
The Cysteine-Containing Cell-Penetrating Peptide AP Enables Efficient Macromolecule Delivery to T Cells and Controls Autoimmune Encephalomyelitis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. DNA Cloning
2.2. Purification of the Recombinant Proteins
2.3. Cell Culture
2.4. In Vitro T Cell Delivery
2.5. Intracellular Delivery Mechanisms
2.6. In Vivo Delivery
2.7. Multiphoton Microscopy of Live Animals
2.8. Peptide Synthesis
2.9. T Cell Differentiation
2.10. EAE Induction
2.11. Statistical Analysis
3. Results
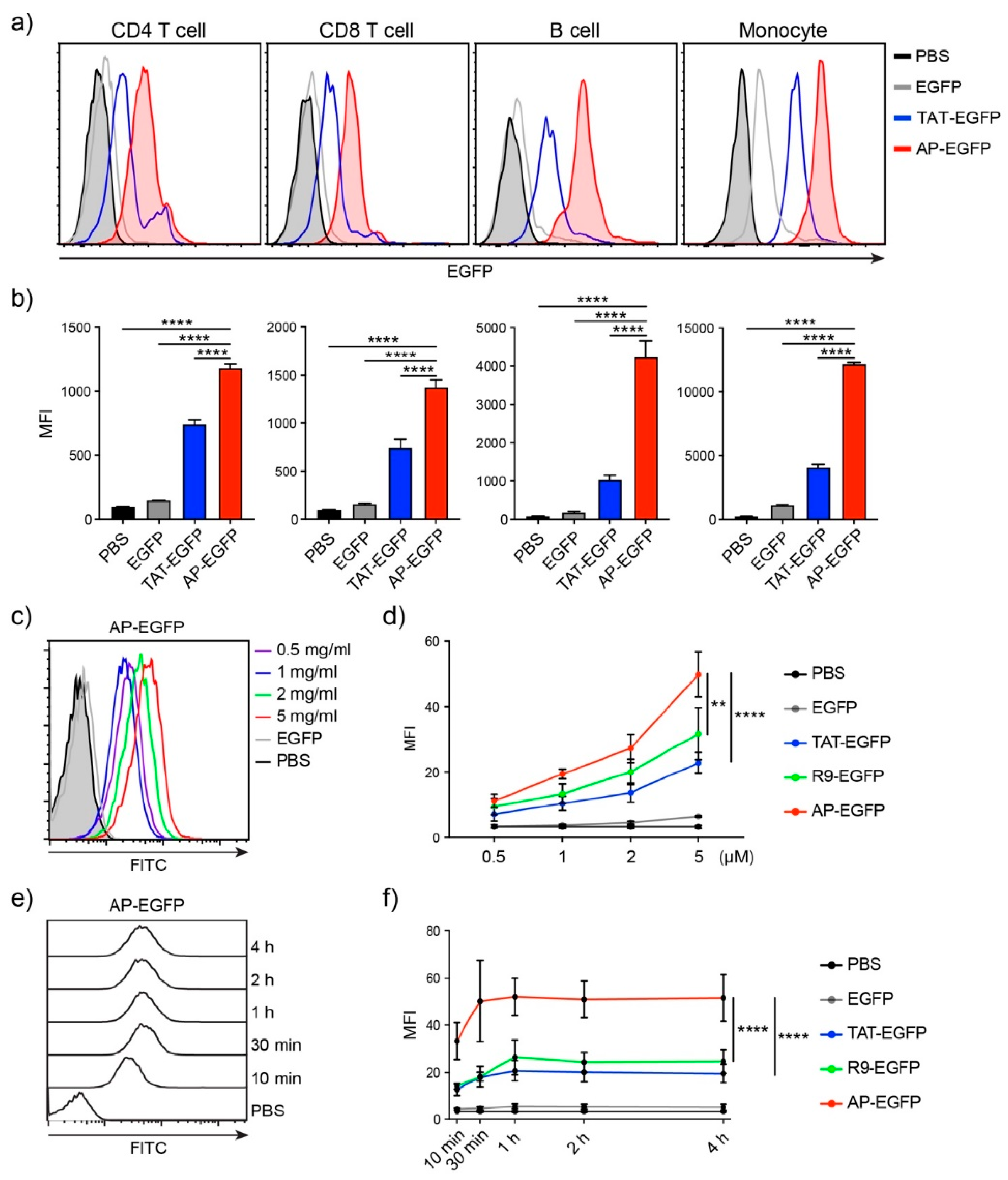
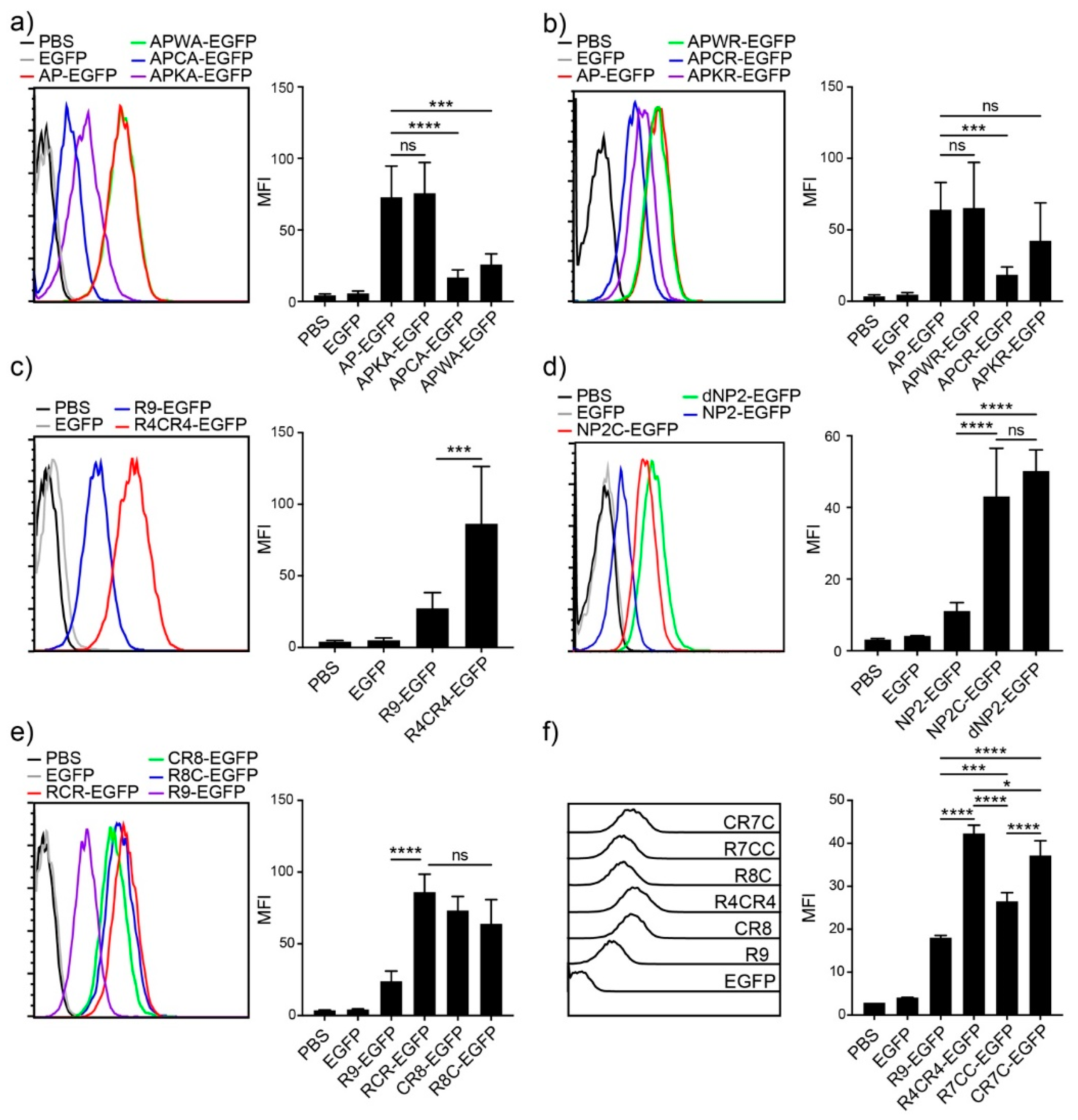
3.1. Efficient Peptide-Based Protein Delivery System, AP to T Cells
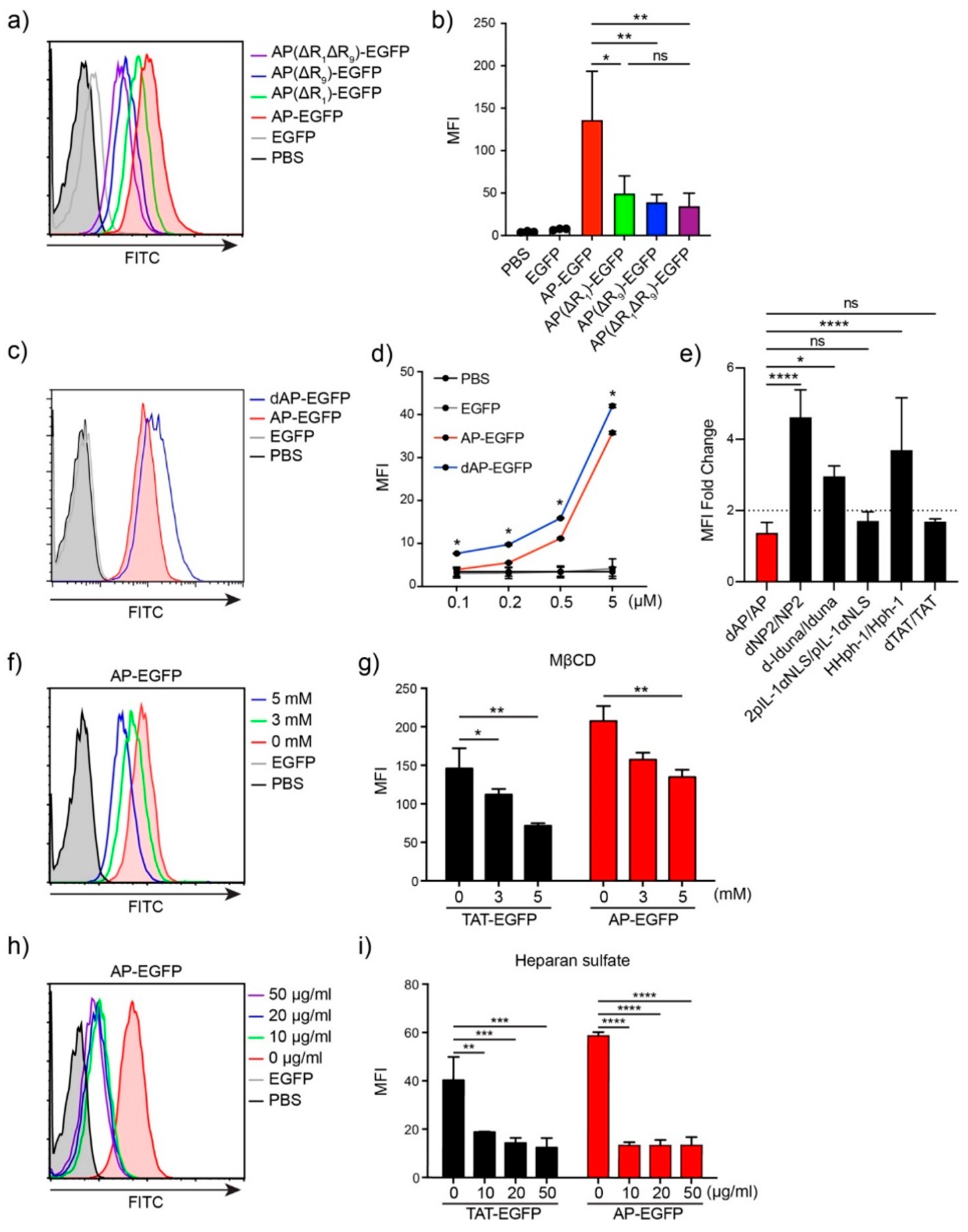
3.2. T Cell Delivery Mechanism of the Optimized AP Peptide
3.3. The Cysteine Effect on the Transduction Efficiency to T Cells
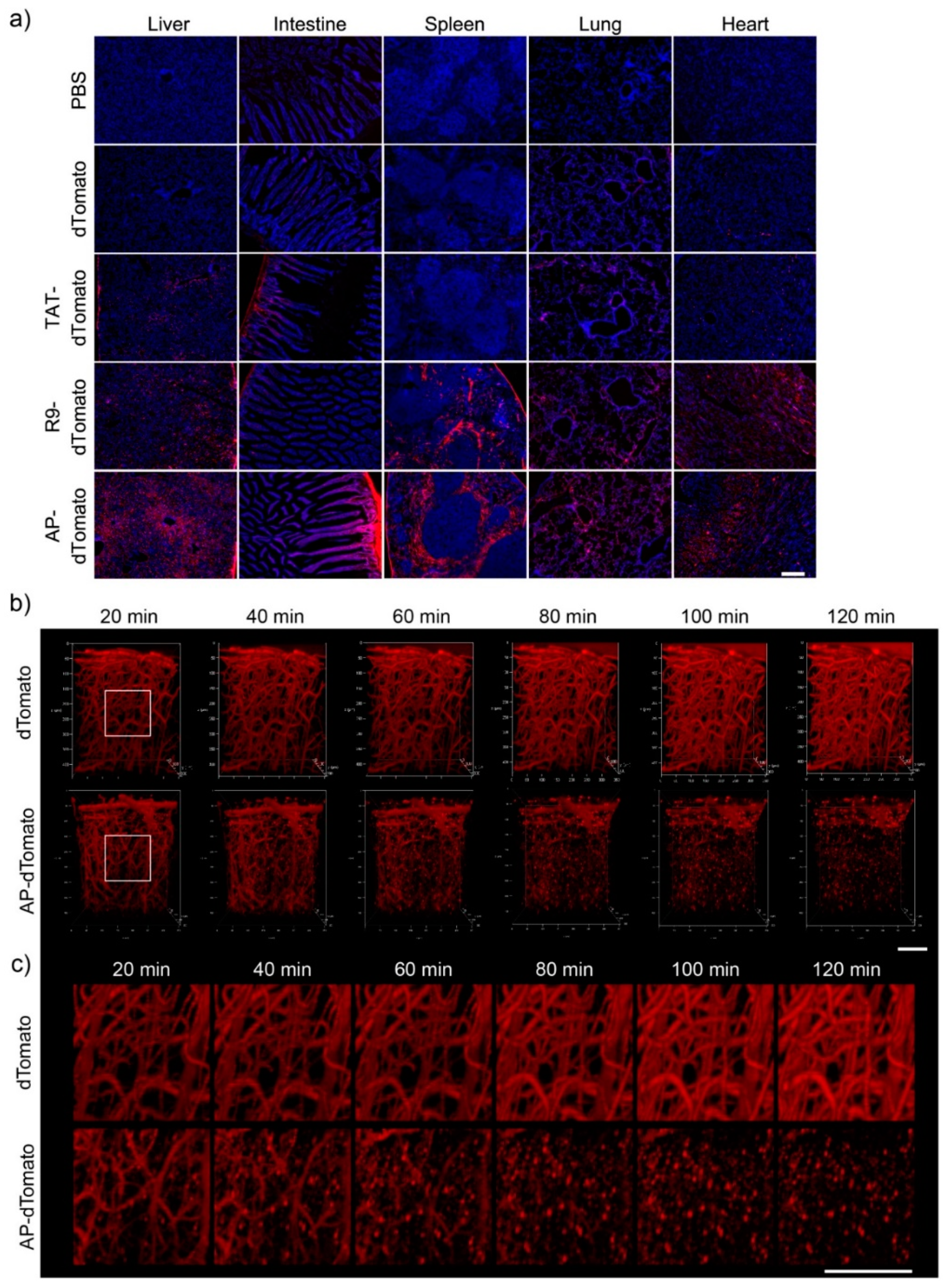
3.4. In Vivo Biodistribution and BBB-Penetration of AP
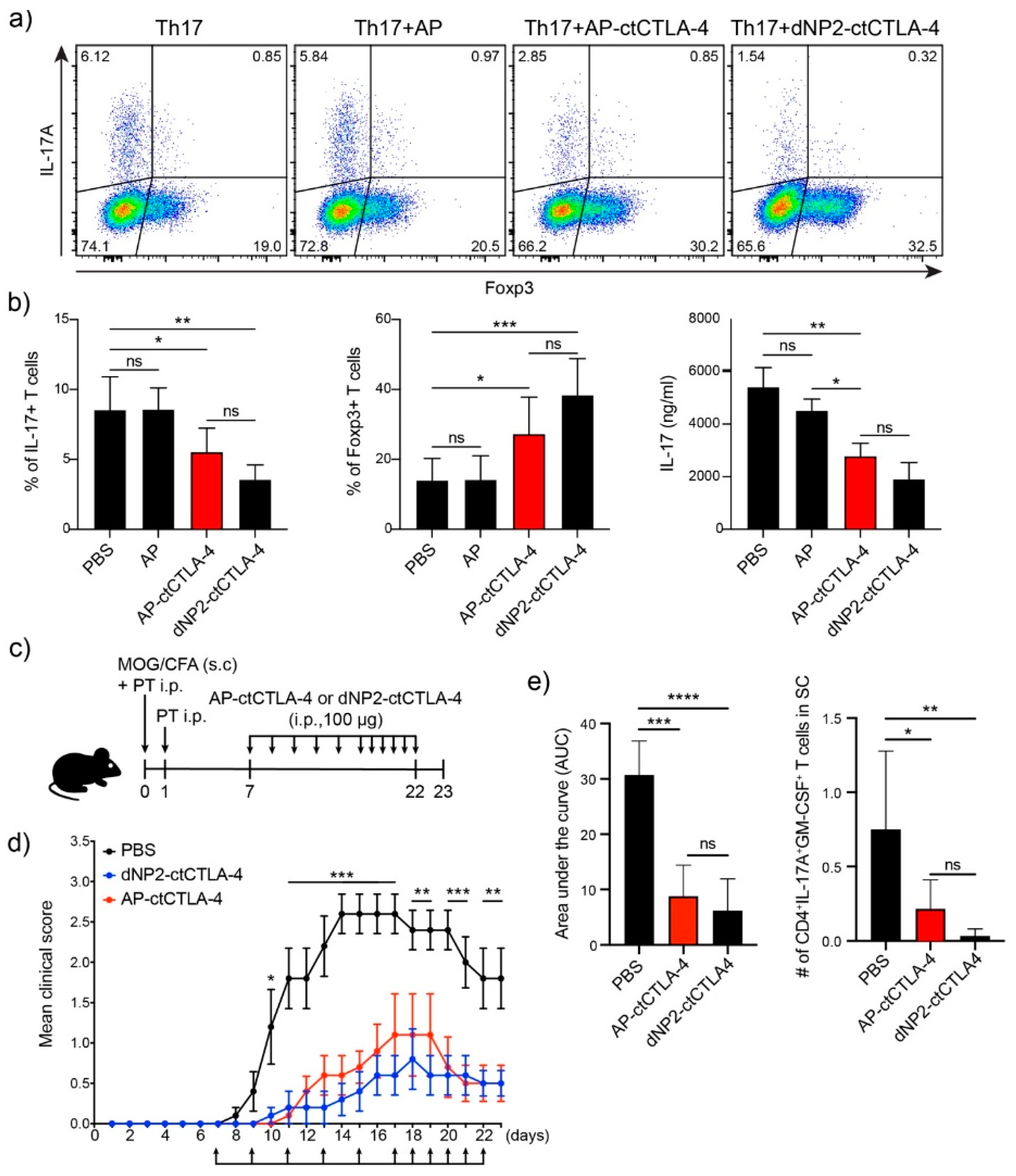
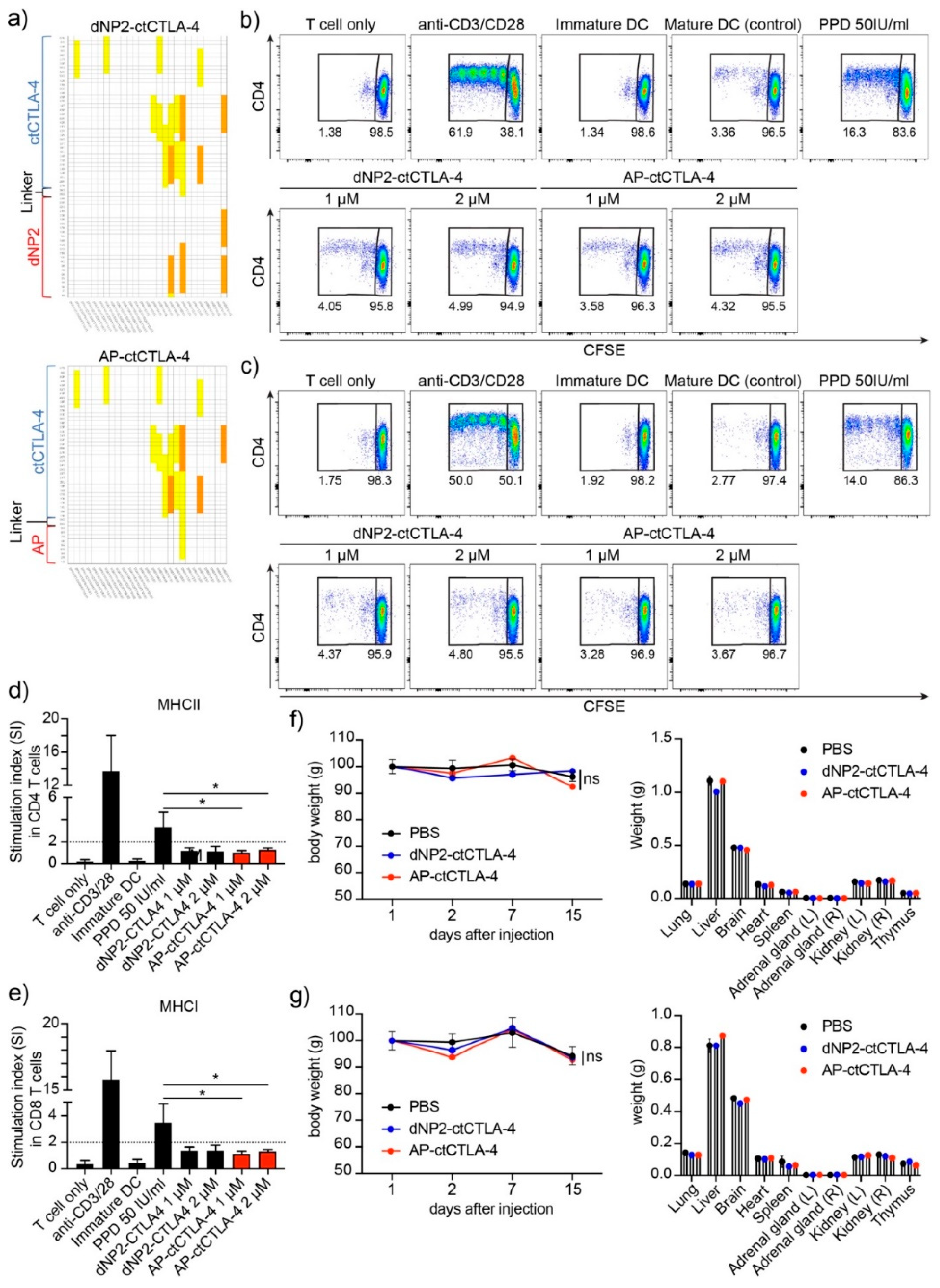
3.5. T Cell Regulation in Th17 Differentiation and EAE by AP-ctCTLA-4
3.6. Immunogenicity and In Vivo Single Dose Toxicity of AP-ctCTLA-4
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chaplin, D.D. Overview of the immune response. J. Allergy Clin. Immunol. 2010, 125, S3–S23. [Google Scholar] [CrossRef]
