
Peptide Inhibitors of Vascular Endothelial Growth Factor A: Current Situation and Perspectives




Abstract
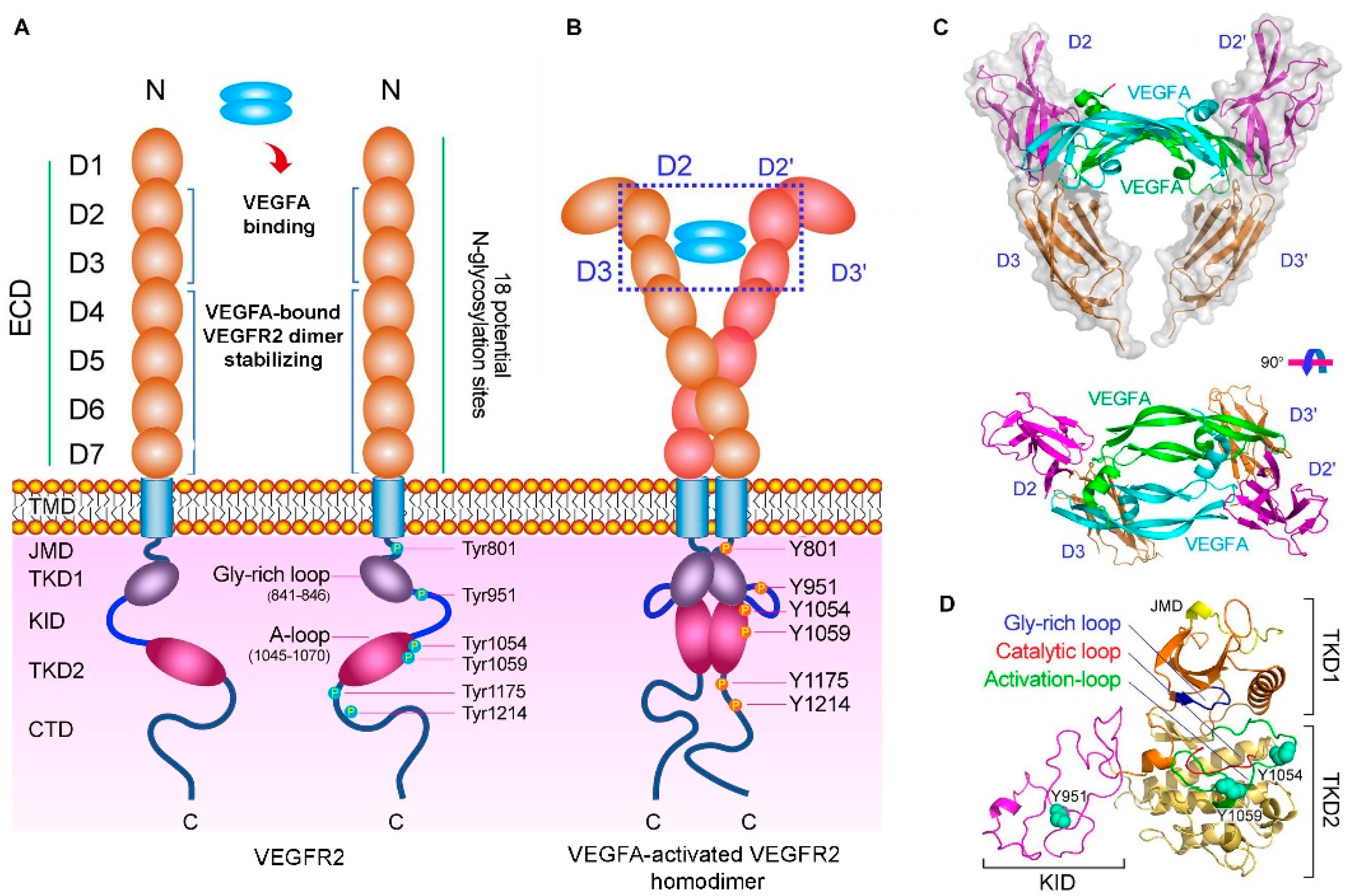
1. Introduction
2. Peptide-Based Therapeutics for VEGFA Binding
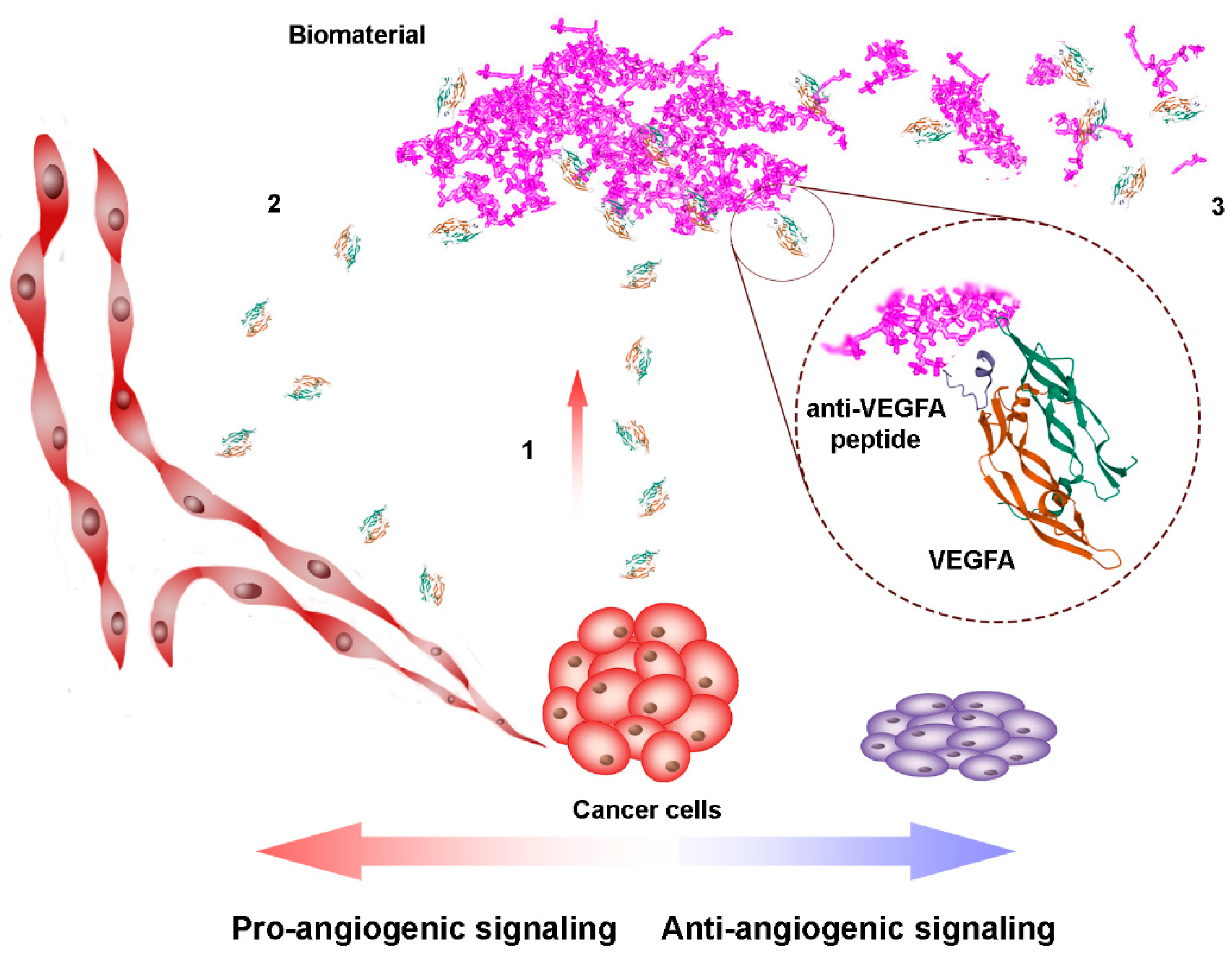
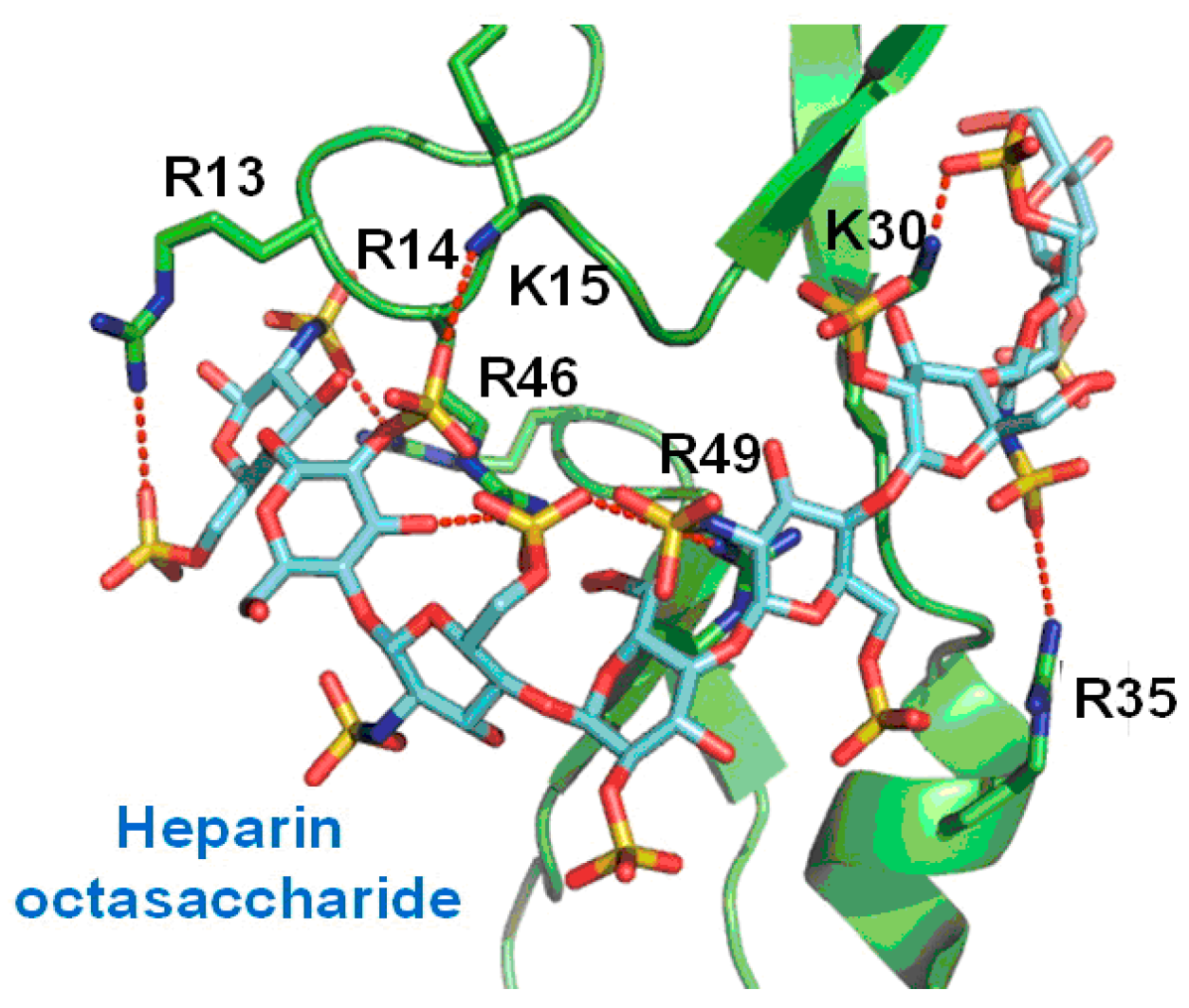
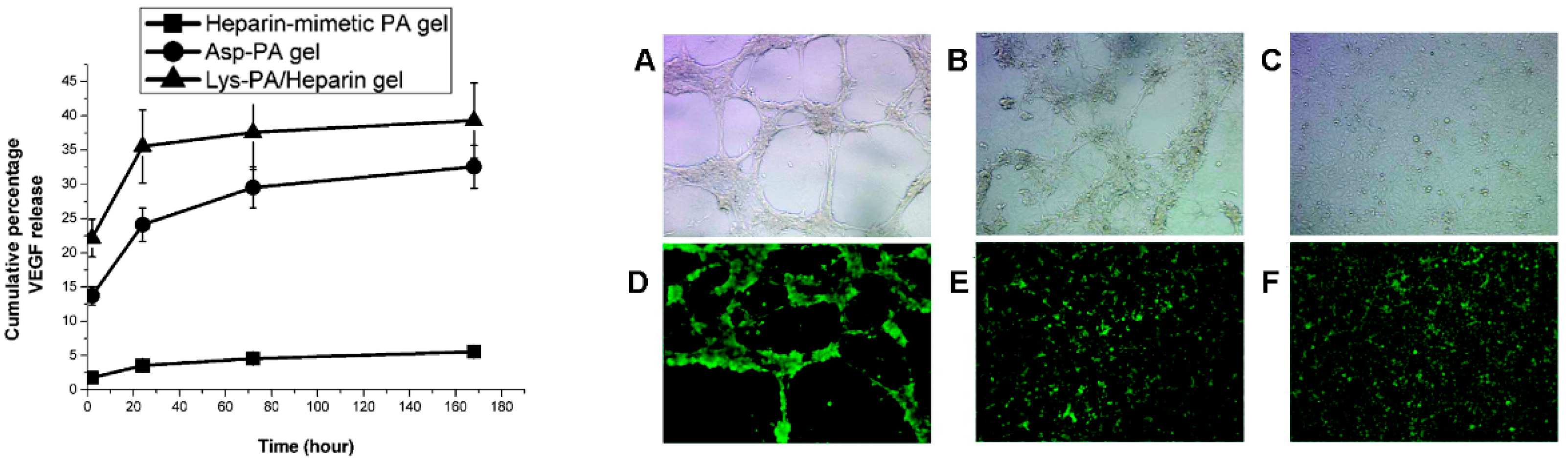
2.1. Peptides as Glycosaminoglycan Mimics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
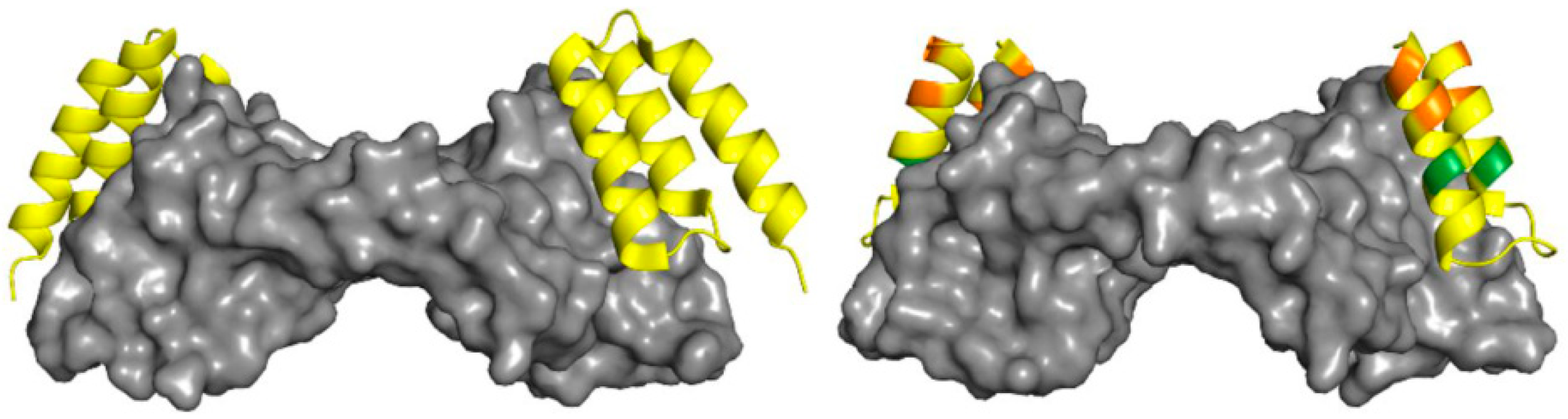{kind=link}
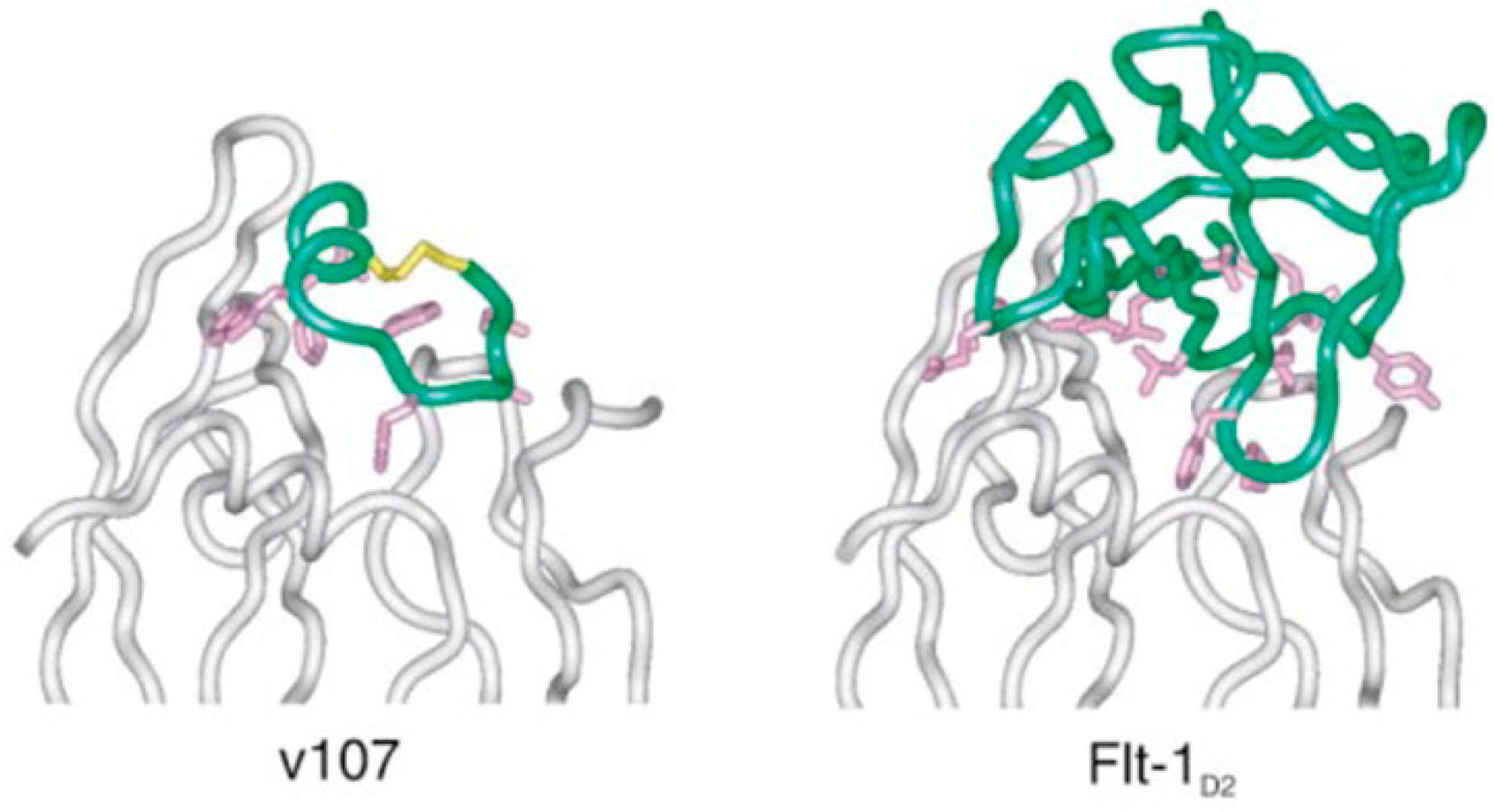{kind=link}
{kind=link}
2.2. Peptides as VEGFR Mimics
| Seq. ID | Sequence | VEGF Binding | Ref. |
|---|---|---|---|
| Je-7 | RTELNVGIDFNWEYPASK | IC50 = 0.1 μM (proliferation assay) | [69] |
| Je-11 | (RTELNVGIDFNWEYPAS)2K | IC50 = 0.5 μM (proliferation assay) | |
| - | EfaylIDFNWEYPASK | IC50 = 1 μM (competition assay) | [70,71,72,73,74,75] |
| - | (EfaylIDFNWEYPAS)2K | IC50 = 0.03 μM (competition assay) | |
| - | RRKRRR | IC50 = 10 μM (inhibition assay) | [76,77,78,79,80] |
| - | rrkrrr | IC50 = 10 μM (inhibition assay) | |
| MCoAA-01 | CGRKRRRGCRRDSDCGACICRYwKVCGSGSDGGV | IC50 = 10.83 ± 0.09 μM (inhibition assay, HUVECs)IC50 = 40.12 ± 0.13 μM (inhibition assay, NT-29) | |
| WHK7 | WHKPFRF | IC50 ~ 0.5 mM (proliferation assay) | [82] |
| WHL7 | WHLPFKC | IC50 ~ 0.5 mM (proliferation assay) | |
| Mini-Z-1 | FNKECLLRYKEAALDPNLNLYQRIAKIVSIDDDC | IC50 = 227 nM (ELISA) | [83] |
| Z-1-2 | VDNKFNKEMHNAYAIEIALLPNLNDQQFHAFIWSLIDDPSQSANLLAEAKKLNDAQAPK | IC50 = 343 nM (ELISA) | |
| Z-3B | VDNKFNKEMQNAYAIEIALLPNLNGSQTFAFITSLRDDPSQSANLLAEAKKLNDAQAPK | KD = 55 nM (Bio-Layer Interferometry) | |
| α/β-VEGF-1 | VDNKFNKEXc[CNZRAIEUALDPNLNDQQFHUKIWZIIXDC] | Ki = 0.11 μM (competitive fluorescence polarization assay) | [68,84] |
| α/β-VEGF-2 | VDNKFNKEXc[CNZRAIEUALDPNLNDUQFHUKIWZIIXDC] | Ki = 0.39 μM (competitive fluorescence polarization assay) | |
| 21 | KFNKEXc[CNZRAIEUALDPNLNDUQFHUKIWZIIXDC] where D, F = β3-residues, X, Z = cyclic β-residues and U = Aib | Ki = 0.15 μM (competitive fluorescence polarization assay) | |
| v107 | GGNEc[CDIARMWEWEC]FERL | IC50 = 0.70 ± 0.06 μM (SPR) | [85] |
| v114 | VEPNc[CDIHVMWEWEC]FERL | IC50 = 0.22 μM (SPR) | |
| v114* | VEPNc[CDIHVnLWEWEC]FERL | Ki = 0.06 μM (competitive fluorescence polarization assay) | [92] |
| - | VXPXc[CDIHVnLWXWEC]FZRX | Ki = 1.6 μM (competitive fluorescence polarization assay) | |
| - | XEXNc[CDIHVnLXEWXC]FZRX, where X = β3-residues, Z = cyclic β-residue | Ki = 4.6 μM (competitive fluorescence polarization assay) | |
| Aib2 | VAibPNc[CDIHVnLWEWEC]FERL | EC50 = 10.0 ± 1.1 μM (proliferation assay) | [94] |
| Aib12 | VEPNc[CDIHVnLWAibWEC]FERL | EC50 = 3.5 ± 0.7 μM (proliferation assay) | |
| kv114* | KAibKKc[CDIHVnLWEWEC]FERL | EC50 = 6.0 ± 0.4 μM (proliferation assay) | |
| VN | VNc[CDIHVnLWEWEC]FERL | EC50 = 4.0 ± 0.5 μM (proliferation assay) | |
| Bi-Lv | X-VEPNCDIHVMWEWECFERL-Tz4-lfrew | - | [95] |
| Tri-Lv | X-VEPNCDIHVMWEWECFERL-Tz4-lfrew-Tz4-eeird | EC50 = 2.6 ± 0.5 nM (ELISA) | |
| Tetra-Lv | X-VEPNCDIHVMWEWECFERL-Tz4-lfrew-Tz4-eeird-Tz4-qfkyr where X = biotin-PEG3 label Az4 = L-azidolysine | EC50 = 0.74 ± 0.05 nM (ELISA) |
3. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kazerounian, S.; Lawler, J. Integration of pro- and anti-angiogenic signals by endothelial cells. J. Cell Commun. Signal. 2018, 12, 171–179. [Google Scholar] [CrossRef]
