
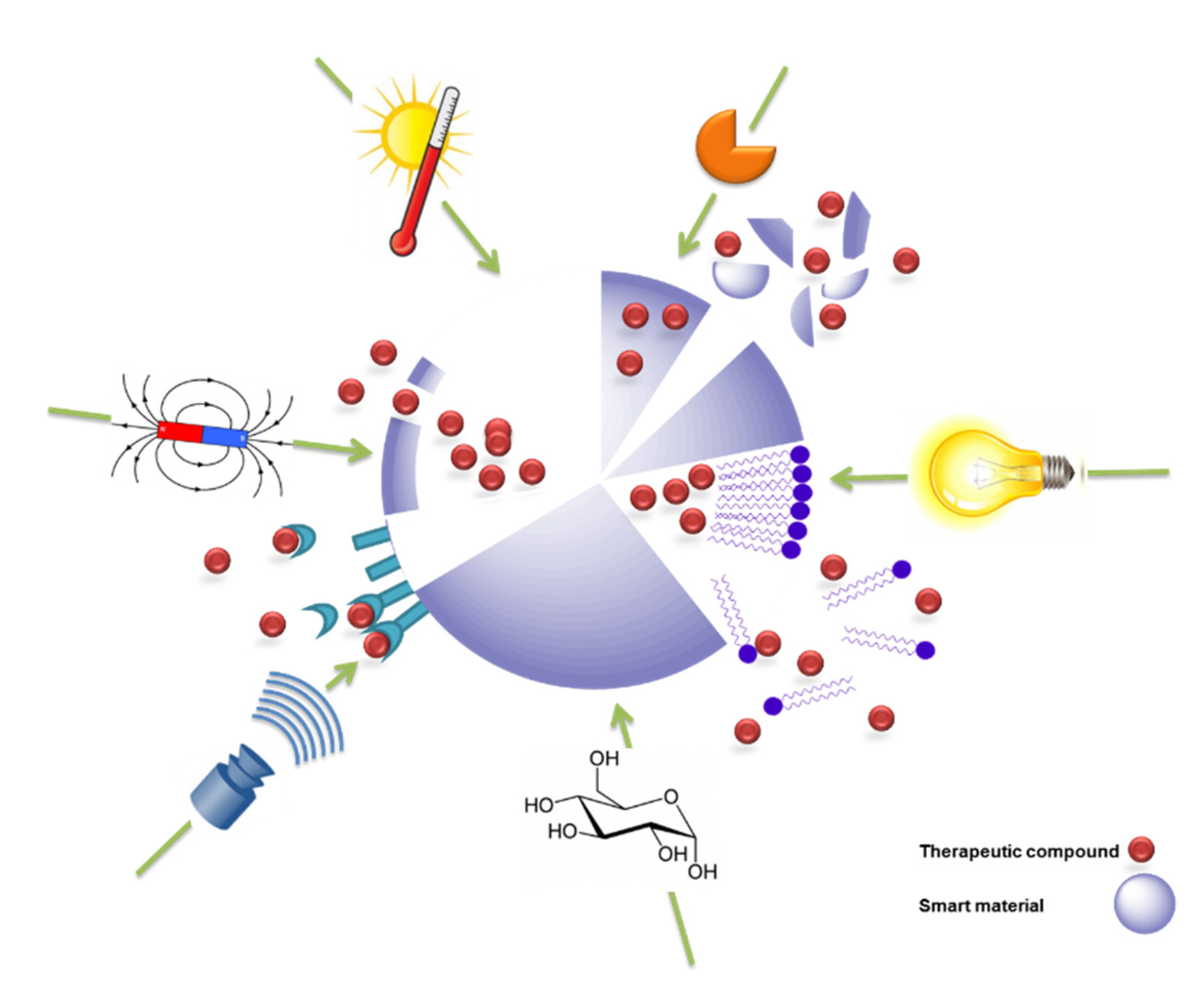
Opportunities and Challenges of Switchable Materials for Pharmaceutical Use

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
- The materials and their stimuli should be compatible with physiological requirements. However, this putatively trivial fact is not considered in numerous publications, suggesting the biomedical use of materials that are responsive only to, e.g., intense irradiation, non-physiological solvents, or high temperatures, or are based on components that are expected to show short-term or long-term toxicity.
- The drug should be effectively incorporated in relevant quantities, with material switching allowing the adjustment of drug-release rates, while premature diffusion-controlled release is suppressed. Typically, drug release should be enhanced upon stimulation. The inhibition of ongoing release from an implanted long-term drug-dosage system might also be of interest, e.g., in case of critical side effects of the medication or to adapt release rates to the progress of healing processes or physiological cycles.
- The in vivo fate of the carrier materials should be considered. While non-degradable large-sized devices may be surgically removed, this is typically not comfortable for patients. In the case of injectables based on particulate carriers, surgical removal is practically impossible, thus setting specific requirements for degradability, suitability for excretion, and/or cellular clearance. Theoretical degradability of some or all bonds in a polymeric construct does not necessarily mean that the material can or will be quantitatively removed in the expected time frame. For instance, a solubility drop of hydrophobic segments after cleavage from amphiphilic copolymers, or crystallization upon increasing the chain mobility of oligomeric degradation products, may create long-lasting residues.
2. Responsiveness to External Stimuli
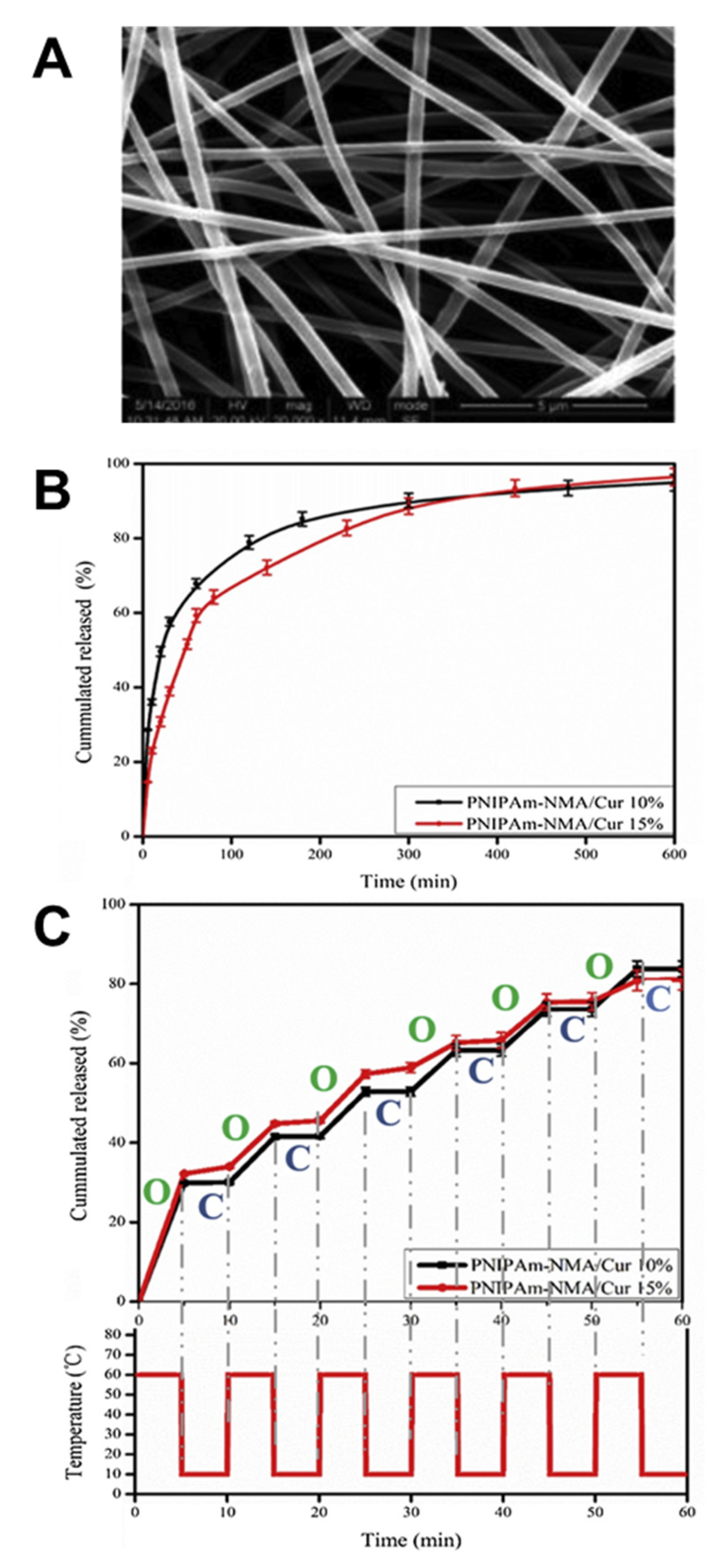
2.1. Temperature-Responsive Materials
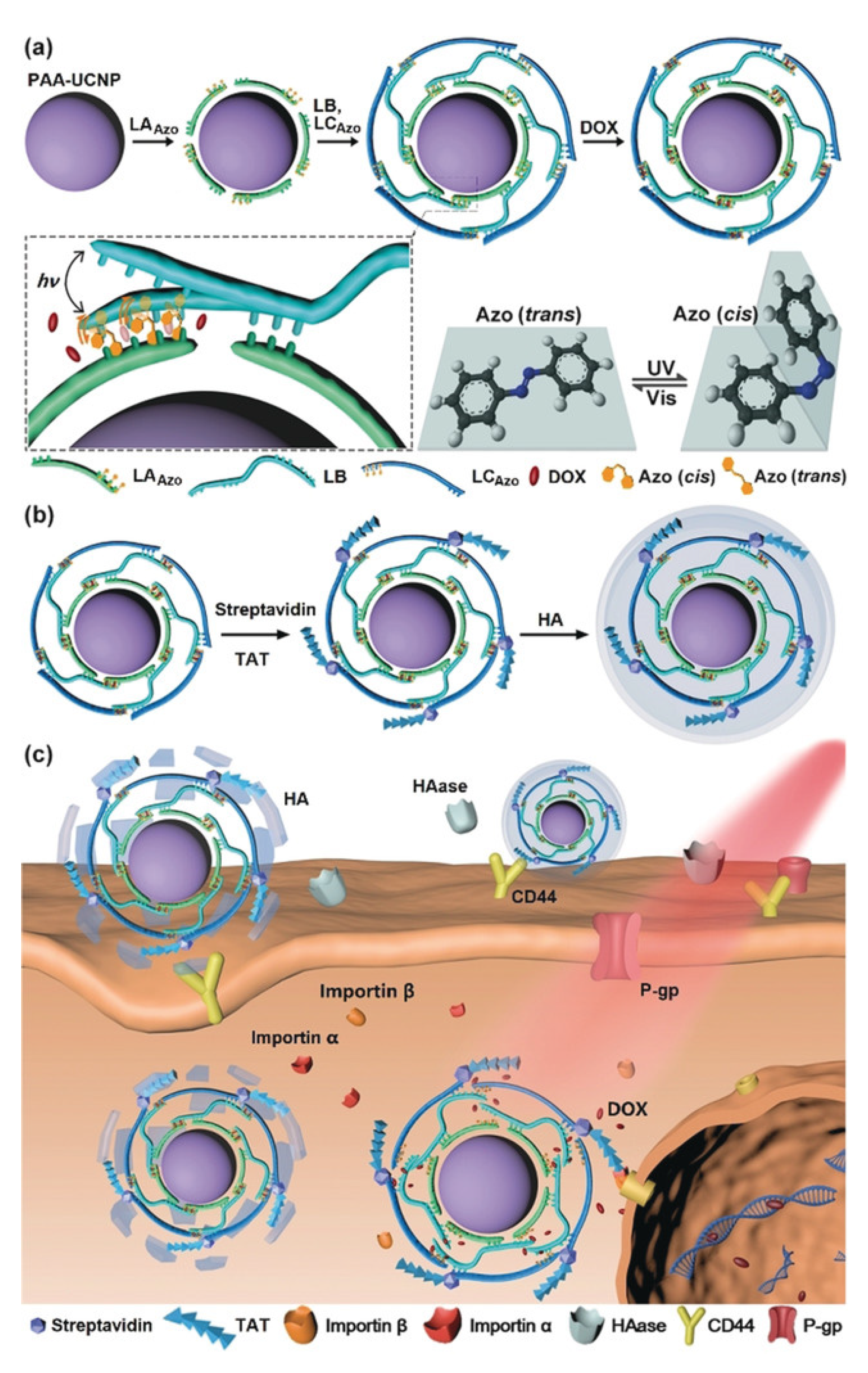
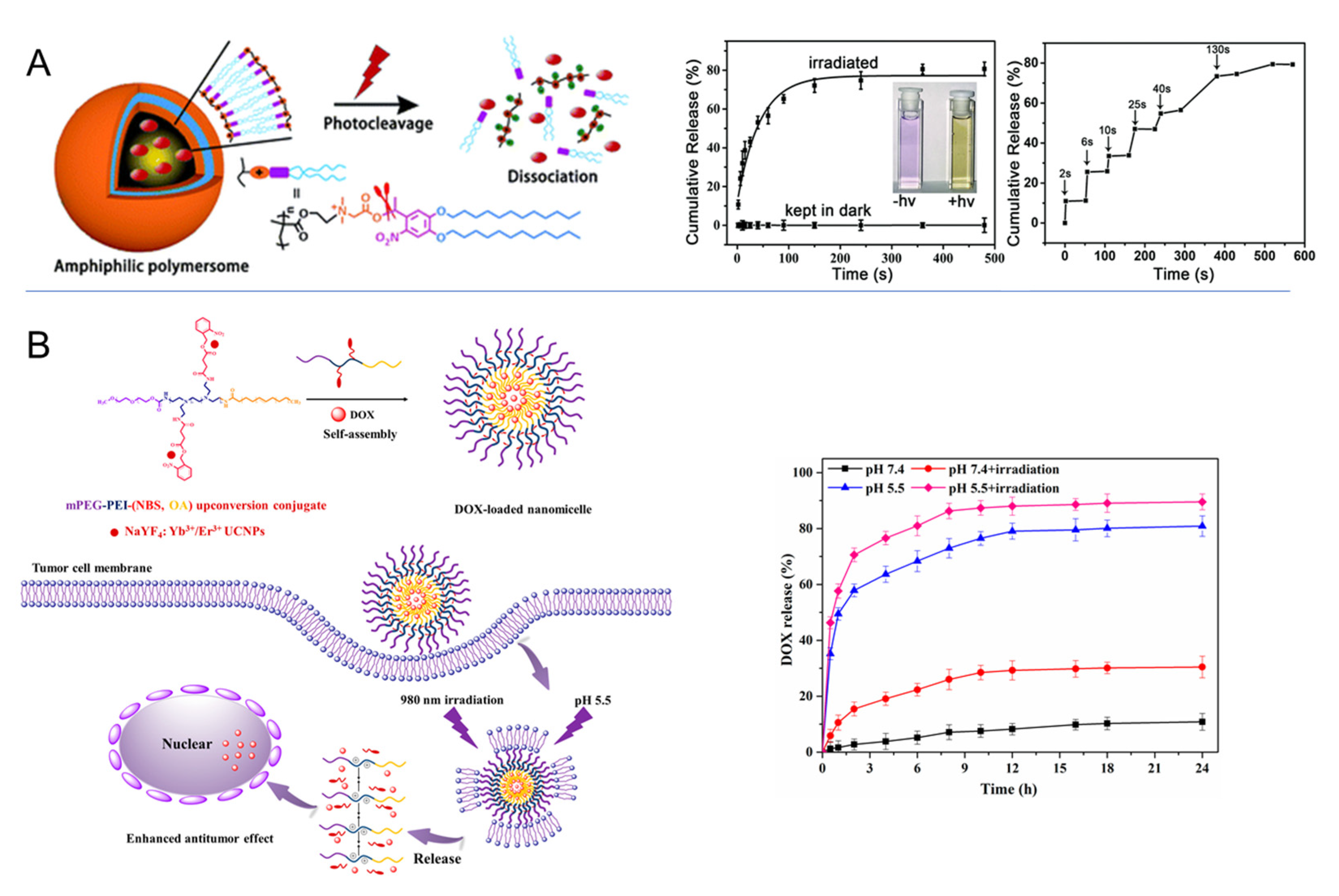
2.2. Light-Responsive Materials
2.3. Magnetic Field-Responsive Materials
2.4. Ultrasound-Responsive Materials
3. Responsiveness to In Vivo Stimuli
3.1. pH- and/or Ion-Responsive Materials
3.2. Enzyme-Responsive Materials

3.3. Systems Switching in Response to Small-Molecule Stimuli—From Physiological Markers to Danger Signals
3.4. Responsiveness to Specific Cells
4. Future Considerations and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wischke, C.; Schwendeman, S.P. Principles of encapsulating hydrophobic drugs in PLA/PLGA microparticles. Int. J. Pharm. 2008, 364, 298–327. [Google Scholar] [CrossRef] [PubMed]
- Amukarimi, S.; Ramakrishna, S.; Mozafari, M. Smart biomaterials—A proposed definition and overview of the field. Curr. Opin. Biomed. Eng. 2021, 19, 100311. [Google Scholar] [CrossRef]
- Grainger, D.W. Connecting drug delivery reality to smart materials design. Int. J. Pharm. 2013, 454, 521–524. [Google Scholar] [CrossRef] [PubMed]
- Cabane, E.; Zhang, X.; Langowska, K.; Palivan, C.G.; Meier, W. Stimuli-Responsive Polymers and Their Applications in Nanomedicine. Biointerphases 2012, 7, 9. [Google Scholar] [CrossRef]
- Kumar, A.; Srivastava, A.; Galaev, I.Y.; Mattiasson, B. Smart polymers: Physical forms and bioengineering applications. Prog. Polym. Sci. 2007, 32, 1205–1237. [Google Scholar] [CrossRef]
- Stuart, M.A.C.; Huck, W.T.S.; Genzer, J.; Müller, M.; Ober, C.; Stamm, M.; Sukhorukov, G.B.; Szleifer, I.; Tsukruk, V.V.; Urban, M.; et al. Emerging applications of stimuli-responsive polymer materials. Nat. Mater. 2010, 9, 101–113. [Google Scholar] [CrossRef]
- Bikram, M.; West, J.L. Thermo-responsive systems for controlled drug delivery. Expert Opin. Drug Deliv. 2008, 5, 1077–1091. [Google Scholar] [CrossRef]
- Nakayama, M.; Okano, T.; Miyazaki, T.; Kohori, F.; Sakai, K.; Yokoyama, M. Molecular design of biodegradable polymeric micelles for temperature-responsive drug release. J. Control. Release 2006, 115, 46–56. [Google Scholar] [CrossRef]
- Bromberg, L.E.; Ron, E.S. Temperature-responsive gels and thermogelling polymer matrices for protein and peptide delivery. Adv. Drug Deliv. Rev. 1998, 31, 197–221. [Google Scholar] [CrossRef]
- Gil, E.S.; Hudson, S.M. Stimuli-reponsive polymers and their bioconjugates. Prog. Polym. Sci. 2004, 29, 1173–1222. [Google Scholar] [CrossRef]
- Fukumura, D.; Jain, R.K. Tumor microenvironment abnormalities: Causes, consequences, and strategies to normalize. J. Cell. Biochem. 2007, 101, 937–949. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Zhu, Q.; Kenkare, P.U. Lower critical solution temperature of linear PNIPA obtained from a Yukawa potential of polymer chains. J. Appl. Polym. Sci. 2000, 78, 1971–1976. [Google Scholar] [CrossRef]
- Pelton, R. Poly(N-isopropylacrylamide) (PNIPAM) is never hydrophobic. J. Colloid Interface Sci. 2010, 348, 673–674. [Google Scholar] [CrossRef] [PubMed]
- Kulshreshtha, A.K.; Vasile, C. Handbook of Polymer Blends and Composites; Water Soluble Polymers in Solution-Phase Behaviour; Rapra Technology Ltd.: Shawbury, UK, 2002. [Google Scholar]
- Zhang, Q.; Weber, C.; Schubert, U.S.; Hoogenboom, R. Thermoresponsive polymers with lower critical solution temperature: From fundamental aspects and measuring techniques to recommended turbidimetry conditions. Mater. Horiz. 2017, 4, 109–116. [Google Scholar] [CrossRef]
- Jeong, B.; Gutowska, A. Lessons from nature: Stimuli-responsive polymers and their biomedical applications. Trends Biotechnol. 2002, 20, 305–311. [Google Scholar] [CrossRef]
- Wischke, C.; Zhang, Y.; Mittal, S.; Schwendeman, S.P. Development of PLGA-Based Injectable Delivery Systems for Hydrophobic Fenretinide. Pharm. Res. 2010, 27, 2063–2074. [Google Scholar] [CrossRef]
- Brown, D.J.A.; Brugger, H.; Boyd, J.; Paal, P. Accidental Hypothermia. N. Engl. J. Med. 2012, 367, 1930–1938. [Google Scholar] [CrossRef]
- Tang, L.; Wang, L.; Yang, X.; Feng, Y.; Li, Y.; Feng, W. Poly(N-isopropylacrylamide)-based smart hydrogels: Design, properties and applications. Prog. Mater. Sci. 2021, 115, 100702. [Google Scholar] [CrossRef]
- Idziak, I.; Avoce, D.; Lessard, D.; Gravel, D.; Zhu, X.X. Thermosensitivity of Aqueous Solutions of Poly(N,N-diethylacrylamide). Macromolecules 1999, 32, 1260–1263. [Google Scholar] [CrossRef]
- Vihola, H.; Laukkanen, A.; Tenhu, H.; Hirvonen, J. Drug release characteristics of physically cross-linked thermosensitive poly(N-vinylcaprolactam) hydrogel particles. J. Pharm. Sci. 2008, 97, 4783–4793. [Google Scholar] [CrossRef]
- Vihola, H.; Marttila, A.; Pakkanen, J.; Andersson, M.; Laukkanen, A.; Kaukonen, A.; Tenhu, H.; Hirvonen, J. Cell–polymer interactions of fluorescent polystyrene latex particles coated with thermosensitive poly(N-isopropylacrylamide) and poly(N-vinylcaprolactam) or grafted with poly(ethylene oxide)-macromonomer. Int. J. Pharm. 2007, 343, 238–246. [Google Scholar] [CrossRef] [PubMed]
- Vihola, H.; Laukkanen, A.; Valtola, L.; Tenhu, H.; Hirvonen, J. Cytotoxicity of thermosensitive polymers poly(N-isopropylacrylamide), poly(N-vinylcaprolactam) and amphiphilically modified poly(N-vinylcaprolactam). Biomaterials 2005, 26, 3055–3064. [Google Scholar] [CrossRef] [PubMed]
- Drozdov, A.D. Equilibrium Swelling of Biocompatible Thermo-Responsive Copolymer Gels. Gels 2021, 7, 40. [Google Scholar] [CrossRef] [PubMed]
- Kishi, R.; Kihara, H.; Miura, T.; Ichijo, H. Microporous poly(vinyl methyl ether) hydrogels prepared by γ-ray irradiation at different heating rates. Radiat. Phys. Chem. 2005, 72, 679–685. [Google Scholar] [CrossRef]
- Burillo, G.; Bucio, E.; Arenas, E.; Lopez, G.P. Temperature and pH-Sensitive Swelling Behavior of Binary DMAEMA/4VP Grafts on Poly(propylene) Films. Macromol. Mater. Eng. 2007, 292, 214–219. [Google Scholar] [CrossRef]
- Weber, C.; Hoogenboom, R.; Schubert, U.S. Temperature responsive bio-compatible polymers based on poly(ethylene oxide) and poly(2-oxazoline)s. Prog. Polym. Sci. 2012, 37, 686–714. [Google Scholar] [CrossRef]