- Kumar, B.V.; Connors, T.J.; Farber, D.L. Human T Cell Development, Localization, and Function throughout Life. Immunity 2018, 48, 202–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenblum, M.D.; Remedios, K.A.; Abbas, A.K. Mechanisms of human autoimmunity. J. Clin. Investig. 2015, 125, 2228–2233. [Google Scholar] [CrossRef] [Green Version]
- Filippi, M.; Bar-Or, A.; Piehl, F.; Preziosa, P.; Solari, A.; Vukusic, S.; Rocca, M.A. Multiple sclerosis. Nat. Rev. Dis. Primers 2018, 4, 43. [Google Scholar] [CrossRef] [PubMed]
- Trautmann, A.; Akdis, M.; Brocker, E.B.; Blaser, K.; Akdis, C.A. New insights into the role of T cells in atopic dermatitis and allergic contact dermatitis. Trends Immunol. 2001, 22, 530–532. [Google Scholar] [CrossRef]
- He, J.; Zhang, X.; Wei, Y.; Sun, X.; Chen, Y.; Deng, J.; Jin, Y.; Gan, Y.; Hu, X.; Jia, R.; et al. Low-dose interleukin-2 treatment selectively modulates CD4(+) T cell subsets in patients with systemic lupus erythematosus. Nat. Med. 2016, 22, 991–993. [Google Scholar] [CrossRef]
- Gong, N.; Sheppard, N.C.; Billingsley, M.M.; June, C.H.; Mitchell, M.J. Nanomaterials for T-cell cancer immunotherapy. Nat. Nanotechnol. 2021, 16, 25–36. [Google Scholar] [CrossRef]
- Page, D.B.; Postow, M.A.; Callahan, M.K.; Allison, J.P.; Wolchok, J.D. Immune modulation in cancer with antibodies. Annu. Rev. Med. 2014, 65, 185–202. [Google Scholar] [CrossRef]
