
Mechanical Activation by Ball Milling as a Strategy to Prepare Highly Soluble Pharmaceutical Formulations in the Form of Co-Amorphous, Co-Crystals, or Polymorphs

 ,
,  , ,
, ,
Abstract
1. Introduction
2. Pharmaceutical Formulations Based on Structural Properties
2.1. Amorphous Pharmaceutical Formulations Prepared by Milling
2.2. Drug Co-Crystals Prepared by Mechanical Activation
2.3. Drug Polymorphs as a Result of the Milling Process
3. Factors Affecting Drug Formulations during the Mechanical Activation Process
3.1. Ball Milling Instruments
3.2. Temperature during the Milling Process
3.3. Phase Transformation Mechanism by Ball Milling and Temperature Effect
3.4. Solvent Effect
3.5. Effect Changing Composition
3.6. Milling Time

{kind=link}
{kind=link}
{kind=link}
| # | Drug 1 | Drug 2 Molar-Ratio | Amorphous Stability (Storage-Conditions) | Mill Type | Volume Cell Material | Balls-Num. Material and Sample Weight | Milling Frequency | Milling Temp. (°C) | Milling Time | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| 1A | Mebendazole | Twenty different amino acids 1:1 | Not reported | Oscillatory ball mill | 25 mL Jar | 2 (d = 12 mm) stainless steel balls 1000 mg | 30 Hz | Not specified | 1, 5, 15, 30, and 60 min | [84] |
| Carvedilol | ||||||||||
| Carbamazepine | ||||||||||
| Simvastatin | ||||||||||
| Indomethacin | ||||||||||
| Furosemide | ||||||||||
| 2A | Furosemide | Arginine | Dry conditions at 25 °C or 40 °C for 15 months of storage | Oscillatory ball mill | 25 mL Jar | 2 (d = 12 mm) stainless steel balls 750 mg | 30 Hz | 5 °C | 180 min | [85] |
| Nitrofurantoin | ||||||||||
| Cimetidine | Citrulline | |||||||||
| Mebendazole | ||||||||||
| 3A | Sulfathiazole | Polyvinylpyrrolidone Xpvp: 0.6 and 0.7 | Storage at 4 °C over a year | Planetary mill | 50 cm3 ZrO2 milling jars | 3 balls (d = 20 mm) ZrO2. 2.5 g | 6.6 Hz | Room temperature | 10 h (15 h total) 10 min pauses after every 20 min | [86] |
| Sulfadimidine | ||||||||||
| 4A | Naproxen | Cimetidine 1:2, 1:1, 2:1 | Dry conditions at 4, 25 and 40 °C for up to 33 days or further extended to 186 days | Oscillatory ball mill | 25 mL stainless steel milling jar | 2 (d = 12 mm) stainless steel balls 1 g of sample per grinding cell | 30 Hz | 4 °C ± 2 °C | 60 min | [87] |
| 5A | γ-Indomethacin | Ranitidine hydrochloride 2:1, 1:1, 1:2 | Dry conditions at 4, 25, and 40 °C up to 30 days | Oscillatory ball mill | 25 mL stainless steel milling jar | 2 (d = 12 mm) stainless steel balls 1 g of sample per grinding cell | 30 Hz | 4 °C ± 2 °C | 60 min | [28] |
| 6A | γ-Indomethacin | None | Not reported | Oscillatory ball mill | 25 mL stainless steel milling jar | 6 (d = 9 mm) stainless steel balls 1 g of sample per grinding cell | 30 Hz | 4 °C ± 2 °C | 6 h | [88] |
| α-Indomethacin | Not reported | immersion in liquid nitrogen | ||||||||
| 7A | Tadalafil | None | Not reported | 6770 SPEX freezer/mill | Stainless steel vessel | Stainless steel rod (no balls) 1 g of sample per grinding cell | 15 Hz | Cryogenic temperature (liquid nitrogen) | 10 min grinding, 3 min cool-down (2 h total) | [26] |
| Not reported | Planetary ball mill | 250 mL zirconium jar | 6 zirconia balls (d = 20 mm) 16 g of sample per grinding cell | 6.6 Hz | Room temperature | 15 min cycles, 5 min breaks (24 h total) | ||||
| 8A | Glibenclamide | None | Not reported | 6770 SPEX freezer/mill | Stainless steel vessel | Stainless steel rod (no balls) 1 g | 15 Hz | Cryogenic temperature (liquid nitrogen) | 6 min grinding, 3 min cool-down (3 h total) | [89] |
| 9A | Trehalose dihydrate | None | Not reported | Spex SamplePrep 6870 freezer/mill | Polycarbonate vials (23.9 cm3) with steel end caps | Magnetic rod (no balls) 1 g | 15 cycles per second | Cryogenic temperature (liquid nitrogen) | 2 min milling, 1 min of cool-down (30 min total) | [90] |
| 10A | Atenolol | Hydrochlorothiazide 1:1, 1:2, and 2:1 | Stored in desiccators at 4 °C and 25 °C for 30 days | 6770 SPEX freezer/mill | Airtight tube | 1 g | 10 Hz | Cryogenic temperature (liquid nitrogen) | 2 min milling, 2 min cool down (48 min total) | [91] |
| 11A | Furosemide | Tryptophan 1:1 | Not reported | Oscillatory ball mill | 25 mL jars | 2 stainless steel balls (d = 12 mm) 500 mg | 30 Hz | 6 °C | 90 min | [92] |
| Indomethacin | Arginine | |||||||||
| 12A | Dexamethasone | None | Not reported | High-energy planetary mill | 43 cm3 ZrO2 milling jars | 7 ZrO2 balls (d = 15 mm) 1.1 g | 6.6 Hz | Room temperature | 15 min milling, 5 min cool down (12 h total) | [27] |
| 13A | α-Lactose | None | Not reported | Planetary ball mill | 12 cm3 stainless steel jar | 50 stainless steel balls (d = 5 mm) 1 g | 6.6 Hz | 30 ± 5% relative humidity and 22 ± 3 °C | 20 min milling, 5 min cool down (1–20 h total) | [93] |
| 14A | α-D-Glucose | None | Not reported | High-energy planetary mill | 45 cm3 ZrO2 milling jar | 7 ZrO2 balls (d = 1.5 cm) 1 g | 5 Hz | −15 °C | 20 min milling 10 min cool down (1 and 14 h total) | [68] |
| 25 °C | ||||||||||
| 15A | Mebendazole | Aspartame 1:1/1:1:1 | Stored in desiccators at 40 °C and 25 °C up to 4 months | Oscillatory ball mill | 25 mL ball milling jars | 2 stainless steel balls (d = 12 mm) 500 mg | 30 Hz | 5 °C (cold room) | 90 min | [94] |
| Tadalafil | Phenylalanine 1:1/1:1:1 | |||||||||
| Piroxicam | ||||||||||
| 16A | α-D-Glucose | None | Not reported | High-energy planetary mill | 45 cm3 ZrO2 milling jar | 7 ZrO2 balls (d = 1.5 cm) 1 g | 5 Hz | −15 °C | 20 min milling, 10 min cool down (1, 14 h total) | [95] |
| β-Glucose | Not reported | 25 °C | ||||||||
| 17A | Carvedilol | 11 different amino acids 1:1 | Stored at 25 °C under dry conditions for up to 2 years | Mixer mill MM400 | 25 mL stainless steel jars | 2 stainless steel balls (d = 12 mm) 1000 mg | 30 Hz | 6 °C (cold room) | 90 min | [31] |
| Carbamazepine | ||||||||||
| Furosemide | ||||||||||
| Indomethacin | ||||||||||
| Mebendazole | ||||||||||
| Simvastatin | ||||||||||
| 18A | Salts of indomethacin | Lysine 1:1 | Stored at 25 °C, and 40 °C under dry conditions up to 36 weeks | Vibrational ball mill | 25 mL milling jars | 2 stainless steel balls (d = 12 mm) 1000 mg | 30 Hz | 6 °C (cold room) | 60 min | [96] |
| 19A | Mebendazole | Tryptophan Xdrug = 0.1, 0.3, and 0.5 | Not reported | Vibrational ball mill | 50 mL stainless steel jars | 2 stainless steel balls (d = 12 mm) | 30 Hz | Room temperature | 60, 120, and 150 min | [97] unpublished data |
| 20A | 18 different drugs | NaTC natural bile acid surfactant sodium taurocholate 1:1 | Stored at 22 ± 2 °C | Oscillatory ball mill | 25 mL stainless steel jar | 1 stainless steel ball (d = 15-mm) 1 g | 25 Hz | Room temperature and −10 ± 2 °C | 180 min. total time, with 10 min. break every 30 min | [37] |
| 120 min, with 7.5 min breaks cooled in liquid nitrogen | ||||||||||
| 21A | Carbamazepine | Arginine | Not reported | Oscillatory ball mill | 25 mL stainless steel jar | 2 stainless steel ball (d = 12 mm) 500 mg | 30 Hz | 6 °C | 90 min | [98] |
| Indomethacin | Phenylalanine | |||||||||
| Tryptophan | ||||||||||
| 22A | (S)-Naproxen | L-arginine | Stored at 25 °C, and 40 °C under dry conditions | Oscillatory ball mill | 25 mL stainless steel jar | 2 stainless steel ball (d = 12 mm) 1 g | 30 Hz | 6 °C | 60 min | [99] |
| 23A | Griseofulvin | Aspartic Ac | Stored at 23–28 °C under dry conditions up to 12 months | High-energy planetary ball mill | Stainless steel crucible | 3 stainless steel balls 2.5 g | 9.3 Hz | Not specified | 6 h, with 0.5 min pauses every 30 min | [100] |
| Lysine | ||||||||||
| Methionine | ||||||||||
| Valine | ||||||||||
| Tryptophan | ||||||||||
| 24A | Naproxen | Tryptophan and proline | Stored at 40 °C under dry conditions up to 332 days | Oscillatory ball mill | 25 mL stainless steel jar | 2 stainless steel ball (d = 12 mm) 1 g | 30 Hz | 6 °C | 90 min | [101] |
| 25A | Mebendazole | None | Stored at 40 °C under dry conditions up 4 weeks or 3 months | Oscillatory ball mill | 25 mL stainless steel jar | 2 stainless steel ball (d = 12 mm) 500 mg | 30 Hz | 5 °C | 90–180 min | [102] |
| Dipeptide 1:1 | ||||||||||
| Aminoacid mixtures 1:1:1 | ||||||||||
| 26A | Oxaprozin | RameβCD 1:1 | Not reported | High-energy vibrational micro mill | Not specified | Not specified | 24 Hz | Not specified | 30 min | [103] |
| RameβCD-Arg. 1:1:1 | ||||||||||
| 27A | Furosemide | Arginine 1:1 | Not reported | Vibrational ball milling | 25 mL stainless steel jar | 2 stainless steel ball (d = 9 mm) 500 mg | 25 Hz | 6 °C | 99 min | [104] |
| γ-Indomethacin | ||||||||||
| γ-Indomethacin + CA | ||||||||||
| 28A | Indomethacin | L-tryptophan 1:1 | Not reported | Oscillatory ball mill | 25 mL stainless steel jar | 2 stainless steel ball (d = 12 mm) 1500 mg | 30 Hz | 6 °C | 0, 5, 15, 30, 45, 60, and 90 min. 3 or 6 h | [105] |
| Furosemide | ||||||||||
| 29A | Naproxen | Naproxen sodium 2:1, 1:1, and 1:2 | Stored at 40 °C under dry conditions up to 2 weeks or 2 months | Oscillatory ball mill | 25 mL stainless steel jar | 2 stainless steel ball (d = 12 mm) 500 mg | 30 Hz | 4 °C | 90 min | [106] |
| 30A | Carvedilol | Glutamic Ac | Not reported | Vibrational ball mill | 25 mL stainless steel jar | 2 stainless steel ball (d = 12 mm) 700 mg | 30 Hz | 6 °C | 60 min | [107] |
| Aspartic Ac | ||||||||||
| 31A | Indomethacine | Arginine | Stored in refrigerator (≈5 °C) | Mixer mill MM400 | 25 mL stainless steel jar | 2 stainless steel ball (d = 12 mm) 500 mg | 30 Hz | Not specified | 60 min, with 10 min pauses; cell would be in liquid nitrogen for 2 min | [36] |
| Phenylalanine | ||||||||||
| Tryptophan | ||||||||||
| 32A | Simvastatin | Lysine | Stored in desiccators at 4 °C | Oscillatory ball mill | 25 mL stainless steel jar | 2 stainless steel ball (d = 15 mm) 500 mg | 30 Hz | Not specified | 60 min. with 10 min. pauses cell would be in liquid nitrogen for 2 min | [108] |
| Serine | ||||||||||
| Glibenclamide | Threonine | |||||||||
| Aspartic acid | ||||||||||
| 33A | Indomethacin | Arginine | Stored at 40 °C under dry conditions | Oscillatory ball mill | 25 mL stainless steel jar | 2 stainless steel ball (d = 12 mm) 500 mg | 30 Hz | 6 °C | 90 min | [98] |
| Tryptophan | ||||||||||
| Carbamazepine | Tyrosine | |||||||||
| Phenylalanine | ||||||||||
| 34A | Indomethacin | Tryptophan | - | Oscillatory mill | 12 mL Stainless steel jar | 2 stainless steel ball (d = 10 mm) 1.2 g | 10.83 Hz | Not specified | 360 min | [109] |
| 35A | Carbamazepine | Citric acid | Stored at 40 °C under dry conditions up to 2 months | Oscillatory ball mill | 25 mL stainless steel jar | 2 stainless steel ball (d = 12 mm) 500 mg | 30 Hz | 4 °C | 90–180 min | [110] |
| 36A | Arginine | Glibenclamide 1:1 | Stored at 4 °C, room temperature, and 40 °C up to 13 months | Oscillatory ball mill | 25 mL milling chambers | 2 stainless steel balls (d = 12 mm) 500 mg | 30 HZ | Not specified | 60 min, chambers were cooled in liquid nitrogen | [111] |
| Serine | ||||||||||
| Quercetin | ||||||||||
| 37A | Glutamic ac | Mebendazole 1:1 and 1:1:1 | Stored at 40 °C and 25 °C in desiccators under dry conditions up to 6 months | Oscillatory ball mill | 25 mL stainless steel jar | 2 stainless steel ball (d = 1.2 cm) 500 mg | 30 Hz | 5 °C (cold room) | 30, 60, and 90 min | [112] |
| L-arginine | ||||||||||
| Glutamic Ac-Arginine | ||||||||||
| Arginine-glutamic ac | ||||||||||
| Glutamic-arginine | ||||||||||
| 38A | Mefenamic acid | Meglumine 1:1, 1:2, and 1:4 | Not reported | Planetary ball mill | Not specified | 5 stainless steel balls (d = 10 mm) | 4.16 Hz | Not specified | 20 min | [113] |
| Indomethacin | PVP 1:1, 1:2, and 1:4 | |||||||||
| 39A | L-methionine | Rutin 1:1, 1:2, 2:1 | Not reported | Planetary ball mill | 45 mL zirconia jar | 8 YTZ balls (d = 10 mm) | 10 Hz | Room temperature | 12 h with a break every 10 min | [114] |
| Naringin hydrate | ||||||||||
| Quercetin dihydrate | ||||||||||
| Hesperidin Chlorothiazide Indapamide Triamterene Nifedipine | ||||||||||
| 40A | Benzamidine | Gliclazide 1:1, 1:5, or 5:1 | Stored in a desiccator at 22 ± 2 °C, and 40 °C under relative humidity up to 180 days | Oscillatory ball mill | 25 mL stainless steel milling jar | Stainless steel ball (d = 15 mm) 0.25 g | 25 Hz | Cromilling inmersing jars in liquid nitrogen for 5 min prior to milling. 7.5 min milling | 180 min, with a cool down period of 15 min after every 30 min | [38] |
| 41A | Arginine | Quercetin 1:1, 1:2 | Not reported | Not specified | 25 mL stainless steel | 1–3 stainless steel ball (d = 18, 15, and 12 mm) | Not specified | 2 h | Not specified | [115] |
| Glutamic acid | ||||||||||
| Aspartic acid | ||||||||||
| Tryptophan | ||||||||||
| Glycin | ||||||||||
| 42A | Candesartan cilexetil | Hydrochlorothiazide | Stored at 4 °C, 30 °C, and 40 °C under dry conditions up to 90 days | Planetary ball mill | 125 mL stainless steel grinding jars | 3 stainless steel grinding balls (d = 10-mm) 2 g | 9.3 Hz | Room temperature | 2.5 h | [116] |
| Hydroxypropyl methylcellulose | ||||||||||
| Acetate succinate (HPMCAS) type M |
| # | Sample | Molar Ratio | Method of Preparation | Milling Type | Instrument Brand | Milling Jar | Balls (# and Material) | Milling Frequency | Milling Temp | Milling Time | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1C | Nicotinamide: L-(+)-Ascorbic acid | 1:1 | Assisted by solvent | Vibrational | Mixer Mill (IST 500) InSolido Technologies | Polymethylmetacrylate | Two stainless steel balls | 30 Hz | NR | 60 min | [66] |
| 2C | Salicylic acid:2-pyridone Salicylic acid: 4-Pyridone | 1:1 | NR | Vibrational | Mixer Mill (IST 500) InSolido Technologies | Polymethylmetacrylate | Two stainless steel balls | 30 Hz | NR | 50 min | [117] |
| 3C | Ciprofloxacin- thymol | 1:2 | Assisted by solvent (EtOH) | NR | Retsch MM200 ball miller, | NR | NR | 20 Hz | NR | 30 min | [118] |
| 4C | Urea- caffeine | 1:1 | NR | Oscillatory ball | Mixer Mill MM400-Retsch GmbH, Haan | Stainless steel jar | One 15 mm stainless steel ball | 25 Hz | Room temperature | 60 min | [119] |
| 5C | Brexpiprazol-Catechol Brexpiprazol-Succinic acid | 1:1 | NR | NR | Nano Ball Mill (Fritsch Premium Line, FRITSCH GmbH, Idar-Oberstein, Germany) using | NR | Stainless steel balls | 8.3 Hz | NR | 120 min | [120] |
| 6C | Quercetin- malonic acid | 1:1 and 1:2 | Solvent drop grinding | NR | NR | NR | NR | NR | NR | 30 min | [121] |
| 7C | Paracetamol-trimethylglycine | 1:1 | NA | Planetary ball | QM-3SP2, Nanjing NTU Instrument Co. | NR | NR | 6.6 Hz | NR | 5 h | [44] |
