
Focus on the Lymphatic Route to Optimize Drug Delivery in Cardiovascular Medicine

 ,
,
Abstract

1. Introduction
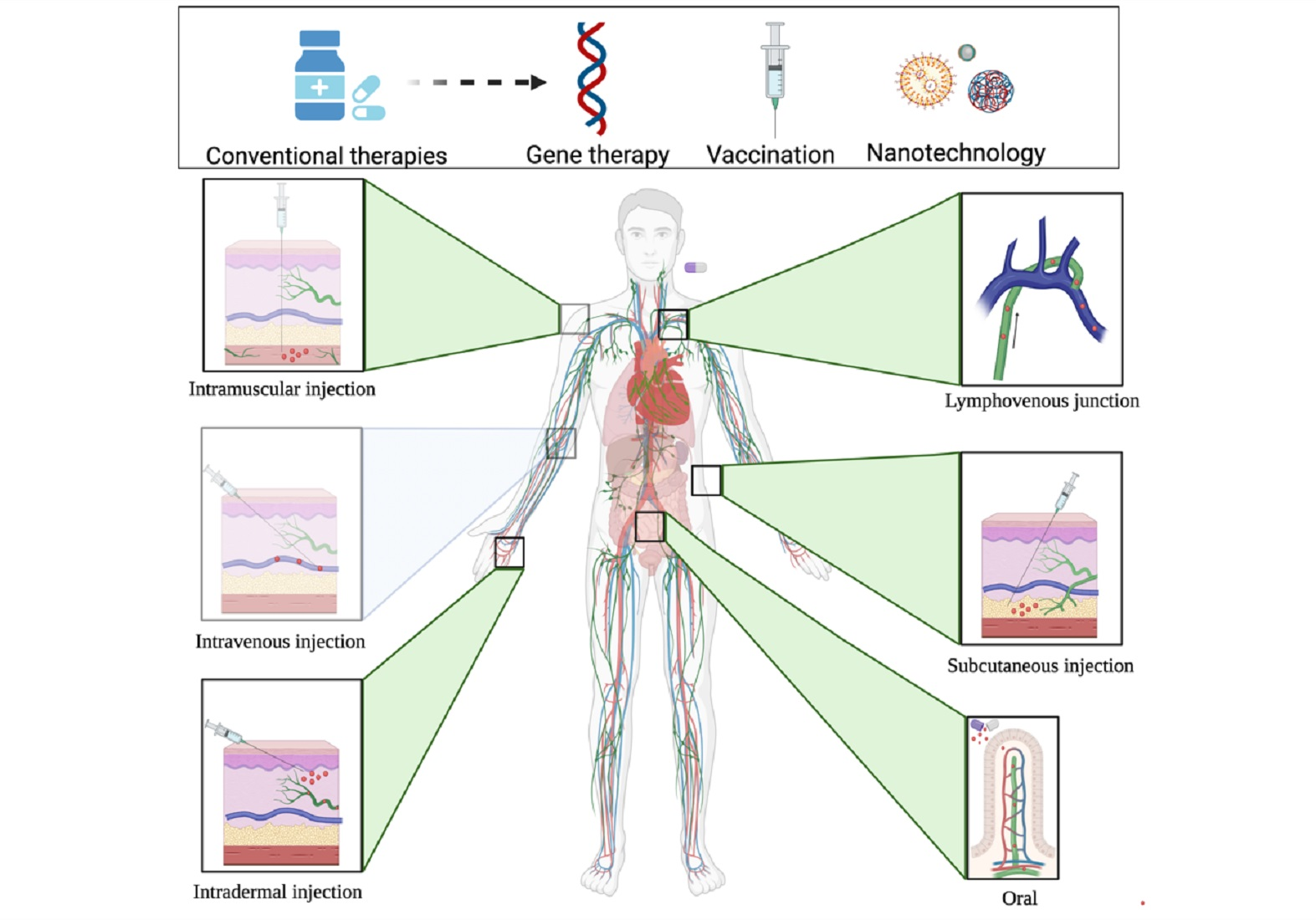
2. Conventional and Novel Therapies to Treat CVD
3. Treating CVD through Various Administration Routes
3.1. Oral Administration
- Diabetes
- Hypertension
- Hypercholesterolemia and hyperlipidemia

{kind=link}
| Condition | Intervention and Identifier | Target | Dose and Outcome |
|---|---|---|---|
| Diabetes | Metformin | From 500 to 850 mg, 2–3 times a day, during the meal [58] | |
| Diabetes | Sulfonylureas Meglitinide | Dosage is very different from one class of medication to another [59] | |
| Diabetes | Acarbose, Miglitol Voglibose | Carbohydrate digesting enzymes in the brush border | 50 mg three times daily (up to 100 mg) [60] |
| Diabetes | Rosiglitazone Pioglitazone | PPAR-α | Rosiglitazone: 4 mg per day (up to 8 mg) Pioglitazone: 15–30 mg per day [61] |
| Diabetes | Sitaglipin Vildaglipin Saxaglipin Linaglipin Aloglipin | DPP4 | 2.5–100 mg once daily depending on the inhibitor used [62] |
| Diabetes | Dapagliflozin Canagliflozin Empagliflozin | SGLTP2 | Dapagliflozin: 2.5–10 mg daily Canagliflozin: 100–300 mg Empagliflozin: 5–25 mg daily [63] |
| Diabetes | AG019 (NCT03751007) or in combination with the anti-CD3 monoclonal antibody teplizumab | 2 or 6 capsules per day for 8 weeks (repeated dose) or for one day (single dose) | |
| Diabetes | Insulin nanocarriers | Protection of insulin from enzymatic degradation Enhancement of stability, intestinal permeability, and bioavailability [35] | |
| Diabetes | Electrostatically-complexed insulin with partially uncapped cationic liposomes | Improved insulin pharmacokinetic profile [64] | |
| Diabetes | Insulin-loaded PLGA | Improved bioavailability and sustained hypoglycemic effect [65] | |
| Diabetes | Exenatide combined to phase-changeable nanoemulsion with medium-chain fatty acid | Enhancement of intestinal absorption and lymphatic transport [66] | |
| HTN | Prazosine Terazosine Doxazosine | Alpha-adrenergic receptor | Prazosine: 3–7.5 mg per day in two doses Terazosine: 1–9 mg per day in the evening at bedtime Doxazosine: 4 mg per day [71] |
| HTN | Clonidine Methyldopa | Alpha-adrenergic receptor (agonists) | Clonidine: 0.1 mg twice daily [72] Methydopa: 250 mg two to three times daily [73] |
| HTN | Carvedilol into nanoemulsion | Beta-adrenergic receptors | Significant improvement in its absorption, permeability, and bioavailability [88,89] |
| HTN | Valsartan, Ramipril and Amlodipine into nanoemulsion | Enhanced solubility, oral bioavailability, and pharmacological outcome [90] | |
| HTN | Felodipine-loaded PLGA nanoparticles | Calcium-channel | Sustained drug release both in vitro and ex vivo [93] |
| MI HF HTN Arrhythmia | ß-blocker | Beta-adrenergic receptors | Acebutol: 200 mg twice daily [74] |
| MI HF HTN | Conversion enzyme inhibitors | Conversion enzyme | Captopril: 100 mg per day [75] |
| MI HF HTN | Valsartan Losartan | Angiotensin II | 20 mg twice a day, up to 160 mg [76] |
| HF HTN | Hydrochlorothiazide Bumetanide | Angiotensin/neprilysin receptor | 49 mg/51 mg twice daily and doubled after 2–4 weeks [77] |
| HF HTN | Sacubitril Valsartan | Calcium channel | 5–10 mg daily [78] 60 mg three times daily [79] |
| HTN Arrhythmia | Amlodipine Diltiazem | Calcium channel | 5–10 mg daily [78] 60 mg three times daily [79] |
| HF | Ivabradine | Bradycardic 5–7.5 mg twice a day [80] | |
| HF MI | Eplerenone Spironolactone | Aldosterone | 50 mg once a day [81] and 12.5–25 mg with each intake [82] |
| HF Arrhythmia | Digoxin | 0.25 mg once daily [83] | |
| HF MI HCL | Statin | HMG-CoA | 10 mg once daily [84] |
| MI | Aspirin | Platelets | 325 mg, then 81 mg per day [85] |
| MI | Clopidogrel Prasugrel Ticagrelor | Platelets | 300 mg, then 75 mg daily with aspirin 60 mg, then 10 mg daily 180 mg, then 90 mg twice a day [86,87] |
| HCL | Ezetimibe | Intestinal cholesterol absorption | 10 mg once daily [99] |
| HLD | Tricor Triglide | Fenofibrates 100–300 mg per day [100] | |
| HCL HLD | Atorvastatin formulated into ethylcellulose nanoparticles | Enhanced atorvastatin’s lymphatic absorption and oral bioavailability [101] | |
| HCL HLD | Atorvastatin formulated into nanocrystals prepared with poloxamer 188 | Improved atorvastatin’s gastric solubility and bioavailability [102] Reduced circulating cholesterol, TG and LDL | |
| HCL HLD | Atorvastatin formulated into polycaprolactone nanoparticles | Enhanced atorvastatin’s bioavailability [103] | |
| HCL HLD | Nanostructured lipid carriers | Enhanced atorvastatin bioavailability by 2.1 fold compared to the commercial product: lipitor® Reduced the serum level of cholesterol, TG and LDL [104] | |
| HCL HLD | Nanoemulsion | Increased the bioavailability of atorvastatin compared to the commercial tablet ozovasTM [105] | |
| HCL HLD | Simvastatin Rosuvastatin Fluvastatin Fibrates Ezetimibe lipid-based nanoparticles | Improved bioavailability via lymphatic uptake [106,107,108,109,110,111,112,113,114] |
3.2. Subcutaneous Injection
| Condition | Intervention and Identifier | Therapy | Target | Stage and Status | Dose and Outcome |
|---|---|---|---|---|---|
| Diabetes | Insulin | Different types of insulin At least 3 injections per day Dosage adapted to the patient [116] | |||
| Diabetes | Exenatide Lixisenatide Liraglutide Exenatide LAR Albiglutide Dulaglutide | GLP-1 analogues [117] Exenatide: 5–10 µg twice a day Lixisenatide: 10–20 µg once daily Liraglutide: 0.6–1.8 mg once daily Exenatide LAR: 2 mg once a week Albiglutide: 30–50 mg once a week Dulaglutide: 0.75–1.5 mg once a week | |||
| Diabetes | Vaccine formed of virus-like particles coupled to IAPP | Against the insoluble IAPP- derived amyloid aggregates | Three doses—10 µg Strong immune response against these aggregates and restored insulin production Diminished the amyloid deposits in the pancreatic islets, reduced the level of the pro-inflammatory cytokine IL-1β, and reprieved the onset of amyloid-induced hyperglycemia [118] | ||
| Diabetes | IL-1β epitope peptide | Against IL-1β | Three doses—50 µg Enhancement glucose tolerance, improved insulin sensitivity, restored β-cell mass, reduced β-cell apoptosis, and enhanced β-cell proliferation, as well as downregulation of IL-1β expression and inhibition of the inflammatory activity [119,120] | ||
| Diabetes | hIL1bQb vaccine (NCT00924105) | Against IL-1β | Six doses—300 µg Mediated a dose-dependent IL-1β-specific antibody response More studies are required to precisely investigate the clinical efficiency of this vaccine [121] | ||
| Diabetes | Neutralizing antibodies against DPP4 | The GLP-1 and GIP inhibitor, DPP4 | Five doses—2–20 µg Increased pancreatic and plasma insulin level and improved postprandial blood glucose level [122] | ||
| HTN | hR32 vaccine | Renin-derived peptide | Five doses—500 µg Reduced systolic blood pressure by 15 mmHg [123] | ||
| HTN | Angiotensin I vaccine (PMD3117) | Three or four doses—100 µg The vaccine failed to reduce the blood pressure [124] | |||
| HTN | AngI-R vaccine | Modifiedendogenous angiotensin I peptide | Four doses—50 µg 15 mmHg reduction in systolic blood pressure and reduced angiotensin I/II level [125] | ||
| HTN | ATRQβ-001 | Angiotensin II type I receptors | Two doses—100 µg Protective role against target organ damage induced by hypertension [126] | ||
| HTN | ATR12181 vaccine | Angiotensin II type I receptors | Nine doses—0.1 mg Attenuated the development of hemodynamic alterations of hypertension [127] | ||
| HTN | CYT006-AngQb vaccine | Against angiotensin II | 100 or 300 µg Reduction in blood pressure and reduced ambulatory daytime blood pressure [128] | ||
| HF HTN | Ang II-KLH vaccine | Angiotensin II | Three doses—5 µg Suppressed post-MI cardiac remodeling and improved cardiac function [129] | ||
| MI | Celecoxib loaded in nanoparticles | Promoted vascularization in the ischemic myocardium and delayed HF progression [130] | |||
| MI | Chitosan-hyaluronic acid based hydrogel containing deferoxamine-PLGA nanoparticles | Persistent neovascularization in mice [131] | |||
| HCL | Alirocumab Evolocumab | PCSK9 | One dose every two weeks [132,133] | ||
| HCL | Inclisiran | PCSK9 | Two doses per year [134] | ||
| HoFH HeFH severe HCL | Mipomersen (NCT00607373) (NCT00706849) (NCT00770146) (NCT00794664) | ASO | ApoB | Approved | 200 mg once/week. Phase III: reduction in LDL-C [135] |
| ASCVD HCL HeFH | Inclisiran (NCT03399370) (NCT03400800) (NCT03397121) | siRNA | PCSK9 | Approved | 284 mg inclisiran, injected on day 1, day 90 and then twice/year Phase III: reduction in LDL-C level [134,136] |
| FCS | Volanesorsen (NCT02211209) | ASO | ApoC3 | Approved | 300 mg once/week Phase III: reduction in mean plasma APOC3 and TG level [137] |
| Elevated LP(a) | ISIS-APO(a)Rx (NCT02160899) | ASO | APO(a) | Phase II (Complete) | Multiple escalating (100–300 mg) doses, injected on a weekly interval for 4 weeks each Phase I/II: reduction in plasma Lp(a) concentration [138] |
| Elevated LP(a) CVD | AKCEA-APO(a)-LRx (NCT03070782) (NCT02414594) (NCT04023552) | GalNAc3 conjugated-ASO | APO(a) | Phase III (Recruiting) | 80 mg administered monthly Phase I/II: reduction in plasma Lp(a) [138] |
| HTG CVD FCS | AKCEA-APOCIII-LRx (NCT02900027) (NCT03385239) (NCT04568434) | GalNAc3 conjugated-ASO | APOC3 | Phase III (Recruiting) | Multiple dosing injected as once/4 weeks for up to 49 weeks Phase II: reduction in ApoC3 and TG levels [139] |
| HTG FH HLP | Vupanorsen (NCT02709850) (NCT04459767) (NCT04516291) | ASO | ANGPTL3 | Phase IIb (Active, Not recruiting) | Multiple escalating dosing (60–160 mg, once/2 or 4 weeks) Phase I: reduction in TG and LDL-C levels [140] |
| HCL | Neutralizing antibodies against PCSK9 | PCSK9 | Three doses—5–50 µg Long-lasting reduction in the level of total cholesterol, VLDL and chylomicron [141] | ||
| HCL | AT04A | PCSK9 | Five doses Strong and persistent anti-PCSK9 antibody production, reduced plasma cholesterol level, attenuated progression of atherosclerosis and reduced vascular and systemic inflammation [142] | ||
| HCL | AT04A | PCSK9 | Four doses—15 µg and 75 µg Reduced serum LDL-C level and elevated anti-PCSK9 antibody titer [143] | ||
HCL | A peptide representing the mouse ANGPTL3 | Angiopoietin-like proteins 3 (ANGPTL3) | Three doses—5 µg Reduced steady-state plasma TGs and promoted LPL activity |
- Diabetes
- Hypertension
- Myocardial infarction
- Hypercholesterolemia and hyperlipidemia
3.3. Intradermal Injection
- Diabetes
| Condition | Intervention and Identifier | Target | Dose and Outcome |
|---|---|---|---|
| Diabetes | Proinsulin peptide vaccine C19-A3 | CD4 T cells | Three equal doses—10–100 µg Vaccine was well tolerated [177] |
| Diabetes | C19-A3 (NCT02837094) | CD4 T cells | Three doses—10 ug In vitro and ex vivo studies of in human skin reported rapid diffusion of the injected particles through the skin layers and preferential uptake by Langerhans cells in the epidermis, which have a primary role in the tolerance mechanism [178] |