- Lucca, L.E.; Dominguez-Villar, M. Modulation of regulatory T cell function and stability by co-inhibitory receptors. Nat. Rev. Immunol. 2020, 20, 680–693. [Google Scholar] [CrossRef]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Uhlen, M.; Karlsson, M.J.; Hober, A.; Svensson, A.S.; Scheffel, J.; Kotol, D.; Zhong, W.; Tebani, A.; Strandberg, L.; Edfors, F.; et al. The human secretome. Sci Signal. 2019, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, L.; Wang, M.; Lin, S.; Jian, R.; Li, X.; Chan, J.; Dong, G.; Fang, H.; Robinson, A.E.; Consortium, G.T.; et al. A Quantitative Proteome Map of the Human Body. Cell 2020, 183, 269–283.e219. [Google Scholar] [CrossRef] [PubMed]
- Niamsuphap, S.; Fercher, C.; Kumble, S.; Huda, P.; Mahler, S.M.; Howard, C.B. Targeting the undruggable: Emerging technologies in antibody delivery against intracellular targets. Expert Opin. Drug Deliv. 2020, 17, 1189–1211. [Google Scholar] [CrossRef]
- Lim, S.; Koo, J.H.; Choi, J.M. Use of Cell-Penetrating Peptides in Dendritic Cell-Based Vaccination. Immune Netw. 2016, 16, 33–43. [Google Scholar] [CrossRef] [Green Version]
- Frankel, A.D.; Pabo, C.O. Cellular uptake of the tat protein from human immunodeficiency virus. Cell 1988, 55, 1189–1193. [Google Scholar] [CrossRef]
- Green, M.; Loewenstein, P.M. Autonomous functional domains of chemically synthesized human immunodeficiency virus tat trans-activator protein. Cell 1988, 55, 1179–1188. [Google Scholar] [CrossRef]
- Xie, J.; Bi, Y.; Zhang, H.; Dong, S.; Teng, L.; Lee, R.J.; Yang, Z. Cell-Penetrating Peptides in Diagnosis and Treatment of Human Diseases: From Preclinical Research to Clinical Application. Front. Pharm. 2020, 11, 697. [Google Scholar] [CrossRef]