| 8C | Meloxicam- benzoic acid | 1:1 | LAG | NR | Retsch CryoMill | NR | NR | 25 Hz | Room temperature | 30 min | [122] |
| 9C | Acetazolamide and 4-hydroxybenzoic acid | 1:1 | LAG | Planetary ball | QM-3SP04, gear type | 25 mL stainless steel milling jars | NR | 25 Hz | NR | 30 min | [123] |
| 10C | Furosemide-urea and carbamazepine-indomethacin | 1:1 | LAG | NR | Retsch MM400 ball mill | 50 mL jar, with two 5 mm stainless steel balls and drops of acetone. | NR | NR | NR | 60 min | [51] |
| 11C | Ciprofloxacin-nicotinic and isonicotinic acids | 1:1 | Assisted or not by solvent (EtOH) | NR | Retsch MM 400 mixer mill | 10 mL stainless-steel jars | 1 stainless steel ball of 7 mm diameter, 100, 500 mg sample | 30 and 15 Hz | NR | 30 min | [124] |
| 12C | Pyrazinamide-diflunisal | 1:1 | LAG | Oscillatory ball mill | Mixer Mill MM400 | 25 mL stainless steel milling jars | NR | 15 Hz | Room temperature | 60 min | [125] |
| 13C | Acetazolamide–4-aminobenzoic acid | 1:1 | With solvent | Planetary ball | Fritsch micro mill model Pulverisette 7 | 12 mL agate grinding jars | Ten 5 mm agate balls | 8.3 Hz | NR | 30 min | [67] |
| 14C | Acetazolamide-nicotinamide-2-pyridone | 1:1:1 | LAG with ethyl acetate and tetrahydrofuran solvents | Planetary ball | QM-3SP04, gear type | 25 mL stainless steel milling jars | NR | 15 Hz | NR | 60 min | [126] |
| 15C | β-Lapachone-resorcinol | 1:1 | LAG | NR | Retsh Mixer Mill (Model MW 200) | Stainless steel jar together | A stainless steel ball | 20 Hz | NR | 20 min | [127] |
| 16C | Norfloxacin-nicotinic acid | NR | NT and LAG | Ocillatory ball system | Mixer Mill MM 400, Retsch GmbH and Co | Stainless steel jars | 7 mm diameter stainless steel ball | 15 Hz | NR | 30 min | [128] |
| 17C | Chlorothiazide, D-proline, L-proline | 1:1 | NT and LAG | Oscillatory ball | Retsch (MM400, Retsch) | NR | NR | 30 Hz | NR | 30 min | [129] |
| 18C | Praziquantel, poloxamer F-127, and sucrose stearate | 20:1, 10:1, 10:2, and 10:3 | NT | High-energy vibrational ball | Mixer Mill MM 200, Retch, GmbH | 10 mL volume stainless steel grinding jars | Two 7 mm stainless steel grinding balls | 25 Hz | 28.10–30.34 °C | 30 or 90 min | [130] |
| 19C | Ferulic acid, urea, nicotinamide, and isonicotinamide (INA) | 1:1 and 1:2 | LAG | NR | Retsch Mixer Mill (model MM301) | Stainless steel grinding jar | One 7 mm stainless steel ball | 20 Hz | NR | 20 min | [131] |
| 20C | Ketoconazole, fumaric acid, and succinic acid | 1:1.1 and 1:1 | NT and LAG | Oscillatory ball | Retsch MM 400 | 25 mL stainless steel jars | One stainless steel ball | 19 Hz | NR | 60 min | [132] |
| 21C | Itraconazole: 4-aminobenzoic acid Itraconazole: 4-hydroxybenzamide | 1:1 2:1 1:2 | LAG | Planetary micro | Fritsch planetary micro mill, Pulverisette 7 | 12 mL agate grinding jars | Ten 5 mm agate balls | 8.3 Hz | NR | 40 min | [133] |
| 22C | S-ibuprofen: nicotinamide | 1:1 | N.R | Oscillatory ball | MM400—Retsch | 10 mL ZrO2 milling jars | One ball, 10 mm | 30 Hz | NR | 60 and 10 min and 5 min pauses | [134] |
| 23C | Pyrazinamide: 4-aminosalicylic acid | 1:1 | LAG | Planetary ball | QM3SP04, gear type, Nanjing University Instrument Factory | 20 mL stainless steel grinding tank | N.R | 20 Hz | Room temperature | 40 min | [135] |
| 24C | Theophylline: 4-aminobenzoic acid | 1:1 | N.R | N.R | MM 400, Retsch, Germany | 10 mL jar 25 mL jar | One ball, 8.74 mm, One ball, 13.72 mm | 30 Hz | N.R | Period times: 2,5,10, 15, 20, and 25 min | [136] |
| 25C | Betulin-terephthalic acid | 1:1 2:1 | Assisted by solvent | NR | SPEX 8000 mixer mill (CertiPrep Inc., Metuchen, NJ, USA) | 60 mL steel jar | Steel balls 6 mm | NR | NR | Pre-milled: 5 min After solvent: 10 min | [137] |
| 26C | 5-Fluorocytosine:5-fluorouracil | 1:1 | NT SDG | Oscillatory | Mixer Mill MM400 RETSCH | 25-mL stainless steel milling jar | Two 7 mm stainless steel balls | 25 Hz | Room temperature | 90 min SDG: 60 min | [138] |
| 27C | Nicotinamide:adipic acid (polymorph, form 2) | 1:1 | Assisted by solvent (acetonitrile) | NR | Retsch MM400 mill (in-house modified) | Stainless steel milling jar | Two 7 mm stainless steel balls | 30 Hz | NR | 60–90 min | [139] |
| # | Sample | Obtained Polymorph | Mill Type | Milling Cell | Ball (#, Material) Sample Weight | Milling Frequency | Milling Temperature | Milling Time and Solvent | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| 1P | Ranitidine hydrochloride | Ranitidine hydrochloride, form 2 | Oscillatory ball mill (mixer mill MM301, Retsch GmbH and Co., Weinheim, Germany) | 25 mL Stainless steel | 2 stainless steel balls (d = 12 mm) 1 g s | 30 Hz | 12 ± 3 °C | 180 min, stop every 30 min to scrape and remix powder | [74] |
| Ranitidine, form 2 (with traces of form 1) | 35 °C | 120 min, stop every 30 min to scrape and remix powder | |||||||
| Ranitidine, form 2 | 240 min, stop every 30 min to scrape and remix powder | ||||||||
| 2P | Chlorhexidine dihydrochloride | 2-step polymorphism produces ChxHC form 2 as a precursor of form 3 | High-energy planetary mill (Pulverisette 7; Fritsch, Idar-Oberstein) | 43 cm3 ZrO2 | 7 ZrO2 balls (d = 15 mm) 1 g | 6.6 Hz | Room temperature | 12 h (15 min milling periods with 5 min rests) | [140] |
| 3P | Γ-sorbitol | A form sorbitol | High-energy planetary micro-mill (Pulverisette 7; Fritsch, Idar-Oberstein) | 45 cm3 zirconium | 7 zirconium balls (d = 15 mm) 1 g of sample | 6.6 Hz | Room temperature | 10 h | [34] |
| 4P | Rivastigmine (RHT form 2) | RHT form I | Retsch planetary ball mill PM100 | 50 mL stainless steel | 3 stainless steel balls (d = 20 mm) 1 g | 6.6 Hz | Room temperature | 3 h (stopping at 15 min, 30 min, 1 h and 2 h) | [141] |
| 5P | o-Aminobenzoic acid (mixture of FII and FIII forms) | FIII form | Oscillatory ball mill (Mixer mill MM400, Retsch GmbH and Co., Germany) | 25 mL stainless steel | One stainless steel ball (d = 15 mm) 0.5 g 30 μL of solvent | 25 Hz | Room temperature | 2.5 h (30 min milling periods with 15 min pauses) Solvent: valeric acid (FIV and FIII) | [54] |
| FII form | |||||||||
| m-Aminobenzoic acid (FIII form) | FIV form | ||||||||
| FIV and FIII | |||||||||
| Carbamazepine | FIV form | ||||||||
| p-aminobenzoic acid | β-PABA | 1 stainless steel ball (d = 15 mm) 0.5 g 30 μL of solvent | Cryogenic temperature (immersed in liquid N2 for 5 min prior to miling every 7.5 min) | 2.5 h (7.5 min milling and 2.5 min pauses in liquid nitrogen) Solvent: valeric acid, 10% acetamide or ethanol. (FI) | |||||
| o-Aminobenzoic acid (mixture of FII and FIII forms) | FI form (FII converts to FIII and subsequently FIII converts to FI.) | ||||||||
| FI form | |||||||||
| 6P | Dexamethasone | DEX form A and B | High-energy planetary mill (Pulverisette 7, Fritsch, Idar-Oberstein) | 43 cm3 ZrO2 | 7 ZrO2 balls (d = 15 mm) 1.1 g | 6.6 Hz | Room temperature | 12 h (15 min milling periods, with 5 min rests) | [27] |
| 7P | Sofosbuvir (anhydrous form 1) | Form A or B | Vibrational ball mill (MM400, RETSCH) | 5 mL stainless steel | 2 stainless steel balls (d = 5 mm) 50 mg 10 μL of Solvent | 25 Hz | Room temperature | 30 min Solvent: water or methanol | [79] |
| Form A | 30 min Solvent: anisole, n-butyl acetate, or ethyl acetate | ||||||||
| Form A (form 1 changes to form V) | 30 min Solvent: anisole | ||||||||
| Form A | 60 min, solvent: tetrahydrofuran | ||||||||
| Form A (form 1 changes into form B and then forms A) | 20 min, solvent: butyl acetate or ethyl acetate | ||||||||
| 8P | Sulindac (form II) | Form II and form I | High-energy planetary mill (Pulverisette 7eFritsch) | 43 cm3 ZrO2 | 7 ZrO2 balls (d = 15 mm) 1 g | 6.6 Hz | Room temperature | 5 min | [69] |
| Form I | 600 min (10 min milling, with 5 min pauses) | ||||||||
| Mixture of form II and form I | 20 min (10 min milling periods, with 5 min pauses) | ||||||||
| 9P | Γ-sorbitol | A form sorbitol | High-energy planetary mill (Pulverisette 7-Fritsch) | 43 cm3 ZrO2 | 7 ZrO2 balls (d = 15 mm) | 6.6 Hz | Room temperature (dry nitrogen atmosphere) | 10 h | [75] |
| Mannitol (β) | α Mannitol | ||||||||
| Mannitol (δ) | α Mannitol | ||||||||
| 10P | Famotidine (form B) | Form A (form B to A transformation ratio increased with milling time) | Oscillatory ball mill (Mixer Mill MM301, Retsch GmbH and Co., Germany) | 25 mL stainless steel | 2 stainless steel balls (d = 12 mm) 0.2 g | 15 Hz | 130 °C | 10 min | [142] |
| 110 °C | 20 min | ||||||||
| 110 °C | 30 min | ||||||||
| 11P | Gabapentin (GBP) form I | GBP form II | Oscillatory ball mill (Mixer Mill MM301, Retsch GmbH and Co., Germany) | 25 mL stainless steel | 2 stainless steel balls (d = 15 mm) 0.2 g of sample | 20 Hz | Room temperature | 120 min | [76] |
| GBP form II | GBP form III | 105 min | |||||||
| GBP form IV | 120 min | ||||||||
| GBP form III | GBP form II | 15 min | |||||||
| GBP form III (produced by the coexistence of form I and II after 15 min milling) | 60 min | ||||||||
| GBP form IV | 105 min | ||||||||
| GBP form IV | GBP form II | 2 min | |||||||
| GBP form III | 30 min | ||||||||
| GBP form IV | 105 min | ||||||||
| 12P | Ciprofloxacin salicylate (monohydrate) | Form I (after 4 min of neat grinding) From 2 (after 9.5 min of neat grinding) | Fritsch planetary micro mill, model Pulverisette 7 | 12 mL agate | 10 agate balls (d = 5 mm) 0.1 g 60 μL of solvent | 8.3 Hz | NR | 50 min, solvent: water, and the use of water/organic solvents decreases the time of existence for form I | [143] |
| Ciprofloxacin salicylate (3.67 hydrate) | Form II (after 17 min of neat grinding) | ||||||||
| Anhydrous ciprofloxacin salicylate | From I | ||||||||
| 13P | γ-sorbitol | Form α (complete transformation) | High-energy planetary mill (Pulveri- sette, 7-Fritsch) | 43 cm3 ZrO2 | 7 ZrO2 balls (d = 15 mm) | 6.6 Hz | Room temperature | 180 min (10 min milling periods, with 5 min rests) | [144] |
| 14P | Ethenzamide: ethylmalonic acid (Co-crystal) | Form l (SDG with n-hexane) Form ll (after neat grinding or SDG with toluene or cyclohexane) | Oscillatory ball mill (Mixer Mill MM301, Retsch GmbH and Co., Germany) | 10 mL stainless steel | 1 stainless steel ball (d = 7 mm) 0.1 g of EA and 0.0799 g of EMA (1:1 molar ratio) 0.05 mL of solvent | 20 Hz | Room temperature | 15 min, solvent: toluene, cyclohexane, or n-hexane | [145] |
| 15P | Caffeine: glutaric acid (co-crystal) | Form l (after neat grinding and SDG with n-hexane, cyclohexane or heptane) | Oscillatory ball mill (Mixer Mill, Retsch GmbH and Co., Germany) | Stainless steel (volume NR) | 2 stainless stell balls (d = NR) 0.75 g (1:1 molar ratio) | 30 Hz | Room temperature | 60 min Solvent: n-hexane, cyclohexane, or heptane | [146] |
4. Evaluation of Physicochemical Properties of Co-Amorphous, Co-Crystals, and Polymorphs Induced by Mechanical Activation
4.1. Evaluation of Solubility Enhancements as an Effect of the Milling Process
- (a)
- Solubility for co-amorphous systems after ball milling
- (b)
- Solubility of co-crystals after grinding
4.2. FT-IR Spectroscopic Evaluation of Intermolecular Interactions Induced by Ball Milling
- (c)
- Structural characterization of amorphous systems by spectroscopy techniques
- (d)
- Structural characterization of co-crystals by spectroscopy techniques
- (e)
- Spectroscopic studies reported for polymorphs obtained by ball milling
4.3. Thermal Analysis Techniques to Study Phase Transitions Induced by Grinding
- (f)
- Thermal analysis of ball-milled co-amorphous systems
- (g)
- Phase transitions reported for co-crystals prepared by milling
- (h)
- Phase transitions of polymorphs resulting from mechanical activation
| # | Sample | Polymorph Identified | Transition Temperature (°C) | Milling Temperature | Conditions and Milling Time | Ref. |
|---|---|---|---|---|---|---|
| 1P | Ranitidine hydrochloride | Form 1 | Tm = 142.73 | 12 ± 3 °C and 35 °C | 0 to 160 °C, 10 K/min | [74] |
| Form 2 | Tm = 145.01 | |||||
| 2P | Chlorhexidine dihydrochloride | Form 2 | Tc2 = 124 | Room temperature | 5 °C/min | [140] |
| Form 3 | Tc3 = 157 | |||||
| Form 3 | Tm3 = 256 | |||||
| 3P | Γ-sorbitol | Form A | Decrease in melting temperature (value not reported) | Room temperature | NR | [34] |
| 4P | Rivastigmine (RHT form II) | Form II | Tm1 = 97.5, Tm2 = 124.5 | Room temperature | 10 °C/min from 0 to 150 °C | [141] |
| Exo peak = 105.5 | ||||||
| Form I | Tm = 123.5 | |||||
| 6P | Dexamethasone | Form A | Tm = 242 | Room temperature | 5 °C/min | [27] |
| Form B | Tm = 250 | |||||
| 7P | Sofosbuvir (anhydrous form 1) | Form 1 | Tm = 96.57 | Room temperature | 0 to 300 °C, 5 °C/min | [79] |
| Form A | Tm = 117.90 | |||||
| Form B | Tm = 124.83 | |||||
| Form V | Tm = 71.54 | |||||
| 8P | Sulindac (form II) | II → I | Endo peak = 160 | Room temperature | 5 °C/min | [69] |
| 9P | Γ-sorbitol | Γ-sorbitol | Tm = 98.5 | Room temperature with dry nitrogen atmosphere | 5 °C/min | [75] |
| A-form | Tm = 85 | |||||
| 12P | Sulfamerazine | Form I | Tm = 236 | Room temperature | 100 mL/min | [166] |
| Form II | Tm = 212–214 |
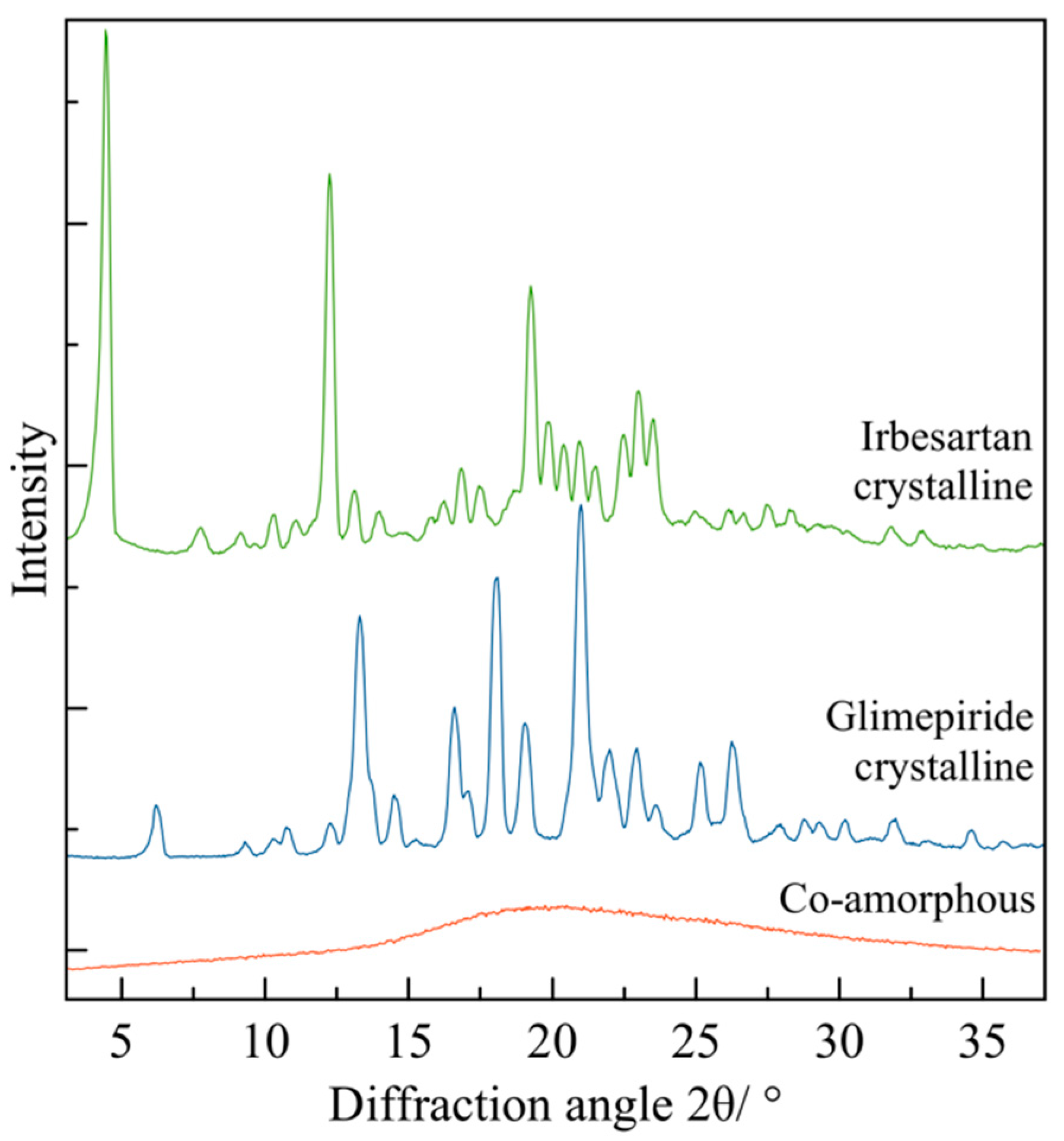
4.4. Identification of Amorphous and Crystalline Phases by Powder X-ray Diffraction (PXRD)
- (i)
- Measurement of structural stability on co-amorphous systems during storage by XRD
| # | Sample | XRD Interpretation | Storage Time (Days) | Storage Conditions * | Ref. |
|---|---|---|---|---|---|
| 2A | Furosemide-arginine, furosemide-citrulline nitrofurantoin-arginine, nitrofurantoin-citrulline (1:1) | Remained amorphous | 450 | 25 °C, (dry conditions, silica gel) | [85] |
| Furosemide-arginine, furosemide-citrulline, nitrofurantoin-arginine | Remained amorphous | 450 | 40 °C, (dry conditions, silica gel) | ||
| Nitrofurantoin-citrulline | Recrystallization of Nitrofurantoin | 450 | 40 °C, (dry conditions, silica gel) | ||
| 3A | Sulfathiazole-polyvinylpyrrolidone sulfadimidine-polyvinylpyrrolidone | Diffused halo → amorphous state | 365 | 4 °C with desiccant | [86] |
| 4A | Naproxen-cimetidine (1:1) | Halo, most stable sample | 186 | 4 °C, 25 °C and 40 °C, dry conditions (silica gel) | [87] |
| Naproxen-cimetidine (2:1) | Halo, stable | 33 | 4 °C, dry conditions (silica gel) | ||
| Naproxen-cimetidine (2:1) | Crystalline naproxen (in excess) peaks | 33 | 25 °C and 40 °C, dry conditions (silica gel) | ||
| Naproxen-cimetidine (1:2) | Traces of crystalline cimetidine | 33 | 4 °C, 25 °C and 40 °C, dry conditions (silica gel) | ||
| 5A | γ-indomethacin–ranitidine hydrochloride (1:1) | Halo, highest stability | 30 | 4 °C and 25 °C, dry conditions (silica gel) | [28] |