- Hoogenboom, R.; Thijs, H.M.; Jochems, M.J.; van Lankvelt, B.M.; Fijten, M.W.; Schubert, U.S. Tuning the LCST of poly(2-oxazoline)s by varying composition and molecular weight: Alternatives to poly(N-isopropylacrylamide)? Chem. Commun. 2008, 44, 5758–5760. [Google Scholar] [CrossRef]
- Bloksma, M.M.; Weber, C.; Perevyazko, I.Y.; Kuse, A.; Baumgärtel, A.; Vollrath, A.; Hoogenboom, R.; Schubert, U.S. Poly(2-cyclopropyl-2-oxazoline): From Rate Acceleration by Cyclopropyl to Thermoresponsive Properties. Macromolecules 2011, 44, 4057–4064. [Google Scholar] [CrossRef]
- Sahn, M.; Stafast, L.M.; Dirauf, M.; Bandelli, D.; Weber, C.; Schubert, U.S. LCST behavior of poly(2-ethyl-2-oxazoline) containing diblock and triblock copolymers. Eur. Polym. J. 2018, 100, 57–66. [Google Scholar] [CrossRef]
- Dargaville, T.R.; Park, J.-R.; Hoogenboom, R. Poly(2-oxazoline) Hydrogels: State-of-the-Art and Emerging Applications. Macromol. Biosci. 2018, 18, 1800070. [Google Scholar] [CrossRef]
- Muljajew, I.; Huschke, S.; Ramoji, A.; Cseresnyés, Z.; Hoeppener, S.; Nischang, I.; Foo, W.; Popp, J.; Figge, M.T.; Weber, C.; et al. Stealth Effect of Short Polyoxazolines in Graft Copolymers: Minor Changes of Backbone End Group Determine Liver Cell-Type Specificity. ACS Nano 2021, 15, 12298–12313. [Google Scholar] [CrossRef] [PubMed]
- Baskan, T.; Tuncaboylu, D.C.; Okay, O. Tough interpenetrating Pluronic F127/polyacrylic acid hydrogels. Polymer 2013, 54, 2979–2987. [Google Scholar] [CrossRef]
- Yu, L.; Chang, G.; Zhang, H.; Ding, J. Temperature-induced spontaneous sol–gel transitions of poly(D,L-lactic acid-co-glycolic acid)-b-poly(ethylene glycol)-b-poly(D,L-lactic acid-co-glycolic acid) triblock copolymers and their end-capped derivatives in water. J. Polym. Sci. Part A Polym. Chem. 2007, 45, 1122–1133. [Google Scholar] [CrossRef]
- Missirlis, D.; Hubbell, J.A.; Tirelli, N. Thermally-induced glass formation from hydrogel nanoparticles. Soft Matter 2006, 2, 1067–1075. [Google Scholar] [CrossRef] [PubMed]
- Nair, A.B.; Chaudhary, S.; Shah, H.; Jacob, S.; Mewada, V.; Shinu, P.; Aldhubiab, B.; Sreeharsha, N.; Venugopala, K.N.; Attimarad, M.; et al. Intranasal Delivery of Darunavir-Loaded Mucoadhesive In Situ Gel: Experimental Design, In Vitro Evaluation, and Pharmacokinetic Studies. Gels 2022, 8, 342. [Google Scholar] [CrossRef]
- Fathalla, Z.; Mustafa, W.W.; Abdelkader, H.; Moharram, H.; Sabry, A.M.; Alany, R.G. Hybrid thermosensitive-mucoadhesive in situ forming gels for enhanced corneal wound healing effect of L-carnosine. Drug Deliv. 2022, 29, 374–385. [Google Scholar] [CrossRef]
- Alemdar, M.; Tuncaboylu, D.C.; Batu, H.K.; Temel, B.A. Pluronic based injectable smart gels with coumarin functional amphiphilic copolymers. Eur. Polym. J. 2022, 177, 111378. [Google Scholar] [CrossRef]
- Seçer, S.; Tuncaboylu, D.C. Supramolecular poloxamer-based in situ gels with hyaluronic acid and cyclodextrins. Int. J. Polym. Mater. Polym. Biomater. 2022, 71, 647–655. [Google Scholar] [CrossRef]
- Jeong, B.; Bae, Y.H.; Lee, D.S.; Kim, S.W. Biodegradable block copolymers as injectable drug-delivery systems. Nature 1997, 388, 860–862. [Google Scholar] [CrossRef]
- Jeong, B.; Choi, Y.; Bae, Y.; Zentner, G.; Kim, S. New biodegradable polymers for injectable drug delivery systems. J. Control. Release 1999, 62, 109–114. [Google Scholar] [CrossRef]
- Cespi, M.; Bonacucina, G.; Tiboni, M.; Casettari, L.; Cambriani, A.; Fini, F.; Perinelli, D.R.; Palmieri, G.F. Insights in the rheological properties of PLGA-PEG-PLGA aqueous dispersions: Structural properties and temperature-dependent behaviour. Polymer 2021, 213, 123216. [Google Scholar] [CrossRef]
- Jeong, B.; Bae, Y.H.; Kim, S.W. In situ gelation of PEG-PLGA-PEG triblock copolymer aqueous solutions and degradation thereof. J. Biomed. Mater. Res. 2000, 50, 171–177. [Google Scholar] [CrossRef]
- Yu, L.; Zhang, Z.; Ding, J. Influence of LA and GA Sequence in the PLGA Block on the Properties of Thermogelling PLGA-PEG-PLGA Block Copolymers. Biomacromolecules 2011, 12, 1290–1297. [Google Scholar] [CrossRef] [PubMed]
- Lai, M.-C.; Chang, K.-C.; Hsu, S.-C.; Chou, M.-C.; Hung, W.-I.; Hsiao, Y.-R.; Lee, H.-M.; Hsieh, M.-F.; Yeh, J.-M. In situ gelation of PEG-PLGA-PEG hydrogels containing high loading of hydroxyapatite: In vitro and in vivo characteristics. Biomed. Mater. 2014, 9, 015011. [Google Scholar] [CrossRef] [PubMed]
- Pan, A.; Wang, Z.; Chen, B.; Dai, W.; Zhang, H.; He, B.; Wang, X.; Wang, Y.; Zhang, Q. Localized co-delivery of collagenase and trastuzumab by thermosensitive hydrogels for enhanced antitumor efficacy in human breast xenograft. Drug Deliv. 2018, 25, 1495–1503. [Google Scholar] [CrossRef]
- Ono, K.; Sumiya, M.; Yoshinobu, N.; Dode, T.; Katayama, T.; Ueda, N.; Nagahama, K. Angiogenesis Promotion by Combined Administration of DFO and Vein Endothelial Cells Using Injectable, Biodegradable, Nanocomposite Hydrogel Scaffolds. ACS Appl. Bio Mater. 2022, 5, 471–482. [Google Scholar] [CrossRef]
- Matthes, R.; Frey, H. Polyethers Based on Short-Chain Alkyl Glycidyl Ethers: Thermoresponsive and Highly Biocompatible Materials. Biomacromolecules 2022, 23, 2219–2235. [Google Scholar] [CrossRef]
- Chen, G.; Hoffman, A.S. Graft copolymers that exhibit temperature-induced phase transitions over a wide range of pH. Nature 1995, 373, 49–52. [Google Scholar] [CrossRef]
- Neradovic, D.; Hinrichs, W.L.J.; Bosch, J.J.K.-V.D.; Hennink, W.E. Poly(N-isopropylacrylamide) with hydrolyzable lactic acid ester side groups: A new type of thermosensitive polymer. Macromol. Rapid Commun. 1999, 20, 577–581. [Google Scholar] [CrossRef]
- Chang, K.; Rubright, N.C.; Lowery, P.D.; Taite, L.J. Structural optimization of highly branched thermally responsive polymers as a means of controlling transition temperature. J. Polym. Sci. Part A Polym. Chem. 2013, 51, 2068–2078. [Google Scholar] [CrossRef]
- Cao, M.; Liu, Y.; Zhang, X.; Li, F.; Zhong, M. Expanding the toolbox of controlled/living branching radical polymerization through simulation-informed reaction design. Chem 2022, 8, 1460–1475. [Google Scholar] [CrossRef]
- Bae, Y.H.; Okano, T.; Kim, S.W. “On–Off” Thermocontrol of Solute Transport. I. Temperature Dependence of Swelling of N-Isopropylacrylamide Networks Modified with Hydrophobic Components in Water. Pharm. Res. 1991, 8, 531–537. [Google Scholar] [CrossRef] [PubMed]
- Bae, Y.H.; Okano, T.; Kim, S.W.; Kirn, S.W. “On–Off” Thermocontrol of Solute Transport. II. Solute Release from Thermosensitive Hydrogels. Pharm. Res. 1991, 8, 624–628. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.; Wei, H.; Wu, D.-Q.; Yang, B.; Chen, N.; Cheng, S.-X.; Zhang, X.-Z.; Zhuo, R.-X. Thermo-responsive shell cross-linked PMMA-b-P(NIPAAm-co-NAS) micelles for drug delivery. Int. J. Pharm. 2011, 420, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Zhao, W.; Wang, Y.; Wang, X.; Long, S.; Yang, J. Novel PNIPAm-based electrospun nanofibres used directly as a drug carrier for “on-off” switchable drug release. Colloids Surf. B Biointerfaces 2019, 182, 110347. [Google Scholar] [CrossRef]
- Yoshida, T.; Aoyagi, T.; Kokufuta, E.; Okano, T. Newly designed hydrogel with both sensitive thermoresponse and biodegradability. J. Polym. Sci. Part A Polym. Chem. 2003, 41, 779–787. [Google Scholar] [CrossRef]
- Lai, J.-Y.; Hsieh, A.-C. A gelatin-g-poly(N-isopropylacrylamide) biodegradable in situ gelling delivery system for the intracameral administration of pilocarpine. Biomaterials 2012, 33, 2372–2387. [Google Scholar] [CrossRef]
- Capella, V.; Rivero, R.E.; Liaudat, A.C.; Ibarra, L.E.; Roma, D.A.; Alustiza, F.; Mañas, F.; Barbero, C.A.; Bosch, P.; Rivarola, C.R.; et al. Cytotoxicity and bioadhesive properties of poly-N-isopropylacrylamide hydrogel. Heliyon 2019, 5, e01474. [Google Scholar] [CrossRef]
- Yang, L.; Lou, J.; Yuan, J.; Deng, J. A review of shape memory polymers based on the intrinsic structures of their responsive switches. RSC Adv. 2021, 11, 28838–28850. [Google Scholar] [CrossRef]