- Lim, S.; Kim, W.J.; Kim, Y.H.; Lee, S.; Koo, J.H.; Lee, J.A.; Yoon, H.; Kim, D.H.; Park, H.J.; Kim, H.M.; et al. dNP2 is a blood-brain barrier-permeable peptide enabling ctCTLA-4 protein delivery to ameliorate experimental autoimmune encephalomyelitis. Nat. Commun. 2015, 6, 8244. [Google Scholar] [CrossRef] [Green Version]
- Kim, G.R.; Kim, W.J.; Lim, S.; Lee, H.G.; Koo, J.H.; Nam, K.H.; Kim, S.M.; Park, S.D.; Choi, J.M. In Vivo Induction of Regulatory T Cells Via CTLA-4 Signaling Peptide to Control Autoimmune Encephalomyelitis and Prevent Disease Relapse. Adv. Sci. 2021. [Google Scholar] [CrossRef]
- Kim, W.J.; Koo, J.H.; Cho, H.J.; Lee, J.U.; Kim, J.Y.; Lee, H.G.; Lee, S.; Kim, J.H.; Oh, M.S.; Suh, M.; et al. Protein tyrosine phosphatase conjugated with a novel transdermal delivery peptide, astrotactin 1-derived peptide recombinant protein tyrosine phosphatase (AP-rPTP), alleviates both atopic dermatitis-like and psoriasis-like dermatitis. J. Allergy Clin. Immunol. 2018, 141, 137–151. [Google Scholar] [CrossRef] [Green Version]
- Vercauteren, D.; Vandenbroucke, R.E.; Jones, A.T.; Rejman, J.; Demeester, J.; De Smedt, S.C.; Sanders, N.N.; Braeckmans, K. The use of inhibitors to study endocytic pathways of gene carriers: Optimization and pitfalls. Mol. Ther. 2010, 18, 561–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Sayed, A.; Harashima, H. Endocytosis