- Potente, M.; Holger Gerhardt, H.; Carmeliet, P. Basic and Therapeutic Aspects of Angiogenesis. Cell 2011, 146, 873–887. [Google Scholar] [CrossRef]
- Masłowska, K.; Halik, P.K.; Tymecka, D.; Misicka, A.; Gniazdowska, E. The Role of VEGF Receptors as Molecular Target in Nuclear Medicine for Cancer Diagnosis and Combination Therapy. Cancers 2021, 13, 1072. [Google Scholar] [CrossRef] [PubMed]
- Shim, J.W.; Madsen, J.R. VEGF Signaling in Neurological Disorders. Int. J. Mol. Sci. 2018, 19, 275. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Agents for the Prevention and Treatment of Age-Related Macular Degeneration and Macular Edema: A Literature and Patent Review. Expert Opin. Ther. Pat. 2019, 29, 761–767. [Google Scholar] [CrossRef] [PubMed]
- Shaik, F.; Cuthbert, G.A.; Homer-Vanniasinkam, S.; Muench, S.P.; Ponnambalam, S.; Harrison, M.A. Structural Basis for Vascular Endothelial Growth Factor Receptor Activation and Implications for Disease Therapy. Biomolecules 2020, 10, 1673. [Google Scholar] [CrossRef]
- Peach, C.J.; Mignone, V.W.; Arruda, M.A.; Alcobia, D.C.; Hill, S.J.; Kilpatrick, L.E.; Woolard, J. Molecular Pharmacology of VEGF-A Isoforms: Binding and Signalling at VEGFR2. Int. J. Mol. Sci. 2018, 19, 1264. [Google Scholar] [CrossRef]
- Muller, Y.A.; Christinger, H.W.; Keyt, B.A.; de Vos, A.M. The Crystal Structure of Vascular Endothelial Growth Factor (VEGF) Refined to 1.93 Å Resolution: Multiple Copy Flexibility and Receptor Binding. Structure 1997, 5, 1325–1338. [Google Scholar] [CrossRef]
- Simons, M.; Gordon, E.; Claesson-Welsh, L. Mechanisms and Regulation of Endothelial VEGF Receptor Signalling. Nat. Rev. Mol. Cell Biol. 2016, 17, 611–625. [Google Scholar] [CrossRef] [PubMed]
- Markovic-Mueller, S.; Stuttfeld, E.; Asthana, M.; Weinert, T.; Bliven, S.; Goldie, K.N.; Kisko, K.; Capitani, G.; Ballmer-Hofer, K. Structure of the Full-length VEGFR-1 Extracellular Domain in Complex with VEGF-A. Structure 2017, 25, 341–352. [Google Scholar] [CrossRef]
- Wang, X.; Bove, A.M.; Simone, G.; Ma, B. Molecular Bases of VEGFR-2-Mediated Physiological Function and Pathological Role. Front. Cell Dev. Biol. 2020, 8, 599281. [Google Scholar] [CrossRef] [PubMed]
- Koch, S. Neuropilin signalling in angiogenesis. Biochem. Soc. Trans. 2012, 40, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Domingues, A.; Fantin, A. Neuropilin 1 Regulation of Vascular Permeability Signaling. Biomolecules 2021, 11, 666. [Google Scholar] [CrossRef]
- Teran, M.; Nugent, M.A. Synergistic Binding of Vascular Endothelial Growth Factor-A and Its Receptors to Heparin Selectively Modulates Complex Affinity. J. Biol. Chem. 2015, 290, 16451–16462. [Google Scholar] [CrossRef] [PubMed]
- Kargozar, S.; Baino, F.; Hamzehlou, S.; Hamblin, M.R.; Mozafari, M. Nanotechnology for Angiogenesis: Opportunities and Challenges. Chem. Soc. Rev. 2020, 49, 5008. [Google Scholar] [CrossRef]
- Liang, X.; Xu, F.; Li, X.; Ma, C.; Zhang, Y.; Xu, W. VEGF Signal System: The Application of Antiangiogenesis. Cur. Med. Chem. 2014, 21, 894–910. [Google Scholar] [CrossRef]
- Ferrara, N. VEGF as a Therapeutic Target in Cancer. Oncology 2005, 69, 11–16. [Google Scholar] [CrossRef]