| γ-indomethacin–ranitidine hydrochloride (2:1) | Small crystalline peaks of indomethacin (indo in excess) | 30 | 25 °C and 40 °C, dry conditions (silica gel) | ||
| γ-indomethacin–ranitidine hydrochloride (1:2) | Progressive increase in peak intensity as temperature increased. | 30 | 4 °C, 25 °C and 40 °C, dry conditions (silica gel) | ||
| 6A | γ-indomethacin | γ-form, crystallized | <1 | 22 °C over P2O5 | [88] |
| α-indomethacin | α-form crystallized to γ-form | 4 | |||
| 7A | Tadafil | Amorphous | 365 | 4 °C with desiccant | [26] |
| 8A | Glibenclamide (GCM) | Broad halo, amorphous state | 210 | 25 °C, 10% RH, dry conditions | [89] |
| 9A | Trehalose dihydrate | Recrystallised material is trehalose dihydrate | 2 | 25 °C | [90] |
| 10A | Atenolol-hydrochlorothiazide (1:1) | Amorphous, stable | 30 | 4 °C and 25 °C, in desiccator | [91] |
| Atenolol-hydrochlorothiazide (1:2) | Amorphous, stable | 30 | 4 °C, in desiccator | ||
| Atenolol-hydrochlorothiazide (1:2) | Traces of crystals | 30 | 25 °C, in desiccator | ||
| 12A | Dexamethasone | Form A converts to form B | 7 | 150 °C | [27] |
| 14A | α-D-glucose | Absence of Bragg peaks → amorphization | Immediate analysis after 14 hrs of milling | −15 °C | [68] |
| Well-defined Bragg peaks → crystalline state | Immediate analysis after 14 hrs of milling | 25 °C | |||
| 15A | Mebendazole-ASPA | Amorphous | 120 days | 25 °C and 40 °C (silica gel) | [94] |
| Tadalafil-ASPA | Amorphous | 120 days | 25 °C and 40 °C (silica gel) | ||
| Piroxicam-ASPA | Amorphous | 120 days | 25 °C and 40 °C (silica gel) | ||
| 16A | β-D-Glucose | Bragg peaks restore immediately after the end of the milling process | 1 h | 25 °C | [95] |
| 17A | Carvedilol, carbamazepine, furosemide, indomethacin, mebendazole-amino acids | Recrystallization → Meb-Lys, Meb-Ile, Meb-Leu, Car-Val, Sim-Lys, Ind-Ile, Ind-Val | 140 | 25 °C, 5% RH (P2O5) | [31] |
| Recrystallization peaks → Fur-Met, Fur-Val, Ind-Leu | 140–365 | ||||
| Amorphous → Arg-Fur, Arg-Ind, His-Fur, Lys-Fur, Lys-Ind, Car-Ile, Car-Leu, Car-Met, Car-Phe, Car-Trp, Meb-Met, Meb-Phe, Meb-Trp, Sim-Phe, Cbz-Trp, Sim-Trp | 365–730 | ||||
| 18A | Indomethacin-lysine | Amorphous halo | 252 days | DMB, 25 °C (P2O5) and 40 °C (silica gel), dry conditions | [96] |
| Recrystallization → within 25 days it turned into same crystalline form of LAG | 10 days | DMB, 25 °C, 75% RH | |||
| Crystalline form | 252 days | LAG, 25° and 40 °C | |||
| 23A | Griseofulvin-tryptophan | Amorphous state, no recrystalization detected | 365 | Silica gel (13–32% RH), vacuum, 23–28 °C | [100] |
| 25A | Mebendazole-tryptophan-phenylalanine | Remained amorphous | 90 | 40 °C, 2% RH (silica gel) | [102] |
| Mebendazole-tryptophanphenylalanine | Remained amorphous | ||||
| Mebendazole-phenylalanine-tryptophan | Remained amorphous | ||||
| Mebendazole-aspartate-tyrosine | Remained amorphous | ||||
| Mebendazole-histidine-glycine | Remained amorphous | ||||
| Mebendazole-proline-tryptophan | Remained amorphous | ||||
| Mebendazole-prolinetryptophan | Remained amorphous | ||||
| Mebendazole-tryptophan | Remained amorphous | ||||
| Mebendazole-proline | Recrystallized | ||||
| All samples | Remained amorphous | 90 | 25 °C, 2% RH (silica gel) | ||
| 29A | Naproxen-NAP(Na) (2:1) | Recrystallization peaks are visible | 7 | 40 °C, silica gel | [106] |
| Naproxen-NAP(Na) (1:1) | Remained amorphous | 60 | |||
| 32A | Simvastatin-lysine | Amorphous | 150 | 4 °C and 0% RH | [108] |
| Recrystallization | 90 | 40 °C and 0% RH | |||
| Recrystallization | 56 | Ambient temperature and 60% RH | |||
| Glibenclamide-threonine | Recrystallization | 40 | 40 °C and 0% RH | ||
| Glibenclamide-serine-threonine | Recrystallization | 90 | |||
| Glibenclamide-serine | Amorphous | 180 | |||
| Glibenclamide-serine | Amorphous | 180 | 4 °C and 0% RH | ||
| Glibenclamide-threonine | Recrystallization | 44 | |||
| Glibenclamide-serine-threonine | Recrystallization | 90 | |||
| Glibenclamide-serine | Recrystallization | 150 | Ambient temperature and 60% RH | ||
| Glibenclamide-threonine | Recrystallization | 26 | |||
| Glibenclamide-serine-threonine | Recrystallization | 90 | |||
| 33A | Indomethacin, carbamazepine, L-arginine, L-phenylalanine, L-tryptophan and L-tyrosine | Remained amorphous (halo) | 180 | 40 °C, dry conditions (silica gel) | [169] |
| 35A | Carbamazepine-arginine (1:1, 1:2, 1:3, 1:4) carbamazepine-Citric acid-arginine (1:1:1, 1:1:2, 1:1:3) | Amorphous | 60 | 40 °C, silica gel | [110] |
| 36A | Mebendazole (Meb)-glutamate-arginine (crystalline salt), meb-arginine-glutamate (amorphous salt), meb-glutamatearginine, meb-arginineglutamate (dipeptide) | Remained amorphous | 180 | 25 °C, dry conditions (silica gel), 2% RH | [112] |
| Meb-glutamate-arginine meb-arginine-glutamate | Recrystallization | 180 | 40 °C, dry conditions (silica gel), 2% RH | ||
| Meb-glutamatearginine meb-arginineglutamate | Remained amorphous | 180 | |||
| 38A | Glibenclamide-serine glibenclamide-arginine | Samples after storage were similar to the patterns exhibited before the test | 180 | 40 °C and 75% RH | [170] |
| 39A | Rutin-naringin hydrate (all molar ratios), rutin-hesperidin (all molar ratios), rutin-methionine (1:1), rutin-quercetin dihydrate (1:1, 2:1) | Remained amorphous | 12 h | Dry and wet conditions | [114] |
| Rutin-methionine (1:2 and 2:1) | Small peaks | 12 h | Dry conditions | ||
| Rutin-quercetin dihydrate (1:2) | Small peaks | 12 h | Dry and wet conditions | ||
| 40A | Gliclazide (Glz)-nifedipine | Crystallized to a physical mixture | 3 | Ambient temperature, 56% RH | [38] |
| Glz-indapamide, Glz-triamterene, Glz-hydrochlorothiazide | Remained amorphous | 180 | |||
| Glz-chlorothiazide | Recrystallized | 30 | |||
| Glz-indapamide, Glz-triamterene, Glz-hydrochlorothiazide | Remained amorphous | 120 | Ambient temperature, 98% RH | ||
| Glz-hydrochlorothiazide | New peaks | 30 | |||
| Glz-triamterene | Small peaks | 120 | |||
| Glz-benzamidine | New pattern assigned to the salt | 30 | |||
| 42C | Cilexetil-hydrochlorothiazide | Recrystallization | 30 | 4 °C, 0% RH | [116] |
| Cilexetil-hydrochlorothiazide-hydroxypropylmethylcellulose acetate succinate type M (HPMCAS) | 60 | ||||
| Cilexetil-hydrochlorothiazide | 15 | 40 °C, 75% RH | |||
| Cilexetil-hydrochlorothiazide-HPMCAS (CH50) | Small reflections | 90 | |||
| Cilexetil-hydrochlorothiazide-HPMCAS (CH70) | 30 | ||||
| 43C | Glibenclamide-quercetin | Remained amorphous | 120 | 4 °C, 0% RH | [111] |
| Recrystallization | 390 | ||||
| 10 | Room temperature, 60% RH | ||||
| 120 | 40 °C, 0% RH |
- (j)
- Measurement of structural stability on co-crystals after milling by XRD
- (k)
- Structural stability on polymorphs after mechanical activation by XRD
5. Characterization by Microscopy
6. Concluding Remarks and Future Works
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Takagi, T.; Ramachandran, C.; Bermejo, M.; Yamashita, S.; Yu, L.X.; Amidon, G.L. A Provisional Biopharmaceutical Classification of the Top 200 Oral Drug Products in the United States, Great Britain, Spain, and Japan. Mol. Pharm. 2006, 3, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Thayer, A.M. Finding Solutions. Chem. Eng. News 2010, 88, 13–18. [Google Scholar] [CrossRef]
- Kalepu, S.; Nekkanti, V. Insoluble Drug Delivery Strategies: Review of Recent Advances and Business Prospects. Acta Pharm. Sin. B 2015, 5, 442–453. [Google Scholar] [CrossRef]
- Dengale, S.J.; Grohganz, H.; Rades, T.; Löbmann, K. Recent Advances in Co-Amorphous Drug Formulations. Adv. Drug Deliv. Rev. 2016, 100, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Mizoguchi, R.; Waraya, H.; Hirakura, Y. Application of Co-Amorphous Technology for Improving the Physicochemical Properties of Amorphous Formulations. Mol. Pharm. 2019, 16, 2142–2152. [Google Scholar] [CrossRef]
- Martínez, L.M.; Videa, M.; López Silva, T.; Castro, S.; Caballero, A.; Lara-Díaz, V.J.; Castorena-Torres, F. Two-Phase Amorphous-Amorphous Solid Drug Dispersion with Enhanced Stability, Solubility and Bioavailability Resulting from Ultrasonic Dispersion of an Immiscible System. Eur. J. Pharm. Biopharm. 2017, 119, 243–252. [Google Scholar] [CrossRef]
- Vo, C.L.N.; Park, C.; Lee, B.J. Current Trends and Future Perspectives of Solid Dispersions Containing Poorly Water-Soluble Drugs. Eur. J. Pharm. Biopharm. 2013, 85, 799–813. [Google Scholar] [CrossRef]
- Zhang, X.; Xing, H.; Zhao, Y.; Ma, Z. Pharmaceutical Dispersion Techniques for Dissolution and Bioavailability Enhancement of Poorly Water-Soluble Drugs. Pharmaceutics 2018, 10, 74. [Google Scholar] [CrossRef]
- Tran, P.H.L.; Tran, T.T.D. Nano-Sized Solid Dispersions for Improving the Bioavailability of Poorly Water-Soluble Drugs. Curr. Pharm. Des. 2020, 26, 4917–4924. [Google Scholar] [CrossRef]
- Dutt, B.; Choudhary, M.; Vikaas, B. Cocrystallization: An Innovative Route toward Better Medication. J. Rep. Pharm. Sci. 2020, 9, 256–270. [Google Scholar] [CrossRef]
- Berry, D.J.; Steed, J.W. Pharmaceutical Cocrystals, Salts and Multicomponent Systems; Intermolecular Interactions and Property Based Design. Adv. Drug Deliv. Rev. 2017, 117, 3–24. [Google Scholar] [CrossRef] [PubMed]
- Blagden, N.; de Matas, M.; Gavan, P.T.; York, P. Crystal Engineering of Active Pharmaceutical Ingredients to Improve Solubility and Dissolution Rates. Adv. Drug Deliv. Rev. 2007, 59, 617–630. [Google Scholar] [CrossRef] [PubMed]
- Llinàs, A.; Goodman, J.M. Polymorph Control: Past, Present and Future. Drug Discov. Today 2008, 13, 198–210. [Google Scholar] [CrossRef] [PubMed]
- Douroumis, D.; Ross, S.A.; Nokhodchi, A. Advanced Methodologies for Cocrystal Synthesis. Adv. Drug Deliv. Rev. 2017, 117, 178–195. [Google Scholar] [CrossRef]
- Braga, D.; Maini, L.; Grepioni, F. Mechanochemical Preparation of Co-Crystals. Chem. Soc. Rev. 2013, 42, 7638–7648. [Google Scholar] [CrossRef] [PubMed]
- Einfal, T.; Planinšek, O.; Hrovat, K. Methods of Amorphization and Investigation of the Amorphous State. Acta Pharm. 2013, 63, 305–334. [Google Scholar] [CrossRef]
- Loh, Z.H.; Samanta, A.K.; Sia Heng, P.W. Overview of Milling Techniques for Improving the Solubility of Poorly Water-Soluble Drugs. Asian J. Pharm. Sci. 2015, 10, 255–274. [Google Scholar] [CrossRef]
- Korhonen, O.; Pajula, K.; Laitinen, R. Rational Excipient Selection for Co-Amorphous Formulations. Expert Opin. Drug Deliv. 2017, 14, 551–569. [Google Scholar] [CrossRef]
- Han, J.; Wei, Y.; Lu, Y.; Wang, R.; Zhang, J.; Gao, Y.; Qian, S. Co-Amorphous Systems for the Delivery of Poorly Water-Soluble Drugs: Recent Advances and an Update. Expert Opin. Drug Deliv. 2020, 17, 1411–1436. [Google Scholar] [CrossRef]
- Kanaujia, P.; Poovizhi, P.; Ng, W.K.; Tan, R.B.H. Amorphous Formulations for Dissolution and Bioavailability Enhancement of Poorly Soluble APIs. Powder Technol. 2015, 285, 2–15. [Google Scholar] [CrossRef]
- Martínez-Jiménez, C.; Cruz-Angeles, J.; Videa, M.; Martínez, L.M. Co-Amorphous Simvastatin-Nifedipine with Enhanced Solubility for Possible Use in Combination Therapy of Hypertension and Hypercholesterolemia. Molecules 2018, 23, 2161. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Angeles, J.; Videa, M.; Martínez, L.M. Highly Soluble Glimepiride and Irbesartan Co-Amorphous Formulation with Potential Application in Combination Therapy. AAPS PharmSciTech 2019, 20, 144. [Google Scholar] [CrossRef] [PubMed]
- Martínez, L.M.; Videa, M.; López-Silva, G.A.; de los Reyes, C.A.; Cruz-Angeles, J.; González, N. Stabilization of Amorphous Paracetamol Based Systems Using Traditional and Novel Strategies. Int. J. Pharm. 2014, 477, 294–305. [Google Scholar] [CrossRef]
- Martínez, L.M.; Videa, M.; Sosa, N.G.; Ramírez, J.H.; Castro, S. Long-Term Stability of New Co-Amorphous Drug Binary Systems: Study of Glass Transitions as a Function of Composition and Shelf Time. Molecules 2016, 21, 1712. [Google Scholar] [CrossRef] [PubMed]
- Chavan, R.B.; Thipparaboina, R.; Kumar, D.; Shastri, N.R. Co Amorphous Systems: A Product Development Perspective. Int. J. Pharm. 2016, 515, 403–415. [Google Scholar] [CrossRef] [PubMed]
- Wlodarski, K.; Sawicki, W.; Paluch, K.J.; Tajber, L.; Grembecka, M.; Hawelek, L.; Wojnarowska, Z.; Grzybowska, K.; Talik, E.; Paluch, M. The Influence of Amorphization Methods on the Apparent Solubility and Dissolution Rate of Tadalafil. Eur. J. Pharm. Sci. 2014, 62, 132–140. [Google Scholar] [CrossRef]
- Oliveira, P.F.M.; Willart, J.-F.; Siepmann, J.; Siepmann, F.; Descamps, M. Using Milling To Explore Physical States: The Amorphous and Polymorphic Forms of Dexamethasone. Cryst. Growth Des. 2018, 18, 1748–1757. [Google Scholar] [CrossRef]
- Chieng, N.; Aaltonen, J.; Saville, D.; Rades, T. Physical Characterization and Stability of Amorphous Indomethacin and Ranitidine Hydrochloride Binary Systems Prepared by Mechanical Activation. Eur. J. Pharm. Biopharm. 2009, 71, 47–54. [Google Scholar] [CrossRef]
- Baláž, P.; Achimovičová, M.; Baláž, M.; Billik, P.; Cherkezova-Zheleva, Z.; Criado, J.M.; Delogu, F.; Dutková, E.; Gaffet, E.; Gotor, F.J.; et al. Hallmarks of Mechanochemistry: From Nanoparticles to Technology. Chem. Soc. Rev. 2013, 42, 7571. [Google Scholar] [CrossRef]
- Yu, L. Amorphous Pharmaceutical Solids: Preparation, Characterization and Stabilization. Adv. Drug Deliv. Rev. 2001, 48, 27–42. [Google Scholar] [CrossRef]
- Kasten, G.; Löbmann, K.; Grohganz, H.; Rades, T. Co-Former Selection for Co-Amorphous Drug-Amino Acid Formulations. Int. J. Pharm. 2019, 557, 366–373. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Zhang, Q.; Wang, J.R.; Lin, K.L.; Mei, X. Amino Acids as Co-Amorphous Excipients for Tackling the Poor Aqueous Solubility of Valsartan. Pharm. Dev. Technol. 2017, 22, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Gao, H.; Babu, S.; Garad, S. Co-Amorphous Formation of High-Dose Zwitterionic Compounds with Amino Acids to Improve Solubility and Enable Parenteral Delivery. Mol. Pharm. 2018, 15, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Descamps, M.; Willart, J.F.; Dudognon, E.; Caron, V. Transformation of Pharmaceutical Compounds upon Milling and Comilling: The Role of Tg. J. Pharm. Sci. 2006, 96, 1398–1407. [Google Scholar] [CrossRef]
- Wu, W.; Ueda, H.; Löbmann, K.; Rades, T.; Grohganz, H. Organic Acids as Co-Formers for Co-Amorphous Systems—Influence of Variation in Molar Ratio on the Physicochemical Properties of the Co-Amorphous Systems. Eur. J. Pharm. Biopharm. 2018, 131, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Ojarinta, R.; Heikkinen, A.T.; Sievänen, E.; Laitinen, R. Dissolution Behavior of Co-Amorphous Amino Acid-Indomethacin Mixtures: The Ability of Amino Acids to Stabilize the Supersaturated State of Indomethacin. Eur. J. Pharm. Biopharm. 2017, 112, 85–95. [Google Scholar] [CrossRef]