| Diabetes | PIpepTolDC vaccine (NCT04590872) | Tolerogenic DC Vaccine | One dose and another after 28 days No results yet, but, it is believed to be able to produce proinsulin-specific Treg [179] |
3.4. Intramuscular Injection
| Condition | Intervention and Identifier | Target | Dose and Outcome |
|---|---|---|---|
| Diabetes | Preproinsulin-encoding plasmid DNA | Pancreatic islets | 40% higher survival rate as compared to the control group [185] |
| HTN | CoVaccine HT (NCT00702221) | Against angiotensin II | Three doses Terminated in 2016 due to dose-limiting adverse effects |
| HTN | AGMG0201 vaccine | Against angiotensin II | High or low dose (0.2 mg plasmid DNA and 0.5 or 0.25 mg Ang II-KLH conjugate) Ongoing |
| ACS HF CVD | Inactivated influenza vaccine | Less frequent hospitalization from ACS, hospitalization from HF and stroke [186] | |
| MI | Influenza vaccine | Risk of cardiovascular-related death was significantly lower [187] | |
| CVD MI | Pneumococcal vaccines | Reduced incidence of cardiovascular events and mortality Reduced risk of MI in the elderly [188] | |
| MI HF Stroke | Influenza vaccine (NCT02831608) | The primary endpoints: death, new MI and stent thrombosis Secondary endpoints: patients with hospitalization for HF |
- Diabetes
- Heart failure
- Hypercholesterolemia and hyperlipidemia
3.5. Intramyocardial Injection
- Myocardial infarction
| Condition | Intervention and Identifier | Therapy | Target | Stage and Status | Dose and Outcome |
|---|---|---|---|---|---|
| HF | Ad5.hAC6 (NCT007) | Ad5 | AC6 | Phase I/II (Completed) | Single administration of escalating doses (3.2 × 109 vp to 1012 vp) Phase II: Reduced HF admission rate. Enhanced left ventricular function beyond the optimal HF therapy following a single administration [209] |
| HF | Ad5.hAC6 (NCT03360448) | Ad5 | AC6 | Phase III (withdrawn) | Phase III: withdrawn for re-evaluation |
| HF | MYDICAR (NCT00454818) | AAV1 | SERCA2a | Phase I/II (Completed) | Single administration of escalating doses (1.4 × 1011–1 × 1013 DRP of AAV1/SERCA2a) Phase I/II (CUPID): high-dose treatment resulted in increased time and reduced frequency of cardiovascular events within a year and reduced cardiovascular hospitalizations [210] |
| HF | MYDICAR (NCT01643330) | AAV1 | SERCA2a | Phase IIb (completed) | Single infusion of 1 × 1013 DRP of AAV1/SERCA2a Phase IIb (CUPID-2b): no improvement was observed at the tested dose in patients with HF during the follow-up period [201] |
| HF | MYDICAR (NCT01966887) | AAVI | SERCA2a | Phase II (Terminated) | 1 × 1013 DRP of AAV1/SERCA2a as a single intracoronary infusion Phase II: no improvement observed in the ventricular remodeling.The study terminated driven by the CUPID-2 trial neutral outcome [211] |
| HF | SRD-001 (NCT04703842) | AAVI | SERCA2a | Phase I/II (Active, not recruiting) | Single administration of 3 × 1013 vg CUPID-3: aims to investigate the safety and efficacy of SRD-001 in anti-AAV1 neutralizing antibody-negative subjects with HFrEF |
| HF CVD | INXN-4001 (NCT03409627) | Non-viral, triple effector plasmid | SDF-1α, S100A1, VEGF-165 | Phase I (Completed) | Single 80 mg dose, given in 40 mL or 80 mL at a rate of 20 mL/min Phase I: an improvement in the quality of life in 50% of patients was reported [212] |
| HF | ACRX-100 (NCT01082094) | Plasmid DNA | SDF-1 | Phase I (Completed) | Single escalating doses, injected at multiple sites Preclinical studies: enhanced vasculogenesis and improved cardiac function reported with all doses [213] |
| HF | JVS-100 (NCT01643590) | Plasmid DNA | SDF-1 | Phase II (Completed) | Single injection of escalating doses (15 and 30 mg) Phase II (STOP-HF): JVS-100 showed potential to improve cardiac function through reducing left ventricular remodeling and improving ejection fraction [214] |
| HF | JVS-100 (NCT01961726) | Plasmid DNA | SDF-1 | Phase I/II (Unknown) | Single injection of escalating doses (30 and 45 mg) Phase I (RETRO-HF): JVS-100 showed promising signs of clinical efficacy [215] |
| HF | AZD8601 (NCT02935712) (NCT03370887) | mRNA | VEGF-A165 | Phase IIa (Active, not recruiting) | Single injection of escalating doses (3 mg and 30 mg) Preclinical studies: promoted angiogenesis, improved cardiac function and enhanced survival were reported [216] Phase I: ID injection of AZD8601 was well tolerated and enhanced the basal skin blood flow [217] |
| HF | NAN-101 (NCT04179643) | AAV | I-1c | Phase I (Recruiting) | Single escalating doses (3 × 1013 vg–3 × 1014 vg) of NAN-101 Preclinical studies: enhancement in left ventricular ejection fraction and improved cardiac performance [218] |
| AMI IHD | VM202RY (NCT01422772) (NCT03404024) | DNA plasmid | HGF-X7 | Phase II (Recruiting) | Single escalating (0.5–3 mg) doses, administered into multiple sites Phase I: improved myocardial function and wall thickness [219,220] |
| MI Angina pectoris | AdVEGF-D (NCT01002430) | AV | VEGF-D | Phase I/IIa (Completed) | Single escalating (1 × 109–1 × 1011 Vpu) doses, injected into multiple sites in the endocardium Phase 1/IIa: AdVEGF-D improved myocardial perfusion reserve in the injected region [220] |
| MI | Ad-HGF (NCT02844283) | AV | HGF | Phase I/II (Unknown) | Single dose Preclinical studies: Ad-HGF preserved cardiac function, reduced infarct size, and improved post-MI cardiac remodeling [221]; fractional repeated dosing significantly improved cardiac function compared with single injection [222] |
| MI | L-type Ca2+ channels’ AID peptide and antioxidant molecule (curcumin) in poly nanoparticles | Reduced the elevated level of ROS and the intracellular Ca2+ [223] | |||
| LPLD | Alipogene tiparvovec (NCT00891306) | AAV | LPL | Approved | 1 × 1012 GC/kg Phase II/III: reduction in mean total plasma and chylomicron TG level [224] |
3.6. Intravenous Injection
| Condition | Intervention and Identifier | Therapy | Target | Stage and Status | Dose and Outcome |
|---|---|---|---|---|---|
| HTN | NO-releasing nanoparticles | Reduction in the mean arterial blood pressure [235] | |||
| HF Arrhythmia | Digoxin | Dose: 0.25 mg once daily [83] | |||
| MI HF HTN Arrhythmia | ß-blocker | Beta-adrenergic receptors | Acebutol: 200 mg twice daily [74] | ||
| HF | Mesoporous silicon vector (Nanoconstruct) | Able to internalize, accumulate, and traffic within the cardiomyocytes [236] | |||
| HF | Combination of biocompatible magnetic nanoparticles and low-frequency magnetic stimulation | Cardio-myocytes | Managed the drug release by controlling the applied frequencies [237] | ||
| HF | S100A1-loaded nanoparticles, decorated with N-acetylglucosamine | Regulated Ca2+ release and restored contractile function in the isolated failing cardiomyocytes [238] | |||
| HF | Biodegradable nanoparticles conjugated with myocyte-targeting peptide and PDT-enabling photosensitizer | PDT | Cardio-myocytes | Induced cell-specific death upon application of laser light, leaving adjacent and surrounding cells completely intact [239] | |
| MI | Unfractionated heparin | Anticoagulant 60 IU/kg for initial bolus 12 IU/kg/h for maintenance [240] | |||
| MI | Aspirin | Platelets | 325 mg, then 81 mg per day [85] | ||
| MI | Human recombinant VEGF-165 | Significant improvement in the infarcted zone perfusion and cardiac function for up to six weeks post-MI [241]. | |||
| MI | Nanoparticles containing siRNA | Anti-inflammatory effect in the infarcted heart and reduction of the post-MI heart failure [242] | |||
| MI | Magnetic nanoparticles-loaded cells | Robust improvement in the left ventricular and cardiac function [243] | |||
| MI | Insulin-like growth factor electrostatically-complexed with PLGA nanoparticles | Higher incidence in preventing cardiomyocytes’ apoptosis, reducing infarct size, and enhancing left ventricular function [244] | |||
| MI | Pitavastatin in PLGA nanoparticles | Cardioprotective effect against ischemia-reperfusion injury [245] | |||
| HoFH | AAV8.TBG.HldlR (NCT02651675) | AAV | hLDLR | Phase I/II (Completed) | Single dose Preclinical studies: reduction in total cholesterol [246,247] |
| Elevated LDL-C | ALN-PCS02 (NCT01437059) | siRNA | PCSK9 | Phase I (Completed) | Single escalating (15 and 400 μg/kg) doses Phase I: reduction in the level of circulating PCSK9 protein and LDL-C [248] |
- Diabetes
- Hypertension
- Heart failure
- Myocardial infarction
3.7. Intraperitoneal Injection
- Diabetes
- Myocardial infarction
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kaptoge, S.; Pennells, L.; De Bacquer, D.; Cooney, M.T.; Kavousi, M.; Stevens, G.; Riley, L.M.; Savin, S.; Khan, T.; Altay, S.; et al. World Health Organization cardiovascular disease risk charts: Revised models to estimate risk in 21 global regions. Lancet Glob. Health 2019, 7, e1332–e1345. [Google Scholar] [CrossRef]
- Virani, S.S.; Alonso, A.; Aparicio, H.J.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Cheng, S.; Delling, F.N. Heart disease and stroke statistics—2021 update: A report from the American Heart Association. Circulation 2021, 143, e254–e743. [Google Scholar] [CrossRef]
- Tiwari, G.; Tiwari, R.; Sriwastawa, B.; Bhati, L.; Pandey, S.; Pandey, P.; Bannerjee, S.K. Drug delivery systems: An updated review. Int. J. Pharm. Investig. 2012, 2, 2. [Google Scholar] [CrossRef] [PubMed]
- Ryan, G.M.; Kaminskas, L.M.; Porter, C.J. Nano-chemotherapeutics: Maximising lymphatic drug exposure to improve the treatment of lymph-metastatic cancers. J. Control. Release 2014, 193, 241–256. [Google Scholar] [CrossRef]
- Maisel, K.; Sasso, M.S.; Potin, L.; Swartz, M.A. Exploiting lymphatic vessels for immunomodulation: Rationale, opportunities, and challenges. Adv. Drug Deliv. Rev. 2017, 114, 43–59. [Google Scholar] [CrossRef] [PubMed]
- Pal, I.; Ramsey, J.D. The role of the lymphatic system in vaccine trafficking and immune response. Adv. Drug Deliv. Rev. 2011, 63, 909–922. [Google Scholar] [CrossRef] [PubMed]
- Sleeman, J.P. The relationship between tumors and the lymphatics: What more is there to know? Lymphology 2006, 39, 62–68. [Google Scholar]
- Porter, C.J.; Charman, S.A. Lymphatic transport of proteins after subcutaneous administration. J. Pharm. Sci. 2000, 89, 297–310. [Google Scholar] [CrossRef]
- Zhang, X.-Y.; Lu, W.-Y. Recent advances in lymphatic targeted drug delivery system for tumor metastasis. Cancer Biol. Med. 2014, 11, 247–254. [Google Scholar] [CrossRef]
- Yáñez, J.A.; Wang, S.W.; Knemeyer, I.W.; Wirth, M.A.; Alton, K.B. Intestinal lymphatic transport for drug delivery. Adv. Drug Deliv. Rev. 2011, 63, 923–942. [Google Scholar] [CrossRef] [PubMed]
- Asellius, G. De Lactibus Sive Lacteis Venis; JB Bidellium: Milan, Italy, 1627. [Google Scholar]
- Cueni, L.N.; Detmar, M. The lymphatic system in health and disease. Lymphat. Res. Biol. 2008, 6, 109–122. [Google Scholar] [CrossRef]
- Milasan, A.; Farhat, M.; Martel, C. Extracellular Vesicles as Potential Prognostic Markers of Lymphatic Dysfunction. Front. Physiol. 2020, 11, 476. [Google Scholar] [CrossRef]
- Lemole, G.M. The role of lymphstasis in atherogenesis. Ann. Thorac. Surg. 1981, 31, 290–293. [Google Scholar] [CrossRef]