- Park, J.E.; Chen, H.H.; Winer, J.; Houck, K.A.; Ferrara, N. Placenta Growth Factor. Potentiation of Vascular Endothelial Growth Factor Bioactivity, In Vitro and In Vivo, and High Affinity Binding to Flt-1 but not to Flk-1/KDR. J. Biol. Chem. 1994, 269, 25646–25654. [Google Scholar] [CrossRef]
- Olofsson, B.; Korpelainen, E.; Pepper, M.S.; Mandriota, S.J.; Aase, K.; Kumar, V.; Gunji, Y.; Jeltsch, M.M.; Shibuya, M.; Alitalo, K.; et al. Vascular Endothelial Growth Factor B (VEGF-B) Binds to VEGF Receptor-1 and Regulates Plasminogen Activator Activity in Endothelial Cells. Proc. Natl. Acad. Sci. USA 1998, 95, 11709–11714. [Google Scholar] [CrossRef]
- Zirlik, K.; Duyster, J. Anti-Angiogenics: Current Situation and Future Perspectives. Oncol. Res. Treat. 2018, 41, 166–171. [Google Scholar] [CrossRef]
- Gao, F.; Yang, C. Anti-VEGF/VEGFR2 Monoclonal Antibodies and their Combinations with PD-1/PD-L1 Inhibitors in Clinic. Curr. Cancer Drug Targets 2020, 20, 3–18. [Google Scholar] [CrossRef]
- Pozarowska, D.; Pozarowski, P. The Era of Anti-Vascular Endothelial Growth Factor (VEGF) Drugs in Ophthalmology, VEGF and Anti-VEGF Therapy. Cent. Eur. J. Immunol. 2016, 41, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, P.K. Antivascular Endothelial Growth Factor Agents and Their Development: Therapeutic Implications in Ocular Diseases. Am. J. Ophthalmol. 2006, 142, 660–668. [Google Scholar] [CrossRef] [PubMed]
- Shahsuvaryan, M.L. Therapeutic Potential of Ranibizumab in Corneal Neovascularization. Trends Pharm. Sci. 2017, 38, 667–668. [Google Scholar] [CrossRef] [PubMed]
- Holz, F.G.; Dugel, P.U.; Weissgerber, G.; Hamilton, R.; Silva, R.; Bandello, F.; Larsen, M.; Weichselberger, A.; Wenzel, A.; Schmidt, A.; et al. Single-Chain Antibody Fragment VEGF Inhibitor RTH258 for Neovascular Age-Related Macular Degeneration. Ophtalmology 2016, 123, 1080–1089. [Google Scholar] [CrossRef] [PubMed]
- Sarwar, S.; Clearfield, E.; Soliman, M.K.; Sadiq, M.A.; Baldwin, A.J.; Hanout, M.; Agarwal, A.; Sepah, Y.J.; Do, D.V.; Nguyen, Q.D. Aflibercept for Neovascular Age-Related Macular Degeneration. Cochrane Database Syst. Rev. 2016, 2, CD011346. [Google Scholar] [CrossRef] [PubMed]
- Holash, J.; Davis, S.; Papadopoulos, N.; Croll, S.D.; Ho, L.; Russell, M.; Boland, P.; Leidich, R.; Hylton, D.; Burova, E.; et al. VEGF-Trap: A VEGF Blocker with Potent Antitumor Effects. Proc. Natl. Acad. Sci. USA 2002, 99, 11393–11398. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Li, T.; Wu, Z.; Wu, Q.; Ke, X.; Luo, D.; Wang, H. Novel VEGF Decoy Receptor Fusion Protein Conbercept Targeting Multiple VEGF Isoforms Provide Remarkable Anti-Angiogenesis Effect In Vivo. PLoS ONE 2013, 8, e70544. [Google Scholar] [CrossRef]
- Rust, R.; Gantner, C.; Schwab, M.E. Pro- and Antiangiogenic Therapies: Current Status and Clinical Implications. FASEB J. 2019, 33, 34–48. [Google Scholar] [CrossRef]
- Abdullah, S.E.; Perez-Soler, R. Mechanisms of Resistance to Vascular Endothelial Growth Factor Blockade. Cancer 2012, 118, 3455–3467. [Google Scholar] [CrossRef]
- García-Quintanilla, L.; Luaces-Rodríguez, A.; Gil-Martínez, M.; Mondelo-García, C.; Maroña, O.; Mangas-Sanjuan, V.; González-Barcia, M.; Zarra-Ferro, I.; Aguiar, P.; Otero-Espinar, F.J.; et al. Pharmacokinetics of Intravitreal Anti-VEGF Drugs in Age-Related Macular Degeneration. Pharmaceutics 2019, 11, 365. [Google Scholar] [CrossRef]