- Wischke, C.; Lendlein, A. Method for Preparation, Programming, and Characterization of Miniaturized Particulate Shape-Memory Polymer Matrices. Langmuir 2014, 30, 2820–2827. [Google Scholar] [CrossRef]
- Friess, F.; Nöchel, U.; Lendlein, A.; Wischke, C. Polymer Micronetworks with Shape-Memory as Future Platform to Explore Shape-Dependent Biological Effects. Adv. Healthc. Mater. 2014, 3, 1986–1990. [Google Scholar] [CrossRef] [PubMed]
- Friess, F.; Lendlein, A.; Wischke, C. Switching microobjects from low to high aspect ratios using a shape-memory effect. Soft Matter 2021, 17, 9326–9331. [Google Scholar] [CrossRef] [PubMed]
- Wischke, C.; Hofmann, D. Predictive Shapes of Ellipsoid PPDL-PTHF Copolymer Particles Prepared by the Phantom Stretching Technique. Polymers 2022, 14, 3762. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Chen, Z.; Gao, Y.; Xing, Y.; Zhou, Y.; Luo, Y.; Xu, W.; Chen, Z.; Gao, X.; Gupta, K.; et al. Shape memory micro-anchors with magnetic guidance for precision micro-vascular deployment. Biomaterials 2022, 283, 121426. [Google Scholar] [CrossRef]
- Lendlein, A.; Behl, M.; Hiebl, B.; Wischke, C. Shape-memory polymers as a technology platform for biomedical applications. Expert Rev. Med. Devices 2010, 7, 357–379. [Google Scholar] [CrossRef]
- Basak, S. Redesigning the modern applied medical sciences and engineering with shape memory polymers. Adv. Compos. Hybrid Mater. 2021, 4, 223–234. [Google Scholar] [CrossRef]
- Kılıç, H.; Ceylan Tuncaboylu, D.; Argun, A.; Ozturk Civelek, D. Design of Biocompatible Multifunctional Hydrogels with Stearyl Methacrylate and Vinylpyrrolidone. ACS Appl. Polym. Mater. 2022, 4, 1717–1727. [Google Scholar] [CrossRef]
- Wischke, C.; Neffe, A.T.; Steuer, S.; Engelhardt, E.; Lendlein, A. AB-polymer Networks with Cooligoester and Poly(n-butyl acrylate) Segments as a Multifunctional Matrix for Controlled Drug Release. Macromol. Biosci. 2010, 10, 1063–1072. [Google Scholar] [CrossRef]
- Wischke, C.; Neffe, A.T.; Steuer, S.; Lendlein, A. Comparing techniques for drug loading of shape-memory polymer networks—Effect on their functionalities. Eur. J. Pharm. Sci. 2010, 41, 136–147. [Google Scholar] [CrossRef]
- Maity, N.; Mansour, N.; Chakraborty, P.; Bychenko, D.; Gazit, E.; Cohn, D. A Personalized Multifunctional 3D Printed Shape Memory-Displaying, Drug Releasing Tracheal Stent. Adv. Funct. Mater. 2021, 31, 2108436. [Google Scholar] [CrossRef]
- Tuncaboylu, D.C.; Friess, F.; Wischke, C.; Lendlein, A. A multifunctional multimaterial system for on-demand protein release. J. Control. Release 2018, 284, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Fei, G.; Xia, H.; Han, J.; Zhao, Y. Spatial and temporal control of shape memory polymers and simultaneous drug release using high intensity focused ultrasound. J. Mater. Chem. 2012, 22, 7692–7696. [Google Scholar] [CrossRef]
- Alvarez-Lorenzo, C.; Bromberg, L.; Concheiro, A. Light-sensitive Intelligent Drug Delivery Systems. Photochem. Photobiol. 2009, 85, 848–860. [Google Scholar] [CrossRef] [PubMed]
- Xie, S.; Natansohn, A.; Rochon, P. Recent developments in aromatic azo polymers research. Chem. Mater. 1993, 5, 403–411. [Google Scholar] [CrossRef]
- Fischer, E.; Hirshberg, Y. Formation of coloured forms of spirans by low-temperature irradiation. J. Chem. Soc. 1952, 4522–4524. [Google Scholar]
- Higashiguchi, K.; Taira, G.; Kitai, J.-I.; Hirose, T.; Matsuda, K. Photoinduced Macroscopic Morphological Transformation of an Amphiphilic Diarylethene Assembly: Reversible Dynamic Motion. J. Am. Chem. Soc. 2015, 137, 2722–2729. [Google Scholar] [CrossRef]
- Irie, M.; Mohri, M. Thermally irreversible photochromic systems. Revers. Photocyclization Diarylethene Deriv. J. Org. Chem. 1988, 53, 803–808. [Google Scholar] [CrossRef]
- Meier, H.; Zeller, K.-P. The Wolff Rearrangement of α-Diazo Carbonyl Compounds. Angew. Chem. Int. Ed. Engl. 1975, 14, 32–43. [Google Scholar] [CrossRef]
- Wolff, L. Ueber Diazoanhydride. Liebigs Ann. Chem. 1902, 325, 129. [Google Scholar] [CrossRef]
- Li, J.; Cao, Y.; You, Q.; Zhang, Y.; Shi, H.; Yang, Z.; He, L.; Wang, J.; Ni, C.; Chen, Y.; et al. Shape memory supramolecular networks for the photoregulated adsorption and release of model molecules. Mater. Express 2013, 3, 310–318. [Google Scholar] [CrossRef]
- Liu, N.; Wu, S.; Tian, X.; Li, X. Fabrication of injectable hydrogels from an anticancer peptide for local therapeutic delivery and synergistic photothermal–chemotherapy. J. Mater. Chem. B 2022, 10, 5165–5173. [Google Scholar] [CrossRef] [PubMed]
- Moreno, V.; Meroño, C.; Baeza, A.; Usategui, A.; Ortiz-Romero, P.; Pablos, J.; Vallet-Regí, M. UVA-Degradable Collagenase Nanocapsules as a Potential Treatment for Fibrotic Diseases. Pharmaceutics 2021, 13, 499. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.; Liu, W.; Wang, Z.; Zheng, P.; Liu, W.; Zhao, J.; Zhong, Y.; Zhang, Y.; Lin, J.; Xue, W.; et al. Near-infrared laser-controlled nitric oxide-releasing gold nanostar/hollow polydopamine Janus nanoparticles for synergistic elimination of methicillin-resistant Staphylococcus aureus and wound healing. Acta Biomater. 2022, 143, 428–444. [Google Scholar] [CrossRef]
- Veeren, A.; Ogunyankin, M.O.; Shin, J.E.; Zasadzinski, J.A. Liposome-Tethered Gold Nanoparticles Triggered by Pulsed NIR Light for Rapid Liposome Contents Release and Endosome Escape. Pharmaceutics 2022, 14, 701. [Google Scholar] [CrossRef] [PubMed]
- Roy, B.; Mengji, R.; Roy, S.; Pal, B.; Jana, A.; Singh, N.D.P. NIR-Responsive Lysosomotropic Phototrigger: An “AIE + ESIPT” Active Naphthalene-Based Single-Component Photoresponsive Nanocarrier with Two-Photon Uncaging and Real-Time Monitoring Ability. ACS Appl. Mater. Interfaces 2022, 14, 4862–4870. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W.; Chen, W.-H.; Xu, X.-D.; Li, C.; Zhang, J.; Zhuo, R.-X.; Zhang, X.-Z. Design of a Cellular-Uptake-Shielding “Plug and Play” Template for Photo Controllable Drug Release. Adv. Mater. 2011, 23, 3526–3530. [Google Scholar] [CrossRef]
- Yang, Z.; Wang, X.; Liang, G.; Yang, A.; Li, J. Photocontrolled chondrogenic differentiation and long-term tracking of mesenchymal stem cells in vivo by upconversion nanoparticles. J. Mater. Chem. B 2022, 10, 518–536. [Google Scholar] [CrossRef]
- Wang, X.; Chen, Z.; Yang, Y.; Guo, H.; Yang, Y.; Tang, C.-Y.; Li, X.; Law, W.-C. Near-infrared and pH responsive molecular machine for controlled encapsulation and release of drugs. Polym. Test. 2022, 112, 107631. [Google Scholar] [CrossRef]
- Wang, D.; Zhao, W.; Wei, Q.; Zhao, C.; Zheng, Y. Photoswitchable Azobenzene/Cyclodextrin Host-Guest Complexes: From UV- to Visible/Near-IR-Light-Responsive Systems. ChemPhotoChem 2018, 2, 403–415. [Google Scholar] [CrossRef]
- Wang, Y.; Yu, J.; Wang, Z.; Iqbal, S.; Zhang, W.; Zhang, Z.; Zhou, N.; Zhu, X. Real-time near-infrared fluorescence reporting the azoreductase-triggered drug release. Polym. Chem. 2020, 11, 734–743. [Google Scholar] [CrossRef]
- Ge, L.; Qiao, C.; Tang, Y.; Zhang, X.; Jiang, X. Light-Activated Hypoxia-Sensitive Covalent Organic Framework for Tandem-Responsive Drug Delivery. Nano Lett. 2021, 21, 3218–3224. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Song, G.; He, Y.; Zhang, X.; Liu, Y.; Ju, H. A DNA–Azobenzene Nanopump Fueled by Upconversion Luminescence for Controllable Intracellular Drug Release. Angew. Chem. Int. Ed. 2019, 58, 18207–18211. [Google Scholar] [CrossRef] [PubMed]
- Mandl, G.A.; Rojas-Gutierrez, P.A.; Capobianco, J.A. A NIR-responsive azobenzene-based supramolecular hydrogel using upconverting nanoparticles. Chem. Commun. 2018, 54, 5847–5850. [Google Scholar] [CrossRef] [PubMed]
- Schimka, S.; Klier, D.T.; de Guereñu, A.L.; Bastian, P.; Lomadze, N.; Kumke, M.U.; Santer, S. Photo-isomerization of azobenzene containing surfactants induced by near-infrared light using upconversion nanoparticles as mediator. J. Phys. Condens. Matter 2019, 31, 125201. [Google Scholar] [CrossRef] [PubMed]