- Martel, C.; Li, W.; Fulp, B.; Platt, A.M.; Gautier, E.L.; Westerterp, M.; Bittman, R.; Tall, A.R.; Chen, S.-H.; Thomas, M.J. Lymphatic vasculature mediates macrophage reverse cholesterol transport in mice. J. Clin. Investig. 2013, 123, 1571–1579. [Google Scholar] [CrossRef]
- Milasan, A.; Smaani, A.; Martel, C. Early rescue of lymphatic function limits atherosclerosis progression in Ldlr−/− mice. Atherosclerosis 2019, 283, 106–119. [Google Scholar] [CrossRef]
- Yeo, K.P.; Lim, H.Y.; Thiam, C.H.; Azhar, S.H.; Tan, C.; Tang, Y.; See, W.Q.; Koh, X.H.; Zhao, M.H.; Phua, M.L.; et al. Efficient aortic lymphatic drainage is necessary for atherosclerosis regression induced by ezetimibe. Sci. Adv. 2020, 6, eabc2697. [Google Scholar] [CrossRef] [PubMed]
- Singla, B.; Lin, H.P.; Chen, A.; Ahn, W.; Ghoshal, P.; Cherian-Shaw, M.; White, J.; Stansfield, B.K.; Csányi, G. Role of R-spondin 2 in arterial lymphangiogenesis and atherosclerosis. Cardiovasc. Res. 2021, 117, 1489–1509. [Google Scholar] [CrossRef] [PubMed]
- Milasan, A.; Jean, G.; Dallaire, F.; Tardif, J.C.; Merhi, Y.; Sorci-Thomas, M.; Martel, C. Apolipoprotein A-I Modulates Atherosclerosis Through Lymphatic Vessel-Dependent Mechanisms in Mice. J. Am. Heart Assoc. 2017, 6, e006892. [Google Scholar] [CrossRef] [PubMed]
- Milasan, A.; Ledoux, J.; Martel, C. Lymphatic network in atherosclerosis: The underestimated path. Future Sci. OA 2015, 1, fso 61. [Google Scholar] [CrossRef]
- Milasan, A.; Tessandier, N.; Tan, S.; Brisson, A.; Boilard, E.; Martel, C. Extracellular vesicles are present in mouse lymph and their level differs in atherosclerosis. J. Extracell. Vesicles 2016, 5, 31427. [Google Scholar] [CrossRef]
- Milasan, A.; Dallaire, F.; Mayer, G.; Martel, C. Effects of LDL Receptor Modulation on Lymphatic Function. Sci. Rep. 2016, 6, 27862. [Google Scholar] [CrossRef]
- Libby, P.; Ridker, P.M.; Hansson, G.K. Progress and challenges in translating the biology of atherosclerosis. Nature 2011, 473, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Henri, O.; Pouehe, C.; Houssari, M.; Galas, L.; Nicol, L.; Edwards-Lévy, F.; Henry, J.-P.; Dumesnil, A.; Boukhalfa, I.; Banquet, S.; et al. Selective Stimulation of Cardiac Lymphangiogenesis Reduces Myocardial Edema and Fibrosis Leading to Improved Cardiac Function Following Myocardial Infarction. Circulation 2016, 133, 1484–1497. [Google Scholar] [CrossRef] [PubMed]
- Klotz, L.; Norman, S.; Vieira, J.M.; Masters, M.; Rohling, M.; Dubé, K.N.; Bollini, S.; Matsuzaki, F.; Carr, C.A.; Riley, P.R. Cardiac lymphatics are heterogeneous in origin and respond to injury. Nature 2015, 522, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Vieira, J.M.; Norman, S.; Del Campo, C.V.; Cahill, T.J.; Barnette, D.N.; Gunadasa-Rohling, M.; Johnson, L.A.; Greaves, D.R.; Carr, C.A.; Jackson, D.G. The cardiac lymphatic system stimulates resolution of inflammation following myocardial infarction. J. Clin. Investig. 2018, 128, 3402–3412. [Google Scholar] [CrossRef] [PubMed]
- Vuorio, T.; Ylä-Herttuala, E.; Laakkonen, J.P.; Laidinen, S.; Liimatainen, T.; Ylä-Herttuala, S. Downregulation of VEGFR3 signaling alters cardiac lymphatic vessel organization and leads to a higher mortality after acute myocardial infarction. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef]
- Trevaskis, N.L.; Kaminskas, L.M.; Porter, C.J.H. From sewer to saviour—targeting the lymphatic system to promote drug exposure and activity. Nat. Rev. Drug Discov. 2015, 14, 781–803. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Liang, W.; Xiao, X.; Qian, Y.J. Nanotechnology, an alternative with promising prospects and advantages for the treatment of cardiovascular diseases. Int. J. Nanomed. 2018, 13, 7349. [Google Scholar] [CrossRef]
- Wong, M.S.; Hawthorne, W.J.; Manolios, N. Gene therapy in diabetes. Self Nonself 2010, 1, 165–175. [Google Scholar] [CrossRef]
- Phillips, M.I. Gene therapy for hypertension: Sense and antisense strategies. Expert Opin. Biol. Ther. 2001, 1, 655–662. [Google Scholar] [CrossRef]
- Tromp, T.R.; Stroes, E.S.; Hovingh, G.K. Gene-based therapy in lipid management: The winding road from promise to practice. Expert Opin. Investig. Drugs 2020, 29, 483–493. [Google Scholar] [CrossRef] [PubMed]
- Kieserman, J.M.; Myers, V.D.; Dubey, P.; Cheung, J.Y.; Feldman, A.M. Current landscape of heart failure gene therapy. J. Am. Heart Assoc. 2019, 8, e012239. [Google Scholar] [CrossRef] [PubMed]
- Shimamura, M.; Nakagami, H.; Taniyama, Y.; Morishita, R. Gene therapy for peripheral arterial disease. Expert Opin. Biol. Ther. 2014, 14, 1175–1184. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Lu, Z.; Yang, J.; Zhang, L.; Li, Y.; Zhang, X. Drug Delivery System in the Treatment of Diabetes Mellitus. Front. Bioeng. Biotechnol. 2020, 8, 880. [Google Scholar] [CrossRef] [PubMed]
- Avery, O.T.; MacLeod, C.M.; McCarty, M. Studies on the chemical nature of the substance inducing transformation of pneumococcal types: Induction of transformation by a desoxyribonucleic acid fraction isolated from pneumococcus type III. J. Exp. Med. 1944, 79, 137–158. [Google Scholar] [CrossRef]
- Meyerson, S.L.; Skelly, C.L.; Curi, M.A.; Schwartz, L.B. Gene therapy for cardiovascular disease. Semin. Cardiothorac. Vasc. Anesth. 2000, 4, 289–300. [Google Scholar]
- Bulcha, J.T.; Wang, Y.; Ma, H.; Tai, P.W.; Gao, G. Viral vector platforms within the gene therapy landscape. Signal Transduct. Target. Ther. 2021, 6, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Su, C.-H.; Wu, Y.-J.; Wang, H.-H.; Yeh, H.-I. Nonviral gene therapy targeting cardiovascular system. Am. J. Physiol. Heart Circ. Physiol. 2012, 303, H629–H638. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.; Lächelt, U.; Bartek, J.; Wagner, E.; Moghimi, S.M. Polyplex Evolution: Understanding Biology, Optimizing Performance. Mol. Ther. 2017, 25, 1476–1490. [Google Scholar] [CrossRef]
- Scimia, M.C.; Gumpert, A.M.; Koch, W.J. Cardiovascular gene therapy for myocardial infarction. Expert Opin. Biol. Ther. 2014, 14, 183–195. [Google Scholar] [CrossRef]
- Cannatà, A.; Ali, H.; Sinagra, G.; Giacca, M. Gene therapy for the heart lessons learned and future perspectives. Circ. Res. 2020, 126, 1394–1414. [Google Scholar] [CrossRef]
- Nakagami, H.; Morishita, R. Recent advances in therapeutic vaccines to treat hypertension. Hypertension 2018, 72, 1031–1036. [Google Scholar] [CrossRef] [PubMed]
- Siriwardena, A.N. Increasing Evidence That Influenza Is a Trigger for Cardiovascular Disease; Oxford University Press: Oxford, UK, 2012. [Google Scholar]
- Singanayagam, A.; Elder, D.; Chalmers, J.D. Is community-acquired pneumonia an independent risk factor for cardiovascular disease? Eur. Respir. J. 2012, 39, 187–196. [Google Scholar] [CrossRef]
- Deng, Y.; Zhang, X.; Shen, H.; He, Q.; Wu, Z.; Liao, W.; Yuan, M. Application of the Nano-Drug Delivery System in Treatment of Cardiovascular Diseases. Front. Bioeng. Biotechnol. 2019, 7, 489. [Google Scholar] [CrossRef] [PubMed]
- Sezer, A.D. Application of Nanotechnology in Drug Delivery; BoD–Books on Demand: London, UK, 2014. [Google Scholar]
- Zhang, J.; Xie, Z.; Zhang, N.; Zhong, J. Nanosuspension drug delivery system: Preparation, characterization, postproduction processing, dosage form, and application. In Nanostructures for Drug Delivery; Elsevier: Amsterdam, The Netherlands, 2017; pp. 413–443. [Google Scholar]
- Fox, C.B.; Kim, J.; Le, L.V.; Nemeth, C.L.; Chirra, H.D.; Desai, T.A. Micro/nanofabricated platforms for oral drug delivery. J. Control. Release 2015, 219, 431–444. [Google Scholar] [CrossRef] [PubMed]
- Trevaskis, N.L.; McEvoy, C.L.; McIntosh, M.P.; Edwards, G.A.; Shanker, R.M.; Charman, W.N.; Porter, C.J. The role of the intestinal lymphatics in the absorption of two highly lipophilic cholesterol ester transfer protein inhibitors (CP524,515 and CP532,623). Pharm. Res. 2010, 27, 878–893. [Google Scholar] [CrossRef]
- Vinarov, Z.; Abdallah, M.; Agundez, J.A.G.; Allegaert, K.; Basit, A.W.; Braeckmans, M.; Ceulemans, J.; Corsetti, M.; Griffin, B.T.; Grimm, M.; et al. Impact of gastrointestinal tract variability on oral drug absorption and pharmacokinetics: An UNGAP review. Eur. J. Pharm. Sci. 2021, 162, 105812. [Google Scholar] [CrossRef]
- Brocks, D.R.; Davies, N.M. Lymphatic drug absorption via the enterocytes: Pharmacokinetic simulation, modeling, and considerations for optimal drug development. J. Pharm. Pharm. Sci. 2018, 21, 254s–270s. [Google Scholar] [CrossRef]
- Jenkins, P.; Howard, K.; Blackball, N.; Thomas, N.; Davis, S.; O’hagan, D.T. Microparticulate absorption from the rat intestine. J. Control. Release 1994, 29, 339–350. [Google Scholar] [CrossRef]
- Charman, W.; Stella, V.J. Estimating the maximal potential for intestinal lymphatic transport of lipophilic drug molecules. Int. J. Pharm. 1986, 34, 175–178. [Google Scholar] [CrossRef]
- Cifarelli, V.; Eichmann, A. The Intestinal Lymphatic System: Functions and Metabolic Implications. Cell. Mol. Gastroenterol. Hepatol. 2019, 7, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.J.; Newberry, R.D. Microanatomy of the intestinal lymphatic system. Ann. N. Y. Acad. Sci. 2010, 1207, E21–E28. [Google Scholar] [CrossRef]
- Baena-Díez, J.M.; Peñafiel, J.; Subirana, I.; Ramos, R.; Elosua, R.; Marín-Ibañez, A.; Guembe, M.J.; Rigo, F.; Tormo-Díaz, M.J.; Moreno-Iribas, C.; et al. Risk of Cause-Specific Death in Individuals With Diabetes: A Competing Risks Analysis. Diabetes Care 2016, 39, 1987–1995. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Rangel, E.; Inzucchi, S.E. Metformin: Clinical use in type 2 diabetes. Diabetologia 2017, 60, 1586–1593. [Google Scholar] [CrossRef]
- Sola, D.; Rossi, L.; Schianca, G.P.C.; Maffioli, P.; Bigliocca, M.; Mella, R.; Corlianò, F.; Fra, G.P.; Bartoli, E.; Derosa, G. Sulfonylureas and their use in clinical practice. Arch. Med. Sci. 2015, 11, 840–848. [Google Scholar] [CrossRef] [PubMed]
- van de Laar, F.A. Alpha-glucosidase inhibitors in the early treatment of type 2 diabetes. Vasc. Health Risk Manag. 2008, 4, 1189–1195. [Google Scholar] [CrossRef]
- Lebovitz, H.E. Thiazolidinediones: The Forgotten Diabetes Medications. Curr. Diabetes Rep. 2019, 19, 151. [Google Scholar] [CrossRef]
- Gallwitz, B. Clinical Use of DPP-4 Inhibitors. Front. Endocrinol. 2019, 10, 389. [Google Scholar] [CrossRef]
- Neuen, B.L.; Cherney, D.Z.; Jardine, M.J.; Perkovic, V. Sodium-glucose cotransporter inhibitors in type 2 diabetes: Thinking beyond glucose lowering. CMAJ 2019, 191, E1128–E1135. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.S.; Kwag, D.S.; Hwang, H.S.; Lee, E.S.; Bae, Y.H. Immense insulin intestinal uptake and lymphatic transport using bile acid conjugated partially uncapped liposome. Mol. Pharm. 2018, 15, 4756–4763. [Google Scholar] [CrossRef]
- Jain, S.; Rathi, V.V.; Jain, A.K.; Das, M.; Godugu, C. Folate-decorated PLGA nanoparticles as a rationally designed vehicle for the oral delivery of insulin. Nanomedicine 2012, 7, 1311–1337. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.Y.; Chen, K.H.; Miao, Y.B.; Chen, H.L.; Lin, K.J.; Chen, C.T.; Yeh, C.N.; Chang, Y.; Sung, H.W. Phase-Changeable Nanoemulsions for Oral Delivery of a Therapeutic Peptide: Toward Targeting the Pancreas for Antidiabetic Treatments Using Lymphatic Transport. Adv. Funct. Mater. 2019, 29, 1809015. [Google Scholar] [CrossRef]