- Stewart, M.W. Extended Duration Vascular Endothelial Growth Factor Inhibition in the Eye: Failures, Successes, and Future Possibilities. Pharmaceutics 2018, 10, 21. [Google Scholar] [CrossRef]
- Ghosh, J.G.; Nguyen, A.A.; Bigelow, C.E.; Poor, S.; Qiu, Y.; Rangaswamy, N.; Ornberg, R.; Jackson, B.; Mak, H.; Mak, H.; et al. Long-acting Protein Drugs for the Treatment of Ocular Diseases. Nat. Commun. 2017, 8, 14837. [Google Scholar] [CrossRef]
- Chirio, D.; Peira, E.; Sapino, S.; Chiniamo, G.; Oliaro-Bosso, S.; Adinolfi, S.; Dianzani, C.; Baratta, F.; Gallarate, M.A. New Bevacizumab Carrier for Intravitreal Administration: Focus on Stability. Pharmaceutics 2021, 13, 560. [Google Scholar] [CrossRef]
- Pelay-Gimeno, M.; Glas, A.; Koch, O. Structure-Based Design of Inhibitors of Protein-Protein Interactions: Mimicking Peptide Binding Epitopes. Angew. Chem. Int. Ed. 2015, 54, 8896–8927. [Google Scholar] [CrossRef]
- Guryanov, I.; Fiorucci, S.; Tennikova, T. Receptor-Ligand Interactions: Advanced Biomedical Applications. Mater. Sci. Eng. C 2016, 68, 890–903. [Google Scholar] [CrossRef] [PubMed]
- Guryanov, I.; Real-Fernández, F.; Sabatino, G.; Prisco, N.; Korzhikov-Vlakh, V.; Biondi, B.; Papini, A.M.; Korzhikova-Vlakh, E.; Rovero, P.; Tennikova, T. Modeling Interaction Between gp120 HIV Protein and CCR5 Receptor. J. Pept. Sci. 2019, 25, e3142. [Google Scholar] [CrossRef] [PubMed]
- Zane, D.; Feldman, P.L.; Sawyer, T.; Sobol, Z.; Hawes, J. Development and Regulatory Challenges for Peptide Therapeutics. Int. J. Toxicol. 2021, 40, 108–124. [Google Scholar] [CrossRef]
- Muttenthaler, M.; King, G.F.; Adams, D.J.; Alewood, P.F. Trends in Peptide Drug Discovery. Nat. Rev. Drug Discov. 2021, 20, 309–325. [Google Scholar] [CrossRef] [PubMed]
- Courtois, F.; Agrawal, N.J.; Lauer, T.M.; Trout, B.L. Rational Design of Therapeutic mAbs Against Aggregation Through Protein Engineering and Incorporation of Glycosylation Motifs Applied to Bevacizumab. MAbs 2016, 8, 99–112. [Google Scholar] [CrossRef] [PubMed]
- Zheng, K.; Bantog, C.; Bayer, R. The Impact of Glycosylation on Monoclonal Antibody Conformation and Stability. MAbs 2011, 3, 568–576. [Google Scholar] [CrossRef]
- Rouet, R.; Lowe, D.; Christ, D. Stability Engineering of the Human Antibody Repertoire. FEBS Lett. 2014, 588, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Zashikhina, N.; Sharoyko, V.; Antipchik, M.; Tarasenko, I.; Anufrikov, Y.; Lavrentieva, A.; Tennikova, T.; Korzhikova-Vlakh, E. Novel Formulations of C-Peptide with Long-Acting Therapeutic Potential for Treatment of Diabetic Complications. Pharmaceutics 2019, 11, 27. [Google Scholar] [CrossRef] [PubMed]
- Belair, D.; Le, N.; Murphy, W.L. Design of Growth Factor Sequestering Biomaterials. Chem. Commun. 2014, 50, 15651–15668. [Google Scholar] [CrossRef]
- Shi, D.; Sheng, A.; Chi, L. Glycosaminoglycan-Protein Interactions and Their Roles in Human Disease. Front. Mol. Biosci. 2021, 8, 639666. [Google Scholar] [CrossRef]