- Gniado, K.; MacFhionnghaile, P.; McArdle, P.; Erxleben, A. The Natural Bile Acid Surfactant Sodium Taurocholate (NaTC) as a Coformer in Coamorphous Systems: Enhanced Physical Stability and Dissolution Behavior of Coamorphous Drug-NaTc Systems. Int. J. Pharm. 2018, 535, 132–139. [Google Scholar] [CrossRef]
- Aljohani, M.; MacFhionnghaile, P.; McArdle, P.; Erxleben, A. Investigation of the Formation of Drug-Drug Cocrystals and Coamorphous Systems of the Antidiabetic Drug Gliclazide. Int. J. Pharm. 2019, 561, 35–42. [Google Scholar] [CrossRef]
- Bansal, S.; Bansal, M.; Kumria, R. Nanocrystals: Current Strategies and Trends. Int. J. Res. Pharm. Biomed. Sci. 2012, 4, 10. [Google Scholar]
- Babu, N.J.; Nangia, A. Solubility Advantage of Amorphous Drugs and Pharmaceutical Cocrystals. Cryst. Growth Des. 2011, 11, 2662–2679. [Google Scholar] [CrossRef]
- Kumari, N.; Ghosh, A. Cocrystallization: Cutting Edge Tool for Physicochemical Modulation of Active Pharmaceutical Ingredients. Curr. Pharm. Des. 2020, 26, 4858–4882. [Google Scholar] [CrossRef] [PubMed]
- Elder, D.P.; Holm, R.; De Diego, H.L. Use of Pharmaceutical Salts and Cocrystals to Address the Issue of Poor Solubility. Int. J. Pharm. 2013, 453, 88–100. [Google Scholar] [CrossRef] [PubMed]
- Karimi-Jafari, M.; Padrela, L.; Walker, G.M.; Croker, D.M. Creating Cocrystals: A Review of Pharmaceutical Cocrystal Preparation Routes and Applications. Cryst. Growth Des. 2018, 18, 6370–6387. [Google Scholar] [CrossRef]
- Zhao, Z.; Liu, G.; Lin, Q.; Jiang, Y. Co-Crystal of Paracetamol and Trimethylglycine Prepared by a Supercritical CO2 Anti-Solvent Process. Chem. Eng. Technol. 2018, 41, 1122–1131. [Google Scholar] [CrossRef]
- Koide, T.; Takeuchi, Y.; Otaki, T.; Yamamoto, K.; Shimamura, R.; Ohashi, R.; Inoue, M.; Fukami, T.; Izutsu, K. ichi Quantification of a Cocrystal and Its Dissociated Compounds in Solid Dosage Form Using Transmission Raman Spectroscopy. J. Pharm. Biomed. Anal. 2020, 177, 112886. [Google Scholar] [CrossRef]
- Neurohr, C.; Revelli, A.L.; Billot, P.; Marchivie, M.; Lecomte, S.; Laugier, S.; Massip, S.; Subra-Paternault, P. Naproxen-Nicotinamide Cocrystals Produced by CO2 Antisolvent. J. Supercrit. Fluids 2013, 83, 78–85. [Google Scholar] [CrossRef]
- Müllers, K.C.; Paisana, M.; Wahl, M.A. Simultaneous Formation and Micronization of Pharmaceutical Cocrystals by Rapid Expansion of Supercritical Solutions (RESS). Pharm. Res. 2015, 32, 702–713. [Google Scholar] [CrossRef]
- Kudo, S.; Takiyama, H. Production Method of Carbamazepine/Saccharin Cocrystal Particles by Using Two Solution Mixing Based on the Ternary Phase Diagram. J. Cryst. Growth 2014, 392, 87–91. [Google Scholar] [CrossRef]
- Zhou, J.; Li, L.; Zhang, H.; Xu, J.; Huang, D.; Gong, N.; Han, W.; Yang, X.; Zhou, Z. Crystal Structures, Dissolution and Pharmacokinetic Study on a Novel Phosphodiesterase-4 Inhibitor Chlorbipram Cocrystals. Int. J. Pharm. 2020, 576, 118984. [Google Scholar] [CrossRef]
- Merah, A.; Abidi, A.; Chaffai, N.; Bataille, B.; Gherraf, N. Role of Hydroxypropylmethylcellulose (HPMC 4000) in the Protection of the Polymorphs of Piroxicam Extended Release Tablets. Arab. J. Chem. 2017, 10, S1243–S1253. [Google Scholar] [CrossRef]
- Al Rahal, O.; Majumder, M.; Spillman, M.J.; van de Streek, J.; Shankland, K. Co-Crystal Structures of Furosemide: Urea and Carbamazepine: Indomethacin determined from powder X-ray diffraction data. Crystals 2020, 10, 42. [Google Scholar] [CrossRef]
- Nugrahani, I.; Utami, D.; Ayuningtyas, L.; Garmana, A.N.; Oktaviary, R. New Preparation Method Using Microwave, Kinetics, In Vitro Dissolution-Diffusion, and Anti-Inflammatory Study of Diclofenac- Proline Co–Crystal. ChemistrySelect 2019, 4, 13396–13403. [Google Scholar] [CrossRef]
- Kuang, W.; Ji, S.; Wang, X.; Zhang, J.; Lan, P. Relationship between Crystal Structures and Physicochemical Properties of Lamotrigine Cocrystal. Powder Technol. 2021, 380, 18–25. [Google Scholar] [CrossRef]
- Kamali, N.; Gniado, K.; McArdle, P.; Erxleben, A. Application of Ball Milling for Highly Selective Mechanochemical Polymorph Transformations. Org. Process Res. Dev. 2018, 22, 796–802. [Google Scholar] [CrossRef]
- Chieng, N.; Rades, T.; Aaltonen, J. An Overview of Recent Studies on the Analysis of Pharmaceutical Polymorphs. J. Pharm. Biomed. Anal. 2011, 55, 618–644. [Google Scholar] [CrossRef]
- Cruz-Cabeza, A.J.; Bernstein, J. Conformational Polymorphism. Chem. Rev. 2014, 114, 2170–2191. [Google Scholar] [CrossRef]
- Cruz-Cabeza, A.J.; Reutzel-Edens, S.M.; Bernstein, J. Facts and Fictions about Polymorphism. Chem. Soc. Rev. 2015, 44, 8619–8635. [Google Scholar] [CrossRef]
- Zvoníček, V.; Skořepová, E.; Dušek, M.; Žvátora, P.; Šoóš, M. Ibrutinib Polymorphs: Crystallographic Study. Cryst. Growth Des. 2018, 18, 1315–1326. [Google Scholar] [CrossRef]
- Stahly, G.P. Diversity in Single- and Multiple-Component Crystals. the Search for and Prevalence of Polymorphs and Cocrystals. Cryst. Growth Des. 2007, 7, 1007–1026. [Google Scholar] [CrossRef]
- Morissette, S.L.; Soukasene, S.; Levinson, D.; Cima, M.J.; Almarsson, Ö. Elucidation of Crystal Form Diversity of the HIV Protease Inhibitor Ritonavir by High-Throughput Crystallization. Proc. Natl. Acad. Sci. USA 2003, 100, 2180–2184. [Google Scholar] [CrossRef]
- Lee, J.; Boerrigter, S.X.M.; Jung, Y.W.; Byun, Y.; Yuk, S.H.; Byrn, S.R.; Lee, E.H. Organic Vapor Sorption Method of Isostructural Solvates and Polymorph of Tenofovir Disoproxil Fumarate. Eur. J. Pharm. Sci. 2013, 50, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Campeta, A.M.; Chekal, B.P.; Abramov, Y.A.; Meenan, P.A.; Henson, M.J.; Shi, B.; Singer, R.A.; Horspool, K.R. Development of a Targeted Polymorph Screening Approach for a Complex Polymorphic and Highly Solvating API. J. Pharm. Sci. 2010, 99, 3874–3886. [Google Scholar] [CrossRef] [PubMed]
- Beckmann, W.; Nickisch, K.; Budde, U. Development of a Seeding Technique for the Crystallization of the Metastable a Modification of Abecarnil. Org. Process Res. Dev. 1998, 2, 298–304. [Google Scholar] [CrossRef]
- Zaccaro, J.; Matic, J.; Myerson, A.S.; Garetz, B.A. Nonphotochemical, Laser-Induced Nucleation of Supersaturated Aqueous Glycine Produces Unexpected γ-Polymorph. Cryst. Growth Des. 2001, 1, 5–8. [Google Scholar] [CrossRef]
- Pasquali, I.; Bettini, R.; Giordano, F. Supercritical Fluid Technologies: An Innovative Approach for Manipulating the Solid-State of Pharmaceuticals. Adv. Drug Deliv. Rev. 2008, 60, 399–410. [Google Scholar] [CrossRef]
- Stolar, T.; Lukin, S.; Tireli, M.; Sović, I.; Karadeniz, B.; Kereković, I.; Matijašić, G.; Gretić, M.; Katančić, Z.; Dejanović, I.; et al. Control of Pharmaceutical Cocrystal Polymorphism on Various Scales by Mechanochemistry: Transfer from the Laboratory Batch to the Large-Scale Extrusion Processing. ACS Sustain. Chem. Eng. 2019, 7, 7102–7110. [Google Scholar] [CrossRef]
- Manin, A.N.; Drozd, K.V.; Surov, A.O.; Churakov, A.V.; Volkova, T.V.; Perlovich, G.L. Identification of a Previously Unreported Co-Crystal Form of Acetazolamide: A Combination of Multiple Experimental and Virtual Screening Methods. Phys. Chem. Chem. Phys. 2020, 22, 20867–20879. [Google Scholar] [CrossRef]
- Dujardin, N.; Willart, J.F.; Dudognon, E.; Danède, F.; Descamps, M. Mechanism of Solid State Amorphization of Glucose upon Milling. J. Phys. Chem. B 2013, 117, 1437–1443. [Google Scholar] [CrossRef]
- Latreche, M.; Willart, J.F.; Guerain, M.; Hédoux, A.; Danède, F. Using Milling to Explore Physical States: The Amorphous and Polymorphic Forms of Sulindac. J. Pharm. Sci. 2019, 108, 2635–2642. [Google Scholar] [CrossRef]
- Stoler, E.; Warner, J.C. Non-Covalent Derivatives: Cocrystals and Eutectics. Molecules 2015, 20, 14833–14848. [Google Scholar] [CrossRef]
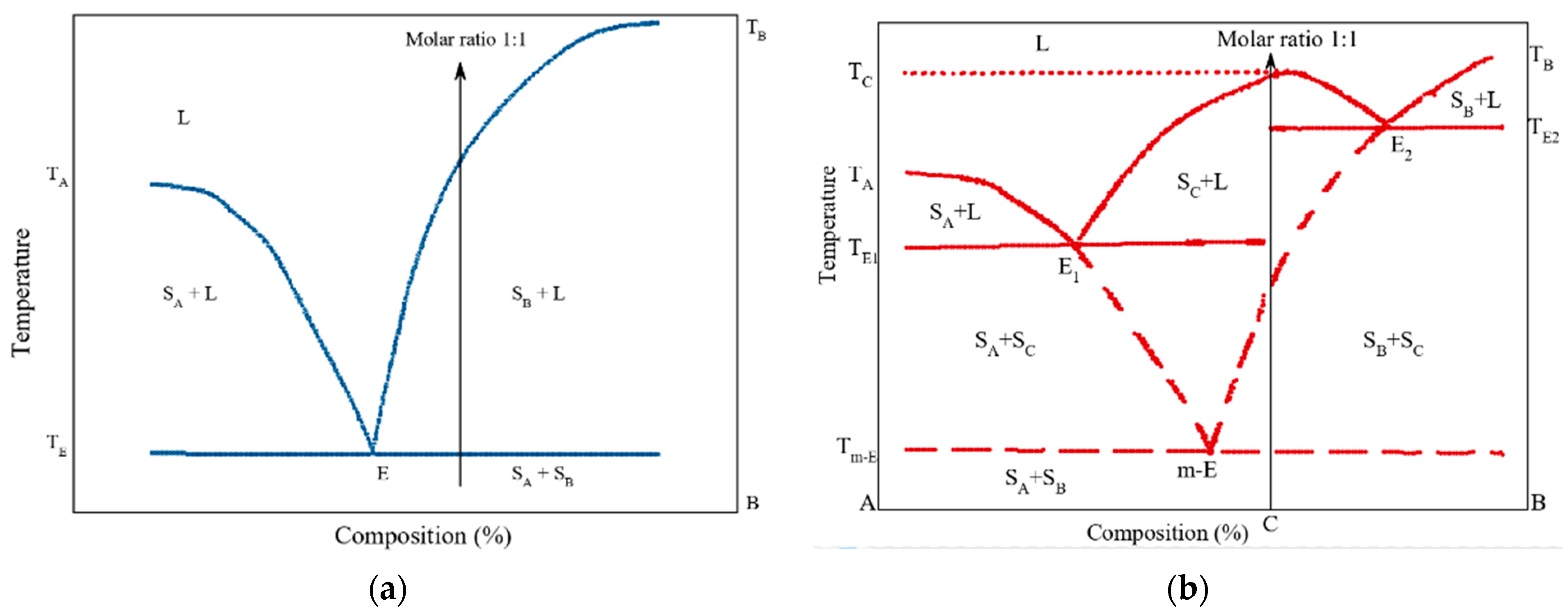
- Yamashita, H.; Hirakura, Y.; Yuda, M.; Teramura, T.; Terada, K. Detection of Cocrystal Formation Based on Binary Phase Diagrams Using Thermal Analysis. Pharm. Res. 2013, 30, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, H.; Hirakura, Y.; Yuda, M.; Terada, K. Coformer Screening Using Thermal Analysis Based on Binary Phase Diagrams. Pharm. Res. 2014, 31, 1946–1957. [Google Scholar] [CrossRef] [PubMed]
- Ren, R.; Yang, Z.; Shaw, L.L. Polymorphic Transformation and Powder Characteristics of TiO2 during High Energy Milling. J. Mater. Sci. 2000, 35, 6015–6026. [Google Scholar] [CrossRef]
- Chieng, N.; Zujovic, Z.; Bowmaker, G.; Rades, T.; Saville, D. Effect of Milling Conditions on the Solid-State Conversion of Ranitidine Hydrochloride Form 1. Int. J. Pharm. 2006, 327, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Willart, J.F.; Lefebvre, J.; Danède, F.; Comini, S.; Looten, P.; Descamps, M. Polymorphic Transformation of the Γ-Form of D-Sorbitol upon Milling: Structural and Nanostructural Analyses. Solid State Commun. 2005, 135, 519–524. [Google Scholar] [CrossRef]
- Lin, S.Y.; Hsu, C.H.; Ke, W.T. Solid-State Transformation of Different Gabapentin Polymorphs upon Milling and Co-Milling. Int. J. Pharm. 2010, 396, 83–90. [Google Scholar] [CrossRef]
- Friščić, T.; Trask, A.V.; Jones, W.; Motherwell, W.D.S. Screening for Inclusion Compounds and Systematic Construction of Three-Component Solids by Liquid-Assisted Grinding. Angew. Chemie—Int. Ed. 2006, 45, 7546–7550. [Google Scholar] [CrossRef]
- Greco, K.; Bogner, R. Solution-Mediated Phase Transformation: Significance During Dissolution and Implications for Bioavailability. J. Pharm. Sci. 2012, 101, 2996–3018. [Google Scholar] [CrossRef]
- Chatziadi, A.; Skořepová, E.; Rohlíček, J.; Dušek, M.; Ridvan, L.; Šoóš, M. Mechanochemically Induced Polymorphic Transformations of Sofosbuvir. Cryst. Growth Des. 2020, 20, 139–147. [Google Scholar] [CrossRef]
- Trask, A.V.; Shan, N.; Motherwell, W.D.S.; Jones, W.; Feng, S.; Tan, R.B.H.; Carpenter, K.J. Selective Polymorph Transformation via Solvent-Drop Grinding. Chem. Commun. 2005, 880–882. [Google Scholar] [CrossRef]
- Bouvart, N.; Palix, R.M.; Arkhipov, S.G.; Tumanov, I.A.; Michalchuk, A.A.L.; Boldyreva, E.V. Polymorphism of Chlorpropamide on Liquid-Assisted Mechanical Treatment: Choice of Liquid and Type of Mechanical Treatment Matter. CrystEngComm 2018, 20, 1797–1803. [Google Scholar] [CrossRef]
- Fischer, F.; Heidrich, A.; Greiser, S.; Benemann, S.; Rademann, K.; Emmerling, F. Polymorphism of Mechanochemically Synthesized Cocrystals: A Case Study. Cryst. Growth Des. 2016, 16, 1701–1707. [Google Scholar] [CrossRef]
- Gu, C.H.; Li, H.; Gandhi, R.B.; Raghavan, K. Grouping Solvents by Statistical Analysis of Solvent Property Parameters: Implication to Polymorph Screening. Int. J. Pharm. 2004, 283, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Kasten, G.; Grohganz, H.; Rades, T.; Löbmann, K. Development of a Screening Method for Co-Amorphous Formulations of Drugs and Amino Acids. Eur. J. Pharm. Sci. 2016, 95, 28–35. [Google Scholar] [CrossRef]
- Wu, W.; Löbmann, K.; Rades, T.; Grohganz, H. On the Role of Salt Formation and Structural Similarity of Co-Formers in Co-Amorphous Drug Delivery Systems. Int. J. Pharm. 2018, 535, 86–94. [Google Scholar] [CrossRef]
- Caron, V.; Tajber, L.; Corrigan, O.I.; Healy, A.M. A Comparison of Spray Drying and Milling in the Production of Amorphous Dispersions of Sulfathiazole/Polyvinylpyrrolidone and Sulfadimidine/Polyvinylpyrrolidone. Mol. Pharm. 2011, 8, 532–542. [Google Scholar] [CrossRef]
- Allesø, M.; Chieng, N.; Rehder, S.; Rantanen, J.; Rades, T.; Aaltonen, J. Enhanced Dissolution Rate and Synchronized Release of Drugs in Binary Systems through Formulation: Amorphous Naproxen-Cimetidine Mixtures Prepared by Mechanical Activation. J. Control. Release 2009, 136, 45–53. [Google Scholar] [CrossRef]
- Karmwar, P.; Graeser, K.; Gordon, K.C.; Strachan, C.J.; Rades, T. Investigation of Properties and Recrystallisation Behaviour of Amorphous Indomethacin Samples Prepared by Different Methods. Int. J. Pharm. 2011, 417, 94–100. [Google Scholar] [CrossRef]
- Wojnarowska, Z.; Grzybowska, K.; Adrjanowicz, K.; Kaminski, K.; Paluch, M.; Hawelek, L.; Wrzalik, R.; Dulski, M.; Sawicki, W.; Mazgalski, J.; et al. Study of the Amorphous Glibenclamide Drug: Analysis of the Molecular Dynamics of Quenched and Cryomilled Material. Mol. Pharm. 2010, 7, 1692–1707. [Google Scholar] [CrossRef]
- Megarry, A.J.; Booth, J.; Burley, J. Amorphous Trehalose Dihydrate by Cryogenic Milling. Carbohydr. Res. 2011, 346, 1061–1064. [Google Scholar] [CrossRef]
- Moinuddin, S.M.; Ruan, S.; Huang, Y.; Gao, Q.; Shi, Q.; Cai, B.; Cai, T. Facile Formation of Co-Amorphous Atenolol and Hydrochlorothiazide Mixtures via Cryogenic-Milling: Enhanced Physical Stability, Dissolution and Pharmacokinetic Profile. Int. J. Pharm. 2017, 532, 393–400. [Google Scholar] [CrossRef]
- Jensen, K.T.; Larsen, F.H.; Löbmann, K.; Rades, T.; Grohganz, H. Influence of Variation in Molar Ratio on Co-Amorphous Drug-Amino Acid Systems. Eur. J. Pharm. Biopharm. 2016, 107, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Badal Tejedor, M.; Pazesh, S.; Nordgren, N.; Schuleit, M.; Rutland, M.W.; Alderborn, G.; Millqvist-Fureby, A. Milling Induced Amorphisation and Recrystallization of α-Lactose Monohydrate. Int. J. Pharm. 2018, 537, 140–147. [Google Scholar] [CrossRef]
- Wu, W.; Löbmann, K.; Schnitzkewitz, J.; Knuhtsen, A.; Pedersen, D.S.; Grohganz, H.; Rades, T. Aspartame as a Co-Former in Co-Amorphous Systems. Int. J. Pharm. 2018, 549, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Dujardin, N.; Willart, J.F.; Dudognon, E.; Hédoux, A.; Guinet, Y.; Paccou, L.; Chazallon, B.; Descamps, M. Solid State Vitrification of Crystalline α and β-D-Glucose by Mechanical Milling. Solid State Commun. 2008, 148, 78–82. [Google Scholar] [CrossRef]
- Kasten, G.; Nouri, K.; Grohganz, H.; Rades, T.; Löbmann, K. Performance Comparison between Crystalline and Co-Amorphous Salts of Indomethacin-Lysine. Int. J. Pharm. 2017, 533, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Martinez, L.M.; Cruz, J. Preparación de Formulaciones Farmacéuticas Amorfas Usando Metodologías Alternativas Emergentes de Amorfización. 2018. Available online: https://www.researchgate.net/publication/363611674_PREPARACION_DE_FORMULACIONES_FARMACEUTICAS_AMORFAS_USANDO_METODOLOGIAS_ALTERNATIVAS_EMERGENTES_DE_AMORFIZACION (accessed on 1 March 2021).