of gene delivery vectors: From clathrin-dependent to lipid raft-mediated endocytosis. Mol. Ther. 2013, 21, 1118–1130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farber, D.L. Form and function for T cells in health and disease. Nat. Rev. Immunol. 2020, 20, 83–84. [Google Scholar] [CrossRef] [PubMed]
- Stewart, M.P.; Langer, R.; Jensen, K.F. Intracellular Delivery by Membrane Disruption: Mechanisms, Strategies, and Concepts. Chem. Rev. 2018, 118, 7409–7531. [Google Scholar] [CrossRef] [PubMed]
- Pelchen-Matthews, A.; Armes, J.E.; Griffiths, G.; Marsh, M. Differential endocytosis of CD4 in lymphocytic and nonlymphocytic cells. J. Exp. Med. 1991, 173, 575–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cevaal, P.M.; Ali, A.; Czuba-Wojnilowicz, E.; Symons, J.; Lewin, S.R.; Cortez-Jugo, C.; Caruso, F. In Vivo T Cell-Targeting Nanoparticle Drug Delivery Systems: Considerations for Rational Design. ACS Nano 2021, 15, 3736–3753. [Google Scholar] [CrossRef]
- Guidotti, G.; Brambilla, L.; Rossi, D. Cell-Penetrating Peptides: From Basic Research to Clinics. Trends Pharm. Sci. 2017, 38, 406–424. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Park, H.J.; Lim, S.; Koo, J.H.; Lee, H.G.; Choi, J.O.; Oh, J.H.; Ha, S.J.; Kang, M.J.; Lee, C.M.; et al. Regulation of chitinase-3-like-1 in T cell elicits Th1 and cytotoxic responses to inhibit lung metastasis. Nat. Commun. 2018, 9, 503. [Google Scholar] [CrossRef]