- Koehler, L.; Ruiz-Gόmez, G.; Balamurugan, K.; Rother, S.; Freyse, J.; Mӧller, S.; Schnabelrauch, M.; Kӧnling, S.; Djordjevic, S.; Scharnweber, D.; et al. Dual Action of Sulfated Hyaluronan on Angiogenic Processes in Relation to Vascular Endothelial Growth Factor-A. Sci. Rep. 2019, 9, 18143. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Liu, X.; Liu, Z.; Gu, H.; Li, D.; Chen, H. Glycosaminoglycans (GAGs) and GAG Mimetics Regulate the Behaviorof Stem Cell Differentiation. Colloids Surf. B Biointerfaces 2017, 150, 175–182. [Google Scholar] [CrossRef]
- Hassan, N.; Greve, B.; Espinoza-Sánchez, N.A.; Gӧtte, M. Cell-Surface Heparan Sulfate Proteoglycans as Multifunctional Integrators of Signaling in Cancer. Cell. Signal. 2021, 77, 109822. [Google Scholar] [CrossRef] [PubMed]
- Grünewald, F.S.; Prota, A.E.; Giese, A.; Ballmer-Hofer, K. Structure–Function Analysis of VEGF Receptor Activation and the Role of Coreceptors in Angiogenic Signaling. Biochim. Biophys. Acta 2010, 1804, 567–580. [Google Scholar] [CrossRef]
- Capila, I.; Linhardt, R.J. Heparin–Protein Interactions. Angew. Chem. Int. Ed. 2002, 41, 390–412. [Google Scholar] [CrossRef]
- Fairbrother, W.J.; Champe, M.A.; Christinger, H.W.; Keyt, B.A.; Starovasnik, M.A. Solution Structure of the Heparin-Binding Domain of Vascular Endothelial Growth Factor. Structure 1998, 6, 637–648. [Google Scholar] [CrossRef]
- He, C.; Ji, H.; Qian, Y.; Wang, Q.; Liu, X.; Zhao, W.; Zhao, C. Heparin-Based and Heparin-Inspired Hydrogels: Size-Effect, Gelation and Biomedical Applications. J. Mater. Chem. 2019, 7, 1186–1208. [Google Scholar] [CrossRef]
- Norrby, K. 2.5 kDa and 5.0 kDa Heparin Fragments Specifically Inhibit Microvessel Sprouting and Network Formation in VEGF(165)-Mediated Mammalian Angiogenesis. Int. J. Exp. Pathol. 2000, 81, 191–198. [Google Scholar] [CrossRef]
- Cole, C.L.; Hansen, S.U.; Barath, M.; Rushton, G.; Gardiner, J.M.; Avizienyte, E.; Jayson, G.C. Synthetic Heparan Sulfate Oligosaccharides Inhibit Endothelial Cell Functions Essential for Angiogenesis. PLoS ONE. 2010, 5, e11644. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.L.; Esposito, E.; Hayakawa, K.; Mandaville, E.; Smith, R.A.A.; Guo, S.; Niu, W.; Wong, P.T.-H.; Cool, S.M.; Lo, E.H.; et al. Vascular Endothelial Growth Factor 165-Binding Heparan Sulfate Promotes Functional Recovery from Cerebral Ischemia. Stroke 2020, 51, 2844–2853. [Google Scholar] [CrossRef] [PubMed]
- Hachim, D.; Whittaker, T.E.; Kim, H.; Stevens, M.M. Glycosaminoglycan-Based Biomaterials for Growth Factor and Cytokine Delivery: Making the Right Choices. J. Control. Release 2019, 313, 131–147. [Google Scholar] [CrossRef] [PubMed]
- Jeong, M.-C.; Jin, B.; Kim, Y. Relationship between Structural Flexibility and Function in the C-Terminal Region of the Heparin-Binding Domain of VEGFA165. Biochemistry 2013, 52, 8823–8832. [Google Scholar] [CrossRef]
- Maynard, H.D.; Hubbell, J.A. Discovery of a Sulfated Tetrapeptide that Binds to Vascular Endothelial Growth Factor. Acta Biomater. 2005, 1, 451–459. [Google Scholar] [CrossRef]
- Kim, S.H.; Kiick, K.L. Heparin-Mimetic Sulfated Peptides with Modulated Affinities for Heparin-Binding Peptides and Growth Factors. Peptides 2007, 28, 2125–2136. [Google Scholar] [CrossRef]
- Yasa, O.; Uysal, O.; Ekiz, M.S.; Guler, M.O.; Ayse, B.; Tekinay, A.B. Presentation of Functional Groups on Self-Assembled Supramolecular Peptide Nanofibers Mimicking Glycosaminoglycans for Directed Mesenchymal Stem Cell Differentiation. J. Mater. Chem. B 2017, 5, 4890–4900. [Google Scholar] [CrossRef]
