
Effects of 1α,25-Dihydroxyvitamin D3 on the Pharmacokinetics of Procainamide and Its Metabolite N-Acetylprocainamide, Organic Cation Transporter Substrates, in Rats with PBPK Modeling Approach

 , , , and
, , , and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Experimental Animals and Treatment of Rats with 1,25(OH)2D3
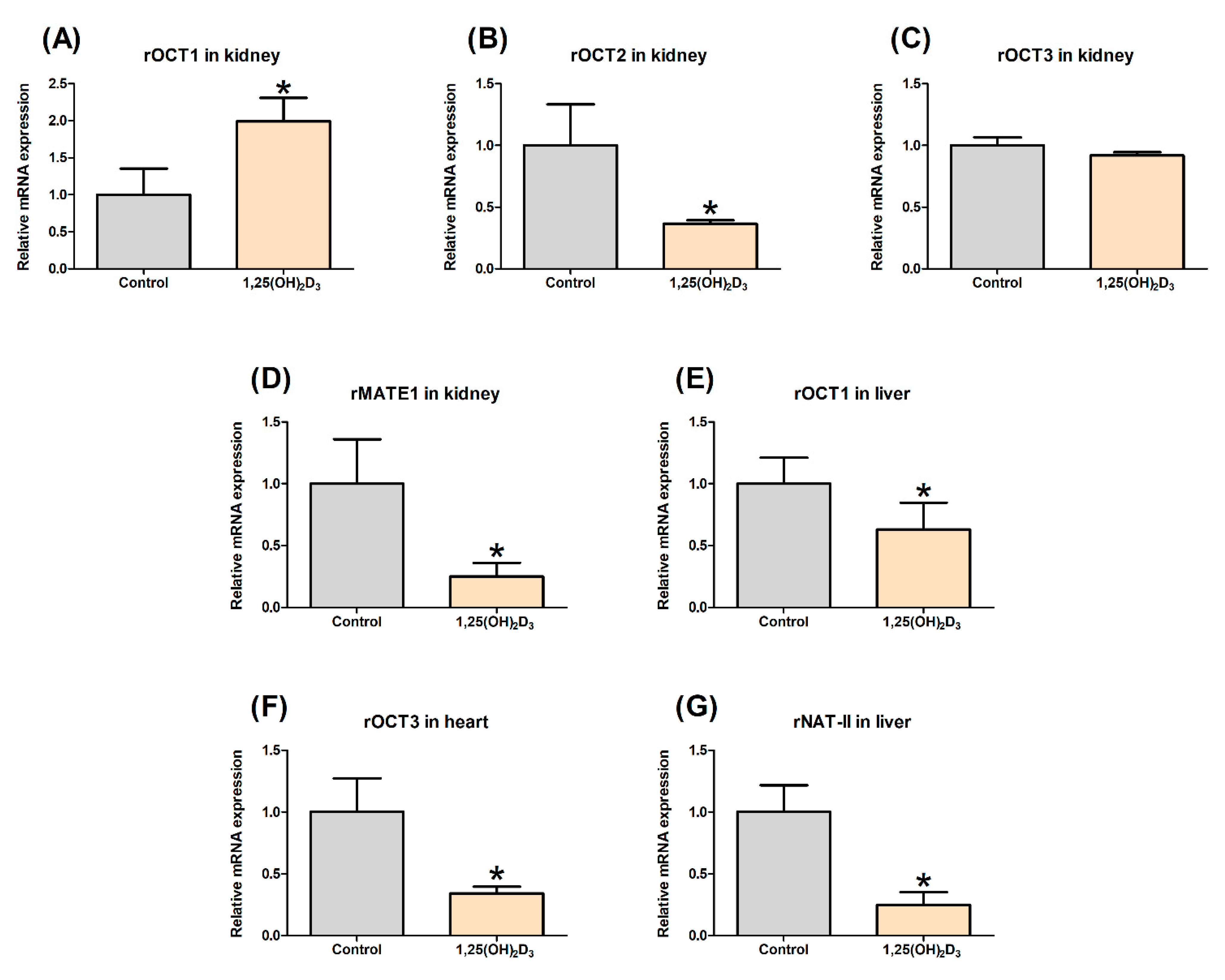