- Weinstain, R.; Slanina, T.; Kand, D.; Klán, P. Visible-to-NIR-Light Activated Release: From Small Molecules to Nanomaterials. Chem. Rev. 2020, 120, 13135–13272. [Google Scholar] [CrossRef]
- Bochet, C.G. Photolabile protecting groups and linkers. J. Chem. Soc. Perkin Trans. 1 2002, 2, 125–142. [Google Scholar] [CrossRef]
- Yan, B.; Boyer, J.-C.; Habault, D.; Branda, N.R.; Zhao, Y. Near Infrared Light Triggered Release of Biomacromolecules from Hydrogels Loaded with Upconversion Nanoparticles. J. Am. Chem. Soc. 2012, 134, 16558–16561. [Google Scholar] [CrossRef]
- Zhou, Y.; Chen, R.; Yang, H.; Bao, C.; Fan, J.; Wang, C.; Lin, Q.; Zhu, L. Light-responsive polymersomes with a charge-switch for targeted drug delivery. J. Mater. Chem. B 2020, 8, 727–735. [Google Scholar] [CrossRef]
- Tsai, M.-F.; Lo, Y.-L.; Soorni, Y.; Su, C.-H.; Sivasoorian, S.S.; Yang, J.-Y.; Wang, L.-F. Near-Infrared Light-Triggered Drug Release from Ultraviolet- and Redox-Responsive Polymersome Encapsulated with Core–Shell Upconversion Nanoparticles for Cancer Therapy. ACS Appl. Bio Mater. 2021, 4, 3264–3275. [Google Scholar] [CrossRef]
- Zhang, J.; Tang, X.; Huang, C.; Liu, Z.; Ye, Y. Oleic Acid Copolymer as A Novel Upconversion Nanomaterial to Make Doxorubicin-Loaded Nanomicelles with Dual Responsiveness to pH and NIR. Pharmaceutics 2020, 12, 680. [Google Scholar] [CrossRef]
- Zhang, Y.; Fang, C.; Carvalho, W.S.P.; Gao, Y.; Serpe, M.J. Triggered Small-Molecule Release from Dual-Stimuli Responsive Microgels. ACS Appl. Polym. Mater. 2021, 3, 410–417. [Google Scholar] [CrossRef]
- Stefanachi, A.; Leonetti, F.; Pisani, L.; Catto, M.; Carotti, A. Coumarin: A Natural, Privileged and Versatile Scaffold for Bioactive Compounds. Molecules 2018, 23, 250. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Yan, B.; Tremblay, L.; Zhao, Y. Both Core- and Shell-Cross-Linked Nanogels: Photoinduced Size Change, Intraparticle LCST, and Interparticle UCST Thermal Behaviors. Langmuir 2011, 27, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Jin, Q.; Mitschang, F.; Agarwal, S. Biocompatible Drug Delivery System for Photo-Triggered Controlled Release of 5-Fluorouracil. Biomacromolecules 2011, 12, 3684–3691. [Google Scholar] [CrossRef]
- Karthik, S.; Jana, A.; Selvakumar, M.; Venkatesh, Y.; Paul, A.; Shah, S.S.; Singh, N.D.P. Coumarin polycaprolactone polymeric nanoparticles: Light and tumor microenvironment activated cocktail drug delivery. J. Mater. Chem. B 2017, 5, 1734–1741. [Google Scholar] [CrossRef]
- Jiang, J.; Tong, X.; Zhao, Y. A New Design for Light-Breakable Polymer Micelles. J. Am. Chem. Soc. 2005, 127, 8290–8291. [Google Scholar] [CrossRef]
- Yan, B.; Boyer, J.-C.; Branda, N.R.; Zhao, Y. Near-Infrared Light-Triggered Dissociation of Block Copolymer Micelles Using Upconverting Nanoparticles. J. Am. Chem. Soc. 2011, 133, 19714–19717. [Google Scholar] [CrossRef]
- Kemp, S.J.; Ferguson, R.M.; Khandhar, A.P.; Krishnan, K.M. Monodisperse magnetite nanoparticles with nearly ideal saturation magnetization. RSC Adv. 2016, 6, 77452–77464. [Google Scholar] [CrossRef]
- Cao, D.; Li, H.; Pan, L.; Li, J.; Wang, X.; Jing, P.; Cheng, X.; Wang, W.; Wang, J.; Liu, Q. High saturation magnetization of γ-Fe2O3 nano-particles by a facile one-step synthesis approach. Sci. Rep. 2016, 6, 32360. [Google Scholar] [CrossRef]
- Sharifianjazi, F.; Moradi, M.; Parvin, N.; Nemati, A.; Rad, A.J.; Sheysi, N.; Abouchenari, A.; Mohammadi, A.; Karbasi, S.; Ahmadi, Z.; et al. Magnetic CoFe2O4 nanoparticles doped with metal ions: A review. Ceram. Int. 2020, 46 Pt B, 18391–18412. [Google Scholar] [CrossRef]
- Bulte, J.W.M.; Kraitchman, D.L. Iron oxide MR contrast agents for molecular and cellular imaging. NMR Biomed. 2004, 17, 484–499. [Google Scholar] [CrossRef] [PubMed]
- Mengesha, A.; Hoerres, A.; Mahajan, P. Cytocompatibility of oleic acid modified iron oxide nanoparticles. Mater. Lett. 2022, 323, 132528. [Google Scholar] [CrossRef]
- Senyei, A.E.; Widder, K.J.; Czerlinski, G. Magnetic guidance of drug-carrying microspheres. J. Appl. Phys. 1978, 49, 3578–3583. [Google Scholar] [CrossRef]
- Mosbach, K.; Schröder, U. Preparation and application of magnetic polymers for targeting of drugs. FEBS Lett. 1979, 102, 112–116. [Google Scholar] [CrossRef]
- Janikowska, A.; Matuszak, J.; Lyer, S.; Schreiber, E.; Unterweger, H.; Zaloga, J.; Groll, J.; Alexiou, C.; Cicha, I. A novel human artery model to assess the magnetic accumulation of SPIONs under flow conditions. Sci. Rep. 2017, 7, 42314. [Google Scholar] [CrossRef] [PubMed]
- Neuberger, T.; Schöpf, B.; Hofmann, H.; Hofmann, M.; von Rechenberg, B. Superparamagnetic nanoparticles for biomedical applications: Possibilities and limitations of a new drug delivery system. J. Magn. Magn. Mater. 2005, 293, 483–496. [Google Scholar] [CrossRef]
- Huang, S.-H.; Juang, R.-S. Biochemical and biomedical applications of multifunctional magnetic nanoparticles: A review. J. Nanoparticle Res. 2011, 13, 4411–4430. [Google Scholar] [CrossRef]
- Pradhan, P.; Giri, J.; Banerjee, R.; Bellare, J.; Bahadur, D. Cellular interactions of lauric acid and dextran-coated magnetite nanoparticles. J. Magn. Magn. Mater. 2007, 311, 282–287. [Google Scholar] [CrossRef]
- Khandhar, A.P.; Ferguson, R.M.; Arami, H.; Kemp, S.J.; Krishnan, K.M. Tuning Surface Coatings of Optimized Magnetite Nanoparticle Tracers for In Vivo Magnetic Particle Imaging. IEEE Trans. Magn. 2015, 51, 1–4. [Google Scholar] [CrossRef]
- Xuan, M.; Shao, J.; Zhao, J.; Li, Q.; Dai, L.; Li, J. Magnetic Mesoporous Silica Nanoparticles Cloaked by Red Blood Cell Membranes: Applications in Cancer Therapy. Angew. Chem. Int. Ed. 2018, 57, 6049–6053. [Google Scholar] [CrossRef]
- Nelson, N.R.; Port, J.D.; Pandey, M.K. Use of Superparamagnetic Iron Oxide Nanoparticles (SPIONs) via Multiple Imaging Modalities and Modifications to Reduce Cytotoxicity: An Educational Review. J. Nanotheranostics 2020, 1, 105–135. [Google Scholar] [CrossRef]
- Nizamov, T.R.; Garanina, A.; Uvarova, V.; Naumenko, V.; Shchetinin, I.; Savchenko, A. The use of iron oxide magnetic nanospheres and nanocubes for targeted doxorubicin delivery into 4t1 mouse breast carcinoma cells. Bull. Russ. State Med Univ. 2018, 6, 135–144. [Google Scholar] [CrossRef]
- Gallo, J.M.; Hafeli, U. Correspondence re: A. S. Lübbe et al., Preclinical Experiences with Magnetic Drug Targeting: Tolerance and Efficacy. Cancer Res., 56: 4694–4701, 1996; and Clinical Experiences with Magnetic Drug Targeting: A Phase I Study with 4′-Epidoxorubicin in 14 Patients with Advanced Solid Tumors. Cancer Res., 56: 4686–4693, 1996: Letter. Cancer Res. 1997, 57, 3063–3064. [Google Scholar] [PubMed]
- Zhou, Y.; Han, Y.; Li, G.; Yang, S.; Xiong, F.; Chu, F. Preparation of Targeted Lignin–Based Hollow Nanoparticles for the Delivery of Doxorubicin. Nanomaterials 2019, 9, 188. [Google Scholar] [CrossRef] [PubMed]
- Alexiou, C.; Tietze, R.; Schreiber, E.; Jurgons, R.; Richter, H.; Trahms, L.; Rahn, H.; Odenbach, S.; Lyer, S. Cancer therapy with drug loaded magnetic nanoparticles—Magnetic drug targeting. J. Magn. Magn. Mater. 2011, 323, 1404–1407. [Google Scholar] [CrossRef]
- Mejías, R.; Pérez-Yagüe, S.; Gutiérrez, L.; Cabrera, L.I.; Spada, R.; Acedo, P.; Serna, C.J.; Lázaro, F.J.; Villanueva, A.; Morales, M.D.P.; et al. Dimercaptosuccinic acid-coated magnetite nanoparticles for magnetically guided in vivo delivery of interferon gamma for cancer immunotherapy. Biomaterials 2011, 32, 2938–2952. [Google Scholar] [CrossRef] [PubMed]
- Lübbe, A.S.; Bergemann, C.; Riess, H.; Schriever, F.; Reichardt, P.; Possinger, K.; Matthias, M.; Dörken, B.; Herrmann, F.; Gürtler, R.; et al. Clinical experiences with magnetic drug targeting: A phase I study with 4’-epidoxorubicin in 14 patients with advanced solid tumors. Cancer Res. 1996, 56, 4686–4693. [Google Scholar]
- Magnetic-Targeted Doxorubicin in Treating Patients with Cancer Metastatic to the Liver. 2001. Available online: https://ClinicalTrials.gov/show/NCT00041808 (accessed on 21 October 2022).