- Kocabey, S.; Ceylan, H.; Tekinay, A.B.; Guler, M.O. Glycosaminoglycan Mimetic Peptide Nanofibers Promote Mineralization by Osteogenic Cells. Acta Biomater. 2013, 9, 9075–9085. [Google Scholar] [CrossRef] [PubMed]
- Mammadov, B.; Toksoz, S.; Aydin, B.; Yagci, R.; Tekinay, A.B.; Guler, M.O. Heparin Mimetic Peptide Nanofibers Promote Angiogenesis. Biomacromolecules 2011, 12, 3508–3519. [Google Scholar] [CrossRef]
- Uzunalli, G.; Mammadov, R.; Yesildal, F.; Alhan, D.; Ozturk, S.; Ozgurtas, T.; Guler, M.O.; Tekinay, A.B. Angiogenic Heparin-Mimetic Peptide Nanofiber Gel Improves Regenerative Healing of Acute Wounds. ACS Biomater. Sci. Eng. 2017, 3, 1296–1303. [Google Scholar] [CrossRef]
- Lee, S.S.; Fyrner, T.; Chen, F.; Álvarez, Z.; Sleep, E.; Chun, D.; Weiner, J.A.; Cook, R.W.; Freshman, R.D.; Schallmo, M.S.; et al. Sulfated Glycopeptide Nanostructures for Multipotent Protein Activation. Nat. Nanotecnol. 2017, 12, 821–831. [Google Scholar] [CrossRef]
- Kendall, R.L.; Thomas, K.A. Inhibition of Vascular Endothelial Cell Growth Factor Activity by an Endogenously Encoded Soluble Receptor. Proc. Natl. Acad. Sci. USA 1993, 90, 10705–10709. [Google Scholar] [CrossRef] [PubMed]
- Hornig, C.; Barleon, B.; Ahmad, S.; Vuorela, P.; Ahmed, A.; Weich, H.A. Release and Complex Formation of Soluble VEGFR-1 from Endothelial Cells and Biological Fluids. Lab. Investig. 2000, 80, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, V.; Gautier, B.; Huguenot, F.; Leproux, P.; Garbay, C.; Vidala, M.; Inguimbert, N. Total Chemical Synthesis of the D2 Domain of Human VEGF Receptor 1. J. Pept. Sci. 2009, 15, 417–422. [Google Scholar] [CrossRef]
- Checco, J.W.; Gellman, S.H. Iterative Nonproteinogenic Residue Incorporation Yields α/β-Peptides with a Helix–Loop–Helix Tertiary Structure and High Affinity for VEGF. ChemBioChem 2017, 18, 291–299. [Google Scholar] [CrossRef]
- Piossek, C.; Schneider-Mergener, J.; Schirner, M.; Vakalopoulou, E.; Germeroth, L.; Thierauch, K.-H. Vascular Endothelial Growth Factor (VEGF) Receptor II-derived Peptides Inhibit VEGF. J. Biol. Chem. 1999, 274, 5612–5619. [Google Scholar] [CrossRef]
- Piossek, C.; Thierauch, K.-H.; Schneider-Mergener, J.; Volkmer-Engert, R.; Bachmann, M.F.; Korff, T.; Augustin, H.G.; Germeroth, L. Potent Inhibition of Angiogenesis by D,L-Peptides Derived from Vascular Endothelial Growth Factor Receptor 2. Thromb. Haemostasis 2003, 90, 501–510. [Google Scholar] [CrossRef]
- Toepke, M.W.; Impellitteri, N.A.; Lan Levengood, S.K.; Boeldt, D.S.; Bird, I.M.; Murphy, W.L. Regulating Specific Growth Factor Signaling Using Immobilized Branched Ligands. Adv. Healthc. Mater. 2012, 1, 457–460. [Google Scholar] [CrossRef]
- Belair, D.G.; Khalil, A.S.; Miller, M.J.; Murphy, W.L. Serum-Dependence of Affinity-Mediated VEGF Release from Biomimetic Microspheres. Biomacromolecules 2014, 15, 2038–2048. [Google Scholar] [CrossRef]
- Belair, D.G.; Murphy, W.L. Specific VEGF Sequestering to Biomaterials: Influence of Serum Stability. Acta Biomater. 2013, 9, 8823–8831. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Impellitteri, N.A.; Toepke, M.W.; Lan Levengood, S.K.; Murphy, W.L. Specific VEGF Sequestering and Release Using Peptide-Functionalized Hydrogel Microspheres. Biomaterials 2012, 33, 3475–3484. [Google Scholar] [CrossRef] [PubMed]