- Lee, H.G.; Kim, L.K.; Choi, J.M. NFAT-Specific Inhibition by dNP2-VIVITAmeliorates Autoimmune Encephalomyelitisby Regulation of Th1 and Th17. Mol. Ther. Methods Clin. Dev. 2020, 16, 32–41. [Google Scholar] [CrossRef] [Green Version]
- Koo, J.H.; Kim, S.H.; Jeon, S.H.; Kang, M.J.; Choi, J.M. Macrophage-preferable delivery of the leucine-rich repeat domain of NLRX1 ameliorates lethal sepsis by regulating NF-kappaB and inflammasome signaling activation. Biomaterials 2021, 274, 120845. [Google Scholar] [CrossRef]
- Simon, M.J.; Gao, S.; Kang, W.H.; Banta, S.; Morrison, B., 3rd. TAT-mediated intracellular protein delivery to primary brain cells is dependent on glycosaminoglycan expression. Biotechnol. Bioeng. 2009, 104, 10–19. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, D.J.; Kim, D.T.; Steinman, L.; Fathman, C.G.; Rothbard, J.B. Polyarginine enters cells more efficiently than other polycationic homopolymers. J. Pept. Res. 2000, 56, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Tunnemann, G.; Ter-Avetisyan, G.; Martin, R.M.; Stockl, M.; Herrmann, A.; Cardoso, M.C. Live-cell analysis of cell penetration ability and toxicity of oligo-arginines. J. Pept. Sci. 2008, 14, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Koo, J.H.; Yoon, H.; Kim, W.J.; Lim, S.; Park, H.J.; Choi, J.M. Cell membrane penetrating function of the nuclear localization sequence in human cytokine IL-1alpha. Mol. Biol. Rep. 2014, 41, 8117–8126. [Google Scholar] [CrossRef]
