-2 trial. Lancet 2017, 390, 1758–1768. [Google Scholar] [CrossRef]
- Matencio, A.; Caldera, F.; Cecone, C.; Lopez-Nicolas, J.M.; Trotta, F. Cyclic Oligosaccharides as Active Drugs, an Updated Review. Pharmaceuticals 2020, 13, 281. [Google Scholar] [CrossRef]
- Motoyama, K.; Nishiyama, R.; Maeda, Y.; Higashi, T.; Kawaguchi, Y.; Futaki, S.; Ishitsuka, Y.; Kondo, Y.; Irie, T.; Era, T.; et al. Cholesterol-Lowering Effect of Octaarginine-Appended beta-Cyclodextrin in Npc1-Trap-CHO Cells. Biol. Pharm. Bull. 2016, 39, 1823–1829. [Google Scholar] [CrossRef] [PubMed]
- Yasmin, N.; Ishitsuka, Y.; Fukaura, M.; Yamada, Y.; Nakahara, S.; Ishii, A.; Kondo, Y.; Takeo, T.; Nakagata, N.; Motoyama, K.; et al. In Vitro and In Vivo Evaluation of 6-O-alpha-Maltosyl-beta-Cyclodextrin as a Potential Therapeutic Agent Against Niemann-Pick Disease Type C. Int. J. Mol. Sci. 2019, 20, 1152. [Google Scholar] [CrossRef]
- Kulkarni, A.; Caporali, P.; Dolas, A.; Johny, S.; Goyal, S.; Dragotto, J.; Macone, A.; Jayaraman, R.; Fiorenza, M.T. Linear Cyclodextrin Polymer Prodrugs as Novel Therapeutics for Niemann-Pick Type C1 Disorder. Sci. Rep. 2018, 8, 9547. [Google Scholar] [CrossRef] [PubMed]
- Carradori, D.; Chen, H.; Werner, B.; Shah, A.S.; Leonardi, C.; Usuelli, M.; Mezzenga, R.; Platt, F.; Leroux, J.C. Investigating the Mechanism of Cyclodextrins in the Treatment of Niemann-Pick Disease Type C Using Crosslinked 2-Hydroxypropyl-beta-cyclodextrin. Small 2020, 16, e2004735. [Google Scholar] [CrossRef] [PubMed]
- Puglisi, A.; Yagci, Y. Cyclodextrin-Based Macromolecular Systems as Cholesterol-Mopping Therapeutic Agents in Niemann-Pick Disease Type C. Macromol. Rapid Commun. 2019, 40, e1800557. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Cyclodextrin | Concentration | Method | Intramolecular Target | Findings |
|---|---|---|---|---|---|
| Connexin | αCD, βCD, γCD | 5–20 mM | Transport-specific fractionation of unilamellar liposomes | Pore (specific residues not determined) | Reversible complete pore block in sucrose and urea permeation Characteristics of block depends on the size of CD relative to pore diameter |
| GABAA receptor | βCD | 0.15–1.5 mM | Patch-clamp | Not examined | Increased ligand-induced conductance Decelerated deactivation kinetics Decreased rate and extent of desensitization |
| TASK-1, TASK-3 | αCD, MβCD | 5 mM | Patch-clamp, molecular docking | Residues in the extracellular cavity close to the entrance of the pore (A chain: Arg68 and Gly97; B chain: Glu37, Lys70, Trp184, Gly203, Asp204 and Lys210) | Ionic currents reduced by ~40% Cholesterol-complexing filipin exerted no effects Direct binding between MβCD or αCD and the channel |
| KV1.3 | MβCD, iαCD, iγCD | 1–5 mM | Patch-clamp, molecular docking | Residues at the extracellular entrance of the pore (Thr372, Gly396, Asp397 and His399 of one subunit; Gly396 and His399 of one neighboring subunit; Gly396 and Asp397 of the opposing subunit) | Dose-dependent and partially reversible ~40% current reduction, apparent within 15 s and completed in 90 s Cholesterol-depleting HPβCD and HPγCD exerted no effects No correlation with changes in membrane fluidity, hydration or lipid order Direct binding between MβCD and the channel |