- Harrison, G.A. Insulin in alcoholic solution by the mouth. Br. Med. J. 1923, 2, 1204. [Google Scholar] [CrossRef] [PubMed]
- Sonaje, K.; Lin, Y.-H.; Juang, J.-H.; Wey, S.-P.; Chen, C.-T.; Sung, H.-W. In vivo evaluation of safety and efficacy of self-assembled nanoparticles for oral insulin delivery. Biomaterials 2009, 30, 2329–2339. [Google Scholar] [CrossRef]
- Wu, Z.M.; Zhou, L.; Guo, X.D.; Jiang, W.; Ling, L.; Qian, Y.; Luo, K.Q.; Zhang, L.J. HP55-coated capsule containing PLGA/RS nanoparticles for oral delivery of insulin. Int. J. Pharm. 2012, 425, 1–8. [Google Scholar] [CrossRef]
- Jin, Y.; Song, Y.; Zhu, X.; Zhou, D.; Chen, C.; Zhang, Z.; Huang, Y. Goblet cell-targeting nanoparticles for oral insulin delivery and the influence of mucus on insulin transport. Biomaterials 2012, 33, 1573–1582. [Google Scholar] [CrossRef] [PubMed]
- Cohn, J.N.; Archibald, D.G.; Ziesche, S.; Franciosa, J.A.; Harston, W.E.; Tristani, F.E.; Dunkman, W.B.; Jacobs, W.; Francis, G.S.; Flohr, K.H. Effect of vasodilator therapy on mortality in chronic congestive heart failure. Results of a Veterans Administration Cooperative Study. N. Engl. J. Med. 1986, 314, 1547–1552. [Google Scholar] [CrossRef]
- MacDougall, A.I.; Addis, G.J.; MacKay, N.; Dymock, I.W.; Turpie, A.G.; Ballingall, D.L.; MacLennan, W.J.; Whiting, B.; MacArthur, J.G. Treatment of hypertension with clonidine. Br. Med. J. 1970, 3, 440–442. [Google Scholar] [CrossRef]
- Mah, G.T.; Tejani, A.M.; Musini, V.M. Methyldopa for primary hypertension. Cochrane Database Syst. Rev. 2009, 4, CD003893. [Google Scholar] [CrossRef]
- Bangalore, S.; Steg, G.; Deedwania, P.; Crowley, K.; Eagle, K.A.; Goto, S.; Ohman, E.M.; Cannon, C.P.; Smith, S.C.; Zeymer, U.; et al. β-Blocker use and clinical outcomes in stable outpatients with and without coronary artery disease. JAMA 2012, 308, 1340–1349. [Google Scholar] [CrossRef]
- Lazar, H.L. Role of angiotensin-converting enzyme inhibitors in the coronary artery bypass patient. Ann. Thorac. Surg. 2005, 79, 1081–1089. [Google Scholar] [CrossRef]
- Güleç, S. Valsartan after myocardial infarction. Anadolu Kardiyol. Derg. 2014, 14, S9–S13. [Google Scholar] [CrossRef] [PubMed]
- Hubers, S.A.; Brown, N.J. Combined Angiotensin Receptor Antagonism and Neprilysin Inhibition. Circulation 2016, 133, 1115–1124. [Google Scholar] [CrossRef] [PubMed]
- Fares, H.; DiNicolantonio, J.J.; O’Keefe, J.H.; Lavie, C.J. Amlodipine in hypertension: A first-line agent with efficacy for improving blood pressure and patient outcomes. Open Heart 2016, 3, e000473. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez Padial, L.; Barón-Esquivias, G.; Hernández Madrid, A.; Marzal Martín, D.; Pallarés-Carratalá, V.; de la Sierra, A. Clinical Experience with Diltiazem in the Treatment of Cardiovascular Diseases. Cardiol. Ther. 2016, 5, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Badu-Boateng, C.; Jennings, R.; Hammersley, D. The therapeutic role of ivabradine in heart failure. Ther. Adv. Chronic Dis. 2018, 9, 199–207. [Google Scholar] [CrossRef]
- Pitt, B.; Remme, W.; Zannad, F.; Neaton, J.; Martinez, F.; Roniker, B.; Bittman, R.; Hurley, S.; Kleiman, J.; Gatlin, M.; et al. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N. Engl. J. Med. 2003, 348, 1309–1321. [Google Scholar] [CrossRef]
- Pitt, B.; Zannad, F.; Remme, W.J.; Cody, R.; Castaigne, A.; Perez, A.; Palensky, J.; Wittes, J. The Effect of Spironolactone on Morbidity and Mortality in Patients with Severe Heart Failure. N. Engl. J. Med. 1999, 341, 709–717. [Google Scholar] [CrossRef]
- Campbell, T.J.; MacDonald, P.S. Digoxin in heart failure and cardiac arrhythmias. Med. J. Aust. 2003, 179, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Ramkumar, S.; Raghunath, A.; Raghunath, S. Statin Therapy: Review of Safety and Potential Side Effects. Acta Cardiol. Sin. 2016, 32, 631–639. [Google Scholar] [CrossRef]
- Jneid, H.; Bhatt, D.L.; Corti, R.; Badimon, J.J.; Fuster, V.; Francis, G.S. Aspirin and clopidogrel in acute coronary syndromes: Therapeutic insights from the CURE study. Arch. Intern. Med. 2003, 163, 1145–1153. [Google Scholar] [CrossRef] [PubMed]
- Tran, H.; Mehta, S.R.; Eikelboom, J.W. Clinical update on the therapeutic use of clopidogrel: Treatment of acute ST-segment elevation myocardial infarction (STEMI). Vasc. Health Risk Manag. 2006, 2, 379–387. [Google Scholar] [CrossRef][Green Version]
- Welsh, R.C.; Sidhu, R.S.; Cairns, J.A.; Lavi, S.; Kedev, S.; Moreno, R.; Cantor, W.J.; Stankovic, G.; Meeks, B.; Yuan, F.; et al. Outcomes Among Clopidogrel, Prasugrel, and Ticagrelor in ST-Elevation Myocardial Infarction Patients Who Underwent Primary Percutaneous Coronary Intervention From the TOTAL Trial. Can. J. Cardiol. 2019, 35, 1377–1385. [Google Scholar] [CrossRef]
- Date, A.A.; Desai, N.; Dixit, R.; Nagarsenker, M. Self-nanoemulsifying drug delivery systems: Formulation insights, applications and advances. Nanomedicine 2010, 5, 1595–1616. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Zhai, X.; Xue, K.; Hu, L.; Yang, X.; Li, G.; Si, L. Intestinal absorption and intestinal lymphatic transport of sirolimus from self-microemulsifying drug delivery systems assessed using the single-pass intestinal perfusion (SPIP) technique and a chylomicron flow blocking approach: Linear correlation with oral bioavailabilities in rats. Eur. J. Pharm. Sci. 2011, 43, 132–140. [Google Scholar]
- Nekkanti, V.; Wang, Z.; Betageri, G.V. Pharmacokinetic evaluation of improved oral bioavailability of valsartan: Proliposomes versus self-nanoemulsifying drug delivery system. AAPS PharmSciTech 2016, 17, 851–862. [Google Scholar] [CrossRef]
- Shafiq, S.; Shakeel, F.; Talegaonkar, S.; Ahmad, F.J.; Khar, R.K.; Ali, M. Development and bioavailability assessment of ramipril nanoemulsion formulation. Eur. J. Pharm. Biopharm. 2007, 66, 227–243. [Google Scholar] [CrossRef]
- Chhabra, G.; Chuttani, K.; Mishra, A.K.; Pathak, K. Design and development of nanoemulsion drug delivery system of amlodipine besilate for improvement of oral bioavailability. Drug Dev. Ind. Pharm. 2011, 37, 907–916. [Google Scholar] [CrossRef]
- Shah, U.; Joshi, G.; Sawant, K.J. Improvement in antihypertensive and antianginal effects of felodipine by enhanced absorption from PLGA nanoparticles optimized by factorial design. Mater. Sci. Eng. C 2014, 35, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Dudhipala, N.; Veerabrahma, K. Pharmacokinetic and pharmacodynamic studies of nisoldipine-loaded solid lipid nanoparticles developed by central composite design. Drug Dev. Ind. Pharm. 2015, 41, 1968–1977. [Google Scholar] [CrossRef] [PubMed]
- Ranpise, N.S.; Korabu, S.S.; Ghodake, V.N. Second generation lipid nanoparticles (NLC) as an oral drug carrier for delivery of lercanidipine hydrochloride. Colloids Surf. B Biointerfaces 2014, 116, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, P.B.; Gurram, A.K.; Deshpande, A.; Shavi, G.V.; Musmade, P.; Arumugam, K.; Averineni, R.K.; Mutalik, S.; Reddy, M.S.; Udupa, N. A novel nanoproliposomes of lercanidipine: Development, in vitro and preclinical studies to support its effectiveness in hypertension therapy. Life Sci. 2016, 162, 125–137. [Google Scholar] [CrossRef]
- Kim, Y.I.; Fluckiger, L.; Hoffman, M.; Lartaud-Idjouadiene, I.; Atkinson, J.; Maincent, T. The antihypertensive effect of orally administered nifedipine-loaded nanoparticles in spontaneously hypertensive rats. Br. J. Pharmacol. 1997, 120, 399–404. [Google Scholar] [CrossRef] [PubMed]
- Nelson, R.H. Hyperlipidemia as a risk factor for cardiovascular disease. Prim. Care Clin. Off. Pract. 2013, 40, 195–211. [Google Scholar] [CrossRef] [PubMed]
- Vavlukis, M.; Vavlukis, A. Adding ezetimibe to statin therapy: Latest evidence and clinical implications. Drugs Context 2018, 7, 212534. [Google Scholar] [CrossRef]
- Tziomalos, K.; Athyros, V.G. Fenofibrate: A novel formulation (Triglide) in the treatment of lipid disorders: A review. Int. J. Nanomed. 2006, 1, 129–147. [Google Scholar] [CrossRef]
- Shaker, M.A.; Elbadawy, H.M.; Al Thagfan, S.S.; Shaker, M.A. Enhancement of atorvastatin oral bioavailability via encapsulation in polymeric nanoparticles. Int. J. Pharm. 2021, 592, 120077. [Google Scholar] [CrossRef]
- Sharma, M.; Mehta, I. Surface stabilized atorvastatin nanocrystals with improved bioavailability, safety and antihyperlipidemic potential. Sci. Rep. 2019, 9, 16105. [Google Scholar] [CrossRef]
- Kumar, N.; Chaurasia, S.; Patel, R.R.; Khan, G.; Kumar, V.; Mishra, B. Atorvastatin calcium loaded PCL nanoparticles: Development, optimization, in vitro and in vivo assessments. RSC Adv. 2016, 6, 16520–16532. [Google Scholar] [CrossRef]
- Elmowafy, M.; Ibrahim, H.M.; Ahmed, M.A.; Shalaby, K.; Salama, A.; Hefesha, H. Atorvastatin-loaded nanostructured lipid carriers (NLCs): Strategy to overcome oral delivery drawbacks. Drug Deliv. 2017, 24, 932–941. [Google Scholar] [CrossRef]
- Jain, K.; Kumar, R.S.; Sood, S.; Gowthamarajan, K. Enhanced oral bioavailability of atorvastatin via oil-in-water nanoemulsion using aqueous titration method. J. Pharm. Sci. Res. 2013, 5, 18. [Google Scholar]
- Tiwari, R.; Pathak, K. Nanostructured lipid carrier versus solid lipid nanoparticles of simvastatin: Comparative analysis of characteristics, pharmacokinetics and tissue uptake. Int. J. Pharm. 2011, 415, 232–243. [Google Scholar] [CrossRef]
- Dudhipala, N.; Veerabrahma, K. Improved anti-hyperlipidemic activity of Rosuvastatin Calcium via lipid nanoparticles: Pharmacokinetic and pharmacodynamic evaluation. Eur. J. Pharm. Biopharm. 2017, 110, 47–57. [Google Scholar] [CrossRef]
- El-Helw, A.-R.M.; Fahmy, U.A. Improvement of fluvastatin bioavailability by loading on nanostructured lipid carriers. Int. J. Nanomed. 2015, 10, 5797. [Google Scholar] [CrossRef]
- Chen, Y.; Lu, Y.; Chen, J.; Lai, J.; Sun, J.; Hu, F.; Wu, W. Enhanced bioavailability of the poorly water-soluble drug fenofibrate by using liposomes containing a bile salt. Int. J. Pharm. 2009, 376, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Mohsin, K.; Alamri, R.; Ahmad, A.; Raish, M.; Alanazi, F.K.; Hussain, M.D. Development of self-nanoemulsifying drug delivery systems for the enhancement of solubility and oral bioavailability of fenofibrate, a poorly water-soluble drug. Int. J. Nanomed. 2016, 11, 2829. [Google Scholar]
- Tran, T.H.; Ramasamy, T.; Truong, D.H.; Choi, H.-G.; Yong, C.S.; Kim, J.O. Preparation and characterization of fenofibrate-loaded nanostructured lipid carriers for oral bioavailability enhancement. AAPS Pharmscitech 2014, 15, 1509–1515. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, Y.O.; Mahajan, U.B.; Agnihotri, V.V.; Nilange, M.S.; Mahajan, H.S.; Sharma, C.; Ojha, S.; Patil, C.R.; Goyal, S.N. Ezetimibe-Loaded Nanostructured Lipid Carrier Based Formulation Ameliorates Hyperlipidaemia in an Experimental Model of High Fat Diet. Molecules 2021, 26, 1485. [Google Scholar] [CrossRef]