- Koo, J.H.; Yoon, H.; Kim, W.J.; Cha, D.; Choi, J.M. Cell-Penetrating Function of the Poly(ADP-Ribose) (PAR)-Binding Motif Derived from the PAR-Dependent E3 Ubiquitin Ligase Iduna. Int. J. Mol. Sci. 2018, 19, 779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.M.; Shin, J.H.; Sohn, M.H.; Harding, M.J.; Park, J.H.; Tobiasova, Z.; Kim, D.Y.; Maher, S.E.; Chae, W.J.; Park, S.H.; et al. Cell-permeable Foxp3 protein alleviates autoimmune disease associated with inflammatory bowel disease and allergic airway inflammation. Proc. Natl. Acad. Sci. USA 2010, 107, 18575–18580. [Google Scholar] [CrossRef] [Green Version]
- Okuda, A.; Tahara, S.; Hirose, H.; Takeuchi, T.; Nakase, I.; Ono, A.; Takehashi, M.; Tanaka, S.; Futaki, S. Oligoarginine-Bearing Tandem Repeat Penetration-Accelerating Sequence Delivers Protein to Cytosol via Caveolae-Mediated Endocytosis. Biomacromolecules 2019, 20, 1849–1859. [Google Scholar] [CrossRef]
- Mitchell, M.J.; Billingsley, M.M.; Haley, R.M.; Wechsler, M.E.; Peppas, N.A.; Langer, R. Engineering precision nanoparticles for drug delivery. Nat. Rev. Drug Discov. 2021, 20, 101–124. [Google Scholar] [CrossRef]
- Xie, J.; Shen, Z.; Anraku, Y.; Kataoka, K.; Chen, X. Nanomaterial-based blood-brain-barrier (BBB) crossing strategies. Biomaterials 2019, 224, 119491. [Google Scholar] [CrossRef] [PubMed]