- Löbmann, K.; Laitinen, R.; Strachan, C.; Rades, T.; Grohganz, H. Amino Acids as Co-Amorphous Stabilizers for Poorly Water-Soluble Drugs—Part 2: Molecular Interactions. Eur. J. Pharm. Biopharm. 2013, 85, 882–888. [Google Scholar] [CrossRef]
- Kasten, G.; Lobo, L.; Dengale, S.; Grohganz, H.; Rades, T.; Löbmann, K. In Vitro and in Vivo Comparison between Crystalline and Co-Amorphous Salts of Naproxen-Arginine. Eur. J. Pharm. Biopharm. 2018, 132, 192–199. [Google Scholar] [CrossRef]
- França, M.T.; Marcos, T.M.; Pereira, R.N.; Stulzer, H.K. Could the Small Molecules Such as Amino Acids Improve Aqueous Solubility and Stabilize Amorphous Systems Containing Griseofulvin? Eur. J. Pharm. Sci. 2020, 143, 105178. [Google Scholar] [CrossRef]
- Jensen, K.T.; Löbmann, K.; Rades, T.; Grohganz, H. Improving Co-Amorphous Drug Formulations by the Addition of the Highly Water Soluble Amino Acid, Proline. Pharmaceutics 2014, 6, 416–435. [Google Scholar] [CrossRef]
- Wu, W.; Löbmann, K.; Schnitzkewitz, J.; Knuhtsen, A.; Pedersen, D.S.; Rades, T.; Grohganz, H. Dipeptides as Co-Formers in Co-Amorphous Systems. Eur. J. Pharm. Biopharm. 2019, 134, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Mennini, N.; Maestrelli, F.; Cirri, M.; Mura, P. Analysis of Physicochemical Properties of Ternary Systems of Oxaprozin with Randomly Methylated-ß-Cyclodextrin and L-Arginine Aimed to Improve the Drug Solubility. J. Pharm. Biomed. Anal. 2016, 129, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Petry, I.; Löbmann, K.; Grohganz, H.; Rades, T.; Leopold, C.S. In Situ Co-Amorphisation of Arginine with Indomethacin or Furosemide during Immersion in an Acidic Medium—A Proof of Concept Study. Eur. J. Pharm. Biopharm. 2018, 133, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Jensen, K.T.; Larsen, F.H.; Cornett, C.; Löbmann, K.; Grohganz, H.; Rades, T. Formation Mechanism of Coamorphous Drug-Amino Acid Mixtures. Mol. Pharm. 2015, 12, 2484–2492. [Google Scholar] [CrossRef] [PubMed]
- Ueda, H.; Peter Bøtker, J.; Edinger, M.; Löbmann, K.; Grohganz, H.; Müllertz, A.; Rades, T.; Østergaard, J. Formulation of Co-Amorphous Systems from Naproxen and Naproxen Sodium and in Situ Monitoring of Physicochemical State Changes during Dissolution Testing by Raman Spectroscopy. Int. J. Pharm. 2020, 587, 119662. [Google Scholar] [CrossRef] [PubMed]
- Mishra, J.; Löbmann, K.; Grohganz, H.; Rades, T. Influence of Preparation Technique on Co-Amorphization of Carvedilol with Acidic Amino Acids. Int. J. Pharm. 2018, 552, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Laitinen, R.; Löbmann, K.; Grohganz, H.; Strachan, C.; Rades, T. Amino Acids as Co-Amorphous Excipients for Simvastatin and Glibenclamide: Physical Properties and Stability. Mol. Pharm. 2014, 11, 2381–2389. [Google Scholar] [CrossRef]
- Walker, G.; Römann, P.; Poller, B.; Löbmann, K.; Grohganz, H.; Rooney, J.S.; Huff, G.S.; Smith, G.P.S.; Rades, T.; Gordon, K.C.; et al. Probing Pharmaceutical Mixtures during Milling: The Potency of Low-Frequency Raman Spectroscopy in Identifying Disorder. Mol. Pharm. 2017, 14, 4675–4684. [Google Scholar] [CrossRef]
- Ueda, H.; Wu, W.; Löbmann, K.; Grohganz, H.; Müllertz, A.; Rades, T. Application of a Salt Coformer in a Co-Amorphous Drug System Dramatically Enhances the Glass Transition Temperature: A Case Study of the Ternary System Carbamazepine, Citric Acid, and l -Arginine. Mol. Pharm. 2018, 15, 2036–2044. [Google Scholar] [CrossRef]
- Sormunen, H.; Ruponen, M.; Laitinen, R. The Effect of Co-Amorphization of Glibenclamide on Its Dissolution Properties and Permeability through an MDCKII-MDR1 Cell Layer. Int. J. Pharm. 2019, 570, 118653. [Google Scholar] [CrossRef]
- Wu, W.; Grohganz, H.; Rades, T.; Löbmann, K. Comparison of Co-Former Performance in Co-Amorphous Formulations: Single Amino Acids, Amino Acid Physical Mixtures, Amino Acid Salts and Dipeptides as Co-Formers. Eur. J. Pharm. Sci. 2021, 156, 105582. [Google Scholar] [CrossRef] [PubMed]
- Slámová, M.; Prausová, K.; Epikaridisová, J.; Brokešová, J.; Kuentz, M.; Patera, J.; Zámostný, P. Effect of Co-Milling on Dissolution Rate of Poorly Soluble Drugs. Int. J. Pharm. 2021, 597, 120312. [Google Scholar] [CrossRef] [PubMed]
- Fujioka, S.; Kadota, K.; Yoshida, M.; Shirakawa, Y. Improvement in the Elution Behavior of Rutin via Binary Amorphous Solid with Flavonoid Using a Mechanochemical Process. Food Bioprod. Process. 2020, 123, 274–283. [Google Scholar] [CrossRef]
- Hatwar, P.; Pathan, I.B.; Chishti, N.A.H.; Ambekar, W. Pellets Containing Quercetin Amino Acid Co-Amorphous Mixture for the Treatment of Pain: Formulation, Optimization, In-Vitro and In-Vivo Study. J. Drug Deliv. Sci. Technol. 2021, 62, 102350. [Google Scholar] [CrossRef]
- Pinto, J.M.O.; Leão, A.F.; Bazzo, G.C.; Mendes, C.; Madureira, L.M.P.; Caramori, G.F.; Parreira, R.L.T.; Stulzer, H.K. Supersaturating Drug Delivery Systems Containing Fixed-Dose Combination of Two Antihypertensive Drugs: Formulation, in Vitro Evaluation and Molecular Metadynamics Simulations. Eur. J. Pharm. Sci. 2021, 163, 105860. [Google Scholar] [CrossRef] [PubMed]
- Lukin, S.; Stolar, T.; Tireli, M.; Barišić, D.; di Michiel, M.; Užarević, K.; Halasz, I. Solid-State Supramolecular Assembly of Salicylic Acid and 2-Pyridone, 3-Hydroxypyridine or 4-Pyridone. Croat. Chem. Acta 2017, 90, 707–710. [Google Scholar] [CrossRef]
- Shemchuk, O.; Agostino, S.; Fiore, C.; Zannoli, S.; Grepioni, F.; Braga, D. Natural Antimicrobials Meet a Synthetic Antibiotic: Carvacrol/Thymol and Ciprofloxacin Cocrystals as a Promising Solid-State Route to Activity Enhancement. Cryst. Growth Des. 2020, 20, 6796–6803. [Google Scholar] [CrossRef]
- Macfhionnghaile, P.; Crowley, C.M.; McArdle, P.; Erxleben, A. Spontaneous Solid-State Cocrystallization of Caffeine and Urea. Cryst. Growth Des. 2020, 20, 736–745. [Google Scholar] [CrossRef]
- Arabiani, M.R.; Lodagekar, A.; Yadav, B.; Chavan, R.B.; Shastri, N.R.; Purohit, P.Y.; Shelat, P.; Dave, D. Mechanochemical Synthesis of Brexpiprazole Cocrystals to Improve Its Pharmaceutical Attributes. CrystEngComm 2019, 21, 800–806. [Google Scholar] [CrossRef]
- Setyawan, D.; Jovita, R.O.; Iqbal, M.; Paramanandana, A.; Yusuf, H.; Lestari, M.L.A.D. Co-Crystalization of Quercetin and Malonic Acid Using Solvent-Drop Grinding Method. Trop. J. Pharm. Res. 2018, 17, 997–1002. [Google Scholar] [CrossRef]
- Tantardini, C.; Arkipov, S.G.; Cherkashina, K.A.; Kil’met’ev, A.S.; Boldyreva, E.V. Synthesis and Crystal Structure of a Meloxicam Co-Crystal with Benzoic Acid. Struct. Chem. 2018, 29, 1867–1874. [Google Scholar] [CrossRef]
- Wang, Y.; Xue, J.; Qin, J.; Liu, J.; Du, Y. Structure and Spectroscopic Characterization of Pharmaceutical Co-Crystal Formation between Acetazolamide and 4-Hydroxybenzoic Acid. Spectrochim. Acta—Part A Mol. Biomol. Spectrosc. 2019, 219, 419–426. [Google Scholar] [CrossRef] [PubMed]
- De Almeida, A.C.; Torquetti, C.; Ferreira, P.O.; Fernandes, R.P.; dos Santos, E.C.; Kogawa, A.C.; Caires, F.J. Cocrystals of Ciprofloxacin with Nicotinic and Isonicotinic Acids: Mechanochemical Synthesis, Characterization, Thermal and Solubility Study. Thermochim. Acta 2020, 685, 178346. [Google Scholar] [CrossRef]
- Wu, X.; Wang, Y.; Xue, J.; Liu, J.; Qin, J.; Hong, Z.; Du, Y. Solid Phase Drug-Drug Pharmaceutical Co-Crystal Formed between Pyrazinamide and Diflunisal: Structural Characterization Based on Terahertz/Raman Spectroscopy Combining with DFT Calculation. Spectrochim. Acta—Part A Mol. Biomol. Spectrosc. 2020, 234, 118265. [Google Scholar] [CrossRef]
- Fang, J.; Zhang, Z.; Bo, Y.; Xue, J.; Wang, Y.; Liu, J.; Qin, J.; Hong, Z.; Du, Y. Vibrational Spectral and Structural Characterization of Multicomponent Ternary Co-Crystal Formation within Acetazolamide, Nicotinamide and 2-Pyridone. Spectrochim. Acta—Part A Mol. Biomol. Spectrosc. 2021, 245, 118885. [Google Scholar] [CrossRef]
- Liu, C.; Liu, Z.; Chen, Y.; Chen, Z.; Chen, H.; Pui, Y.; Qian, F. Oral Bioavailability Enhancement of β-Lapachone, a Poorly Soluble Fast Crystallizer, by Cocrystal, Amorphous Solid Dispersion, and Crystalline Solid Dispersion. Eur. J. Pharm. Biopharm. 2018, 124, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, P.O.; de Almeida, A.C.; dos Santos, É.C.; Droppa, R.; Ferreira, F.F.; Kogawa, A.C.; Caires, F.J. A Norfloxacin-Nicotinic Acid Cocrystal: Mechanochemical Synthesis, Thermal and Structural Characterization and Solubility Assays. Thermochim. Acta 2020, 694, 178782. [Google Scholar] [CrossRef]
- Teng, R.; Wang, L.; Chen, M.; Fang, W.; Gao, Z.; Chai, Y.; Zhao, P.; Bao, Y. Amino Acid Based Pharmaceutical Cocrystals and Hydrate Cocrystals of the Chlorothiazide: Structural Studies and Physicochemical Properties. J. Mol. Struct. 2020, 1217, 128432. [Google Scholar] [CrossRef]
- Gaggero, A.; Jurišić Dukovski, B.; Radić, I.; Šagud, I.; Škorić, I.; Cinčić, D.; Jug, M. Co-Grinding with Surfactants as a New Approach to Enhance in Vitro Dissolution of Praziquantel. J. Pharm. Biomed. Anal. 2020, 189, 113494. [Google Scholar] [CrossRef]
- Aitipamula, S.; Das, S. Cocrystal Formulations: A Case Study of Topical Formulations Consisting of Ferulic Acid Cocrystals. Eur. J. Pharm. Biopharm. 2020, 149, 95–104. [Google Scholar] [CrossRef]
- Hossain Mithu, M.S.; Ross, S.A.; Hurt, A.P.; Douroumis, D. Effect of Mechanochemical Grinding Conditions on the Formation of Pharmaceutical Cocrystals and Co-Amorphous Solid Forms of Ketoconazole—Dicarboxylic Acid. J. Drug Deliv. Sci. Technol. 2021, 63, 102508. [Google Scholar] [CrossRef]
- Vasilev, N.A.; Surov, A.O.; Voronin, A.P.; Drozd, K.V.; Perlovich, G.L. Novel Cocrystals of Itraconazole: Insights from Phase Diagrams, Formation Thermodynamics and Solubility. Int. J. Pharm. 2021, 599, 120441. [Google Scholar] [CrossRef] [PubMed]
- Guerain, M.; Guinet, Y.; Correia, N.T.; Paccou, L.; Danède, F.; Hédoux, A. Polymorphism and Stability of Ibuprofen/Nicotinamide Cocrystal: The Effect of the Crystalline Synthesis Method. Int. J. Pharm. 2020, 584, 119454. [Google Scholar] [CrossRef]
- Zhang, Z.; Fang, J.; Bo, Y.; Xue, J.; Liu, J.; Hong, Z.; Du, Y. Terahertz and Raman Spectroscopic Investigation of Anti-Tuberculosis Drug-Drug Cocrystallization Involving 4-Aminosalicylic Acid and Pyrazinamide. J. Mol. Struct. 2021, 1227, 129547. [Google Scholar] [CrossRef]
- Shaikh, R.; Shirazian, S.; Guerin, S.; Sheehan, E.; Thompson, D.; Walker, G.M.; Croker, D.M. Understanding Solid-State Processing of Pharmaceutical Cocrystals via Milling: Role of Tablet Excipients. Int. J. Pharm. 2021, 601, 120514. [Google Scholar] [CrossRef] [PubMed]
- Mikhailovskaya, A.V.; Myz, S.A.; Bulina, N.V.; Gerasimov, K.B.; Kuznetsova, S.A.; Shakhtshneider, T.P. Screening and Characterization of Cocrystal Formation between Betulin and Terephthalic Acid. Mater. Today Proc. 2019, 25, 381–383. [Google Scholar] [CrossRef]
- Da Silva, C.C.P.; de Melo, C.C.; Souza, M.S.; Diniz, L.F.; Carneiro, R.L.; Ellena, J. 5-Fluorocytosine/5-Fluorouracil Drug-Drug Cocrystal: A New Development Route Based on Mechanochemical Synthesis. J. Pharm. Innov. 2019, 14, 50–56. [Google Scholar] [CrossRef]
- Germann, L.S.; Arhangelskis, M.; Etter, M.; Dinnebier, R.E.; Friščić, T. Challenging the Ostwald Rule of Stages in Mechanochemical Cocrystallisation. Chem. Sci. 2020, 11, 10092–10100. [Google Scholar] [CrossRef]
- Elisei, E.; Willart, J.F.; Danède, F.; Siepmann, J.; Siepmann, F.; Descamps, M. Crystalline Polymorphism Emerging From a Milling-Induced Amorphous Form: The Case of Chlorhexidine Dihydrochloride. J. Pharm. Sci. 2018, 107, 121–126. [Google Scholar] [CrossRef]
- Amaro, M.I.; Simon, A.; Cabral, L.M.; De Sousa, V.P.; Healy, A.M. Rivastigmine Hydrogen Tartrate Polymorphs: Solid-State Characterisation of Transition and Polymorphic Conversion via Milling. Solid State Sci. 2018, 49, 29–36. [Google Scholar] [CrossRef]
- Cheng, W.T.; Lin, S.Y.; Li, M.J. Raman Microspectroscopic Mapping or Thermal System Used to Investigate Milling-Induced Solid-State Conversion of Famotidine Polymorphs. J. Raman Spectrosc. 2007, 38, 1595–1601. [Google Scholar] [CrossRef]
- Surov, A.O.; Vasilev, N.A.; Churakov, A.V.; Stroh, J.; Emmerling, F.; Perlovich, G.L. Solid Forms of Ciprofloxacin Salicylate: Polymorphism, Formation Pathways, and Thermodynamic Stability. Cryst. Growth Des. 2019, 19, 2979–2990. [Google Scholar] [CrossRef]
- Dupont, A.; Guerain, M.; Danède, F.; Paccou, L.; Guinet, Y.; Hédoux, A.; Willart, J.-F. Kinetics and Mechanism of Polymorphic Transformation of Sorbitol under Mechanical Milling. Int. J. Pharm. 2020, 590, 119902. [Google Scholar] [CrossRef]
- Aitipamula, S.; Chow, P.S.; Tan, R.B.H. Conformational and Enantiotropic Polymorphism of a 1:1 Cocrystal Involving Ethenzamide and Ethylmalonic Acid. CrystEngComm 2010, 12, 3691–3697. [Google Scholar] [CrossRef]
- Trask, A.V.; Motherwell, W.D.S.; Jones, W. Solvent-Drop Grinding: Green Polymorph Control of Cocrystallisation. Chem. Commun. 2004, 4, 890–891. [Google Scholar] [CrossRef] [PubMed]
- Good, D.J.; Naír, R.H. Solubility Advantage of Pharmaceutical Cocrystals. Cryst. Growth Des. 2009, 9, 2252–2264. [Google Scholar] [CrossRef]
- Alhalaweh, A.; Roy, L.; Rodríguez-Hornedo, N.; Velaga, S.P. PH-Dependent Solubility of Indomethacin-Saccharin and Carbamazepine- Saccharin Cocrystals in Aqueous Media. Mol. Pharm. 2012, 9, 2605–2612. [Google Scholar] [CrossRef]
- Bavishi, D.D.; Borkhataria, C.H. Spring and Parachute: How Cocrystals Enhance Solubility. Prog. Cryst. Growth Charact. Mater. 2016, 62, 1–8. [Google Scholar] [CrossRef]
- Pazesh, S.; Lazorova, L.; Berggren, J.; Alderborn, G.; Gråsjö, J. Considerations on the Quantitative Analysis of Apparent Amorphicity of Milled Lactose by Raman Spectroscopy. Int. J. Pharm. 2016, 511, 488–504. [Google Scholar] [CrossRef]
- Soares, F.L.F.; Carneiro, R.L. Green Synthesis of Ibuprofen-Nicotinamide Cocrystals and in-Line Evaluation by Raman Spectroscopy. Cryst. Growth Des. 2013, 13, 1510–1517. [Google Scholar] [CrossRef]
- Mukherjee, A.; Tothadi, S.; Chakraborty, S.; Ganguly, S.; Desiraju, G.R. Synthon Identification in Co-Crystals and Polymorphs with IR Spectroscopy. Primary Amides as a Case Study. CrystEngComm 2013, 15, 4640–4654. [Google Scholar] [CrossRef]
- Saha, S.; Rajput, L.; Joseph, S.; Mishra, M.K.; Ganguly, S.; Desiraju, G.R. IR Spectroscopy as a Probe for C-H⋯X Hydrogen Bonded Supramolecular Synthons. CrystEngComm 2015, 17, 1273–1290. [Google Scholar] [CrossRef]
- Skorupska, E.; Kaźmierski, S.; Potrzebowski, M.J. Solid State NMR Characterization of Ibuprofen:Nicotinamide Cocrystals and New Idea for Controlling Release of Drugs Embedded into Mesoporous Silica Particles. Mol. Pharm. 2017, 14, 1800–1810. [Google Scholar] [CrossRef] [PubMed]
- Apih, T.; Žagar, V.; Seliger, J. NMR and NQR Study of Polymorphism in Carbamazepine. Solid State Nucl. Magn. Reson. 2020, 107, 101653. [Google Scholar] [CrossRef]
- Thomas, L.C. Use of Multiple Heating Rate DSC and Modulated Temperature DSC to Detect and Analyze Temperature-Time-Dependent Transitions in Materials. Am. Lab. 2001, 33, 26–31. Available online: https://www.researchgate.net/publication/286909193_Use_of_multiple_heating_rate_DSC_and_modulated_temperature_DSC_to_detect_and_analyze_temperature-time-dependent_transitions_in_materials (accessed on 1 March 2021).