- Bandyopadhyay, S.; Katare, O.; Singh, B. Optimized self nano-emulsifying systems of ezetimibe with enhanced bioavailability potential using long chain and medium chain triglycerides. Colloids Surf. B Biointerfaces 2012, 100, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Shevalkar, G.; Vavia, P. Solidified nanostructured lipid carrier (S-NLC) for enhancing the oral bioavailability of ezetimibe. J. Drug Deliv. Sci. Technol. 2019, 53, 101211. [Google Scholar] [CrossRef]
- McLennan, D.N.; Porter, C.J.; Charman, S.A. Subcutaneous drug delivery and the role of the lymphatics. Drug Discov. Today Technol. 2005, 2, 89–96. [Google Scholar] [CrossRef] [PubMed]
- American Diabetes, A. 2. Classification and Diagnosis of Diabetes: Standards of Medical Care in Diabetes-2020. Diabetes Care 2020, 43, S14–S31. [Google Scholar] [CrossRef]
- Hinnen, D. Glucagon-Like Peptide 1 Receptor Agonists for Type 2 Diabetes. Diabetes Spectr. 2017, 30, 202–210. [Google Scholar] [CrossRef] [PubMed]
- Roesti, E.S.; Boyle, C.N.; Zeman, D.T.; Sande-Melon, M.; Storni, F.; Cabral-Miranda, G.; Knuth, A.; Lutz, T.A.; Vogel, M.; Bachmann, M.F. Vaccination against amyloidogenic aggregates in pancreatic islets prevents development of type 2 diabetes mellitus. Vaccines 2020, 8, 116. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yu, X.-L.; Zha, J.; Mao, L.-Z.; Chai, J.-Q.; Liu, R.-T. Therapeutic vaccine against IL-1β improved glucose control in a mouse model of type 2 diabetes. Life Sci. 2018, 192, 68–74. [Google Scholar] [CrossRef]
- Zha, J.; Chi, X.-W.; Yu, X.-L.; Liu, X.-M.; Liu, D.-Q.; Zhu, J.; Ji, H.; Liu, R.-T. Interleukin-1β-targeted vaccine improves glucose control and β-cell function in a diabetic KK-Ay mouse model. PLoS ONE 2016, 11, e0154298. [Google Scholar] [CrossRef] [PubMed]
- Cavelti-Weder, C.; Timper, K.; Seelig, E.; Keller, C.; Osranek, M.; Lässing, U.; Spohn, G.; Maurer, P.; Müller, P.; Jennings, G.T. Development of an interleukin-1β vaccine in patients with type 2 diabetes. Mol. Ther. 2016, 24, 1003–1012. [Google Scholar] [CrossRef]
- Pang, Z.; Nakagami, H.; Osako, M.K.; Koriyama, H.; Nakagami, F.; Tomioka, H.; Shimamura, M.; Kurinami, H.; Takami, Y.; Morishita, R. Therapeutic vaccine against DPP4 improves glucose metabolism in mice. Proc. Natl. Acad. Sci. USA 2014, 111, E1256–E1263. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Z.; Chen, X.; Zhou, Y.; Lin, J.; Ding, D.; Yang, S.; Chen, F.; Wang, M.; Zhu, F.; Yu, X. Therapeutic vaccines against human and rat renin in spontaneously hypertensive rats. PLoS ONE 2013, 8, e66420. [Google Scholar]
- Brown, M.J.; Coltart, J.; Gunewardena, K.; Ritter, J.M.; Auton, T.R.; Glover, J.F. Randomized double-blind placebo-controlled study of an angiotensin immunotherapeutic vaccine (PMD3117) in hypertensive subjects. Clin. Sci. 2004, 107, 167–173. [Google Scholar] [CrossRef]
- Hong, F.; Quan, W.Y.; Pandey, R.; Yi, S.; Chi, L.; Xia, L.Z.; Yuan, M.; Ming, L.J. A vaccine for hypertension based on peptide AngI-R: A pilot study. Int. J. Cardiol. 2011, 148, 76–84. [Google Scholar] [CrossRef]
- Chen, X.; Qiu, Z.; Yang, S.; Ding, D.; Chen, F.; Zhou, Y.; Wang, M.; Lin, J.; Yu, X.; Zhou, Z. Effectiveness and safety of a therapeutic vaccine against angiotensin II receptor type 1 in hypertensive animals. Hypertension 2013, 61, 408–416. [Google Scholar] [CrossRef]
- Zhu, F.; Liao, Y.H.; Li, L.D.; Cheng, M.; Wei, F.; Wei, Y.M.; Wang, M. Target organ protection from a novel angiotensin II receptor (AT1) vaccine ATR12181 in spontaneously hypertensive rats. Cell. Mol. Immunol. 2006, 3, 107–114. [Google Scholar]
- Tissot, A.C.; Maurer, P.; Nussberger, J.; Sabat, R.; Pfister, T.; Ignatenko, S.; Volk, H.-D.; Stocker, H.; Müller, P.; Jennings, G.T. Effect of immunisation against angiotensin II with CYT006-AngQb on ambulatory blood pressure: A double-blind, randomised, placebo-controlled phase IIa study. Lancet 2008, 371, 821–827. [Google Scholar] [CrossRef]
- Watanabe, R.; Suzuki, J.-I.; Wakayama, K.; Maejima, Y.; Shimamura, M.; Koriyama, H.; Nakagami, H.; Kumagai, H.; Ikeda, Y.; Akazawa, H. A peptide vaccine targeting angiotensin II attenuates the cardiac dysfunction induced by myocardial infarction. Sci. Rep. 2017, 7, 43920. [Google Scholar] [CrossRef]
- Margulis, K.; Neofytou, E.A.; Beygui, R.E.; Zare, R.N. Celecoxib nanoparticles for therapeutic angiogenesis. ACS Nano 2015, 9, 9416–9426. [Google Scholar] [CrossRef]
- Vignesh, S.; Sivashanmugam, A.; Annapoorna, M.; Janarthanan, R.; Subramania, I.; Jayakumar, R. Injectable deferoxamine nanoparticles loaded chitosan-hyaluronic acid coacervate hydrogel for therapeutic angiogenesis. Colloids Surf. B Biointerfaces 2018, 161, 129–138. [Google Scholar]
- Tomlinson, B.; Hu, M.; Zhang, Y.; Chan, P.; Liu, Z.-M. Alirocumab for the treatment of hypercholesterolemia. Expert Opin. Biol. Ther. 2017, 17, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Kasichayanula, S.; Grover, A.; Emery, M.G.; Gibbs, M.A.; Somaratne, R.; Wasserman, S.M.; Gibbs, J.P. Clinical Pharmacokinetics and Pharmacodynamics of Evolocumab, a PCSK9 Inhibitor. Clin. Pharmacokinet. 2018, 57, 769–779. [Google Scholar] [CrossRef]
- Ray, K.K.; Wright, R.S.; Kallend, D.; Koenig, W.; Leiter, L.A.; Raal, F.J.; Bisch, J.A.; Richardson, T.; Jaros, M.; Wijngaard, P.L.J.; et al. Two Phase 3 Trials of Inclisiran in Patients with Elevated LDL Cholesterol. N. Engl. J. Med. 2020, 382, 1507–1519. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.D.; Raal, F.J.; Catapano, A.L.; Witztum, J.L.; Steinhagen-Thiessen, E.; Tsimikas, S. Mipomersen, an antisense oligonucleotide to apolipoprotein B-100, reduces lipoprotein (a) in various populations with hypercholesterolemia: Results of 4 phase III trials. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 689–699. [Google Scholar] [CrossRef]
- Raal, F.J.; Kallend, D.; Ray, K.K.; Turner, T.; Koenig, W.; Wright, R.S.; Wijngaard, P.L.; Curcio, D.; Jaros, M.J.; Leiter, L.A. Inclisiran for the treatment of heterozygous familial hypercholesterolemia. N. Engl. J. Med. 2020, 382, 1520–1530. [Google Scholar] [CrossRef]
- Witztum, J.L.; Gaudet, D.; Freedman, S.D.; Alexander, V.J.; Digenio, A.; Williams, K.R.; Yang, Q.; Hughes, S.G.; Geary, R.S.; Arca, M. Volanesorsen and triglyceride levels in familial chylomicronemia syndrome. N. Engl. J. Med. 2019, 381, 531–542. [Google Scholar] [CrossRef]
- Viney, N.J.; van Capelleveen, J.C.; Geary, R.S.; Xia, S.; Tami, J.A.; Rosie, Z.Y.; Marcovina, S.M.; Hughes, S.G.; Graham, M.J.; Crooke, R.M. Antisense oligonucleotides targeting apolipoprotein (a) in people with raised lipoprotein (a): Two randomised, double-blind, placebo-controlled, dose-ranging trials. Lancet 2016, 388, 2239–2253. [Google Scholar] [CrossRef]
- Pharma, I. Positive Phase 2 Clinical Data of AKCEA-APOCIII-L(Rx) at ESC Congress 2020; Ionis Pharma: Boston, MA, USA; Carlsbad, CA, USA, 2020. [Google Scholar]
- Graham, M.J.; Lee, R.G.; Brandt, T.A.; Tai, L.-J.; Fu, W.; Peralta, R.; Yu, R.; Hurh, E.; Paz, E.; McEvoy, B.W. Cardiovascular and metabolic effects of ANGPTL3 antisense oligonucleotides. N. Engl. J. Med. 2017, 377, 222–232. [Google Scholar] [CrossRef]
- Kawakami, R.; Nozato, Y.; Nakagami, H.; Ikeda, Y.; Shimamura, M.; Yoshida, S.; Sun, J.; Kawano, T.; Takami, Y.; Noma, T. Development of vaccine for dyslipidemia targeted to a proprotein convertase subtilisin/kexin type 9 (PCSK9) epitope in mice. PLoS ONE 2018, 13, e0191895. [Google Scholar] [CrossRef] [PubMed]
- Landlinger, C.; Pouwer, M.G.; Juno, C.; van der Hoorn, J.W.; Pieterman, E.J.; Jukema, J.W.; Staffler, G.; Princen, H.M.; Galabova, G. The AT04A vaccine against proprotein convertase subtilisin/kexin type 9 reduces total cholesterol, vascular inflammation, and atherosclerosis in APOE* 3Leiden. CETP mice. Eur. Heart J. 2017, 38, 2499–2507. [Google Scholar] [CrossRef]
- Crossey, E.; Amar, M.J.; Sampson, M.; Peabody, J.; Schiller, J.T.; Chackerian, B.; Remaley, A.T. A cholesterol-lowering VLP vaccine that targets PCSK9. Vaccine 2015, 33, 5747–5755. [Google Scholar] [CrossRef] [PubMed]
- Olearczyk, J.; Gao, S.; Eybye, M.; Yendluri, S.; Andrews, L.; Bartz, S.; Cully, D.; Tadin-Strapps, M. Targeting of hepatic angiotensinogen using chemically modified siRNAs results in significant and sustained blood pressure lowering in a rat model of hypertension. Hypertens. Res. 2014, 37, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.-N.; Tain, Y.-L. Targeting the Renin–Angiotensin–Aldosterone System to Prevent Hypertension and Kidney Disease of Developmental Origins. Int. J. Mol. Sci. 2021, 22, 2298. [Google Scholar] [CrossRef]
- Ames, M.K.; Atkins, C.E.; Pitt, B. The renin-angiotensin-aldosterone system and its suppression. J. Vet. Intern. Med. 2019, 33, 363–382. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. Kynamro (Mipomersen Sodium) Injection: Drug Approval Package. 2013. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2013/203568Orig1s000TOC.cfm (accessed on 1 May 2021).
- Fogacci, F.; Ferri, N.; Toth, P.P.; Ruscica, M.; Corsini, A.; Cicero, A.F. Efficacy and safety of mipomersen: A systematic review and meta-analysis of randomized clinical trials. Drugs 2019, 79, 751–766. [Google Scholar] [CrossRef]
- Geary, R.S.; Baker, B.F.; Crooke, S.T. Clinical and preclinical pharmacokinetics and pharmacodynamics of mipomersen (Kynamro®): A second-generation antisense oligonucleotide inhibitor of apolipoprotein B. Clin. Pharmacokinet. 2015, 54, 133–146. [Google Scholar] [CrossRef]
- Santos, R.D.; Duell, P.B.; East, C.; Guyton, J.R.; Moriarty, P.M.; Chin, W.; Mittleman, R.S. Long-term efficacy and safety of mipomersen in patients with familial hypercholesterolaemia: 2-year interim results of an open-label extension. Eur. Heart J. 2015, 36, 566–575. [Google Scholar] [CrossRef] [PubMed]
- Esan, O.; Wierzbicki, A.S. Volanesorsen in the Treatment of Familial Chylomicronemia Syndrome or Hypertriglyceridaemia: Design, Development and Place in Therapy. Drug Des. Dev. Ther. 2020, 14, 2623. [Google Scholar] [CrossRef]
- European Medicines Agency. Waylivra (Volanesorsen): Public Assessment Report. 2019. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/waylivra (accessed on 1 April 2021).
- Gaudet, D.; Brisson, D.; Tremblay, K.; Alexander, V.J.; Singleton, W.; Hughes, S.G.; Geary, R.S.; Baker, B.F.; Graham, M.J.; Crooke, R.M. Targeting APOC3 in the familial chylomicronemia syndrome. N. Engl. J. Med. 2014, 371, 2200–2206. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. Waylivra, INN-Volanesorsen. 2019. Available online: https://www.ema.europa.eu/en/documents/assessment-report/waylivra-epar-public-assessment-report_en.pdf (accessed on 1 May 2021).
- Dyrbuś, K.; Gąsior, M.; Penson, P.; Ray, K.K.; Banach, M. Inclisiran—New hope in the management of lipid disorders? J. Clin. Lipidol. 2020, 14, 16–27. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. Leqvio (Inclisiran): An Overview of Leqvio and Why It Is Authorised in the EU. 2020. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/leqvio (accessed on 1 May 2021).
- European Medicines Agency. Leqvio: Assessment Report. 2020. Available online: https://www.ema.europa.eu/en/documents/assessment-report/leqvio-epar-public-assessment-report_en.pdf (accessed on 1 May 2021).