- Safety and Efficacy of Doxorubicin Adsorbed to Magnetic Beads. 2001. Available online: https://ClinicalTrials.gov/show/NCT00054951 (accessed on 21 October 2022).
- Hour, F.Q.; Moghadam, A.J.; Shakeri-Zadeh, A.; Bakhtiyari, M.; Shabani, R.; Mehdizadeh, M. Magnetic targeted delivery of the SPIONs-labeled mesenchymal stem cells derived from human Wharton’s jelly in Alzheimer’s rat models. J. Control. Release 2020, 321, 430–441. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, H.; Zhang, T.; Wang, Y.; Jiao, W.; Lu, X.; Gao, X.; Xie, M.; Shan, Q.; Wen, N.; et al. Magnetic nanomaterials-mediated cancer diagnosis and therapy. Prog. Biomed. Eng. 2021, 4, 012005. [Google Scholar] [CrossRef]
- Liu, S.; Chiu-Lam, A.; Rivera-Rodriguez, A.; DeGroff, R.; Savliwala, S.; Sarna, N.; Rinaldi-Ramos, C.M. Long circulating tracer tailored for magnetic particle imaging. Nanotheranostics 2021, 5, 348–361. [Google Scholar] [CrossRef]
- Villalobos-Manzo, R.; Ríos-Castro, E.; Hernández-Hernández, J.M.; Oza, G.; Medina, M.A.; Tapia-Ramírez, J. Identification of Transferrin Receptor 1 (TfR1) Overexpressed in Lung Cancer Cells, and Internalization of Magnetic Au-CoFe2O4 Core-Shell Nanoparticles Functionalized with Its Ligand in a Cellular Model of Small Cell Lung Cancer (SCLC). Pharmaceutics 2022, 14, 1715. [Google Scholar] [CrossRef] [PubMed]
- Schleich, N.; Po, C.; Jacobs, D.; Ucakar, B.; Gallez, B.; Danhier, F.; Préat, V. Comparison of active, passive and magnetic targeting to tumors of multifunctional paclitaxel/SPIO-loaded nanoparticles for tumor imaging and therapy. J. Control. Release 2014, 194, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Lu, G.; Wen, X.; Li, F.; Ji, X.; Li, Q.; Wu, M.; Cheng, Q.; Yu, Y.; Tang, J.; et al. Magnetic nanoparticles coated with polyphenols for spatio-temporally controlled cancer photothermal/immunotherapy. J. Control. Release 2020, 326, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Żuk, M.; Podgórski, R.; Ruszczyńska, A.; Ciach, T.; Majkowska-Pilip, A.; Bilewicz, A.; Krysiński, P. Multifunctional Nanoparticles Based on Iron Oxide and Gold-198 Designed for Magnetic Hyperthermia and Radionuclide Therapy as a Potential Tool for Combined HER2-Positive Cancer Treatment. Pharmaceutics 2022, 14, 1680. [Google Scholar] [CrossRef]
- Maier-Hauff, K.; Ulrich, F.; Nestler, D.; Niehoff, H.; Wust, P.; Thiesen, B.; Jordan, A. Efficacy and safety of intratumoral thermotherapy using magnetic iron-oxide nanoparticles combined with external beam radiotherapy on patients with recurrent glioblastoma multiforme. J. Neuro-Oncol. 2011, 103, 317–324. [Google Scholar] [CrossRef]
- Luengo, Y.; Díaz-Riascos, Z.V.; García-Soriano, D.; Teran, F.J.; Artés-Ibáñez, E.J.; Ibarrola, O.; Somoza, A.; Miranda, R.; Schwartz, S.; Abasolo, I.; et al. Fine Control of In Vivo Magnetic Hyperthermia Using Iron Oxide Nanoparticles with Different Coatings and Degree of Aggregation. Pharmaceutics 2022, 14, 1526. [Google Scholar] [CrossRef]
- Myrovali, E.; Maniotis, N.; Samaras, T.; Angelakeris, M. Spatial focusing of magnetic particle hyperthermia. Nanoscale Adv. 2020, 2, 408–416. [Google Scholar] [CrossRef]
- Zrínyi, M. Intelligent polymer gels controlled by magnetic fields. Colloid Polym. Sci. 2000, 278, 98–103. [Google Scholar] [CrossRef]
- Czaun, M.; Hevesi, L.; Takafuji, M.; Ihara, H. A novel approach to magneto-responsive polymeric gels assisted by iron nanoparticles as nano cross-linkers. Chem. Commun. 2008, 18, 2124–2126. [Google Scholar] [CrossRef]
- Liu, T.Y.; Hu, S.H.; Liu, T.Y.; Liu, D.M.; Chen, S.Y. Magnetic-Sensitive Behavior of Intelligent Ferrogels for Controlled Release of Drug. Langmuir 2006, 22, 5974–5978. [Google Scholar] [CrossRef]
- Oliveira, H.; Pérez-Andrés, E.; Thevenot, J.; Sandre, O.; Berra, E.; Lecommandoux, S. Magnetic field triggered drug release from polymersomes for cancer therapeutics. J. Control. Release 2013, 169, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; Thum, C.; Zhang, Q.; Tu, W.; Pelaz, B.; Parak, W.J.; Zhang, Y.; Schneider, M. Inhibition of the cancer-associated TASK 3 channels by magnetically induced thermal release of Tetrandrine from a polymeric drug carrier. J. Control. Release 2016, 237, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.-H.; Chen, Y.-Y.; Liu, T.-C.; Tung, T.-H.; Liu, D.-M.; Chen, S.-Y. Remotely nano-rupturable yolk/shell capsules for magnetically-triggered drug release. Chem. Commun. 2011, 47, 1776–1778. [Google Scholar] [CrossRef] [PubMed]
- Álvarez, E.; Estévez, M.; Gallo-Cordova, A.; González, B.; Castillo, R.R.; Morales MD, P.; Vallet-Regí, M. Superparamagnetic Iron Oxide Nanoparticles Decorated Mesoporous Silica Nanosystem for Combined Antibiofilm Therapy. Pharmaceutics 2022, 14, 163. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Zhao, B.; Guo, W.; Li, Y.; Min, J.; Miao, W. Penetration and photodynamic ablation of drug-resistant biofilm by cationic Iron oxide nanoparticles. J. Control. Release 2022, 348, 911–923. [Google Scholar] [CrossRef] [PubMed]
- Hammad, M.; Nica, V.; Hempelmann, R. On-command controlled drug release by diels-Alder reaction using Bi-magnetic core/shell nano-carriers. Colloids Surf. B Biointerfaces 2017, 150, 15–22. [Google Scholar] [CrossRef]
- Wang, L.; Razzaq, M.Y.; Rudolph, T.; Heuchel, M.; Nöchel, U.; Mansfeld, U.; Jiang, Y.; Gould, O.E.C.; Behl, M.; Kratz, K.; et al. Reprogrammable, magnetically controlled polymeric nanocomposite actuators. Mater. Horiz. 2018, 5, 861–867. [Google Scholar] [CrossRef]
- Wang, H.; Zhu, Z.; Jin, H.; Wei, R.; Bi, L.; Zhang, W. Magnetic soft robots: Design, actuation, and function. J. Alloy Compd. 2022, 922, 166219. [Google Scholar] [CrossRef]
- Wei, Q.; Becherer, T.; Angioletti-Uberti, S.; Dzubiella, J.; Wischke, C.; Neffe, A.T.; Lendlein, A.; Ballauff, M.; Haag, R. Protein Interactions with Polymer Coatings and Biomaterials. Angew. Chem. Int. Ed. 2014, 53, 8004–8031. [Google Scholar] [CrossRef]
- Lu, S.; Zhao, P.; Deng, Y.; Liu, Y. Mechanistic Insights and Therapeutic Delivery through Micro/Nanobubble-Assisted Ultrasound. Pharmaceutics 2022, 14, 480. [Google Scholar] [CrossRef]
- Kohl, Y.; Kaiser, C.; Bost, W.; Stracke, F.; Fournelle, M.; Wischke, C.; Thielecke, H.; Lendlein, A.; Kratz, K.; Lemor, R. Preparation and biological evaluation of multifunctional PLGA-nanoparticles designed for photoacoustic imaging. Nanomed. Nanotechnol. Biol. Med. 2011, 7, 228–237. [Google Scholar] [CrossRef] [PubMed]
- Roovers, S.; Segers, T.; Lajoinie, G.; Deprez, J.; Versluis, M.; De Smedt, S.C.; Lentacker, I. The Role of Ultrasound-Driven Microbubble Dynamics in Drug Delivery: From Microbubble Fundamentals to Clinical Translation. Langmuir 2019, 35, 10173–10191. [Google Scholar] [CrossRef] [PubMed]
- Rapoport, N.; Gao, Z.; Kennedy, A. Multifunctional Nanoparticles for Combining Ultrasonic Tumor Imaging and Targeted Chemotherapy. J. Natl. Cancer Inst. 2007, 99, 1095–1106. [Google Scholar] [CrossRef] [PubMed]
- Ji, G.; Yang, J.; Chen, J. Preparation of novel curcumin-loaded multifunctional nanodroplets for combining ultrasonic development and targeted chemotherapy. Int. J. Pharm. 2014, 466, 314–320. [Google Scholar] [CrossRef] [PubMed]
- Lea-Banks, H.; Hynynen, K. Sub-millimetre precision of drug delivery in the brain from ultrasound-triggered nanodroplets. J. Control. Release 2021, 338, 731–741. [Google Scholar] [CrossRef] [PubMed]
- Munshi, N.; Rapoport, N.; Pitt, W.G. Ultrasonic activated drug delivery from Pluronic P-105 micelles. Cancer Lett. 1997, 118, 13–19. [Google Scholar] [CrossRef]
- Husseini, G.A.; Rapoport, N.Y.; Christensen, D.A.; Pruitt, J.D.; Pitt, W.G. Kinetics of ultrasonic release of doxorubicin from pluronic P105 micelles. Colloids Surf. B Biointerfaces 2002, 24, 253–264. [Google Scholar] [CrossRef]
- Xuan, J.; Pelletier, M.; Xia, H.; Zhao, Y. Ultrasound-Induced Disruption of Amphiphilic Block Copolymer Micelles. Macromol. Chem. Phys. 2011, 212, 498–506. [Google Scholar] [CrossRef]
- Chen, W.; Liu, P. Dendritic polyurethane-based prodrug as unimolecular micelles for precise ultrasound-activated localized drug delivery. Mater. Today Chem. 2022, 24, 100819. [Google Scholar] [CrossRef]
- Birlik Demirel, G.; Bayrak, Ş. Ultrasound/redox/pH-responsive hybrid nanoparticles for triple-triggered drug delivery. J. Drug Deliv. Sci. Technol. 2022, 71, 103267. [Google Scholar] [CrossRef]
- McHale, A.P.; Callan, J.F.; Nomikou, N.; Fowley, C.; Callan, B. Sonodynamic Therapy: Concept, Mechanism and Application to Cancer Treatment. In Therapeutic Ultrasound; Escoffre, J.-M., Bouakaz, A., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 429–450. [Google Scholar]