- Kissi, E.O.; Kasten, G.; Löbmann, K.; Rades, T.; Grohganz, H. The Role of Glass Transition Temperatures in Coamorphous Drug-Amino Acid Formulations. Mol. Pharm. 2018, 15, 4247–4256. [Google Scholar] [CrossRef] [PubMed]
- Löbmann, K.; Laitinen, R.; Grohganz, H.; Gordon, K.C.; Strachan, C.; Rades, T. Coamorphous Drug Systems: Enhanced Physical Stability and Dissolution Rate of Indomethacin and Naproxen. Mol. Pharm. 2011, 8, 1919–1928. [Google Scholar] [CrossRef]
- Gordon, M.; Taylor, J. Ideal Copolymers and the Second-Order Transition of Rubbers. J. Appl. Chem. 1952, 2, 493–500. [Google Scholar] [CrossRef]
- Shamblin, S.L.; Huang, E.Y.; Zografi, G. The Effects of Co-Lyophilized Polymeric Additives on the Glass Transition Temperature and Crystallization of Amorphous Sucrose. J. Therm. Anal. 1996, 47, 1567–1579. [Google Scholar] [CrossRef]
- Taylor, L.S.; Zografi, G. Sugar-Polymer Hydrogen Bond Interactions in Lyophilized Amorphous Mixtures. J. Pharm. Sci. 1998, 87, 1615–1621. [Google Scholar] [CrossRef]
- Masuda, T.; Yoshihashi, Y.; Yonemochi, E.; Fujii, K.; Uekusa, H.; Terada, K. Cocrystallization and Amorphization Induced by Drug-Excipient Interaction Improves the Physical Properties of Acyclovir. Int. J. Pharm. 2012, 422, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Yamamura, S.; Gotoh, H.; Sakamoto, Y.; Momose, Y. Physicochemical Properties of Amorphous Salt of Cimetidine and Diflunisal System. Int. J. Pharm. 2002, 241, 213–221. [Google Scholar] [CrossRef]
- Warner, J.C. Entropic Control in Chemistry and Design. Pure Appl. Chem. 2006, 78, 2035–2041. [Google Scholar] [CrossRef]
- Nugrahani, I.; Utami, D.; Ibrahim, S.; Nugraha, Y.P.; Uekusa, H. Zwitterionic Cocrystal of Diclofenac and L-Proline: Structure Determination, Solubility, Kinetics of Cocrystallization, and Stability Study. Eur. J. Pharm. Sci. 2018, 117, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.G.Z.; Gu, C.; Zell, M.T.; Todd Burkhardt, R.; Munson, E.J.; Grant, D.J.W. Crystallization and Transitions of Sulfamerazine Polymorphs. J. Pharm. Sci. 2002, 91, 1089–1100. [Google Scholar] [CrossRef] [PubMed]
- Willart, J.F.; De Gusseme, A.; Hemon, S.; Odou, G.; Danede, F.; Descamps, M. Direct Crystal to Glass Transformation of Trehalose Induced by Ball Milling. Solid State Commun. 2001, 119, 501–505. [Google Scholar] [CrossRef]
- Desprez, S.; Descamps, M. Transformations of Glassy Indomethacin Induced by Ball-Milling. J. Non. Cryst. Solids 2006, 352, 4480–4485. [Google Scholar] [CrossRef]
- Löbmann, K.; Grohganz, H.; Laitinen, R.; Strachan, C.; Rades, T. Amino Acids as Co-Amorphous Stabilizers for Poorly Water Soluble Drugs—Part 1: Preparation, Stability and Dissolution Enhancement. Eur. J. Pharm. Biopharm. 2013, 85, 873–881. [Google Scholar] [CrossRef]
- Sterren, V.B.; Zoppi, A.; Abraham-Miranda, J.; Longhi, M.R. Enhanced Dissolution Profiles of Glibenclamide with Amino Acids Using a Cogrinding Method. Mater. Today Commun. 2021, 26, 102126. [Google Scholar] [CrossRef]
- Tejedor, M.B.; Nordgren, N.; Schuleit, M.; Pazesh, S.; Alderborn, G.; Millqvist-Fureby, A.; Rutland, M.W. Determination of Interfacial Amorphicity in Functional Powders. Langmuir 2017, 33, 920–926. [Google Scholar] [CrossRef]



| # | Solubility Evaluation (UV, HPLC) | Sample | Ratio/Composition | Solubilty Increment (Folds) | Ref. |
|---|---|---|---|---|---|
| 2A | HPLC (IDR) | Furosemide-arginine | 1:1 | 38 | [85] |
| Nitrofurantoin-arginine | 20 | ||||
| 3A | UV (IDR) | Sulfathiazole-polyvinylpyrrolidone | Xpvp = 0.7 | 5.2 | [86] |
| Sulfadimidine-polyvinylpyrrolidone | 26.5 | ||||
| 4A | UV (IDR) | Co-milled naproxen | 1:1 | 4 | [87] |
| Co-milled cimetidine | 2 | ||||
| 7A | HPLC (Solubility) | Tadalafil * | N/A | 1.25 (in H2O) | [26] |
| 0.79 (in 0.1 M HCl) | |||||
| 1.35 (Buffer pH = 6.8) | |||||
| 1.83 (in water) | |||||
| 10A | UV (IDR) | Atenolol-hydrochlorothiazide | 1:1 | 12.5 | [91] |
| 15A | HPLC (Powder dissolution studies) | Mebendazole-ASPA | 1:1 | 8.13 | [94] |
| Tadalafil-ASPA | Similar increase to MEB but less pronounced | ||||
| Piroxicam-ASPA | 32.1–35 | ||||
| 17A | HPLC (IDR) | Fur-Phe, Fur-Pro, Fur-Trp | 1:1 | 0.9–1.0 | [31] |
| Fur-Ile, Fur-Leu, Fur-Met, Fur-Val, Ind-Ile, Ind-Leu, Ind-Met, Ind-Phe, Ind-Pro, Ind-Trp, Ind-Val, Meb-Met, Cbz-Trp | 1.1–3.0 | ||||
| Fur-Arg, Fur-His, Fur-Lys, Ind-Arg, Ind-Lys, Car-Ile, Car-Leu, Car-Met, Car-Phe, Car-Trp, Car-Val, Meb-Ile, Meb-Leu, Meb-Phe, Meb-Trp | 3.1–431.8 | ||||
| 18A | HPLC (IDR) | Indomethacin-lysine | 1:1 | 90 | [96] |
| 14 | |||||
| 23A | HPLC (Kinetic solubility studies) | Griseofulvin-tryptophan | 1:1 | 1.19 | [100] |
| 25A | HPLC (Dissolution tests) | Mebendazole-histidine-glycine | 1:1:1 | 19 | [102] |
| Mebendazole-tryptophan-phenylalanine | 1:1:1 | 46 | |||
| Mebendazole-proline-tryptophan | 1:1:1 | 4.3 | |||
| 29A | UV | Naproxen-NAP(Na) | 1:1 | 2.9 | [106] |
| 30A | UV (IDR) | Carvedilol-L-glutamic acid | 1:1 | 12 | [107] |
| Carvedilol-L-aspartic acid | 13 | ||||
| Carvedilol-L-glutamic acid | 14 | ||||
| Carvedilol-L-aspartic acid | 2 | ||||
| 31A | Dissolution studies | Indomethacin-arginine | 1:1 | 1.4 | [36] |
| Indomethacin-phenylalanine | 1 | ||||
| Indomethacin-tryptophan | 1 | ||||
| 33A | HPLC (IDR) | Carbamazepine-arginine-tryptophan * | 1:1:1 | 1.38 | [98] |
| Carbamazepine-phenylalanine-tryptophan * | 1:1:1 | 1.2 | |||
| Carbamazepine-tryptophan * | 1:1 | 1.08 | |||
| Indomethacin-L-arginine * | 1:1 | 306 | |||
| Indomethacin-L-phenylalanine * | 1:1 | 4.3 | |||
| Indomethacin-L-tryptophan * | 1:1 | 2.4 | |||
| Indomethacin-L-phenylalanine-L-tryptophan * | 1:1:1 | 3.35 | |||
| 35A | UV | Carbamazepine-citric acid | 1:1 | 2.2 | [110] |
| Carbamazepine-citric acid-arginine | 1:1:1 | 2.68 | |||
| Carbamazepine-citric acid-arginine | 1:1:2 | 3.28 | |||
| Carbamazepine-citric acid-arginine | 1:1:3 | 3.4 | |||
| 36A | HPLC | Glibenclamide-serine | 1:1 | 10 | [111] |
| Glibenclamide-quercetin | 1:1 | 20 | |||
| Glibenclamide-arginine | 1:1 | 19 | |||
| Glibenclamide-arginine-sls | 1:1 | 21 | |||
| 37A | HPLC | Mebendazole (Meb)-glutamate-arginine (crystalline salt) * | 1:1:1 | 5.2 | [112] |
| Meb-glutamate-arginine (amorphous salt) * | 1:1:1 | 3.5 | |||
| Meb-arginineglutamate * | 1:1 | 5.16 | |||
| Meb-glutamatearginine * | 1:1 | 4.9 | |||
| 38A | HPLC | Indomethacin-meglumine * | 1:1 | 18.56 | [113] |
| 1:2 | 25.39 | ||||
| 1:4 | 28 | ||||
| Mefenamic acid-meglumine * | 1:1 | 81 | |||
| 1:2 | 108.6 | ||||
| 1:4 | 394.3 | ||||
| Indomethacin-polyvinylpyrrolidone * | 1:1 | 0.3 | |||
| 1:2 | 0.3 | ||||
| 1:4 | 0.48 | ||||
| Mefenamic acid-polyvinylpyrrolidone * | 1:1 | 1.6 | |||
| 1:2 | 4 | ||||
| 1:4 | 10.6 | ||||
| 41A | UV | Quercetin-arginine * | 1:2 | 21 | [115] |
| # | Solubility Evaluation (UV, HPLC) | Sample | Folds | Ref. |
|---|---|---|---|---|
| 3C | In vitro | Ciprofloxacin-thymol (1:2) | 4 | [118] |
| 5C | UV | Brexpiprazol-catechol (1:1) | 2.5 | [120] |
| Brexpiprazol-succinic acid (1:1) | 2.5 | |||
| 6C | UV | Quercetin-malonic acid (1:2) | 1.056 | [121] |
| 7C | UV | Paracetamol-trimethylglycine * (1:1) | 0.82 | [44] |
| 11C | UV | Ciprofloxacin-nicotinic acid (1:1) | 20 (in water) | [124] |
| 1.5 | ||||
| Ciprofloxacin-isonicotinic acid (1:1) | 20 | |||
| 2.5 | ||||
| 13C | HPLC | Acetazolamide-4-aminobenzoic acid * (1:1) | 2.5 | [67] |
| 2.17 | ||||
| 15C | IDR | β-lapachone-resorcinol (1:1) | 2 | [127] |
| 16C | UV | Norfloxacin-nicotinic acid (with EtOH) pH = 3 | No change | [128] |
| Norfloxacin-nicotinic acid (with EtOH) pH = 6.1 | 2 | |||
| Norfloxacin-nicotinic acid (with EtOH) pH = 8.5 | <2 | |||
| 17C | UV (Powder dissolution) | Chlorothiazide-DL-proline (w/acetonitrile-water) | 1.05 | [129] |
| Chlorothiazide-L-proline hydrate (w/acetonitrile-water) | Lower value than the initial drug | |||
| Chlorothiazide-D-proline hydrate (w/acetonitrile-water) | ||||
| 19C | HPLC (In vitro release test) | Ferulic acid-nicotinamide | 2.4 | [131] |
| Ferulic acid-isonicotinamide | 3.1 | |||
| Ferulic acid-urea | 1.1 | |||
| 21C | HPLC | Itraconazole-4-hydroxybenzamide form II (1:2) | 225 | [133] |
| Itraconazole-4-aminobenzoic acid (1:1) | 64 |
| # | Sample | Analytical Technique | Wavenumber (cm−1)/δ (ppm) | Interpretation | Ref. | |
|---|---|---|---|---|---|---|
| Crystalline | Co-Amorphous | |||||
| 4A | Naproxen-cimetidine | Raman | 670 (C-S-C str) | 666 cm−1 | Shift → unknown mechanism of interaction | [87] |
| 1601 (ring str) | 1604 cm−1 | Shift → solid-state interaction of imidazole ring with naproxen | ||||
| 5A | γ-Indomethacin–ranitidine hydrochloride | DRIFTS (FT-IR) | 1717 and 1692 (C=O) | 1723 and 1679 | Broadening and shift | [28] |
| N/A | 1735 cm−1 | Shoulder appearance | ||||
| N/A | 1723 (C=O) | Peak formation → conjugated carbonyl acid system | ||||
| 1692 (C=N) | 1679 cm−1 | Shift → larger C=N double bond character or interaction at benzoyl C=O ocurred | ||||
| 1620 (aci-nitro C=N str) | 1610 | Shift → nitro group forming a bond with indomethacin and indirectly reducing the C=N double bond character | ||||
| N/A | 1579 | Small peak formation → interaction at the amidine moiety | ||||
| 6A | γ/α-Indomethacin | Raman | N/A | 1540 to 1700 and 2930 to 3100 cm−1 | Large spectral differences → variations in molecular conformation and intermolecular bonding of amorphous forms | [88] |
| 8A | Glibenclamide | FT-IR | 3315 (N-H str) | N/A | Abscence of band upon cryomilling | [89] |
| 1714 (C=O str) | N/A | Loss in intensity but clearly apparent | ||||
| N/A | 1637 (C=N str) | New band → conversion of the amide to the imidic acid form | ||||
| 9A | Trehalose dihydrate | Raman | 30–400 (several peaks) | N/A | Presence of only a broad peak (boson) → amorphous material | [90] |
| 443, 835, 906, and 1449 | 433, 843, 912, and 1455 cm−1 | Shift → amorphous transformation | ||||
| 10A | Atenolol-hydrochlorothiazide | FT-IR | 3361 (N-H str) and 3169 (OH str) | 3464 and 3357 cm−1 | Shift | [91] |
| 1636 (C=O str) | 1664 cm−1 | Shift → formation of intermolecular interactions | ||||
| 1317 (-SO2 str) | 1327 cm−1 | Shift → involvement of -SO2 in intermolecular hydrogen bonding | ||||
| 11A | Indomethacin-arginine | FT-IR | 1613 (guanidine group) | 1603 cm−1 | Reduction of signal → possibly extremely weak interactions | [92] |
| 1709 and 1738 cm−1 (C=O) | N/A | Disappearance of peaks → possibly extremely weak interactions | ||||
| ssNMR | 159 ppm (guanidine resonance) and 157 ppm (C5) | N/A | Overlap → not easy to identify salt formation | |||
| Furosemide-arginine | FT-IR | 1670 (C=O) | N/A | Decrease of peak → salt formation | ||
| ssNMR | 169 and 173 ppm (C=O) | 175 ppm | One broad resonance → similar environments in the mixture. π-π interactions involved | |||
| 15A | Piroxicam-ASPA | FT-IR | 1377 | 1392 cm−1 | Shift → possible interaction between components | [94] |
| 16A | α-D-glucose | Raman | 769.2 and 838 | N/A | Presence of only the respective vibrational broadened bands → samples free of mutarotation and show anomeric purity | [95] |
| β-glucose | 896.4 | N/A | ||||
| 18A | Indomethacin-lysine | FT-IR | 1713 (C=O str) | N/A | Disappearance of band → suggests ionization and salt formation | [96] |
| N/A | 1586 and 1561 cm−1 (COO-) | Broad peak → ionized carboxyl group for DMB and LAG, respectively | ||||
| 19A | Mebendazole-tryptophan | FT-IR | 1717 (C=O) | 1727 cm−1 | Shift → loss of hydrogen bonds | [97] |
| Pioglitazona-tryptophan | 2930 (N-H) | 1924 cm−1 | Shift → formation of hydrogen bonds | |||
| 20A | Mefenamic acid-NaTC | FT-IR | 754 and 776 | 747 and 769 cm−1 | Broadening and shift → loss of long-range order | [37] |
| 888 | N/A | Intensity of strong, sharp band decreases | ||||
| 1256 | 1219 cm−1 | Shift and overlapping with band at 1193 cm−1 → changes in the hydrogen bonding network of mefenamic acid on amorphization | ||||
| 1329 | 1319 cm−1 | Shift → changes in the hydrogen bonding network of mefenamic acid on amorphization | ||||
| 1509/1502 | 1507 cm−1 | Split peak becomes a broad centered band | ||||
| 1648 and 1196 | 1662 and 1193 cm−1 | Shift → no evidence for specific API-NaTC interactions; hydrogen bonding interactions can be ruled out | ||||
| 21A | Indomethacin-arginine | FT-IR | N/A | 1590 cm−1 (indol) | Peak structure of individual compounds transformed into a broad plateau with a small peak | [98] |
| 1707 and 1734 | N/A | Disappearance of peaks → carboxylic acid vibrations | ||||
| 1314 and 1219 | 1319 and 1222 cm−1 | Shift (chlorobenzene and indol, respectively) → changes in molecular environment | ||||
| 22A | (S)-naproxen-L-arginine | FT-IR | N/A | 1568 cm−1 (C=O) | New broad peak for the LAG sample → carboxyl group ionized | [99] |
| N/A | 1708 cm−1 | New band appearance | ||||
| N/A | 1543 cm−1 (C=O) | New peak with lower intensity compared to LAG sample (DBM formulation) | ||||
| N/A | 1679 cm−1 | Broad shoulder (DMB) | ||||
| 23A | Griseofulvin-tryptophan | FT-IR | 3401 (NH and OH str), 3011 (CH str) | N/A | Enlargement and broadening of bands | [100] |
| N/A | 3227 cm−1 | New band appearance | ||||
| 1663 (QC, C=O) | 1648 cm−1 | Small displacement → formation of hydrogen bonding interaction | ||||
| 24A | Naproxen-tryptophan | FT-IR | 1369 | N/A | Decrease of C=O band due to interactions with NAP | [101] |
| 1659 | 1664 cm−1 | Band transformed into a peak with decreased intensity → interactions involving CO2- | ||||
| Naproxen-tryptophan-proline | 1650–1750 | 1699 cm−1 | Transformation into a broad peak | |||
| 1581 | 1577 cm−1 (amide) | Shift of small shoulder | ||||
| Naproxen-arginine | 1679 and 1728 cm−1 | N/A | Disappearance → indicates salt formation | |||
| 1540, 1600–1700 | N/A | Reduction of bands (amide and guanidyl) → Supports salt formation | ||||
| Naproxen-arginine-proline | 1550 (amide) | 1556 cm−1 | Shift → co-amorphous system | |||
| 1610 | Disappearance of band → co-amorphous blend | |||||
| 26A | Oxaprozin-randomly-methylated-βCD systems | FT-IR | 1725 | 1718 cm−1 (OXA carbonyl) | Reduction of intensity and shift → strong solid-state interactions between the components | [103] |