- Fowler, A.; Sampson, M.; Remaley, A.T.; Chackerian, B. A VLP-based vaccine targeting ANGPTL3 lowers plasma triglycerides in mice. bioRxiv 2021. [Google Scholar] [CrossRef]
- Brakenhielm, E.; Alitalo, K. Cardiac lymphatics in health and disease. Nat. Rev. Cardiol. 2019, 16, 56–68. [Google Scholar] [CrossRef]
- Ananthakrishnan, P.; Mariani, G.; Moresco, L.; Giuliano, A.E. The anatomy and physiology of lymphatic circulation. In Radioguided Surgery; Springer: New York, NY, USA, 2008; pp. 57–71. [Google Scholar]
- Supersaxo, A.; Hein, W.R.; Steffen, H. Effect of molecular weight on the lymphatic absorption of water-soluble compounds following subcutaneous administration. Pharm. Res. 1990, 7, 167–169. [Google Scholar] [CrossRef]
- Hirano, K.; Hunt, C.A. Lymphatic transport of liposome-encapsulated agents: Effects of liposome size following intraperitoneal administration. J. Pharm. Sci. 1985, 74, 915–921. [Google Scholar] [CrossRef]
- Flessner, M.; Dedrick, R.; Schultz, J.S. Exchange of macromolecules between peritoneal cavity and plasma. Am. J. Physiol. Heart Circ. Physiol. 1985, 248, H15–H25. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.Y.; Thiam, C.H.; Yeo, K.P.; Bisoendial, R.; Hii, C.S.; McGrath, K.C.; Tan, K.W.; Heather, A.; Alexander, J.S.J.; Angeli, V. Lymphatic vessels are essential for the removal of cholesterol from peripheral tissues by SR-BI-mediated transport of HDL. Cell Metab. 2013, 17, 671–684. [Google Scholar] [CrossRef] [PubMed]
- Reddy, S.T.; Rehor, A.; Schmoekel, H.G.; Hubbell, J.A.; Swartz, M.A. In vivo targeting of dendritic cells in lymph nodes with poly (propylene sulfide) nanoparticles. J. Control. Release 2006, 112, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Harvey, A.J.; Kaestner, S.A.; Sutter, D.E.; Harvey, N.G.; Mikszta, J.A.; Pettis, R.J. Microneedle-based intradermal delivery enables rapid lymphatic uptake and distribution of protein drugs. Pharm. Res. 2011, 28, 107–116. [Google Scholar] [CrossRef]
- Hettinga, J.; Carlisle, R. Vaccination into the Dermal Compartment: Techniques, Challenges, and Prospects. Vaccines 2020, 8, 534. [Google Scholar] [CrossRef] [PubMed]
- Moore, J.E.; Bertram, C.D. Lymphatic System Flows. Annu. Rev. Fluid Mech. 2018, 50, 459–482. [Google Scholar] [CrossRef]
- Liao, S.; Padera, T.P. Lymphatic Function and Immune Regulation in Health and Disease. Lymphat. Res. Biol. 2013, 11, 136–143. [Google Scholar] [CrossRef]
- Canzona, F.; Massimo, M.; Tuzi, A.; Maggiori, E.; Grosso, M.G.; Antonaci, L.; Santini, S.; Catizzone, A.R.; Troili, F.; Gallo, A.; et al. Intradermal Therapy (mesotherapy) in Dermatology. J. Dermatol. Ski. Sci. 2020, 2, 22–25. [Google Scholar]
- Hawkridge, A.; Hatherill, M.; Little, F.; Goetz, M.A.; Barker, L.; Mahomed, H.; Sadoff, J.; Hanekom, W.; Geiter, L.; Hussey, G. Efficacy of percutaneous versus intradermal BCG in the prevention of tuberculosis in South African infants: Randomised trial. BMJ 2008, 337, a2052. [Google Scholar] [CrossRef]
- Kroger, C.J.; Clark, M.; Ke, Q.; Tisch, R.M. Therapies to suppress β cell autoimmunity in type 1 diabetes. Front. Immunol. 2018, 9, 1891. [Google Scholar] [CrossRef] [PubMed]
- Clark, M.; Kroger, C.J.; Tisch, R.M. Type 1 diabetes: A chronic anti-self-inflammatory response. Front. Immunol. 2017, 8, 1898. [Google Scholar] [CrossRef]
- Richardson, S.J.; Willcox, A.; Bone, A.J.; Morgan, N.G.; Foulis, A.K. Immunopathology of the human pancreas in type-I diabetes. In Seminars in Immunopathology; Springer: New York, NY, USA, 2011; pp. 9–21. [Google Scholar]
- Harrison, L.C. The prospect of vaccination to prevent type 1 diabetes. Hum. Vaccines 2005, 1, 143–150. [Google Scholar] [CrossRef]
- Smith, E.L.; Peakman, M. Peptide immunotherapy for type 1 diabetes—Clinical advances. Front. Immunol. 2018, 9, 392. [Google Scholar] [CrossRef] [PubMed]
- Thrower, S.L.; James, L.; Hall, W.; Green, K.M.; Arif, S.; Allen, J.S.; Van-Krinks, C.; Lozanoska-Ochser, B.; Marquesini, L.; Brown, S.; et al. Proinsulin peptide immunotherapy in type 1 diabetes: Report of a first-in-man Phase I safety study. Clin. Exp. Immunol. 2009, 155, 156–165. [Google Scholar] [CrossRef]
- Dul, M.; Nikolic, T.; Stefanidou, M.; McAteer, M.; Williams, P.; Mous, J.; Roep, B.; Kochba, E.; Levin, Y.; Peakman, M. Conjugation of a peptide autoantigen to gold nanoparticles for intradermally administered antigen specific immunotherapy. Int. J. Pharm. 2019, 562, 303–312. [Google Scholar] [CrossRef]
- Nikolic, T.; Zwaginga, J.J.; Uitbeijerse, B.S.; Woittiez, N.J.; de Koning, E.J.; Aanstoot, H.-J.; Roep, B.O. Safety and feasibility of intradermal injection with tolerogenic dendritic cells pulsed with proinsulin peptide—For type 1 diabetes. Lancet Diabetes Endocrinol. 2020, 8, 470–472. [Google Scholar] [CrossRef]
- Nicoll, L.H.; Hesby, A. Intramuscular injection: An integrative research review and guideline for evidence-based practice. Appl. Nurs. Res. 2002, 15, 149–162. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, Y.; Mukai, K.; Takaoka, K.; Hirose, T.; Morishita, K.; Yamamoto, T.; Yoshida, Y.; Urai, T.; Nakatani, T. Establishing a new appropriate intramuscular injection site in the deltoid muscle. Hum. Vaccin. Immunother. 2017, 13, 2123–2129. [Google Scholar] [CrossRef]
- Ogston-Tuck, S. Intramuscular injection technique: An evidence-based approach. Nurs. Stand. 2014, 29, 52–59. [Google Scholar] [CrossRef]
- Rodger, M.A.; King, L. Drawing up and administering intramuscular injections: A review of the literature. J. Adv. Nurs. 2000, 31, 574–582. [Google Scholar] [CrossRef] [PubMed]
- Kivelä, R.; Havas, E.; Vihko, V. Localisation of lymphatic vessels and vascular endothelial growth factors-C and -D in human and mouse skeletal muscle with immunohistochemistry. Histochem. Cell Biol. 2007, 127, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Abai, A.M.; Hobart, P.M.; Barnhart, K.M. Insulin delivery with plasmid DNA. Hum. Gene Ther. 1999, 10, 2637–2649. [Google Scholar] [CrossRef]
- Phrommintikul, A.; Kuanprasert, S.; Wongcharoen, W.; Kanjanavanit, R.; Chaiwarith, R.; Sukonthasarn, A. Influenza vaccination reduces cardiovascular events in patients with acute coronary syndrome. Eur. Heart J. 2011, 32, 1730–1735. [Google Scholar] [CrossRef] [PubMed]
- Gurfinkel, E.P.; Mendiz, O.; Mautner, B. Flu vaccination in acute coronary syndromes and planned percutaneous coronary interventions (FLUVACS) study. Eur. Heart J. 2004, 25, 25–31. [Google Scholar] [CrossRef]
- Vlachopoulos, C.V.; Terentes-Printzios, D.G.; Aznaouridis, K.A.; Pietri, P.G.; Stefanadis, C.I. Association between pneumococcal vaccination and cardiovascular outcomes: A systematic review and meta-analysis of cohort studies. Eur. J. Prev. Cardiol. 2015, 22, 1185–1199. [Google Scholar] [CrossRef]
- Monteiro, M.P. Obesity vaccines. Hum. Vaccines Immunother. 2014, 10, 887–895. [Google Scholar] [CrossRef]
- Chawla, R.; Madhu, S.; Makkar, B.; Ghosh, S.; Saboo, B.; Kalra, S. RSSDI-ESI clinical practice recommendations for the management of type 2 diabetes mellitus 2020. Int. J. Diabetes Dev. Ctries. 2020, 40, 1–122. [Google Scholar]
- Hulot, J.-S.; Ishikawa, K.; Hajjar, R.J. Gene therapy for the treatment of heart failure: Promise postponed. Eur. Heart J. 2016, 37, 1651–1658. [Google Scholar] [CrossRef]
- El-Armouche, A.; Eschenhagen, T. β-Adrenergic stimulation and myocardial function in the failing heart. Heart Fail. Rev. 2009, 14, 225. [Google Scholar] [CrossRef] [PubMed]
- Rockman, H.A.; Chien, K.R.; Choi, D.-J.; Iaccarino, G.; Hunter, J.J.; Ross, J.; Lefkowitz, R.J.; Koch, W.J. Expression of a β-adrenergic receptor kinase 1 inhibitor prevents the development of myocardial failure in gene-targeted mice. Proc. Natl. Acad. Sci. USA 1998, 95, 7000–7005. [Google Scholar] [CrossRef]
- Harding, V.B.; Jones, L.R.; Lefkowitz, R.J.; Koch, W.J.; Rockman, H.A. Cardiac βARK1 inhibition prolongs survival and augments β blocker therapy in a mouse model of severe heart failure. Proc. Natl. Acad. Sci. USA 2001, 98, 5809–5814. [Google Scholar] [CrossRef]
- Shah, A.S.; White, D.C.; Emani, S.; Kypson, A.P.; Lilly, R.E.; Wilson, K.; Glower, D.D.; Lefkowitz, R.J.; Koch, W.J. In vivo ventricular gene delivery of a β-adrenergic receptor kinase inhibitor to the failing heart reverses cardiac dysfunction. Circulation 2001, 103, 1311–1316. [Google Scholar] [CrossRef] [PubMed]
- Rengo, G.; Lymperopoulos, A.; Zincarelli, C.; Donniacuo, M.; Soltys, S.; Rabinowitz, J.E.; Koch, W.J. Clinical Perspective. Circulation 2009, 119, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Tevaearai, H.T.; Eckhart, A.D.; Shotwell, K.F.; Wilson, K.; Koch, W.J. Ventricular dysfunction after cardioplegic arrest is improved after myocardial gene transfer of a β-adrenergic receptor kinase inhibitor. Circulation 2001, 104, 2069–2074. [Google Scholar] [CrossRef]
- Raake, P.W.; Schlegel, P.; Ksienzyk, J.; Reinkober, J.; Barthelmes, J.; Schinkel, S.; Pleger, S.; Mier, W.; Haberkorn, U.; Koch, W.J. βARKct cardiac gene therapy ameliorates cardiac function and normalizes the catecholaminergic axis in a clinically relevant large animal heart failure model. Eur. Heart J. 2013, 34, 1437–1447. [Google Scholar] [CrossRef] [PubMed]
- Kawase, Y.; Ly, H.Q.; Prunier, F.; Lebeche, D.; Shi, Y.; Jin, H.; Hadri, L.; Yoneyama, R.; Hoshino, K.; Takewa, Y. Reversal of cardiac dysfunction after long-term expression of SERCA2a by gene transfer in a pre-clinical model of heart failure. J. Am. Coll. Cardiol. 2008, 51, 1112–1119. [Google Scholar] [CrossRef]
- Lyon, A.R.; Bannister, M.L.; Collins, T.; Pearce, E.; Sepehripour, A.H.; Dubb, S.S.; Garcia, E.; O’Gara, P.; Liang, L.; Kohlbrenner, E.; et al. SERCA2a gene transfer decreases sarcoplasmic reticulum calcium leak and reduces ventricular arrhythmias in a model of chronic heart failure. Circulation 2011, 4, 362–372. [Google Scholar] [CrossRef]
- Greenberg, B.; Butler, J.; Felker, G.M.; Ponikowski, P.; Voors, A.A.; Desai, A.S.; Barnard, D.; Bouchard, A.; Jaski, B.; Lyon, A.R. Calcium upregulation by percutaneous administration of gene therapy in patients with cardiac disease (CUPID 2): A randomised, multinational, double-blind, placebo-controlled, phase 2b trial. Lancet 2016, 387, 1178–1186. [Google Scholar] [CrossRef]
- Ylä-Herttuala, S. Endgame: Glybera finally recommended for approval as the first gene therapy drug in the European union. Mol. Ther. 2012, 20, 1831–1832. [Google Scholar] [CrossRef]
- Burnett, J.R.; Hooper, A.J.; Hegele, R.A. Familial Lipoprotein Lipase Deficiency; GeneReviews®: Seattle, WA, USA, 2017.
- Rip, J.; Nierman, M.C.; Sierts, J.A.; Petersen, W.; Den Oever, K.V.; Raalte, D.V.; Ross, C.J.; Hayden, M.R.; Bakker, A.C.; Dijkhuizen, P. Gene therapy for lipoprotein lipase deficiency: Working toward clinical application. Hum. Gene Ther. 2005, 16, 1276–1286. [Google Scholar] [CrossRef]
- Bryant, L.M.; Christopher, D.M.; Giles, A.R.; Hinderer, C.; Rodriguez, J.L.; Smith, J.B.; Traxler, E.A.; Tycko, J.; Wojno, A.P.; Wilson, J.M. Lessons learned from the clinical development and market authorization of Glybera. Hum. Gene Ther. Clin. Dev. 2013, 24, 55–64. [Google Scholar] [CrossRef]
- Senior, M. After Glybera’s Withdrawal, What’s Next for Gene Therapy? Nature Publishing Group: Berlin, Germany, 2017. [Google Scholar]
- Tilemann, L.; Ishikawa, K.; Weber, T.; Hajjar, R.J. Gene therapy for heart failure. Circ. Res. 2012, 110, 777–793. [Google Scholar] [CrossRef]
- Huang, L.-H.; Lavine, K.J.; Randolph, G.J. Cardiac Lymphatic Vessels, Transport, and Healing of the Infarcted Heart. JACC Basic Transl. Sci. 2017, 2, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Hammond, H.K.; Penny, W.F.; Traverse, J.H.; Henry, T.D.; Watkins, M.W.; Yancy, C.W.; Sweis, R.N.; Adler, E.D.; Patel, A.N.; Murray, D.R. Intracoronary gene transfer of adenylyl cyclase 6 in patients with heart failure: A randomized clinical trial. JAMA Cardiol. 2016, 1, 163–171. [Google Scholar] [CrossRef]
- Jessup, M.; Greenberg, B.; Mancini, D.; Cappola, T.; Pauly, D.F.; Jaski, B.; Yaroshinsky, A.; Zsebo, K.M.; Dittrich, H.; Hajjar, R.J. Calcium upregulation by percutaneous administration of gene therapy in cardiac disease (CUPID) a phase 2 trial of intracoronary gene therapy of sarcoplasmic reticulum Ca2+-ATPase in patients with advanced heart failure. Circulation 2011, 124, 304–313. [Google Scholar] [CrossRef]
- Hulot, J.S.; Salem, J.E.; Redheuil, A.; Collet, J.P.; Varnous, S.; Jourdain, P.; Logeart, D.; Gandjbakhch, E.; Bernard, C.; Hatem, S.N. Effect of intracoronary administration of AAV1/SERCA2a on ventricular remodelling in patients with advanced systolic heart failure: Results from the AGENT-HF randomized phase 2 trial. Eur. J. Heart Fail. 2017, 19, 1534–1541. [Google Scholar] [CrossRef] [PubMed]
- Precigen Triple-Gene. Precigen Triple-Gene Provides Six-Month Follow-Up Data from Phase I Study of INXN-4001, a Multigenic Investigational Therapeutic Candidate for Heart Failure. 2020. Available online: https://www.prnewswire.com/news-releases/precigen-triple-gene-provides-six-month-follow-up-data-from-phase-i-study-of-inxn-4001-a-multigenic-investigational-therapeutic-candidate-for-heart-failure-301107258.html (accessed on 1 April 2021).