- Belair, D.G.; Miller, M.J.; Wang, S.; Darjatmoko, S.R.; Binder, B.Y.K.; Sheibani, N.; Murphy, W.L. Differential Regulation of Angiogenesis Using Degradable VEGF-Binding Microspheres. Biomaterials 2016, 93, 27–37. [Google Scholar] [CrossRef]
- Bae, D.-G.; Gho, Y.-S.; Yoon, W.-H.; Chae, C.-B. Arginine-rich Anti-vascular Endothelial Growth Factor Peptides Inhibit Tumor Growth and Metastasis by Blocking Angiogenesis. J. Biol. Chem. 2000, 275, 13588–13596. [Google Scholar] [CrossRef] [PubMed]
- Chittasupho, C.; Kengtrong, K.; Chalermnithiwong, S.; Sarisuta, N. Anti-Angiogenesis by Dual Action of R5K Peptide Conjugated Itraconazole Nanoparticles. AAPS PharmSciTech 2020, 21, 74. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.A.; Bae, D.-G.; Ryoo, J.-W.; Kim, H.-R.; Park, G.-S.; Cho, C.-S.; Chae, C.-B.; Kim, W.-U. Arginine-Rich Anti-Vascular Endothelial Growth Factor (Anti-VEGF) Hexapeptide Inhibits. J. Immunol. 2005, 174, 5846–5855. [Google Scholar] [CrossRef]
- Kim, J.-W.; Kim, T.-D.; Hong, B.S.; Kim, O.Y.; Yoon, W.-H.; Chae, C.-B.; Gho, Y.S. A Serum-Stable Branched Dimeric Anti-VEGF Peptide Blocks Tumor Growth via Anti-Angiogenic Activity. Exp. Mol. Med. 2010, 42, 514–523. [Google Scholar] [CrossRef]
- Chan, L.Y.; Craik, D.J.; Daly, N.L. Dual-Targeting Anti-Angiogenic Cyclic Peptides as Potential Drug Leads for Cancer Therapy. Sci. Rep. 2016, 6, 35347. [Google Scholar] [CrossRef]
- Norton, K.-A.; Han, Z.; Popel, A.S.; Pandey, N.B. Antiangiogenic Cancer Drug Sunitinib Exhibits Unexpected Proangiogenic Effects on Endothelial Cells. OncoTargets Ther. 2014, 7, 1571–1582. [Google Scholar] [CrossRef] [PubMed]
- Erdag, B.; Koray, B.; Balcioglu, K.B.; Kumbasar, A.; Celikbicak, O.; Zeder-Lutz, G.; Altschuh, D.; Salih, B.; Baysal, K. Novel Short Peptides Isolated from Phage Display Library Inhibit Vascular Endothelial Growth Factor Activity. Mol. Biotechnol. 2007, 35, 51–63. [Google Scholar] [CrossRef]
- Fedorova, A.; Zobel, K.; Gill, H.S.; Ogasawara, A.; Flores, J.E.; Tinianow, J.N.; Vanderbilt, A.N.; Wu, P.; Meng, Y.G.; Williams, S.P.; et al. The Development of Peptide-Based Tools for the Analysis of Angiogenesis. Chem. Biol. 2011, 18, 839–845. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Checco, J.W.; Kreitler, D.F.; Thomas, N.C.; Belair, D.G.; Rettko, N.J.; Murphy, W.L.; Katrina, T.; Forest, K.T.; Gellman, S.H. Targeting Diverse Protein–Protein Interaction Interfaces with α/β-Peptides Derived from the Z-Domain Scaffold. Proc. Natl. Acad. Sci. USA 2015, 112, 4552–4557. [Google Scholar] [CrossRef]
- Fairbrother, W.J.; Christinger, H.W.; Cochran, A.G.; Fuh, G.; Keenan, C.J.; Quan, C.; Shriver, S.K.; Tom, J.Y.K.; Wells, J.A.; Cunningham, B.C. Novel Peptides Selected to Bind Vascular Endothelial Growth Factor Target the Receptor-Binding Site. Biochemistry 1998, 37, 17754–17764. [Google Scholar] [CrossRef]
- Pan, B.; Li, B.; Russell, S.J.; Tom, Y.J.K.; Cochran, A.G.; Fairbrother, W.J. Solution Structure of a Phage-Derived Peptide Antagonist in Complex with Vascular Endothelial Growth Factor. J. Mol. Biol. 2002, 316, 769–787. [Google Scholar] [CrossRef]
- Gang Wu, G.; Han, K.; Lv, F. Use of Fast Conformational Sampling to Improve the Characterization of VEGF A–Peptide Interactions. J. Theor. Biol. 2013, 317, 293–300. [Google Scholar]