- Schwarze, S.R.; Ho, A.; Vocero-Akbani, A.; Dowdy, S.F. In vivo protein transduction: Delivery of a biologically active protein into the mouse. Science 1999, 285, 1569–1572. [Google Scholar] [CrossRef]
- Muttenthaler, M.; King, G.F.; Adams, D.J.; Alewood, P.F. Trends in peptide drug discovery. Nat. Rev. Drug Discov. 2021, 20, 309–325. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, W.-J.; Kim, G.-R.; Cho, H.-J.; Choi, J.-M. The Cysteine-Containing Cell-Penetrating Peptide AP Enables Efficient Macromolecule Delivery to T Cells and Controls Autoimmune Encephalomyelitis. Pharmaceutics 2021, 13, 1134. https://doi.org/10.3390/pharmaceutics13081134
Kim W-J, Kim G-R, Cho H-J, Choi J-M. The Cysteine-Containing Cell-Penetrating Peptide AP Enables Efficient Macromolecule Delivery to T Cells and Controls Autoimmune Encephalomyelitis. Pharmaceutics. 2021; 13(8):1134. https://doi.org/10.3390/pharmaceutics13081134
Chicago/Turabian StyleKim, Won-Ju, Gil-Ran Kim, Hyun-Jung Cho, and Je-Min Choi. 2021. "The Cysteine-Containing Cell-Penetrating Peptide AP Enables Efficient Macromolecule Delivery to T Cells and Controls Autoimmune Encephalomyelitis" Pharmaceutics 13, no. 8: 1134. https://doi.org/10.3390/pharmaceutics13081134
APA StyleKim, W.-J., Kim, G.-R., Cho, H.-J., & Choi, J.-M. (2021). The Cysteine-Containing Cell-Penetrating Peptide AP Enables Efficient Macromolecule Delivery to T Cells and Controls Autoimmune Encephalomyelitis. Pharmaceutics, 13(8), 1134. https://doi.org/10.3390/pharmaceutics13081134