2.3. Effect of 1,25(OH)2D3 on the Gene Expression of rOCTs, rMATE1, and rNAT-II in Rat Tissues
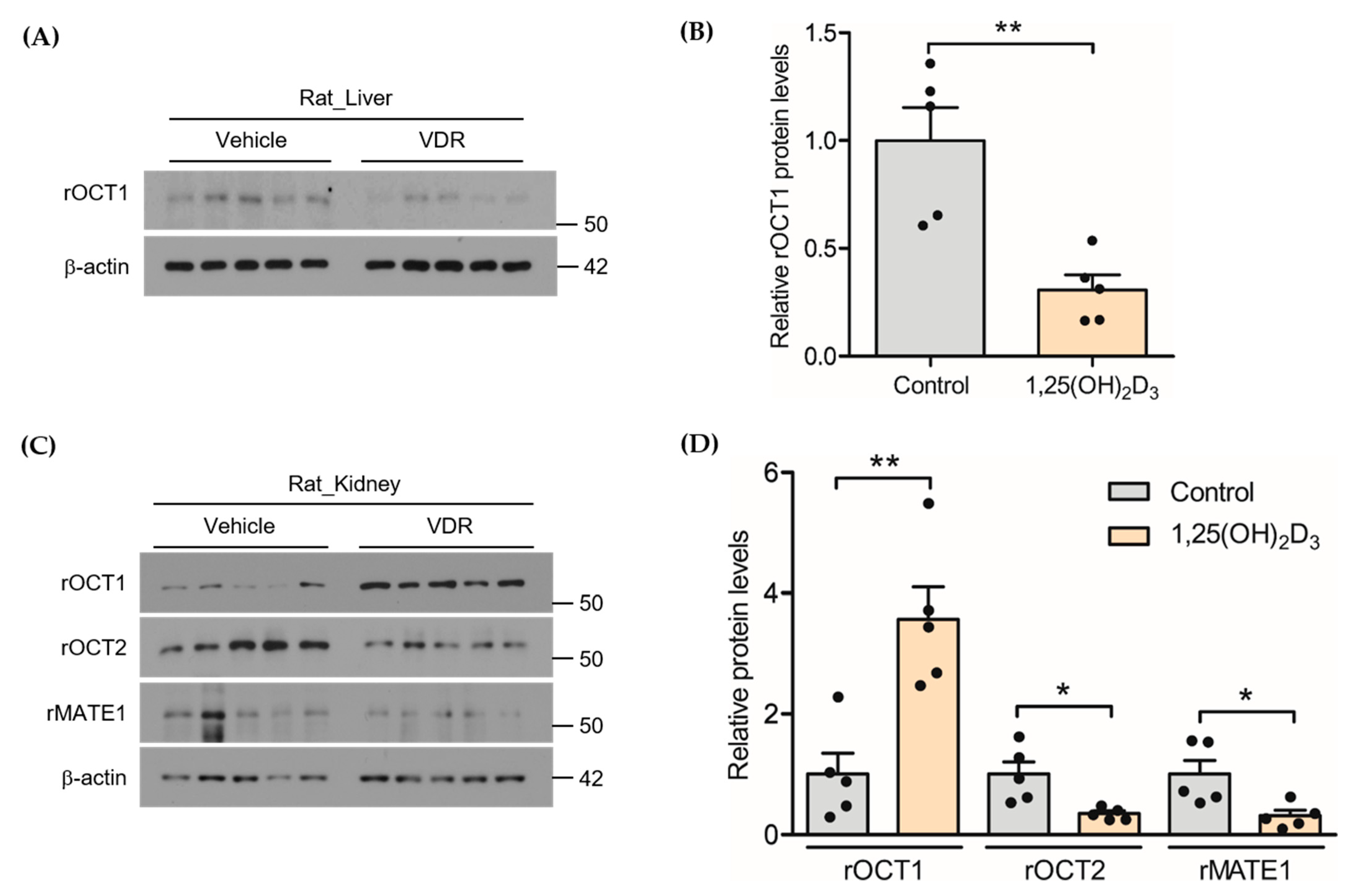
2.4. Effect of 1,25(OH)2D3 on the Protein Expression of rOCTs and rMATE1 in Rat Liver and Kidney
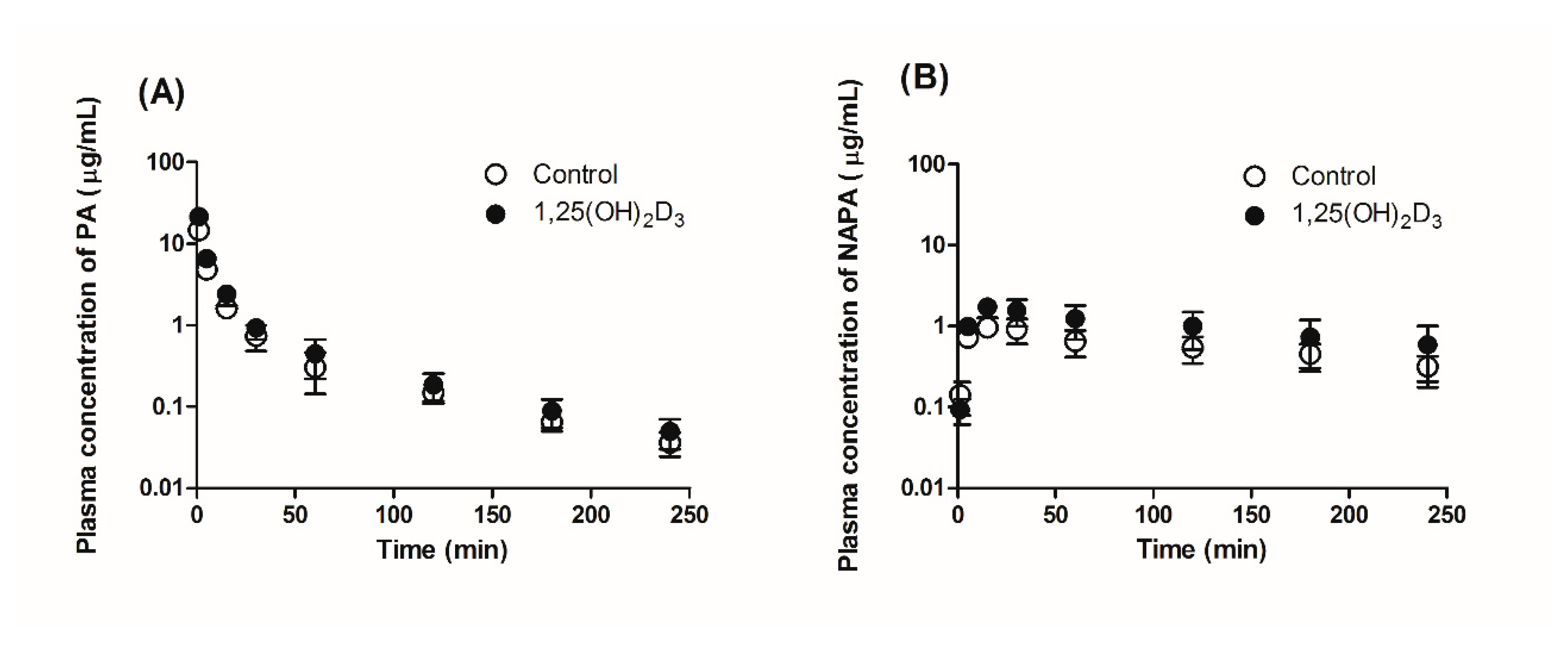
2.5. Effect of 1,25(OH)2D3 on the Pharmacokinetics of PA and NAPA in Rats