- Pan, X.; Bai, L.; Wang, H.; Wu, Q.; Wang, H.; Liu, S.; Xu, B.; Shi, X.; Liu, H. Metal-Organic-Framework-Derived Carbon Nanostructure Augmented Sonodynamic Cancer Therapy. Adv. Mater. 2018, 30, e1800180. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Altman, M.B.; Laszlo, A.; Straube, W.; Zoberi, I.; Hallahan, D.E.; Chen, H. Ultrasound Hyperthermia Technology for Radiosensitization. Ultrasound Med. Biol. 2019, 45, 1025–1043. [Google Scholar] [CrossRef] [PubMed]
- Van Elk, M.; Deckers, R.; Oerlemans, C.; Shi, Y.; Storm, G.; Vermonden, T.; Hennink, W.E. Triggered release of doxorubicin from temperature-sensitive poly(N-(2-hydroxypropyl)-methacrylamide mono/dilactate) grafted liposomes. Biomacromolecules 2014, 15, 1002–1009. [Google Scholar] [CrossRef] [PubMed]
- Gerweck, L.E.; Seetharaman, K. Cellular pH gradient in tumor versus normal tissue: Potential exploitation for the treatment of cancer. Cancer Res. 1996, 56, 1194–1198. [Google Scholar] [PubMed]
- Oh, K.T.; Yin, H.; Lee, E.S.; Bae, Y.H. Polymeric nanovehicles for anticancer drugs with triggering release mechanisms. J. Mater. Chem. 2007, 17, 3987–4001. [Google Scholar] [CrossRef]
- Kocak, G.; Tuncer, C.; Bütün, V. pH-Responsive polymers. Polym. Chem. 2017, 8, 144–176. [Google Scholar] [CrossRef]
- Philippova, O.E.; Hourdet, D.; Audebert, R.; Khokhlov, A.R. pH-Responsive Gels of Hydrophobically Modified Poly(acrylic acid). Macromolecules 1997, 30, 8278–8285. [Google Scholar] [CrossRef]
- Tonge, S.; Tighe, B. Responsive hydrophobically associating polymers: A review of structure and properties. Adv. Drug Deliv. Rev. 2001, 53, 109–122. [Google Scholar] [CrossRef]
- Ofridam, F.; Tarhini, M.; Lebaz, N.; Gagnière, É.; Mangin, D.; Elaissari, A. pH-sensitive polymers: Classification and some fine potential applications. Polym. Adv. Technol. 2021, 32, 1455–1484. [Google Scholar] [CrossRef]
- Cook, J.P.; Riley, D.J. pH induced swelling of PVP microgel particles—A first order phase transition? J. Colloid Interface Sci. 2012, 370, 67–72. [Google Scholar] [CrossRef]
- Ren, Y.; Jiang, X.; Yin, J. Copolymer of poly(4-vinylpyridine)-g-poly(ethylene oxide) respond sharply to temperature, pH and ionic strength. Eur. Polym. J. 2008, 44, 4108–4114. [Google Scholar] [CrossRef]
- Horta, A.; Molina, M.J.; Gómez-Antón, M.R.; Piérola, I.F. The pH inside a swollen polyelectrolyte gel: Poly(N-vinylimidazole). J. Phys. Chem. B 2008, 112, 10123–10129. [Google Scholar] [CrossRef] [PubMed]
- Murugan, E.; Rani, D.G.; Yogaraj, V. Drug delivery investigations of quaternised poly(propylene imine) dendrimer using nimesulide as a model drug. Colloids Surf. B Biointerfaces 2014, 114, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Sideratou, Z.; Tsiourvas, D.; Paleos, C.M. Quaternized Poly(propylene imine) Dendrimers as Novel pH-Sensitive Controlled-Release Systems. Langmuir 1999, 16, 1766–1769. [Google Scholar] [CrossRef]
- Ata, S.; Rasool, A.; Islam, A.; Bibi, I.; Rizwan, M.; Azeem, M.K.; Qureshi, A.U.R.; Iqbal, M. Loading of Cefixime to pH sensitive chitosan based hydrogel and investigation of controlled release kinetics. Int. J. Biol. Macromol. 2020, 155, 1236–1244. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wilhelm, J.; Li, W.; Li, S.; Wang, Z.; Huang, G.; Gao, J. Polycarbonate-based ultra-pH sensitive nanoparticles improve therapeutic window. Nat. Commun. 2020, 11, 5828. [Google Scholar] [CrossRef]
- Simões, M.G.; Hugo, A.; Gómez-Zavaglia, A.; Simões, P.N.; Alves, P. Formulation and Characterization of Stimuli-Responsive Lecithin-Based Liposome Complexes with Poly(acrylic acid)/Poly(N,N-dimethylaminoethyl methacrylate) and Pluronic(®) Copolymers for Controlled Drug Delivery. Pharmaceutics 2022, 14, 735. [Google Scholar] [CrossRef]
- Kang, S.I.; Bae, Y.H. pH-Induced solubility transition of sulfonamide-based polymers. J. Control. Release 2002, 80, 145–155. [Google Scholar] [CrossRef]
- Park, S.Y.; Bae, Y.H. Novel pH-sensitive polymers containing sulfonamide groups. Macromol. Rapid Commun. 1999, 20, 269–273. [Google Scholar] [CrossRef]
- Gupta, P.; Purwar, R. Electrospun pH responsive poly (acrylic acid-co-acrylamide) hydrogel nanofibrous mats for drug delivery. J. Polym. Res. 2020, 27, 296. [Google Scholar] [CrossRef]
- Thomas, J.L.; You, H.; Tirrell, D.A. Tuning the response of a pH-sensitive membrane switch. J. Am. Chem. Soc. 1995, 117, 2949–2950. [Google Scholar] [CrossRef]
- Sauer, M.; Streich, D.; Meier, W. pH-Sensitive Nanocontainers. Adv. Mater. 2001, 13, 1649–1651. [Google Scholar] [CrossRef]
- Fundueanu, G.; Constantin, M.; Turtoi, M.; Bucatariu, S.-M.; Cosman, B.; Anghelache, M.; Voicu, G.; Calin, M. Bio-Responsive Carriers for Controlled Delivery of Doxorubicin to Cancer Cells. Pharmaceutics 2022, 14, 865. [Google Scholar] [CrossRef] [PubMed]
- Bellomo, E.G.; Wyrsta, M.D.; Pakstis, L.; Pochan, D.J.; Deming, T. Stimuli-responsive polypeptide vesicles by conformation-specific assembly. Nat. Mater. 2004, 3, 244–248. [Google Scholar] [CrossRef]
- Deirram, N.; Zhang, C.; Kermaniyan, S.S.; Johnston, A.P.; Such, G.K. pH-Responsive Polymer Nanoparticles for Drug Delivery. Macromol. Rapid Commun. 2019, 40, 1800917. [Google Scholar] [CrossRef]
- Lee, S.; Saito, K.; Lee, H.R.; Lee, M.J.; Shibasaki, Y.; Oishi, Y.; Kim, B.S. Hyperbranched double hydrophilic block copolymer micelles of poly(ethylene oxide) and polyglycerol for pH-responsive drug delivery. Biomacromolecules 2012, 13, 1190–1196. [Google Scholar] [CrossRef]
- Li, Y.; Song, L.; Lin, J.; Ma, J.; Pan, Z.; Zhang, Y.; Hou, Z. Programmed Nanococktail Based on pH-Responsive Function Switch for Self-Synergistic Tumor-Targeting Therapy. ACS Appl. Mater. Interfaces 2017, 9, 39127–39142. [Google Scholar] [CrossRef]
- Feng, X.; Li, D.; Han, J.; Zhuang, X.; Ding, J. Schiff base bond-linked polysaccharide–doxorubicin conjugate for upregulated cancer therapy. Mater. Sci. Eng. C 2017, 76, 1121–1128. [Google Scholar] [CrossRef]
- Storrie, H.; Mooney, D. Sustained delivery of plasmid DNA from polymeric scaffolds for tissue engineering. Adv. Drug Deliv. Rev. 2006, 58, 500–514. [Google Scholar] [CrossRef]
- You, J.-O.; Auguste, D.T. Nanocarrier Cross-Linking Density and pH Sensitivity Regulate Intracellular Gene Transfer. Nano Lett. 2009, 9, 4467–4473. [Google Scholar] [CrossRef]
- Shi, B.; Zhang, H.; Bi, J.; Dai, S. Endosomal pH responsive polymers for efficient cancer targeted gene therapy. Colloids Surf. B Biointerfaces 2014, 119, 55–65. [Google Scholar] [CrossRef] [PubMed]
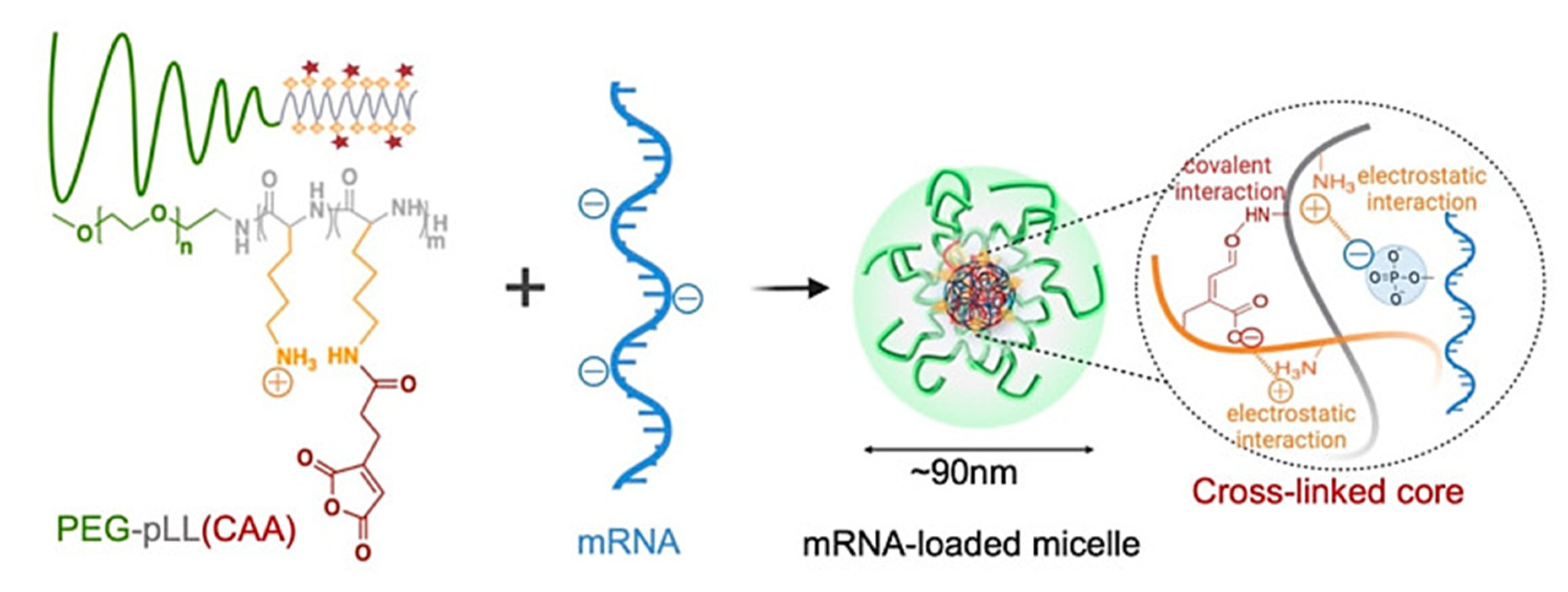
- Yang, W.; Chen, P.; Boonstra, E.; Hong, T.; Cabral, H. Polymeric Micelles with pH-Responsive Cross-Linked Core Enhance In Vivo mRNA Delivery. Pharmaceutics 2022, 14, 1205. [Google Scholar] [CrossRef]
- Varkouhi, A.K.; Lammers, T.; Schiffelers, R.; van Steenbergen, M.J.; Hennink, W.E.; Storm, G. Gene silencing activity of siRNA polyplexes based on biodegradable polymers. Eur. J. Pharm. Biopharm. 2011, 77, 450–457. [Google Scholar] [CrossRef] [PubMed]
- Meyer, M.; Dohmen, C.; Philipp, A.; Kiener, D.; Maiwald, G.; Scheu, C.; Wagner, E. Synthesis and biological evaluation of a bioresponsive and endosomolytic siRNA-polymer conjugate. Mol. Pharm. 2009, 6, 752–762. [Google Scholar] [CrossRef] [PubMed]
- Dohmen, C.; Edinger, D.; Fröhlich, T.; Schreiner, L.; Lächelt, U.; Troiber, C.; Rädler, J.; Hadwiger, P.; Vornlocher, H.-P.; Wagner, E. Nanosized Multifunctional Polyplexes for Receptor-Mediated SiRNA Delivery. ACS Nano 2012, 6, 5198–5208. [Google Scholar] [CrossRef]
- Bouillon, C.; Paolantoni, D.; Rote, J.C.; Bessin, Y.; Peterson, L.W.; Dumy, P.; Ulrich, S. Degradable Hybrid Materials Based on Cationic Acylhydrazone Dynamic Covalent Polymers Promote DNA Complexation through Multivalent Interactions. Chem.-A Eur. J. 2014, 20, 14705–14714. [Google Scholar] [CrossRef]
- de la Cruz-López, K.G.; Castro-Muñoz, L.J.; Reyes-Hernández, D.O.; García-Carrancá, A.; Manzo-Merino, J. Lactate in the Regulation of Tumor Microenvironment and Therapeutic Approaches. Front. Oncol. 2019, 9, 1143. [Google Scholar] [CrossRef]
- Liu, Y.; Ran, Y.; Ge, Y.; Raza, F.; Li, S.; Zafar, H.; Li, F. pH-Sensitive Peptide Hydrogels as a Combination Drug Delivery System for Cancer Treatment. Pharmaceutics 2022, 14, 652. [Google Scholar] [CrossRef]
- Du, J.-Z.; Sun, T.-M.; Song, W.-J.; Wu, J.; Wang, J. A Tumor-Acidity-Activated Charge-Conversional Nanogel as an Intelligent Vehicle for Promoted Tumoral-Cell Uptake and Drug Delivery. Angew. Chem. Int. Ed. 2010, 49, 3621–3626. [Google Scholar] [CrossRef]
- Ulijn, R.V. Enzyme-responsive materials: A new class of smart biomaterials. J. Mater. Chem. 2006, 16, 2217–2225. [Google Scholar] [CrossRef]
- Hu, J.; Zhang, G.; Liu, S. Enzyme-responsive polymeric assemblies, nanoparticles and hydrogels. Chem. Soc. Rev. 2012, 41, 5933–5949. [Google Scholar] [CrossRef] [PubMed]