| 27A | Furosemide-arginine | FT-IR | 1672 and 1562 | N/A | Transformation of bands into shoulders → Salt formation upon co-amorphization | [104] |
| 1591 | 1602 cm−1 | Shift → salt formation upon co-amorphization | ||||
| Indomethacin-arginine | 1714 and 1689 | N/A | Disappearance of bands → salt formation | |||
| N/A | 1680 and 1500 cm−1 | Simultaneous formation of a band plateau → Salt formation | ||||
| N/A | 1589 cm−1 | Formation of a small peak → salt formation | ||||
| 29A | Naproxen-NAP(Na) | FT-IR | 1638–1682 | 1639 cm−1 | Disappearance of peaks and formation of a broaden single peak | [106] |
| 1603 | 1605 cm−1 | Shift | ||||
| 1585–1574 | N/A | Peaks weakened and broadened → formation of intermolecular interactions involving carbonyl groups | ||||
| Raman | N/A | 747 cm−1 | Peak broadened and then disappeared → crystallization of NAP and NAP(Na) | |||
| N/A | 742 cm−1 | Appearance and increase in peak → presence of NAP indicates increasing presence of crystalline NAP | ||||
| N/A | 1383 cm−1 | Small shoulder peak after 10 min → decreased presence of NAP(Na) | ||||
| 31A | Arginine-indomethacin | FT-IR | N/A | 1500–1750 cm−1 | Formation of a plateau | [36] |
| 1321 cm−1 | Presence of peak | |||||
| 32A | Simvastatin-L-lysine | FT-IR | 3442 | 3350 cm−1 (OH) | Broadening → no clear evidence of strong intermolecular interactions between the components | [108] |
| 1356 and 1319 | 1350 and 1312 cm−1 | Shift (aliphatic) → no clear evidence of strong intermolecular interactions between the components | ||||
| Glibenclamide-L-serine | 1519 | 1534 cm−1 | Shift (NH urea group) → intermolecular interaction | |||
| 1584 (C=O) | 1595 cm−1 | Shift and merging → intermolecular interaction | ||||
| 34A | L-tryptophan-indomethacin | Raman | N/A | 1680 cm−1 (C=O) | Appearance and increase in intensity of a broad band → loss of crystalline forms due to changed intermolecular environment | [109] |
| FT-IR | 1661 and 1582 | 1609 cm−1 | Loss of initial bands and formation of broad band | |||
| 495 | 532 cm−1 | Peak shift | ||||
| 35A | Carbamazepine-citric acid-arginine (1:1:1) | FT-IR | 1725, 1659, and 1628, 1568 (C=N) | 1724, 1659, 1630, and 1573 cm−1 | Shift of bands. C=O peak weakened and became a shoulder peak → formation of intermolecular interactions between components | [110] |
| 1659 | 1678 cm−1 | Peak strengthened and shifted → intermolecular interactions | ||||
| Carbamazepine-citric acid-arginine (1:1:2) | 1659 and 1630 | 1678 and 1682 cm−1 | Shift (guanidyl) | |||
| 1568 (C=N) | N/A | Broadening of peak | ||||
| Carbamazepine-citric acid-arginine (1:1:3) | 1659 and 1630 | 1634 and 1636 cm−1 | Shift (guanidyl) → formation of a stronger interaction with the amide group and/or aromatic ring | |||
| 1568 (C=N) | 1559 and 1589 cm−1 | Formation of a doublet → formation of a stronger interaction with the amide group and/or aromatic ring | ||||
| 36A | Glibenclamide-quercetin | FT-IR | 1713 and 1649 (C=O) | 1680 and 1650 cm−1 | Broadening and shift of peaks → amorphization | [111] |
| 38A | Mefenamic acid-meglumine | FT-IR | N/A | 1375 cm−1 | Formation of a new band → chemical interaction between carbonyl group and secondary amino group of the components | [113] |
| 40A | Gliclazide-triamterene | FT-IR | N/A | 3290 (N-H) cm−1 | Formation of new H bonds | [38] |
| 1565 and 1530 (NH2) | 1570 and 1536 cm−1 | Shift → formation of new H bonds | ||||
| 41A | Quercetin-arginine | FT-IR | 3400–3200 (OH) cm−1 | N/A | Loss of intensity → weak intermolecular bonding with the amino acid | [115] |
| 1645 (C=O) | 1654 cm−1 | Shift → intermolecular H-bonding | ||||
| 42A | Candesartan cilexetil-hydrochlorothiazide | FT-IR | N/A | 1732 cm−1 | Visualization of band → occurrence of hydrogen bonds between the components | [116] |
| # | Sample | Analytical Technique | Wavenumber (cm−1) | Interpretation | Ref. | |
|---|---|---|---|---|---|---|
| Crystalline | Co-Crystal | |||||
| 1C | Nicotinamide: L-(+)-ascorbic acid | Raman | 104, 146, 666, 1329 | 93, 133, 631, 1292 cm−1 | Change form I → form II | [66] |
| 4C | Urea-caffeine | ATR-FTIR | 1682 (C=O) | 1707 | Shift → hydrogen bonding | [119] |
| 3341 (N-H) | 3185 | Shift → hydrogen bonding | ||||
| N/A | 809 | Appearance of a new peak → co-crystal | ||||
| 5C | Brexpiprazol-catechol (1:1) | Raman | 1320.8, 1375.7, 1469.6, 1650.4 | 1223.4, 1284.1, 1321.47, 1375.2, 1495.4, 1668.3 | Shift, decrease in C=O str → hydrogen bonding | [120] |
| Brexpiprazol-succinic acid (1:1) | 1320.8, 1375.7, 1469.6, 1650.4 | 1226.8, 1292.2, 1332.6, 1381.6, 1497.4, 1665.7 | Shift, decrease in C=O str → hydrogen bonding | |||
| 6C | Quercetin-malonic acid | FT-IR | 3411 (O-H) | 3427 (1:1) and to 3466 cm−1 (1:2) | Shift → co-crystal formation | [121] |
| 1667 and 1612 (C=O) | 1638 cm−1 (1:2) | Disappearance and shift → co-crystal formation | ||||
| 7C | Paracetamol-trimethylglycine | FT-IR | 1647 (-CONH2), 1595, 1506, 1452 (C6H6), and 804 (-C6H4-) for PCA. 1400 cm−1 (C-N str) and 1323 (-COO-) for TMG. | N/A | No obvious difference in spectra of sample and co-crystal → proton transfer does not occur, no chemical reaction, this confirms co-crystal formation | [44] |
| Raman | 1643 (C=O), 1605 (C=C), 1364 (C-H), 1229 (-OH, aryl), 1161 (N-H), 850 (C6H6, aryl), and 789 (C-O) | 1629, 1607, 1591, 1371, 1224, 1159, 858, and 774 cm−1 | Shift and reduction of band intensities → molecular complex is a co-crystal | |||
| 1454 (C-N) and 882 (-COO-) | 1443 and 886 cm−1 | Shift and reduction of band intensities → molecular complex is a co-crystal | ||||
| 9C | Acetazolamide-4-hydroxybenzoic acid | Raman | N/A | 251 (NH, OH), 1694 and 1738 (sci of, CNH and tor -CH3, and C=O, oop bend of ring) | Appearance of peaks → hydrogen bonding interaction leads to co-crystal formation | [123] |
| 1081 and 1120 | N/A | Weak broad peaks → co-crystal | ||||
| 910, 1383 | 947 (N-H, -CH3) and 1372 (HC=CH, O-H, C-N) cm−1 | Shift → co-crystal formation | ||||
| 1284 | Disappearance → co-crystal formation | |||||
| 11C | Ciprofloxacin-nicotinic acid/EtOH | FT-IR | N/A | 1729 (COOH), 1627 (C=(ketone)), and 3200–2000 (OH) | Presence of bands and OH superimposed by C-H vib, abscence of H bonding → co-crystal formation | [124] |
| 1589 (asym COO-) and 1375 (sym COO-) | N/A | Stretches of COO → co-crystal formation | ||||
| Ciprofloxacin-isonicotinic acid | 1705 (C=O) | 1728 cm−1 | Displacement and increase in intensity | |||
| 1589 (asym COO-) | N/A | Lower intensity and absence of bands attributed to vibrations of H bond → formation of new supramolecular synthons | ||||
| 12C | Pyrazinamide-diflunisal | Raman | N/A | 244 (benzene ring, C-F), 1185 (O-H, HC-CH), 1370 (OH, O=C-O, C-H), 1406 (COH, C-H) and 1750 (C=O, C-O, C-N, C=O, C-C) | Appearance of peaks → hydrogen bonding in COOH-pyridine hetero-synthon leads to co-crystal formation | [125] |
| 807 | N/A | Disappearance → co-crystal formation | ||||
| 458 and 1620 | 449 and 1612 cm−1 (C=O, C-O, C-C, O-H, C=OH) | Shift → co-crystal formation | ||||
| 14C | Acetazolamide, nicotinamide-2-pyridone | Raman | N/A | 475, 857 (CH, NH), 928 and 1716 (C=O, N-H, HO-C=O) | Appearance of bands → hydrogen bonding interaction leads to co-crystal formation | [126] |
| 1014 | N/A | Disappearance → co-crystal formation | ||||
| 1242, 1456 and 1542 | 1260 (O=C-N-H, HC=CH), 1466 (-CH3, O=CNH, N-C-H) and 1559 (C-CH, HC=CH, NCH) cm−1 | Shift → hydrogen bonding interaction leads to co-crystal formation | ||||
| 16C | Norfloxacin-nicotinic acid | FT-IR | 1716 (C=O) | 1728 and 1707 cm−1 | Displacement → New intermolecular interactions | [128] |
| N/A | 365–2492 cm−1 | Presence of a broad band → interactions through carboxyl and aromatic nitrogen groups of Nicotinic acid molecules | ||||
| 17C | Chlorothiazide-L-proline hydrate | FT-IR | N/A | 3337 (NH) cm−1 | Broad peaks → hydrogen bonding | [129] |
| Chlorothiazide-D-proline hydrate | ||||||
| 1332 cm−1 | Shift → formation of hydrogen bond O-H water -Osulfonamide | |||||
| 18C | Praziquantel-poloxamer F-127 and sucrose stearate | ATR-FTIR | 1625 | 1621 cm−1 | Shift → hydrogen bond formation | [130] |
| 20C | Ketoconazole-fumaric acid | FT-IR | 1645 (C=O) | 1700 cm−1 | Shift → strong hydrogen bonding | [132] |
| Ketoconazole-succinic acid | 1714 cm−1 | |||||
| 21C | Itraconazole-4-hydroxybenzamide (1:2) | FT-IR | 1697 (C=O) | 1690 cm−1 | Shift → participation in hydrogen bonding | [133] |
| N/A | 3469 (N-H) cm−1 | More prominent band of form II → higher involvement in hydrogen bonds than form I | ||||
| 3111 (C-H) cm−1 | Sharp peak of form I → asymmetric stretching in both molecules | |||||
| Itraconazole-4-aminobenzoic acid (1:1) | 1689 cm−1 | Shift → participation in hydrogen bonding | ||||
| 23C | Pyrazinamide-4-aminosalicylic acid | Raman | 416, 781, 1055, 1662 | 366, 893, 1000, 1552, 1637 cm−1 | New peaks → formation of a co-crystal | [135] |
| 25C | Betulin-terephthalic acid (w/acetone or isopropanol) | ATR-FTIR | NR | 3300–3600 (OH) and 1020 (C-O) cm−1 | Shift → intermolecular hydrogen bonding | [137] |
| # | Sample | Analytical Technique | Wavenumber (cm−1)/δ (ppm) | Interpretation | Ref. | |
|---|---|---|---|---|---|---|
| Polymorph I | Polymorph II | |||||
| 1P | Ranitidine hydrochloride form 1 | DRIFTS | 1551 (form 1) | 1046 (form 2) | Identification of each band → presence of polymorph | [74] |
| 4P | Rivastigmine (RHT form II) | ATR-FTIR | 1694 (carbamate, form II) | 1725 cm−1 | Band broadening and shift → form II to I | [141] |
| 6P | Dexamethasone | ssNMR | 14–155 ppm (form B) | N/A | Disappearance at high temperatures → change in conformational properties of the molecules and coarsening process. | [27] |
| 10P | Famotidine (form B) | Raman | 3406 (N-H str) and 2897 (C-H sym str) (form B) | 3455 (N-H str), 3422, 2997 cm−1 | Clear observation of bands → polymorphic conversion to form A | [142] |
| 2920 cm−1 (form A) | N/A | Increase in peak intensity → presence of form A | ||||
| 2897 cm−1 | N/A | Decrease in peak intensity → form B dropped off | ||||
| 11P | Gabapentin (GBP) form I, II, III, and IV | FT-IR | 3300 (OH str, form I) | N/A | Disappearance → dehydration | [76] |
| 1660 (C=O, form I) | N/A | Decrease in peak intensity → decrease in hydrogen bonding due to dehydration and polymorphic transformation to II | ||||
| 1624 (carboxylate, form I) | 1620 cm−1 and then to 1615 cm−1 | Shift and decrease in peak intensity → decrease in hydrogen bonding due to dehydration and polymorphic transformation to II | ||||
| N/A | 1301, 709, 2930, 2153, 1615, 1547, and 1165 (form II) | Appearance of peaks → presence of form II | ||||
| N/A | 1699 and 1677 (GBP-lactam) | Appearance of peaks → formation of traces of GBP-lactam due to heating effect | ||||
| N/A | 1644, 1584, 1510, 1462, 1400, 1231, 1160, 1512, 2926, and 2200 (form III) | Appearance of specific peaks → presence of form III | ||||
| N/A | 3150, 1523, 1397, 1377, 1087, 2121, 1621, 1576, and 1431 (form IV) | Appearance of peaks → presence of form IV | ||||
| # | Sample | Molar Ratio/Composition | Glass Transition Temperature (Tg)/(°C) | Milling Temperature | Conditions | Ref. |
|---|---|---|---|---|---|---|
| 2A | Furosemide-arginine | 1:1 | 127 ± 0.5 | 5 °C | 2 °C/min, −10 °C to 180 °C, 50 mL/min | [85] |
| Furosemide-citrulline | 1:1 | 77.1 ± 5.6 | ||||
| Nitrofurantoin-arginine | 1:1 | 139.1 ± 0.2 | ||||
| Nitrofurantoin-citrulline | 1:1 | 49.3 ± 2.1/108.5 ± 0.3 | ||||
| Cimetidine-arginine | 1:1 | 40.4 ± 3.1 | ||||
| Cimetidine-citrulline | 1:1 | 39.5 ± 1.5 | ||||
| Mebendazole-arginine | 1:1 | 53.5 ± 3.3/112.2 ± 0.4 | ||||
| Mebendazole-citrulline | 1:1 | 43.6 ± 1.2/112.1 ± 0.2 | ||||
| 3A | Sulfathiazole-polyvinylpyrrolidone | STZ/PVP Xpvp = 0.4 | 173.2 | Room temperature | 10 °C/min | [86] |
| Sulfadimidine-polyvinylpyrrolidone | SDM/PVP Xpvp = 0.6 | 146.7 | ||||
| 4A | Naproxen-cimetidine | 1:1 | 34.5 | 4 ± 2 °C | 10 K min−1 | [87] |
| 2:1 | 31.5 | |||||
| 1:2 | 40.2 | |||||
| 5A | γ-indomethacin–ranitidine hydrochloride | 1:1 | 32.5 | 4 ± 2 °C | 10 K per min from 0 to 160 °C | [28] |
| 2:1 | 34.3 | |||||
| 1:2 | 29.3 | |||||
| 6A | γ-indomethacin | N/A | 39.23 | 4 ± 2 °C | 10 K min−1 from 0 to 180 °C under nitrogen gas flow 50 mL min−1 | [88] |
| α-indomethacin | N/A | 37.92 | ||||
| 7A | Tadafil | N/A | 147 | Cryogenic temperature (liquid nitrogen) | 10 °C/min under nitrogen atmosphere (60 mL/min) | [26] |
| 8A | Glibenclamide | N/A | 65 | Cryogenic temperature (samples immersed in liquid nitrogen) | 10 K/min from 20 to 190 °C | [89] |
| 9A | Trehalose dihydrate | N/A | 21 | Cryogenic temperature (samples immersed in liquid nitrogen) | 10 °C/min from 0 to 150 °C | [90] |
| 10A | Atenolol-hydrochlorothiazide | 1:1 | 311.44 | Cryogenic temperature (samples immersed in liquid nitrogen) | 10 °C/min, starting at −20 °C | [91] |
| 1:2 | 315.82 | |||||
| 2:1 | Not determined due to fast recrystallization | |||||
| 11A | Indomethacin-tryptophan | 1:1 | Tg ranges from 120 to 45 °C, decreasing as mol% of Ind increases | 6 °C | 2 K/min from −20 to 180 °C | [92] |
| Furosemide-tryptophan | 1:1 | Tg ranges from 138 to 80 °C, decreasing as mol% of Fur increases | ||||
| 12A | Dexamethasone | N/A | 115 < Tg < 120 | Room temperature | 0.663 °C and 50 S, “Heat only” conditions | [27] |
| 13A | α-lactose | N/A | 70 | 30 ± 5% relative humidity and 22 ± 3 °C | From 0 to 240°, 10 °C/min under N2 flow of 50 mL/min | [93] |
| 14A | α-D-glucose | N/A | 38 | −15 °C and 0% relative humidity | 5 °C/min, flushed with highly pure nitrogen gas | [68] |
| 15A | Mebendazole-ASPA | 1:1 | 91 | 5 °C, cold room | −10 °C to 180 °C, 2 °C/min, nitrogen flow was 50 mL/min | [94] |
| Tadalafil-ASPA | 1:1 | 102.9 | ||||
| Piroxicam-ASPA | 1:1 | 76 | ||||
| 16A | α-D-glucose | N/A | 38 | −15 °C and 0% relative humidity | 5 °C/min | [95] |
| β-D-glucose | N/A | 39 | 5 °C/min | |||
| 17A | Carvedilol, carbamazepine, furosemide, indomethacin, mebendazole-amino acids | 1:1 | A single Tg for each formulation | Cold room (+6 °C) | Nitrogen flow of 50 mL/min, 2 °C/min heated to 180 °C | [31] |
| 18A | Indomethacin-lysine | 1:1 | 100 (DMB) | Cold room (+6 °C) | Nitrogen flow of 50 mL/min, 2 °C/min heated to 180 °C | [96] |