- Model, M.I. 425. Arginine and Tetrahydrobiopterin Synergistically Potentiate the Antirestenotic Effect of Vascular Gene Therapy with Inducible Nitric Oxide Synthase. Mol. Ther. 2010, 18, 1. [Google Scholar]
- Chung, E.S.; Miller, L.; Patel, A.N.; Anderson, R.D.; Mendelsohn, F.O.; Traverse, J.; Silver, K.H.; Shin, J.; Ewald, G.; Farr, M.J. Changes in ventricular remodelling and clinical status during the year following a single administration of stromal cell-derived factor-1 non-viral gene therapy in chronic ischaemic heart failure patients: The STOP-HF randomized Phase II trial. Eur. Heart J. 2015, 36, 2228–2238. [Google Scholar] [CrossRef]
- Juventas Therapeutics. Juventas Therapeutics Completes Enrollment of Phase I/II RETRO-HF Trial and Demonstrates Safety for Retrograde Infusion of JVS-100 in Patients with Heart Failure. 2014. Available online: https://www.prnewswire.com/news-releases/juventas-therapeutics-completes-enrollment-of-phase-iii-retro-hf-trial-and-demonstrates-safety-for-retrograde-infusion-of-jvs-100-in-patients-with-heart-failure-270890361.html (accessed on 1 May 2021).
- Anttila, V.; Saraste, A.; Knuuti, J.; Jaakkola, P.; Hedman, M.; Svedlund, S.; Lagerström-Fermér, M.; Kjaer, M.; Jeppsson, A.; Gan, L.-M.; et al. Synthetic mRNA encoding VEGF-A in patients undergoing coronary artery bypass grafting: Design of a phase 2a clinical trial. Mol. Ther. Methods Clin. Dev. 2020, 18, 464–472. [Google Scholar] [CrossRef]
- Gan, L.-M.; Lagerström-Fermér, M.; Carlsson, L.G.; Arfvidsson, C.; Egnell, A.-C.; Rudvik, A.; Kjaer, M.; Collén, A.; Thompson, J.D.; Joyal, J.; et al. Intradermal delivery of modified mRNA encoding VEGF-A in patients with type 2 diabetes. Nat. Commun. 2019, 10, 871. [Google Scholar] [CrossRef]
- Ishikawa, K.; Fish, K.M.; Tilemann, L.; Rapti, K.; Aguero, J.; Santos-Gallego, C.G.; Lee, A.; Karakikes, I.; Xie, C.; Akar, F.G. Cardiac I-1c overexpression with reengineered AAV improves cardiac function in swine ischemic heart failure. Mol. Ther. 2014, 22, 2038–2045. [Google Scholar] [CrossRef]
- Kim, J.S.; Hwang, H.; Cho, K.; Park, E.; Lee, W.; Paeng, J.; Lee, D.; Kim, H.; Sohn, D.; Kim, K. Intramyocardial transfer of hepatocyte growth factor as an adjunct to CABG: Phase I clinical study. Gene Ther. 2013, 20, 717–722. [Google Scholar] [CrossRef]
- Hartikainen, J.; Hassinen, I.; Hedman, A.; Kivelä, A.; Saraste, A.; Knuuti, J.; Husso, M.; Mussalo, H.; Hedman, M.; Rissanen, T.T.; et al. Adenoviral intramyocardial VEGF-DΔNΔC gene transfer increases myocardial perfusion reserve in refractory angina patients: A phase I/IIa study with 1-year follow-up. Eur. Heart J. 2017, 38, 2547–2555. [Google Scholar] [CrossRef]
- Liu, J.; Wu, P.; Wang, Y.; Du, Y. Ad-HGF improves the cardiac remodeling of rat following myocardial infarction by upregulating autophagy and necroptosis and inhibiting apoptosis. Am. J. Transl. Res. 2016, 8, 4605. [Google Scholar]
- Wang, W.; Wang, M.-Q.; Wang, H.; Gao, W.; Zhang, Z.; Zhao, S.; Xu, H.-Z.; Chen, B.; Zhu, M.-X.; Wu, Z.-Z. Effects of adenovirus-mediated hepatocyte growth factor gene therapy on postinfarct heart function: Comparison of single and repeated injections. Hum. Gene Ther. 2016, 27, 643–651. [Google Scholar] [CrossRef]
- Hardy, N.; Viola, H.M.; Johnstone, V.P.; Clemons, T.D.; Cserne Szappanos, H.; Singh, R.; Smith, N.M.; Iyer, K.S.; Hool, L.C. Nanoparticle-mediated dual delivery of an antioxidant and a peptide against the L-Type Ca2+ channel enables simultaneous reduction of cardiac ischemia-reperfusion injury. ACS Nano 2015, 9, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Carpentier, A.C.; Frisch, F.; Labbe, S.M.; Gagnon, R.; de Wal, J.; Greentree, S.; Petry, H.; Twisk, J.; Brisson, D.; Gaudet, D. Effect of alipogene tiparvovec (AAV1-LPLS447X) on postprandial chylomicron metabolism in lipoprotein lipase-deficient patients. J. Clin. Endocrinol. 2012, 97, 1635–1644. [Google Scholar] [CrossRef] [PubMed]
- Anselmo, A.C.; Mitragotri, S. Nanoparticles in the clinic: An update. Bioeng. Transl. Med. 2019, 4, e10143. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Maeda, K.; Takefuji, M.; Kikuchi, R.; Morishita, Y.; Hirashima, M.; Murohara, T. Dynamics of angiogenesis in ischemic areas of the infarcted heart. Sci. Rep. 2017, 7, 7156. [Google Scholar] [CrossRef]
- Post, M.J.; Sato, K.; Murakami, M.; Bao, J.; Tirziu, D.; Pearlman, J.D.; Simons, M. Adenoviral PR39 improves blood flow and myocardial function in a pig model of chronic myocardial ischemia by enhancing collateral formation. Am. J. Physiol. -Regul. Integr. Comp. Physiol. 2006, 290, R494–R500. [Google Scholar] [CrossRef]
- Du, C.; Chen, X.-W.; Wang, Z.-M.; Meng, H.-Y.; Li, Y.-F.; Wei, T.-W.; Wang, L.-S. HGF Treatment Promotes Cardiac Function and Cardiac Repair: Meta-analysis of Pig Models with Myocardial Infarction. Res. Sq. 2020. preprint. [Google Scholar] [CrossRef]
- Korpisalo, P.; Karvinen, H.; Rissanen, T.T.; Kilpijoki, J.; Marjomäki, V.; Baluk, P.; McDonald, D.M.; Cao, Y.; Eriksson, U.; Alitalo, K. VEGF-A and PDGF-B combination gene therapy prolongs angiogenic effects via recruitment of interstitial mononuclear cells and paracrine effects rather than improved pericyte coverage of angiogenic vessels. Circ. Res. 2008, 103, 1092. [Google Scholar] [CrossRef]
- Jin, J.-F.; Zhu, L.-L.; Chen, M.; Xu, H.-M.; Wang, H.-F.; Feng, X.-Q.; Zhu, X.-P.; Zhou, Q. The optimal choice of medication administration route regarding intravenous, intramuscular, and subcutaneous injection. Patient Prefer. Adherence 2015, 9, 923–942. [Google Scholar] [CrossRef] [PubMed]
- 2—Intravenous Drug Administration. In Techniques in the Behavioral and Neural Sciences; Claassen, V., Ed.; Elsevier: Amsterdam, The Netherlands, 1994; Volume 12, pp. 5–22. [Google Scholar]
- Xie, Y.; Bagby, T.R.; Cohen, M.S.; Forrest, M.L. Drug delivery to the lymphatic system: Importance in future cancer diagnosis and therapies. Expert Opin. Drug Deliv. 2009, 6, 785–792. [Google Scholar] [CrossRef]
- Caliph, S.M.; Trevaskis, N.L.; Charman, W.N.; Porter, C.J. Intravenous dosing conditions may affect systemic clearance for highly lipophilic drugs: Implications for lymphatic transport and absolute bioavailability studies. J. Pharm. Sci. 2012, 101, 3540–3546. [Google Scholar] [CrossRef]
- Yadav, P.; McLeod, V.M.; Nowell, C.J.; Selby, L.I.; Johnston, A.P.R.; Kaminskas, L.M.; Trevaskis, N.L. Distribution of therapeutic proteins into thoracic lymph after intravenous administration is protein size-dependent and primarily occurs within the liver and mesentery. J. Control. Release 2018, 272, 17–28. [Google Scholar] [CrossRef]
- Cabrales, P.; Han, G.; Roche, C.; Nacharaju, P.; Friedman, A.J.; Friedman, J.M. Sustained release nitric oxide from long-lived circulating nanoparticles. Free Radic. Biol. Med. 2010, 49, 530–538. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Esparza, G.U.; Segura-Ibarra, V.; Cordero-Reyes, A.M.; Youker, K.A.; Serda, R.E.; Cruz-Solbes, A.S.; Amione-Guerra, J.; Yokoi, K.; Kirui, D.K.; Cara, F.E. A specifically designed nanoconstruct associates, internalizes, traffics in cardiovascular cells, and accumulates in failing myocardium: A new strategy for heart failure diagnostics and therapeutics. Eur. J. Heart Fail. 2016, 18, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Marrella, A.; Iafisco, M.; Adamiano, A.; Rossi, S.; Aiello, M.; Barandalla-Sobrados, M.; Carullo, P.; Miragoli, M.; Tampieri, A.; Scaglione, S. A combined low-frequency electromagnetic and fluidic stimulation for a controlled drug release from superparamagnetic calcium phosphate nanoparticles: Potential application for cardiovascular diseases. J. R. Soc. Interface 2018, 15, 20180236. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, J.T.; Somasuntharam, I.; Gray, W.D.; Shen, M.; Singer, J.M.; Wang, B.; Saafir, T.; Crawford, B.H.; Jiang, R.; Murthy, N. Bioactive nanoparticles improve calcium handling in failing cardiac myocytes. Nanomedicine 2015, 10, 3343–3357. [Google Scholar] [CrossRef]
- Avula, U.M.R.; Kim, G.; Lee, Y.-E.K.; Morady, F.; Kopelman, R.; Kalifa, J. Cell-specific nanoplatform-enabled photodynamic therapy for cardiac cells. Heart Rhythm. 2012, 9, 1504–1509. [Google Scholar] [CrossRef]
- Onwordi, E.N.; Gamal, A.; Zaman, A. Anticoagulant Therapy for Acute Coronary Syndromes. Interv. Cardiol. 2018, 13, 87–92. [Google Scholar] [CrossRef]
- Liu, G.; Li, L.; Huo, D.; Li, Y.; Wu, Y.; Zeng, L.; Cheng, P.; Xing, M.; Zeng, W.; Zhu, C. A VEGF delivery system targeting MI improves angiogenesis and cardiac function based on the tropism of MSCs and layer-by-layer self-assembly. Biomaterials 2017, 127, 117–131. [Google Scholar] [CrossRef] [PubMed]
- Majmudar, M.D.; Keliher, E.J.; Heidt, T.; Leuschner, F.; Truelove, J.; Sena, B.F.; Gorbatov, R.; Iwamoto, Y.; Dutta, P.; Wojtkiewicz, G. Monocyte-directed RNAi targeting CCR2 improves infarct healing in atherosclerosis-prone mice. Circulation 2013, 127, 2038–2046. [Google Scholar] [CrossRef]
- Ottersbach, A.; Mykhaylyk, O.; Heidsieck, A.; Eberbeck, D.; Rieck, S.; Zimmermann, K.; Breitbach, M.; Engelbrecht, B.; Brügmann, T.; Hesse, M. Improved heart repair upon myocardial infarction: Combination of magnetic nanoparticles and tailored magnets strongly increases engraftment of myocytes. Biomaterials 2018, 155, 176–190. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.-Y.; Yang, Y.-J.; Chang, C.-H.; Tang, A.C.; Liao, W.-Y.; Cheng, F.-Y.; Yeh, C.-S.; Lai, J.J.; Stayton, P.S.; Hsieh, P.C. Functionalized nanoparticles provide early cardioprotection after acute myocardial infarction. J. Control. Release 2013, 170, 287–294. [Google Scholar] [CrossRef]
- Nagaoka, K.; Matoba, T.; Mao, Y.; Nakano, Y.; Ikeda, G.; Egusa, S.; Tokutome, M.; Nagahama, R.; Nakano, K.; Sunagawa, K. A new therapeutic modality for acute myocardial infarction: Nanoparticle-mediated delivery of pitavastatin induces cardioprotection from ischemia-reperfusion injury via activation of PI3K/Akt pathway and anti-inflammation in a rat model. PLoS ONE 2015, 10, e0132451. [Google Scholar] [CrossRef]
- Kassim, S.H.; Li, H.; Bell, P.; Somanathan, S.; Lagor, W.; Jacobs, F.; Billheimer, J.; Wilson, J.M.; Rader, D.J. Adeno-associated virus serotype 8 gene therapy leads to significant lowering of plasma cholesterol levels in humanized mouse models of homozygous and heterozygous familial hypercholesterolemia. Hum. Gene Ther. 2013, 24, 19–26. [Google Scholar] [CrossRef]
- Greig, J.A.; Limberis, M.P.; Bell, P.; Chen, S.-J.; Calcedo, R.; Rader, D.J.; Wilson, J.M. Nonclinical pharmacology/toxicology study of AAV8. TBG. mLDLR and AAV8. TBG. hLDLR in a mouse model of homozygous familial hypercholesterolemia. Hum. Gene Ther. Clin. Dev. 2017, 28, 28–38. [Google Scholar] [CrossRef]