- Kenrick, S.A.; Daugherty, P.S. Bacterial Display Enables Efficient and Quantitative Peptide Affinity Maturation. Protein Eng. Des. Sel. 2010, 23, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Bayό-Puxan, N.; Rodríguez-Mias, R.; Goldflam, M.; Kotev, M.; Ciudad, S.; Hipolito, C.J.; Varese, M.; Suga, H.; Campos-Olivas, R.; Barril, X.; et al. Combined Use of Oligopeptides, Fragment Libraries, and Natural Compounds A Comprehensive Approach. ChemMedChem 2016, 11, 928–939. [Google Scholar] [CrossRef]
- Graaf, M.D.; Marquez, B.V.; Yeh, N.-H.; Lapi, S.E.; Moeller, K.D. New Methods for the Site-Selective Placement of Peptides on a Microelectrode Array: Probing VEGF−v107 Binding as Proof of Concept. ACS Chem. Biol. 2016, 11, 2829–2837. [Google Scholar] [CrossRef] [PubMed]
- Peterson, K.J.; Sadowsky, J.D.; Scheef, E.A.; Pal, S.; Kourentzi, K.D.; Willson, R.C.; Bresnick, E.H.; Sheibani, N.; Gellman, S.H. A fluorescence polarization assay for identifying ligands that bind to vascular endothelial growth factor. Anal. Biochem. 2008, 378, 8–14. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Haase, H.S.; Peterson-Kaufman, K.J.; Lan Levengood, S.K.; Checco, J.W.; Murphy, W.L.; Gellman, S.H. Extending Foldamer Design Beyond α-Helix Mimicry αβ-Peptide Inhibitors of Vascular Endothelial Growth Factor Signaling. J. Am. Chem. Soc. 2012, 134, 7652–7655. [Google Scholar] [CrossRef] [PubMed]
- Reille-Seroussi, M.; Gaucher, J.-F.; Desole, C.; Gagey-Eilstein, N.; Brachet, F.; Broutin, I.; Vidal, M.; Broussy, S. Vascular Endothelial Growth Factor Peptide Ligands Explored by Competition Assay and Isothermal Titration Calorimetry. Biochemistry 2015, 54, 5147–5156. [Google Scholar] [CrossRef]
- Guryanov, I.; Korzhikov-Vlakh, V.; Bhattacharya, M.; Biondi, B.; Masiero, G.; Formaggio, F.; Tennikova, T.; Urtti, A. Conformationally Constrained Peptides with High Affinity to Vascular Endothelial Growth Factor. J. Med. Chem. 2021, 64, 10900–10907. [Google Scholar] [CrossRef] [PubMed]
- Coppock, M.B.; Warner, C.R.; Dorsey, B.; Orlicki, J.A.; Sarkes, D.A.; Lai, B.T.; Pitram, S.M.; Rohde, R.D.; Malette, J.; Wilson, J.A.; et al. Protein Catalyzed Capture Agents with Tailored Performance for in Vitro and in Vivo Applications. Pep. Sci. 2017, 108, e22934. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guryanov, I.; Tennikova, T.; Urtti, A. Peptide Inhibitors of Vascular Endothelial Growth Factor A: Current Situation and Perspectives. Pharmaceutics 2021, 13, 1337. https://doi.org/10.3390/pharmaceutics13091337
Guryanov I, Tennikova T, Urtti A. Peptide Inhibitors of Vascular Endothelial Growth Factor A: Current Situation and Perspectives. Pharmaceutics. 2021; 13(9):1337. https://doi.org/10.3390/pharmaceutics13091337
Chicago/Turabian StyleGuryanov, Ivan, Tatiana Tennikova, and Arto Urtti. 2021. "Peptide Inhibitors of Vascular Endothelial Growth Factor A: Current Situation and Perspectives" Pharmaceutics 13, no. 9: 1337. https://doi.org/10.3390/pharmaceutics13091337
APA StyleGuryanov, I., Tennikova, T., & Urtti, A. (2021). Peptide Inhibitors of Vascular Endothelial Growth Factor A: Current Situation and Perspectives. Pharmaceutics, 13(9), 1337. https://doi.org/10.3390/pharmaceutics13091337