- Amir, R.J.; Zhong, S.; Pochan, D.J.; Hawker, C.J. Enzymatically Triggered Self-Assembly of Block Copolymers. J. Am. Chem. Soc. 2009, 131, 13949–13951. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, N.; Ogino, N.; Narita, T.; Kitamura, A.S.; Akiyoshi, K. Enzyme-Responsive Molecular Assembly System with Amylose-Primer Surfactants. J. Am. Chem. Soc. 2006, 129, 458–459. [Google Scholar] [CrossRef] [PubMed]
- Klinger, D.; Aschenbrenner, E.M.; Weiss, C.K.; Landfester, K. Enzymatically degradable nanogels by inverse miniemulsion copolymerization of acrylamide with dextran methacrylates as crosslinkers. Polym. Chem. 2012, 3, 204–216. [Google Scholar] [CrossRef]
- Song, K.; Tang, Z.; Song, Z.; Meng, S.; Yang, X.; Guo, H.; Zhu, Y.; Wang, X. Hyaluronic Acid-Functionalized Mesoporous Silica Nanoparticles Loading Simvastatin for Targeted Therapy of Atherosclerosis. Pharmaceutics 2022, 14, 1265. [Google Scholar] [CrossRef]
- He, Y.; Lei, L.; Cao, J.; Yang, X.; Cai, S.; Tong, F.; Huang, D.; Mei, H.; Luo, K.; Gao, H.; et al. A combinational chemo-immune therapy using an enzyme-sensitive nanoplatform for dual-drug delivery to specific sites by cascade targeting. Sci. Adv. 2021, 7, eaba0776. [Google Scholar] [CrossRef]
- Basel, M.T.; Shrestha, T.B.; Troyer, D.L.; Bossmann, S.H. Protease-Sensitive, Polymer-Caged Liposomes: A Method for Making Highly Targeted Liposomes Using Triggered Release. ACS Nano 2011, 5, 2162–2175. [Google Scholar] [CrossRef]
- Awino, J.K.; Gudipati, S.; Hartmann, A.K.; Santiana, J.J.; Cairns-Gibson, D.F.; Gomez, N.; Rouge, J.L. Nucleic Acid Nanocapsules for Enzyme-Triggered Drug Release. J. Am. Chem. Soc. 2017, 139, 6278–6281. [Google Scholar] [CrossRef]
- Liwinska, W.; Waleka-Bagiel, E.; Stojek, Z.; Karbarz, M.; Zabost, E. Enzyme-triggered- and tumor-targeted delivery with tunable, methacrylated poly(ethylene glycols) and hyaluronic acid hybrid nanogels. Drug Deliv. 2022, 29, 2561–2578. [Google Scholar] [CrossRef]
- Liang, M.; Li, N.; Liu, F.; Zeng, N.; Yu, C.; Li, S. Apurinic/apyrimidinic endonuclease triggered doxorubicin-releasing DNA nanoprism for target therapy. Cell Cycle 2022, 1–8. [Google Scholar] [CrossRef]
- Alkekhia, D.; LaRose, C.; Shukla, A. β-Lactamase-Responsive Hydrogel Drug Delivery Platform for Bacteria-Triggered Cargo Release. ACS Appl. Mater. Interfaces 2022, 14, 27538–27550. [Google Scholar] [CrossRef] [PubMed]
- Hovgaard, L.; Brøndsted, H. Dextran hydrogels for colon-specific drug delivery. J. Control. Release 1995, 36, 159–166. [Google Scholar] [CrossRef]
- Saffran, M.; Kumar, G.S.; Savariar, C.; Burnham, J.C.; Williams, F.; Neckers, D.C. A New Approach to the Oral Administration of Insulin and Other Peptide Drugs. Science 1986, 233, 1081–1084. [Google Scholar] [CrossRef] [PubMed]
- Ghandehari, H.; Kopeckova, P.; Kopecek, J. In vitro degradation of pH-sensitive hydrogels containing aromatic azo bonds. Biomaterials 1997, 18, 861–872. [Google Scholar] [CrossRef]
- Rao, J.; Khan, A. Enzyme Sensitive Synthetic Polymer Micelles Based on the Azobenzene Motif. J. Am. Chem. Soc. 2013, 135, 14056–14059. [Google Scholar] [CrossRef]
- Wang, C.-Y.; Sun, M.; Fan, Z.; Du, J.-Z. Intestine Enzyme-responsive Polysaccharide-based Hydrogel to Open Epithelial Tight Junctions for Oral Delivery of Imatinib against Colon Cancer. Chin. J. Polym. Sci. 2022, 40, 1154–1164. [Google Scholar] [CrossRef]
- Kasuya, Y.; Lu, Z.-R.; Kopečková, P.; Minko, T.; Tabibi, S.; Kopeček, J. Synthesis and characterization of HPMA copolymer–aminopropylgeldanamycin conjugates. J. Control. Release 2001, 74, 203–211. [Google Scholar] [CrossRef]
- Duncan, R. Designing polymer conjugates as lysosomotropic nanomedicines. Biochem. Soc. Trans. 2007, 35, 56–60. [Google Scholar] [CrossRef]
- Šubr, V.; Strohalm, J.; Hirano, T.; Ito, Y.; Ulbrich, K. Poly[N-(2-hydroxypropyl)methacrylamide] conjugates of methotrexate: Synthesis and in vitro drug release. J. Control. Release 1997, 49, 123–132. [Google Scholar] [CrossRef]
- Lu, Z.-R.; Gao, S.-Q.; Kopečková, P.; Kopeček, J. Synthesis of Bioadhesive Lectin-HPMA Copolymer−Cyclosporin Conjugates. Bioconjugate Chem. 1999, 11, 3–7. [Google Scholar] [CrossRef]
- Ferguson, E.L.; Duncan, R. Dextrin−Phospholipase A2: Synthesis and Evaluation as a Bioresponsive Anticancer Conjugate. Biomacromolecules 2009, 10, 1358–1364. [Google Scholar] [CrossRef] [PubMed]
- Seymour, L.W.; Ferry, D.R.; Kerr, D.J.; Rea, D.; Whitlock, M.; Poyner, R.; Boivin, C.; Hesslewood, S.; Twelves, C.; Blackie, R.; et al. Phase II studies of polymer-doxorubicin (PK1, FCE28068) in the treatment of breast, lung and colorectal cancer. Int. J. Oncol. 2009, 34, 1629–1636. [Google Scholar] [CrossRef] [PubMed]
- Veronese, F.M.; Schiavon, O.; Pasut, G.; Mendichi,, R.; Andersson, L.; Tsirk, A.; Ford, J.; Wu, G.; Kneller,, S.; Davies, J.; et al. PEG—Doxorubicin Conjugates: Influence of Polymer Structure on Drug Release, in Vitro Cytotoxicity, Biodistribution, and Antitumor Activity. Bioconjugate Chem. 2005, 16, 775–784. [Google Scholar] [CrossRef] [PubMed]
- Song, S.J.; Choi, J.S. Enzyme-Responsive Amphiphilic Peptide Nanoparticles for Biocompatible and Efficient Drug Delivery. Pharmaceutics 2022, 14, 143. [Google Scholar] [CrossRef]
- Wen, J.; Anderson, S.M.; Du, J.; Yan, M.; Wang, J.; Shen, M.; Lu, Y.; Segura, T. Controlled Protein Delivery Based on Enzyme-Responsive Nanocapsules. Adv. Mater. 2011, 23, 4549–4553. [Google Scholar] [CrossRef]
- Garripelli, V.K.; Kim, J.-K.; Son, S.; Kim, W.J.; Repka, M.A.; Jo, S. Matrix metalloproteinase-sensitive thermogelling polymer for bioresponsive local drug delivery. Acta Biomater. 2011, 7, 1984–1992. [Google Scholar] [CrossRef]
- Ge, J.; Lu, D.; Yang, C.; Liu, Z. A Lipase-Responsive Vehicle Using Amphipathic Polymer Synthesized with the Lipase as Catalyst. Macromol. Rapid Commun. 2011, 32, 546–550. [Google Scholar] [CrossRef]
- Jeong, Y.; Joo, M.K.; Bahk, K.H.; Choi, Y.Y.; Kim, H.-T.; Kim, W.-K.; Lee, H.J.; Sohn, Y.S.; Jeong, B. Enzymatically degradable temperature-sensitive polypeptide as a new in-situ gelling biomaterial. J. Control. Release 2009, 137, 25–30. [Google Scholar] [CrossRef]
- Wu, Q.; Wang, L.; Yu, H.; Wang, J.; Chen, Z. Organization of Glucose-Responsive Systems and Their Properties. Chem. Rev. 2011, 111, 7855–7875. [Google Scholar] [CrossRef]
- Lin, Y.-J.; Mi, F.-L.; Lin, P.-Y.; Miao, Y.-B.; Huang, T.; Chen, K.-H.; Chen, C.-T.; Chang, Y.; Sung, H.-W. Strategies for improving diabetic therapy via alternative administration routes that involve stimuli-responsive insulin-delivering systems. Adv. Drug Deliv. Rev. 2019, 139, 71–82. [Google Scholar] [CrossRef]
- Albin, G.; Horbett, T.A.; Ratner, B.D. Glucose sensitive membranes for controlled delivery of insulin: Insulin transport studies. J. Control. Release 1985, 2, 153–164. [Google Scholar] [CrossRef]
- Traitel, T.; Cohen, Y.; Kost, J. Characterization of glucose-sensitive insulin release systems in simulated in vivo conditions. Biomaterials 2000, 21, 1679–1687. [Google Scholar] [CrossRef]
- Ishihara, K.; Kobayashi, M.; Ishimaru, N.; Shinohara, I. Glucose Induced Permeation Control of Insulin through a Complex Membrane Consisting of Immobilized Glucose Oxidase and a Poly(amine). Polym. J. 1984, 16, 625–631. [Google Scholar] [CrossRef]
- Hassan, C.M.; Doyle, F.J.; Peppas, N.A. Dynamic Behavior of Glucose-Responsive Poly(methacrylic acid-g-ethylene glycol) Hydrogels. Macromolecules 1997, 30, 6166–6173. [Google Scholar] [CrossRef]
- Ito, Y.; Casolaro, M.; Kono, K.; Imanishi, Y. An insulin-releasing system that is responsive to glucose. J. Control. Release 1989, 10, 195–203. [Google Scholar] [CrossRef]
- Huang, H.Y.; Shaw, J.; Yip, C.; Wu, X.Y. Microdomain pH Gradient and Kinetics Inside Composite Polymeric Membranes of pH and Glucose Sensitivity. Pharm. Res. 2008, 25, 1150–1157. [Google Scholar] [CrossRef]
- Cheng, S.-Y.; Gross, J.; Sambanis, A. Hybrid pancreatic tissue substitute consisting of recombinant insulin-secreting cells and glucose-responsive material. Biotechnol. Bioeng. 2004, 87, 863–873. [Google Scholar] [CrossRef]
- Miyata, T.; Jikihara, A.; Nakamae, K.; Hoffman, A.S. Preparation of reversibly glucose-responsive hydrogels by covalent immobilization of lectin in polymer networks having pendant glucose. J. Biomater. Sci. Polym. Ed. 2004, 15, 1085–1098. [Google Scholar] [CrossRef]
- Liu, F.; Song, S.C.; Mix, D.; Baudyš, M.; Kim, S.W. Glucose-Induced Release of Glycosylpoly(ethylene glycol) Insulin Bound to a Soluble Conjugate of Concanavalin A. Bioconjugate Chem. 1997, 8, 664–672. [Google Scholar] [CrossRef]
- Obaidat, A.A.; Park, K. Characterization of Glucose Dependent Gel-Sol Phase Transition of the Polymeric Glucose-Concanavalin A Hydrogel System. Pharm. Res. 1996, 13, 989–995. [Google Scholar] [CrossRef]