| 19A | Mebendazole-tryptophan | Xmeb = 0.1 | 53.5 | Room temperature | −5 °C to 210 °C at 10 °C/min | [97] |
| Pioglitazona-tryptophan | Xpgz = 0.1, 150 min | 44.9 | ||||
| 22A | (S)-naproxen-L-arginine | 1:1 | 91.9 ± 0.2 | 6 °C | Nitrogen flow of 50 mL/min, 2 °C/min from −10 °C to 180 °C | [99] |
| 23A | Griseofulvin-tryptophan | 1:1 | 113.46 | NR | 25 to 300 °C, 5 °C/min | [100] |
| 24A | Naproxen-tryptophan-proline | 1:1:1 | 55.1 ± 3.1 | 6 °C | Nitrogen flow of 20 mL/min, 10 K/min, from −20 to 170 °C | [101] |
| Naproxen-tryptophan | 1:1 | 58.2 ± 0.5 | ||||
| Tryptophan-proline | 1:1 | 67.2 ± 6.8 | ||||
| 25A | Mebendazole-tryptophanphenylalanine | 1:1:1 | 107.5 ± 0.2 | 5 °C | 2 °C/min, heating to 180 °C | [102] |
| Mebendazole-phenylalaninetryptophan | 1:1:1 | 104.6 ± 0.2 | ||||
| Mebendazole-aspartatetyrosine | 1:1:1 | 61.2 ± 0.9 | ||||
| Mebendazole-histidineglycine | 1:1:1 | 34.9 ± 1.2/89 ± 0.6 | ||||
| Mebendazole-prolinetryptophan | 1:1:1 | 6.5 ± 0.2 | ||||
| Mebendazole-tryptophan | 1:1 | 128.7 ± 0.2 | ||||
| Mebendazole-proline | 1:1 | 96.9 ± 0.1 | ||||
| Mebendazole-proline-tryptophan | 1:1:1 | 56.3 ± 0.2 | ||||
| Mebendazole-tryptophan-phenylalanine | 1:1:1 | 119 ± 0.1 | ||||
| 27A | Indomethacin-arginine | 1:1 | 117 ± 4 | 6 °C | Nitrogen gas flow of 50 mL/min, 2 °C/min, from −10 to 180 °C, 0.212 °C and a period of 40 s | [104] |
| 29A | Naproxen-NAP(Na) | 2:1 | 55.8 | 4 °C | 2 °C/min, 0.2120 °C with a period of 40 s | [106] |
| 1:1 | 40 | |||||
| 1:2 | NR | |||||
| 31A | Indomethacin-arginine | 1:1 | 62.9 ± 0.8 | NR | Nitrogen gas flow of 50 mL/min, 10 °C/min to 180 °C | [36] |
| Indomethacin-phenylalanine | 55.3 ± 0.4 | |||||
| Indomethacin-tryptophan | 62.7 ± 7.0 | |||||
| 32A | Simvastatin-lysine | 1:1 | 33.2 ± 0.9 | 6”C | Nitrogen flow of 50 mL/min, 10 °C/min, from −50 °C to 280 °C (depending on the sample) | [108] |
| Glibenclamide-serine | 1:1 | 70.1 ± 1.3 | ||||
| Glibenclamide-threonine | 1:1 | 58.4 ± 1.3 | ||||
| Glibenclamide-serine-threonine | 1:1:1 | 62.5 ± 4.5 | ||||
| 33A | Indomethacin-arginine | 1:1 | 36.7 ± 0.8 | 6 °C | Nitrogen gas flow, 20 mL/min, from −20 to 180 °C, 10 K/min | [98] |
| Indomethacin-phenylalanine | 1:1 | 64.1 ± 1.4 | ||||
| Indomethacin-tryptophan | 1:1 | 47.8 ± 2.9 | ||||
| Indomethacin-phenylalanine-tryptophan | 1:1:1 | 68.7 ± 2.6 | ||||
| Indomethacin-arginine-phenylalanine | 1:1:1 | 63.1 ± 0.8 | ||||
| Carbamazepine-tryptophan | 1:1 | 81 ± 0.6 | Nitrogen gas flow, 20 mL/min, from −20 to 200 °C, 10 K/min | |||
| Carbamazepine-phenylalanine-tryptophan | 1:1:1 | 75.1 ± 1.1 | ||||
| Carbamazepine-arginine-tryptophan | 1:1:1 | 65.4 ± 1.1 | ||||
| 35A | Carbamazepine-citric acid | 1:1 | 38.8 ± 2.7 | 4 °C | Nitrogen gas at 50 mL/min, 2 °C/min from 0 to 150 °C, 0.212 °C with a period of 40 s | [110] |
| Citric acid-arginine | 1:1 | 56.2 ± 0.7 | ||||
| Citric acid-arginine | 1:2 | 106 ± 0.3 | ||||
| Citric acid-arginine | 1:3 | 130.5 ± 0.1 | ||||
| Citric acid-arginine | 1:4 | 119 ± 0.1 | ||||
| Carbamazepine-citric acid-arginine | 1:1:1 | 77.8 ± 1.8 | ||||
| Carbamazepine-citric acid-arginine | 1:1:2 | 105.3 ± 0.2 | ||||
| Carbamazepine-citric acid-arginine | 1:1:3 | 127.8 ± 0.8 | ||||
| 36A | Glibenclamide-quercetin | 1:1 | 85.97 ± 0.29 | Cryomilled | Nitrogen glow of 50 mL/min, 1 °C/min | [111] |
| 37A | Mebendazole-glutamate-arginine (crystalline salt) | 1:1:1 | 37.8 | Cold rooms (5 °C) | Nitrogen gas flow of 50 mL/min, 2 °C/min, 0.212 °C (amplitude), 40 s (period) | [112] |
| Mebendazole-glutamate-arginine (amorphous salt) | 1:1:1 | 37.3 | ||||
| Meb-glutamatearginine | 1:1 | 36.5/77 | ||||
| Meb-arginineglutamate | 1:1 | 36.3/76.3 | ||||
| 42A | Candesartan cilexetil-hydrochlorothiazide | NA | 110 | Room temperature | Nitrogen gas flow, 100 mL/min, 10 °C/min, from 30 to 300 °C | [116] |
| # | Sample | Tm Parent Drug 1 (°C) * | Tm Parent Drug 2 (°C) | Tm of Co-Crystal (°C) | Ref. |
|---|---|---|---|---|---|
| 4C | Urea-caffeine | 135.3 | 235.9 | 132.7 | [119] |
| 5C | Brexpiprazol-catechol | 184.8 | 106.3 | 161.3 | [120] |
| Brexpiprazol-succinic acid | 184.8 | 156.1 | 156.1 | ||
| 6C | Quercetin-malonic acid | 321.92 | 135.07 | 283.02 (1:1) | [121] |
| 266.61 (1:2) | |||||
| 7C | Paracetamol-trimethylglycine | 170.2 | 320.7 | Endo peak = 174.5 °C and 177.4 °C | [44] |
| 11C | Ciprofloxacin-nicotinic acid | 254.8 | 235.1 | 241 | [124] |
| Ciprofloxacin-isonicotinic acid | 268.3 | 267.94 | 242 | ||
| 13C | Acetazolamide (polymorph I)-4-aminobenzoic acid | 269.4 | 190.5 | 208.9 | [67] |
| 15C | β-lapachone-resorcinol | 156 | 110 | 131 | [127] |
| 16C | Norfloxacin-nicotinic acid (Neat grinding) | 222.8 | 237.1 | 230.5 | [128] |
| Norfloxacin-nicotinic acid (LAG) | 236.1 | ||||
| 17C | Chlorothiazide-DL-proline | NR | NR | 212.9 | [129] |
| 18C | Praziquantel-F-127 2B (20:1) | 140.23 | 56.22 | 133.06 | [130] |
| Praziquantel-F-127 4B (10:2) | 135.97 | ||||
| 19C | Ferulic acid-nicotinamide | 172.8 | NR | 124.6 | [131] |
| Ferulic acid-isonicotinamide | 143.9 | ||||
| Ferulic acid-urea | 158.1 | ||||
| 20C | Ketoconazole-fumaric acid | 151 | 294 | 168 | [132] |
| Ketoconazole-succinic acid | 188 | 164 | |||
| 21C | Itraconazole-4-aminobenzoic acid * | 167 | 188.5 | 163.4 | [133] |
| 22C | Ibuprofen-nicotinamide | NR | NR | 80.5 | [134] |
| 24C | Theophylline-4-aminobenzoic acid | 274 | 187 | Endos = 161.2 and 168.2 | [136] |
| # | Sample | Co-Crystal | Characteristic Peaks (° 2θ) | Conditions: Current (mA), Voltage (kV), etc. | Ref. |
|---|---|---|---|---|---|
| 1C | Nicotinamide- L-(+)-ascorbic acid * | Form I polymorph | 1.2, 1.5, 1.9, 2.1, 2.8, 3.2, 3.3 | 7.5 mA, 40 kV | [66] |
| Form II polymorph | 1.5, 1.8, 2.1, 2.7, 3.1, 3.2 | ||||
| 2C | Salicylic acid-2-pyridone * | sal2hyp | 7.8, 11.02, 15.2, 15.8, 16.7, 24.1, 26.8, 28.7 | Exposure time 9 s, time separation between patterns 10 s | [117] |
| Salicylic acid-3-hydroxypiridine * | sal3hyp | 9.2, 20.3, 23.2, 27.5, 31.6 | |||
| Salicylic acid-4-pyridone * | sal4hyp | 1.6, 1.9, 2.0, 2.1, 2.8, 3 | |||
| 3C | Ciprofloxacin-thymol * | N/A | 5.3, 7.1, 7.8, 11.4, 13.2, 15.7, 17.51, 19.4, 20.9 | 40 kV, 40 mA, step size 0.0130° | [118] |
| 4C | Urea-caffeine | N/A | 8.64, 10.82, 13.89, 24.30, 25.08, 25.46 | 35 kV, 25 mA | [119] |
| 5C | Brexpiprazol-catechol | N/A | 8.42, 8.88, 11.83, 12.15, 15.75, 16.22 | 40 kV, 30 mA, step 0.03° | [120] |
| Brexpiprazol-succinic acid | N/A | 3.67, 9.94, 18.47, 22.25, 22.53, 23.98, 24.3 | |||
| 6C | Quercetin-malonic acid | CC1 (1:1) | 16.21, 19.87, 28.88 | 40 kV, 40 mA | [121] |
| CC2 (1:2) | 16.18, 19.86, 28.83 | ||||
| 7C | Paracetamol-trimethylglycine | N/A | 17.50, 23.03 | 40 mA, 40 kV | [44] |
| 8C | Meloxicam-benzoic acid * | N/A | 9.2, 12.9, 15.5, 16.7, 20.2, 25.9, 27.3, 28.7, 29.4, 33.1, 35.0 | 40 kV, 40 mA | [122] |
| 10C | Furosemide-urea * | N/A | 7.9, 10.7, 21.1, 26.1, 30.7 | Step size 0.017°, collection time 18 h | [51] |
| 11C | Ciprofloxacin-nicotinic acid | CIP-NCA/EtOH (1:1) | 9.2, 11.5, 18.5, 19.5, 22.9, 23.4, 26.4, 28.5, 29.4 | 40 kv, 15 mA, 5–50°, step 0.04°, speed 4°/min | [124] |
| Ciprofloxacin-isonicotinic acid | CIP-INCA (without EtOH) | 5.4, 10.6, 19.2, 21.4, 28.4 | |||
| CIP-INCA/EtOH | 5.4, 10.6 | ||||
| 13C | Acetazolamide-4-aminobenzoic acid * | N/A | 6.4, 10.1, 12.1, 12.9, 13.4, 14.1, 15.6, 16.7, 17.2, 17.6, 18.2, 18.3, 19.6, 20.1, 21.4, 22, 23.3, 24.9, 25.6, 26.2, 26.6, 27.8, 29.1 | Ambient conditions | [67] |
| 15C | β-Lapachone-resorcinol * | N/A | 9.9, 10.5, 11.9, 12.9, 16.8, 18.1, 19.1, 21.4, 21.8, 24.9, 28.8 | Speed 1°/min, step size 0.01° | [127] |
| 16C | Norfloxacin-nicotinic acid (with EtOH) | N/A | 5.4, 14.5, 25.4 | Room temperature, 40 kV, 40 mA | [128] |
| 17C | Chlorothiazide-DL-proline * (w/acetonitrile-water) | N/A | 7.3, 20.1, 22.8, 24.12, 25.01 | Ambient temperature, 40 kV, 100 mA, 8°/min | [129] |
| Chlorothiazide-L-proline hydrate * (w/acetonitrile-water) | 8.02, 11.42, 16.4, 23.47, 23.83, 24.95, 25.3 | ||||
| Chlorothiazide-D-proline hydrate* (w/acetonitrile-water) | 8.2, 11.7, 16.2, 16.7, 17.5, 24.03, 25.2, 26.5, 29.2, 30.9 | ||||
| 18C | Praziquantel-F-127 2B (20:1) * | N/A | 8.06, 15.2, 16.4, 16.9, 19.9 | 40 mA, 40 kV, scan rate 0.02°/s | [130] |
| Praziquantel-F-127 4B (10:2) * | 6.08, 7.9, 11.9, 12.5, 15.1, 18.8, 19.8, 22.8, 25.3 | ||||
| 20C | Ketoconazole-fumaric acid * | N/A | 8.03, 12.2, 16.9, 19.3, 20.3, 21.6, 23.9, 25.7, 28.8 | 40 kV, 40 mA, step size 0.02°, counting time set 0.2 s/step | [132] |
| Ketoconazole-succinic acid * | 6.7, 7.9, 12.1, 17.1, 17.7, 19.3, 20.1, 21.2, 23.3, 23.8, 24.3 | ||||
| 21C | Itraconazole-4-hydroxybenzamide form I (1:2) * | N/A | 7.3, 9.4, 9.7, 10.3, 11.1, 12.3, 12.7, 16.2, 16.6, 19.3, 20.4, 21.6, 26, 26.3 | Ambient conditions, rotated at 15 rpm | [133] |
| Itraconazole-4-hydroxybenzamide form II (1:2) * | 5.7, 11.4, 12.9, 18.7, 19.04, 21.01, 22.3, 23.8, 25.2 | ||||
| Itraconazole-4-aminobenzoic acid (1:1) * | 6.1, 10.8, 11.4, 11.9, 13.5, 14, 16.4, 18.8, 19.2, 20.4, 21.2, 21.5, 22, 22.5, 24 | ||||
| 23C | Pyrazinamide-4-aminosalicylic acid | N/A | 5.95, 11.91, 13.06, 13.54, 28.25 | NR | [135] |
| 24C | Theophylline-4-aminobenzoic acid | N/A | 12.3, 14, 15.5, 26.4, 27.5, 28.6 | 40 kV, 40 mV, step size 0.026° and step time of 56 s | [136] |
| 25C | Betulin-rerephthalic acid (w/acetone) * | N/A | 5.08, 8.6, 10.2, 12.8, 14, 14.7, 16, 18.8, 21.3 | Range from 5 to 70° | [137] |
| Betulin-Terephthalic acid (w/isopropanol) * | N/A | 5.1, 8.7, 9.4, 10.2, 12.9, 14.2, 14.6, 16.1, 17.3, 17.9, 18.9, 19.3 |
| # | Sample | Polymorph Identification | Characteristic Peaks (° 2θ) | Ref. |
|---|---|---|---|---|
| 1P | Ranitidine hydrochloride * | Form 1 | 17, 21.8, 24.9 | [74] |
| Form 2 | 20.40, 23.7 | |||
| 2P | Chlorhexidine dihydrochloride * | Form 1 → initial spectrum | 13.9, 18.5, 23.7 | [140] |
| Form 2 → few peaks | 5.2 | |||
| Form 3 → many Bragg peaks | 14.9, 28.3 | |||
| 3P | γ-Sorbitol * | A phase → Sharp peaks, increased milling time | 16.6, 30.9 | [34] |
| γ phase | 11.6, 25.5 | |||
| 4P | Rivastigmine | Form II | 9.5, 11.3, 14.2, 15.5, 19.1, 20 | [141] |
| Form I → Broadeneing of peaks | 5.1, 14.7, 16.5, 17.6, 18.6, 20.4, 21.1 | |||
| 5P | o-Aminobenzoic acid | FI | 10.7, 13.7, 14.35, 16.4, 18.6, 23.5, 24.3, 24.9, 26.2, 27.6, 30.5 | [54] |
| FII | 11.2, 15.4, 22.2, 26.7 | |||
| m-Aminobenzoic acid (FIII form) | FI | 8.6, 17.2, 24.9 | ||
| FIII | 8.3, 16.8, 17.9, 23.7, 23.7, 24.2, 25.9, 26.6, 27.8 | |||
| p-aminobenzoic acid | β-form | 17.2, 17.6, 20, 21.9, 25.5, 27.9 | ||
| α-PABA | 17.1, 19.9, 21.8, 25.3, 27.8 | |||
| 6P | Dexamethasone * | Form A | 7.9, 13.5, 16.0, 17.6 | [27] |
| Form B | 7.5, 16.8, 18.4 | |||
| 7P | Sofosbuvir * | Form I | 5.3, 7.6, 9.0, 9.8, 10.3 | [79] |
| Form A | 6.2, 8.4, 10.5, 12.8,17.4, 17.9, 18.2, 20.3, 21.1 | |||
| Form B | 7.9, 10.3, 12.3, 16.7, 17.1, 19.3, 20, 20.9 | |||
| Form V | 5.6, 6.9, 7.5, 10, 10.8, 13.8, 16.4, 19.7, 25.4 | |||
| 8P | Sulindac * | Form I | 10.8, 17.6 | [69] |
| Form II | 9.3, 16.1 | |||
| 9P | Γ-sorbitol * | Γ-form | 11.7, 25.6 | [75] |
| A-Form | 16.7, 31.1 | |||
| 12P | Sulfamerazine | I | 12.6, 14.8, 16.3, 17.4, 20.5, 22.7, 23, 24.6, 31.2, 32.7 | [166] |
| II | 14.5, 17.0, 19.2, 21.5, 26.6, 27.4, 27.9 |
| # | Co-Crystal | XRD Interpretation | Storage Time (Days) | Storage Conditions * | Ref. |
|---|---|---|---|---|---|
| 1C | Nicotinamide-L-(+)-ascorbic acid | Without changes in peaks → chemically stable | 180 | At shelf | [66] |
| 3C | Ciprofloxacin-thymol | Stable, no changes of crystalline phase | 50 | Open air | [118] |
| 4C | Urea-caffeine | Formation of co-crystal | Within hours | 25 °C, 30% RH | [119] |
| 7C | Paracetamol-trimethylglycine | Physically stable | 90 | 40 and 75% RH | [44] |
| # | Sample | Polymorph Identification | XRD Interpretation | Storage Time (Days) | Storage Conditions | Ref. |
|---|---|---|---|---|---|---|
| 5P | o-aminobenzoic acid | Polymorphs: I, II, III, and IV | FII → reappearance of FII | 9 | 25 °C, 40% and 85% RH | [54] |
| FII → reappearance of FIII | 150 | 25 °C, 85% RH | ||||
| FI → FII | 150 | 25 °C, 85% RH | ||||
| m-aminobenzoic acid | Polymorphs: I, II, III, IV, and V | FIV | 150 | 25 °C, 85% RH | ||
| FI → reappearance of FIII | 3 | 25 °C, 85% RH | ||||
| p-aminobenzoic acid | Polymorphs: α and β | β polymorph | 150 | 25 °C, 85% RH | ||
| 6P | Dexamethasone | Form A | Broaden Bragg peaks, characteristic of form A | Immediate | Freshly milled samples | [27] |
| Form B | Predominantly peaks of form B, peaks of form A decrease | 7 | Heating up to 150 °C | |||
| 7P | Sofosbuvir | Form V | V → transformation to A | 120 | NR | [79] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez, L.M.; Cruz-Angeles, J.; Vázquez-Dávila, M.; Martínez, E.; Cabada, P.; Navarrete-Bernal, C.; Cortez, F. Mechanical Activation by Ball Milling as a Strategy to Prepare Highly Soluble Pharmaceutical Formulations in the Form of Co-Amorphous, Co-Crystals, or Polymorphs. Pharmaceutics 2022, 14, 2003. https://doi.org/10.3390/pharmaceutics14102003
Martínez LM, Cruz-Angeles J, Vázquez-Dávila M, Martínez E, Cabada P, Navarrete-Bernal C, Cortez F. Mechanical Activation by Ball Milling as a Strategy to Prepare Highly Soluble Pharmaceutical Formulations in the Form of Co-Amorphous, Co-Crystals, or Polymorphs. Pharmaceutics. 2022; 14(10):2003. https://doi.org/10.3390/pharmaceutics14102003
Chicago/Turabian StyleMartínez, Luz María, Jorge Cruz-Angeles, Mónica Vázquez-Dávila, Eduardo Martínez, Paulina Cabada, Columba Navarrete-Bernal, and Flor Cortez. 2022. "Mechanical Activation by Ball Milling as a Strategy to Prepare Highly Soluble Pharmaceutical Formulations in the Form of Co-Amorphous, Co-Crystals, or Polymorphs" Pharmaceutics 14, no. 10: 2003. https://doi.org/10.3390/pharmaceutics14102003
APA StyleMartínez, L. M., Cruz-Angeles, J., Vázquez-Dávila, M., Martínez, E., Cabada, P., Navarrete-Bernal, C., & Cortez, F. (2022). Mechanical Activation by Ball Milling as a Strategy to Prepare Highly Soluble Pharmaceutical Formulations in the Form of Co-Amorphous, Co-Crystals, or Polymorphs. Pharmaceutics, 14(10), 2003. https://doi.org/10.3390/pharmaceutics14102003