| Identifier | Phase | Participants | Status | Type | Drug | Dosing |
|---|---|---|---|---|---|---|
| NCT01747135 | 1 | 14 | Completed | Non-randomized, open-label, single-center | HPβCD (VTS-270 /Adrabetadex) | Intrathecal lumbar injection monthly 200 mg escalated to 900 mg |
| NCT02534844 | 2/3 | 51 | Active, not recruiting | Prospective, randomized, double-blind, placebo controlled, multi-center | HPβCD (VTS-270 /Adrabetadex) | Intrathecal lumbar injection 900–1800 mg every 2 weeks |
| NCT02912793 | 1/2 | 12 | Completed | Double-blind, randomized, multi-center | HPβCD (Trappsol® Cyclo™) | Intravenous infusion 1500/2000/2500 mg/kg every 2 weeks |
| NCT02939547 | 1 | 13 | Completed | double-blind, randomized, multi-center | HPβCD (Trappsol® Cyclo™) | Intravenous infusion 1500/2500 mg/kg every 2 weeks |
| NCT03471143 | 1/2 | 3 | Active, not recruiting | Open-label, dose escalation, multi-center | HPβCD (VTS-270 /Adrabetadex) | Intravenous infusion 500/1000 mg/kg twice a week for 6 weeks |
| NCT03687476 | 2 | 0 | Withdrawn (business decision) | Open-label, multi-center | HPβCD (VTS-270 /Adrabetadex) | Intrathecal lumbar injection 200 mg escalated to 900 mg every 2 weeks |
| NCT03887533 | 1/2 | 2 | Terminated (due to COVID-19) | Open-label, randomized, parallel dose, single-center | HPβCD (VTS-270 /Adrabetadex) | Combined intravenous infusion 500/1000 mg/kg monthly + intrathecal lumbar injection 900 mg monthly |
| NCT03893071 | 1 | 12 | Active, not recruiting | Open-label extension study of NCT02939547 | HPβCD (Trappsol® Cyclo™) | Intravenous infusion 1500/2500 mg/kg every 2 weeks |
| NCT04860960 | 3 | 93 | Recruiting | Prospective, randomized, double-blind, placebo controlled, multi-center | HPβCD (Trappsol® Cyclo™) | Intravenous infusion 2000 mg/kg every 2 weeks |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kovacs, T.; Nagy, P.; Panyi, G.; Szente, L.; Varga, Z.; Zakany, F. Cyclodextrins: Only Pharmaceutical Excipients or Full-Fledged Drug Candidates? Pharmaceutics 2022, 14, 2559. https://doi.org/10.3390/pharmaceutics14122559
Kovacs T, Nagy P, Panyi G, Szente L, Varga Z, Zakany F. Cyclodextrins: Only Pharmaceutical Excipients or Full-Fledged Drug Candidates? Pharmaceutics. 2022; 14(12):2559. https://doi.org/10.3390/pharmaceutics14122559
Chicago/Turabian StyleKovacs, Tamas, Peter Nagy, Gyorgy Panyi, Lajos Szente, Zoltan Varga, and Florina Zakany. 2022. "Cyclodextrins: Only Pharmaceutical Excipients or Full-Fledged Drug Candidates?" Pharmaceutics 14, no. 12: 2559. https://doi.org/10.3390/pharmaceutics14122559
APA StyleKovacs, T., Nagy, P., Panyi, G., Szente, L., Varga, Z., & Zakany, F. (2022). Cyclodextrins: Only Pharmaceutical Excipients or Full-Fledged Drug Candidates? Pharmaceutics, 14(12), 2559. https://doi.org/10.3390/pharmaceutics14122559