- Kokufata, E.; Zhang, Y.-Q.; Tanaka, T. Saccharide-sensitive phase transition of a lectin-loaded gel. Nature 1991, 351, 302–304. [Google Scholar] [CrossRef]
- Ye, T.; Yan, S.; Hu, Y.; Ding, L.; Wu, W. Synthesis and volume phase transition of concanavalin A-based glucose-responsive nanogels. Polym. Chem. 2014, 5, 186–194. [Google Scholar] [CrossRef]
- Yin, R.; Wang, K.; Du, S.; Chen, L.; Nie, J.; Zhang, W. Design of genipin-crosslinked microgels from concanavalin A and glucosyloxyethyl acrylated chitosan for glucose-responsive insulin delivery. Carbohydr. Polym. 2014, 103, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Kitano, S.; Kataoka, K.; Koyama, Y.; Okano, T.; Sakurai, Y. Glucose-responsive complex formation between poly(vinyl alcohol) and poly(N-vinyl-2-pyrrolidone) with pendent phenylboronic acid moieties. Die Makromol. Chem. Rapid Commun. 1991, 12, 227–233. [Google Scholar] [CrossRef]
- Kitano, S.; Koyama, Y.; Kataoka, K.; Okano, T.; Sakurai, Y. A novel drug delivery system utilizing a glucose responsive polymer complex between poly (vinyl alcohol) and poly (N-vinyl-2-pyrrolidone) with a phenylboronic acid moiety. J. Control. Release 1992, 19, 161–170. [Google Scholar] [CrossRef]
- Kataoka, K.; Miyazaki, H.; Okano, T.; Sakurai, Y. Sensitive Glucose-Induced Change of the Lower Critical Solution Temperature of Poly[N,N-(dimethylacrylamide)-co-3-(acrylamido)-phenylboronic acid] in Physiological Saline. Macromolecules 1994, 27, 1061–1062. [Google Scholar] [CrossRef]
- Matsumoto, A.; Kurata, T.; Shiino, D.; Kataoka, K. Swelling and Shrinking Kinetics of Totally Synthetic, Glucose-Responsive Polymer Gel Bearing Phenylborate Derivative as a Glucose-Sensing Moiety. Macromolecules 2004, 37, 1502–1510. [Google Scholar] [CrossRef]
- Zhang, X.; Lü, S.; Gao, C.; Chen, C.; Zhang, X.; Liu, M. Highly stable and degradable multifunctional microgel for self-regulated insulin delivery under physiological conditions. Nanoscale 2013, 5, 6498–6506. [Google Scholar] [CrossRef]
- Yao, Y.; Zhao, L.; Yang, J.; Yang, J. Glucose-Responsive Vehicles Containing Phenylborate Ester for Controlled Insulin Release at Neutral pH. Biomacromolecules 2012, 13, 1837–1844. [Google Scholar] [CrossRef]
- Huang, Q.; Yu, H.; Wang, L.; Shen, D.; Chen, X.; Wang, N. Synthesis and testing of polymer grafted mesoporous silica as glucose-responsive insulin release drug delivery systems. Eur. Polym. J. 2021, 157, 110651. [Google Scholar] [CrossRef]
- Shao, Z.; Yin, T.; Jiang, J.; He, Y.; Xiang, T.; Zhou, S. Wound microenvironment self-adaptive hydrogel with efficient angiogenesis for promoting diabetic wound healing. Bioact. Mater. 2023, 20, 561–573. [Google Scholar] [CrossRef] [PubMed]
- Shan, M.; Gong, C.; Li, B.; Wu, G. A pH, glucose, and dopamine triple-responsive, self-healable adhesive hydrogel formed by phenylborate–catechol complexation. Polym. Chem. 2017, 8, 2997–3005. [Google Scholar] [CrossRef]
- Pei, X.; Fang, L.; Chen, W.; Wen, X.; Bai, L.; Ba, X. Facile Fabrication of Multiresponsive Self-Healing Hydrogels with Logic-Gate Responses. Macromol. Chem. Phys. 2021, 222, 2000339. [Google Scholar] [CrossRef]
- Lim, S.L.; Ooi, C.-W.; Low, L.E.; Tan, W.S.; Chan, E.-S.; Ho, K.L.; Tey, B.T. Synthesis of poly(acrylamide)-based hydrogel for bio-sensing of hepatitis B core antigen. Mater. Chem. Phys. 2020, 243, 122578. [Google Scholar] [CrossRef]
- Bae, S.W.; Lee, J.S.; Harms, V.M.; Murphy, W.L. Dynamic, Bioresponsive Hydrogels via Changes in DNA Aptamer Conformation. Macromol. Biosci. 2019, 19, e1800353. [Google Scholar] [CrossRef] [PubMed]
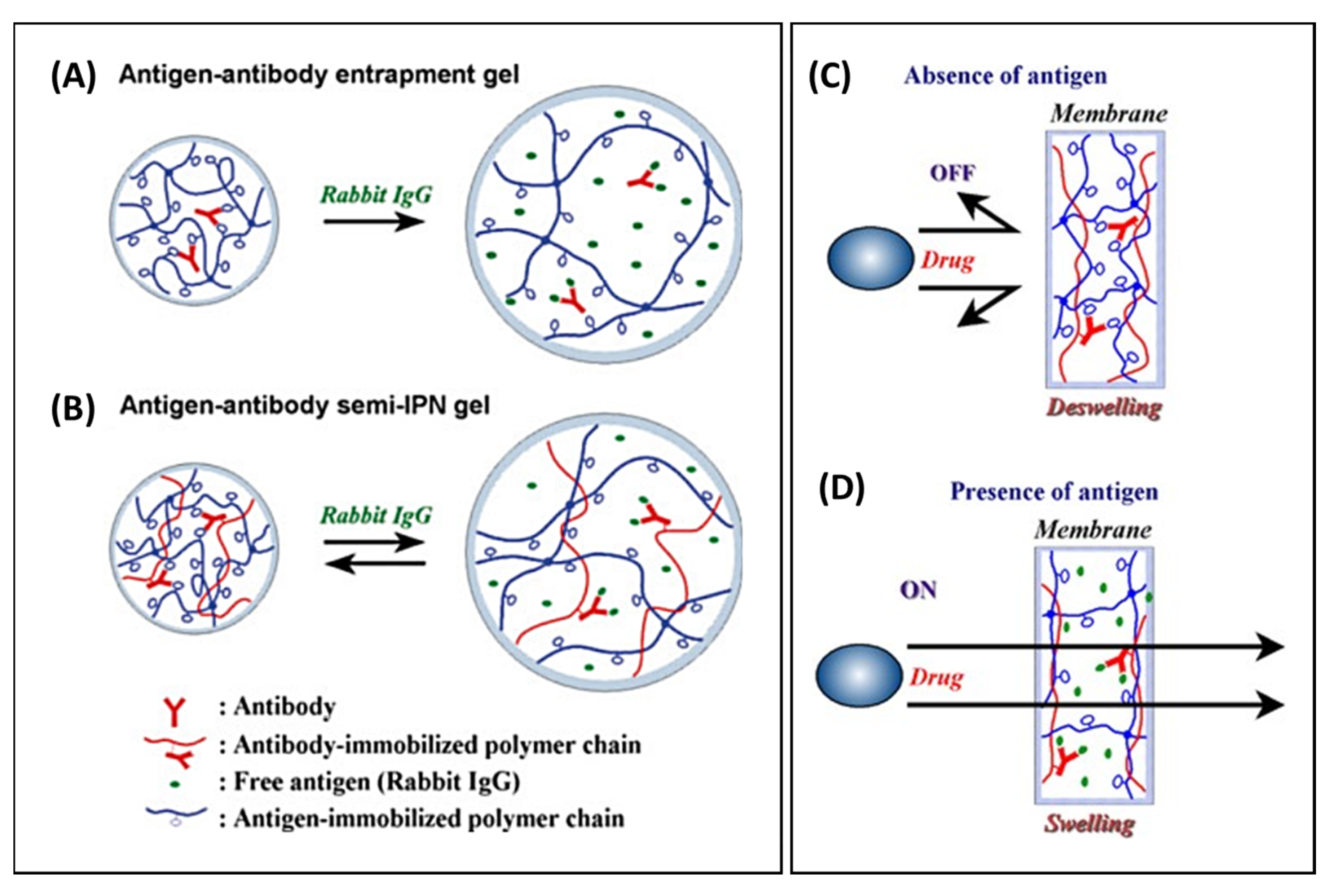
- Miyata, T.; Asami, N.; Uragami, T. A reversibly antigen-responsive hydrogel. Nature 1999, 399, 766–769. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.-R.; Kopečková, P.; Kopeček, J. Antigen Responsive Hydrogels Based on Polymerizable Antibody Fab′ Fragment. Macromol. Biosci. 2003, 3, 296–300. [Google Scholar] [CrossRef]
- Minrath, I.; Arbeiter, D.; Schmitz, K.-P.; Sternberg, K.; Petersen, S. In vitro characterization of polyacrylamide hydrogels for application as implant coating for stimulus-responsive local drug delivery. Polym. Adv. Technol. 2014, 25, 1234–1241. [Google Scholar] [CrossRef]
- Zhang, R.; Bowyer, A.; Eisenthal, R.; Hubble, J. A smart membrane based on an antigen-responsive hydrogel. Biotechnol. Bioeng. 2007, 97, 976–984. [Google Scholar] [CrossRef]
- Miyata, T.; Asami, N.; Okita, Y.; Uragami, T. Controlled permeation of model drugs through a bioconjugated membrane with antigen–antibody complexes as reversible crosslinks. Polym. J. 2010, 42, 834–837. [Google Scholar] [CrossRef][Green Version]
- Miyata, T.; Asami, N.; Uragami, T. Structural design of stimuli-responsive bioconjugated hydrogels that respond to a target antigen. J. Polym. Sci. Part B Polym. Phys. 2009, 47, 2144–2157. [Google Scholar] [CrossRef]
- Song, W.; You, J.; Zhang, Y.; Yang, Q.; Jiao, J.; Zhang, H. Recent Studies on Hydrogels Based on H2O2-Responsive Moieties: Mechanism, Preparation and Application. Gels 2022, 8, 361. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Bae, S.; Hong, D.; Lim, H.; Yoon, J.H.; Hwang, O.; Park, S.; Ke, Q.; Khang, G.; Kang, P.M. H2O2-responsive molecularly engineered polymer nanoparticles as ischemia/reperfusion-targeted nanotherapeutic agents. Sci. Rep. 2013, 3, 2233. [Google Scholar] [CrossRef]
- Yu, F.; Wu, H.; Tang, Y.; Xu, Y.; Qian, X.; Zhu, W. Temperature-sensitive copolymer-coated fluorescent mesoporous silica nanoparticles as a reactive oxygen species activated drug delivery system. Int. J. Pharm. 2018, 536, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Noddeland, H.K.; Kemp, P.; Urquhart, A.J.; Herchenhan, A.; Rytved, K.A.; Petersson, K.; Jensen, L.B. Reactive Oxygen Species-Responsive Polymer Nanoparticles to Improve the Treatment of Inflammatory Skin Diseases. ACS Omega 2022, 7, 25055–25065. [Google Scholar] [CrossRef] [PubMed]
- Jeanmaire, D.; Laliturai, J.; Almalik, A.; Carampin, P.; D’Arcy, R.; Lallana, E.; Evans, R.; Winpenny, R.E.P.; Tirelli, N. Chemical specificity in REDOX-responsive materials: The diverse effects of different Reactive Oxygen Species (ROS) on polysulfide nanoparticles. Polym. Chem. 2014, 5, 1393–1404. [Google Scholar] [CrossRef]
- Ma, B.; Xu, H.; Zhuang, W.; Wang, Y.; Li, G.; Wang, Y. Reactive Oxygen Species Responsive Theranostic Nanoplatform for Two-Photon Aggregation-Induced Emission Imaging and Therapy of Acute and Chronic Inflammation. ACS Nano 2020, 14, 5862–5873. [Google Scholar] [CrossRef]
- Mollazadeh, S.; Mackiewicz, M.; Yazdimamaghani, M. Recent advances in the redox-responsive drug delivery nanoplatforms: A chemical structure and physical property perspective. Mater. Sci. Eng. C 2021, 118, 111536. [Google Scholar] [CrossRef]
- Bej, R.; Dey, P.; Ghosh, S. Disulfide chemistry in responsive aggregation of amphiphilic systems. Soft Matter 2020, 16, 11–26. [Google Scholar] [CrossRef]
- Wu, P.; Gao, J.; Prasad, P.; Dutta, K.; Kanjilal, P.; Thayumanavan, S. Influence of Polymer Structure and Architecture on Drug Loading and Redox-Triggered Release. Biomacromolecules 2022, 23, 339–348. [Google Scholar] [CrossRef]
- Hironaka, K.; Yoshihara, E.; Nabil, A.; Lai, J.J.; Kikuchi, A.; Ebara, M. Conjugation of antibody with temperature-responsive polymer via in situ click reaction to enable biomarker enrichment for increased diagnostic sensitivity. Biomater. Sci. 2021, 9, 4870–4879. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Hamilton, L.E.; Mather, P.T.; Henderson, J.H. Cell-Responsive Shape Memory Polymers. ACS Biomater. Sci. Eng. 2022, 8, 2960–2969. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.-Z.; Xu, X.-L. Endogenous Enzyme-responsive Nanoplatforms for Anti-tumor Therapy. Curr. Drug Targets 2021, 22, 845–855. [Google Scholar] [CrossRef]

















Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tuncaboylu, D.C.; Wischke, C. Opportunities and Challenges of Switchable Materials for Pharmaceutical Use. Pharmaceutics 2022, 14, 2331. https://doi.org/10.3390/pharmaceutics14112331
Tuncaboylu DC, Wischke C. Opportunities and Challenges of Switchable Materials for Pharmaceutical Use. Pharmaceutics. 2022; 14(11):2331. https://doi.org/10.3390/pharmaceutics14112331
Chicago/Turabian StyleTuncaboylu, Deniz Ceylan, and Christian Wischke. 2022. "Opportunities and Challenges of Switchable Materials for Pharmaceutical Use" Pharmaceutics 14, no. 11: 2331. https://doi.org/10.3390/pharmaceutics14112331
APA StyleTuncaboylu, D. C., & Wischke, C. (2022). Opportunities and Challenges of Switchable Materials for Pharmaceutical Use. Pharmaceutics, 14(11), 2331. https://doi.org/10.3390/pharmaceutics14112331