- Fitzgerald, K.; Frank-Kamenetsky, M.; Shulga-Morskaya, S.; Liebow, A.; Bettencourt, B.R.; Sutherland, J.E.; Hutabarat, R.M.; Clausen, V.A.; Karsten, V.; Cehelsky, J. Effect of an RNA interference drug on the synthesis of proprotein convertase subtilisin/kexin type 9 (PCSK9) and the concentration of serum LDL cholesterol in healthy volunteers: A randomised, single-blind, placebo-controlled, phase 1 trial. Lancet 2014, 383, 60–68. [Google Scholar] [CrossRef]
- Nicolau, C.; Le Pape, A.; Soriano, P.; Fargette, F.; Juhel, M.-F. In vivo expression of rat insulin after intravenous administration of the liposome-entrapped gene for rat insulin I. Proc. Natl. Acad. Sci. USA 1983, 80, 1068–1072. [Google Scholar] [CrossRef] [PubMed]
- Shifrin, A.; Auricchio, A.; Yu, Q.; Wilson, J.; Raper, S. Adenoviral vector-mediated insulin gene transfer in the mouse pancreas corrects streptozotocin-induced hyperglycemia. Gene Ther. 2001, 8, 1480–1489. [Google Scholar] [CrossRef]
- Mas, A.; Montané, J.; Anguela, X.M.; Muñoz, S.; Douar, A.M.; Riu, E.; Otaegui, P.; Bosch, F. Reversal of type 1 diabetes by engineering a glucose sensor in skeletal muscle. Diabetes 2006, 55, 1546–1553. [Google Scholar] [CrossRef]
- Oh, T.K.; Li, M.Z.; Kim, S.T. Gene therapy for diabetes mellitus in rats by intramuscular injection of lentivirus containing insulin gene. Diabetes Res. Clin. Pract. 2006, 71, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Muzzin, P.; Eisensmith, R.C.; Copeland, K.C.; Woo, S.L. Hepatic insulin gene expression as treatment for type 1 diabetes mellitus in rats. Mol. Endocrinol. 1997, 11, 833–837. [Google Scholar] [CrossRef][Green Version]
- Thule, P.; Liu, J. Regulated hepatic insulin gene therapy of STZ-diabetic rats. Gene Ther. 2000, 7, 1744–1752. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Park, Y.M.; Woo, S.; Lee, G.T.; Ko, J.Y.; Lee, Y.; Zhao, Z.S.; Kim, H.J.; Ahn, C.W.; Cha, B.S.; Kim, K.S.; et al. Safety and efficacy of adeno-associated viral vector-mediated insulin gene transfer via portal vein to the livers of streptozotocin-induced diabetic Sprague-Dawley rats. J. Gene Med. 2005, 7, 621–629. [Google Scholar] [CrossRef]
- Cheung, A.T.; Dayanandan, B.; Lewis, J.T.; Korbutt, G.S.; Rajotte, R.V.; Bryer-Ash, M.; Boylan, M.O.; Wolfe, M.M.; Kieffer, T.J. Glucose-dependent insulin release from genetically engineered K cells. Science 2000, 290, 1959–1962. [Google Scholar] [CrossRef]
- Encina, G.; Ezquer, F.; Conget, P.; Israel, Y. Insulin is secreted upon glucose stimulation by both gastrointestinal enteroendocrine K-cells and L-cells engineered with the preproinsulin gene. Biol. Res. 2011, 44, 301–305. [Google Scholar] [CrossRef]
- Thulé, P.M.; Campbell, A.G.; Jia, D.; Lin, Y.; You, S.; Paveglio, S.; Olson, D.E.; Kozlowski, M. Long-term glycemic control with hepatic insulin gene therapy in streptozotocin-diabetic mice. J. Gene Med. 2015, 17, 141–152. [Google Scholar] [CrossRef]
- Chatenoud, L.; Primo, J.; Bach, J.-F. CD3 antibody-induced dominant self tolerance in overtly diabetic NOD mice. J. Immunol. 1997, 158, 2947–2954. [Google Scholar] [PubMed]
- von Herrath, M.G.; Coon, B.; Wolfe, T.; Chatenoud, L. Nonmitogenic CD3 antibody reverses virally induced (rat insulin promoter-lymphocytic choriomeningitis virus) autoimmune diabetes without impeding viral clearance. J. Immunol. 2002, 168, 933–941. [Google Scholar] [CrossRef] [PubMed]
- Herold, K.C.; Hagopian, W.; Auger, J.A.; Poumian-Ruiz, E.; Taylor, L.; Donaldson, D.; Gitelman, S.E.; Harlan, D.M.; Xu, D.; Zivin, R.A. Anti-CD3 monoclonal antibody in new-onset type 1 diabetes mellitus. N. Engl. J. Med. 2002, 346, 1692–1698. [Google Scholar] [CrossRef]
- Herold, K.C.; Gitelman, S.E.; Ehlers, M.R.; Gottlieb, P.A.; Greenbaum, C.J.; Hagopian, W.; Boyle, K.D.; Keyes-Elstein, L.; Aggarwal, S.; Phippard, D. Teplizumab (anti-CD3 mAb) treatment preserves C-peptide responses in patients with new-onset type 1 diabetes in a randomized controlled trial: Metabolic and immunologic features at baseline identify a subgroup of responders. Diabetes 2013, 62, 3766–3774. [Google Scholar] [CrossRef]
- Herold, K.C.; Gitelman, S.E.; Masharani, U.; Hagopian, W.; Bisikirska, B.; Donaldson, D.; Rother, K.; Diamond, B.; Harlan, D.M.; Bluestone, J.A. A single course of anti-CD3 monoclonal antibody hOKT3γ1 (Ala-Ala) results in improvement in C-peptide responses and clinical parameters for at least 2 years after onset of type 1 diabetes. Diabetes 2005, 54, 1763–1769. [Google Scholar] [CrossRef]
- Zhang, Y.C.; Bui, J.D.; Shen, L.; Phillips, M.I. Antisense inhibition of β1-adrenergic receptor mRNA in a single dose produces a profound and prolonged reduction in high blood pressure in spontaneously hypertensive rats. Circulation 2000, 101, 682–688. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Li, X.; Zheng, S.; Chen, Y.; Chen, C.; Wang, J.; Tong, H.; Zhou, L.; Yang, J.; Zeng, C. Downregulation of Renal G Protein–Coupled Receptor Kinase Type 4 Expression via Ultrasound-Targeted Microbubble Destruction Lowers Blood Pressure in Spontaneously Hypertensive Rats. J. Am. Heart Assoc. 2016, 5, e004028. [Google Scholar] [CrossRef]
- Paulis, L.; Franke, H.; Simko, F. Gene therapy for hypertension. Expert Opin. Biol. Ther. 2017, 17, 1345–1361. [Google Scholar] [CrossRef] [PubMed]
- Lázár, E.; Sadek, H.A.; Bergmann, O. Cardiomyocyte renewal in the human heart: Insights from the fall-out. Eur. Heart J. 2017, 38, 2333–2342. [Google Scholar] [CrossRef]
- Torchilin, V.; Klibanov, A.; Huang, L.; O’Donnell, S.; Nossiff, N.; Khaw, B. Targeted accumulation of polyethylene glycol-coated immunoliposomes in infarcted rabbit myocardium. FASEB J. 1992, 6, 2716–2719. [Google Scholar] [CrossRef]
- Scott, R.C.; Rosano, J.M.; Ivanov, Z.; Wang, B.; Chong, P.L.-G.; Issekutz, A.C.; Crabbe, D.L.; Kiani, M.F. Targeting VEGF-encapsulated immunoliposomes to MI heart improves vascularity and cardiac function. FASEB J. 2009, 23, 3361–3367. [Google Scholar] [CrossRef] [PubMed]
- Ko, Y.; Hartner, W.; Kale, A.; Torchilin, V. Gene delivery into ischemic myocardium by double-targeted lipoplexes with anti-myosin antibody and TAT peptide. Gene Ther. 2009, 16, 52–59. [Google Scholar] [CrossRef]
- Dvir, T.; Bauer, M.; Schroeder, A.; Tsui, J.H.; Anderson, D.G.; Langer, R.; Liao, R.; Kohane, D.S. Nanoparticles targeting the infarcted heart. Nano Lett. 2011, 11, 4411–4414. [Google Scholar] [CrossRef]
- Pannu, H.K.; Oliphant, M. The subperitoneal space and peritoneal cavity: Basic concepts. Abdom. Imaging 2015, 40, 2710–2722. [Google Scholar] [CrossRef]
- Al Shoyaib, A.; Archie, S.R.; Karamyan, V.T. Intraperitoneal Route of Drug Administration: Should it Be Used in Experimental Animal Studies? Pharm. Res. 2020, 37, 12. [Google Scholar] [CrossRef] [PubMed]
- Michailova, K.N.; Usunoff, K.G. Serosal Membranes (Pleura, Pericardium, Peritoneum): Normal Structure, Development and Experimental Pathology; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2006; Volume 183. [Google Scholar]
- Lee, G.; Han, S.; Inocencio, I.; Cao, E.; Hong, J.; Phillips, A.R.J.; Windsor, J.A.; Porter, C.J.H.; Trevaskis, N.L. Lymphatic Uptake of Liposomes after Intraperitoneal Administration Primarily Occurs via the Diaphragmatic Lymphatics and is Dependent on Liposome Surface Properties. Mol. Pharm. 2019, 16, 4987–4999. [Google Scholar] [CrossRef]
- Torres, I.; Litterst, C.; Guarino, A. Transport of model compounds across the peritoneal membrane in the rat. Pharmacology 1978, 17, 330–340. [Google Scholar] [CrossRef]
- Turner, P.V.; Brabb, T.; Pekow, C.; Vasbinder, M.A. Administration of substances to laboratory animals: Routes of administration and factors to consider. J. Am. Assoc. Lab. Anim. Sci. 2011, 50, 600–613. [Google Scholar] [PubMed]
- Alberts, D.S.; Liu, P.; Hannigan, E.V.; O’Toole, R.; Williams, S.D.; Young, J.A.; Franklin, E.W.; Clarke-Pearson, D.L.; Malviya, V.K.; DuBeshter, B. Intraperitoneal cisplatin plus intravenous cyclophosphamide versus intravenous cisplatin plus intravenous cyclophosphamide for stage III ovarian cancer. N. Engl. J. Med. 1996, 335, 1950–1955. [Google Scholar] [CrossRef]
- Armstrong, D.K.; Bundy, B.; Wenzel, L.; Huang, H.Q.; Baergen, R.; Lele, S.; Copeland, L.J.; Walker, J.L.; Burger, R.A. Intraperitoneal cisplatin and paclitaxel in ovarian cancer. N. Engl. J. Med. 2006, 354, 34–43. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute. NCI Clinical Announcement on Intraperitoneal Chemotherapy for Ovarian Cancer. 2006. Available online: https://ctep.cancer.gov/highlights/docs/clin_annc_010506.pdf (accessed on 1 May 2021).
- Maranhão, R.C.; Guido, M.C.; de Lima, A.D.; Tavares, E.R.; Marques, A.F.; de Melo, M.D.T.; Nicolau, J.C.; Salemi, V.M.; Kalil-Filho, R. Methotrexate carried in lipid core nanoparticles reduces myocardial infarction size and improves cardiac function in rats. Int. J. Nanomed. 2017, 12, 3767. [Google Scholar] [CrossRef] [PubMed]
- Losordo, D.W.; Vale, P.R.; Hendel, R.C.; Milliken, C.E.; Fortuin, F.D.; Cummings, N.; Schatz, R.A.; Asahara, T.; Isner, J.M.; Kuntz, R.E. Phase 1/2 placebo-controlled, double-blind, dose-escalating trial of myocardial vascular endothelial growth factor 2 gene transfer by catheter delivery in patients with chronic myocardial ischemia. Circulation 2002, 105, 2012–2018. [Google Scholar] [CrossRef] [PubMed]
- Reilly, J.P.; Grise, M.A.; Fortuin, F.D.; Vale, P.R.; Schaer, G.L.; Lopez, J.; Van Camp, J.R.; Henry, T.; Richenbacher, W.E.; Losordo, D.W.; et al. Long-term (2-year) clinical events following transthoracic intramyocardial gene transfer of VEGF-2 in no-option patients. J. Interv. Cardiol. 2005, 18, 27–31. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tessier, N.; Moawad, F.; Amri, N.; Brambilla, D.; Martel, C. Focus on the Lymphatic Route to Optimize Drug Delivery in Cardiovascular Medicine. Pharmaceutics 2021, 13, 1200. https://doi.org/10.3390/pharmaceutics13081200
Tessier N, Moawad F, Amri N, Brambilla D, Martel C. Focus on the Lymphatic Route to Optimize Drug Delivery in Cardiovascular Medicine. Pharmaceutics. 2021; 13(8):1200. https://doi.org/10.3390/pharmaceutics13081200
Chicago/Turabian StyleTessier, Nolwenn, Fatma Moawad, Nada Amri, Davide Brambilla, and Catherine Martel. 2021. "Focus on the Lymphatic Route to Optimize Drug Delivery in Cardiovascular Medicine" Pharmaceutics 13, no. 8: 1200. https://doi.org/10.3390/pharmaceutics13081200
APA StyleTessier, N., Moawad, F., Amri, N., Brambilla, D., & Martel, C. (2021). Focus on the Lymphatic Route to Optimize Drug Delivery in Cardiovascular Medicine. Pharmaceutics, 13(8), 1200. https://doi.org/10.3390/pharmaceutics13081200