2.6. Effects of 1,25(OH)2D3 on the Urinary and Fecal Excretion of PA and NAPA in Rats
2.7. Effects of 1,25(OH)2D3 on the Tissue Distribution of PA and NAPA at Steady State
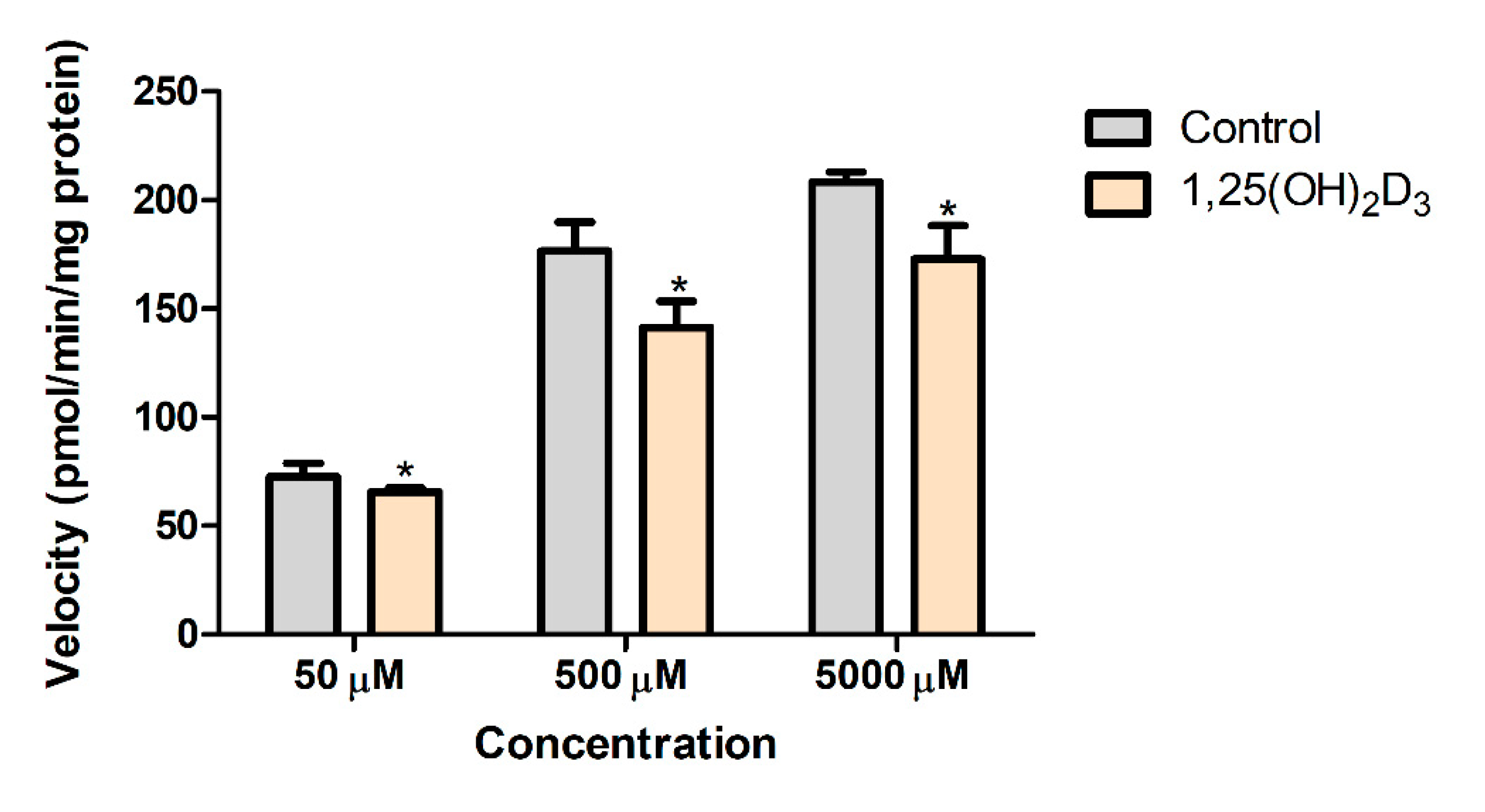
2.8. Effects of 1,25(OH)2D3 on the In Vitro Metabolic Conversion of PA into NAPA in Rat Liver S9 Fractions
2.9. Determination of the Free Fraction of PA in Plasma and Incubation Mixture of Rat Liver S9 Fractions
2.10. UHPLC Analysis
2.11. Application of Physiologically-Based Pharmacokinetic Modeling for PA and NAPA in the Absence and Presence of 1,25(OH)2D3 Treatment in Rats
2.12. Statistical Analysis
3. Results
3.1. Effects of 1,25(OH)2D3 on the mRNA/Protein Expression of OCT and MATE Transporters in Rats
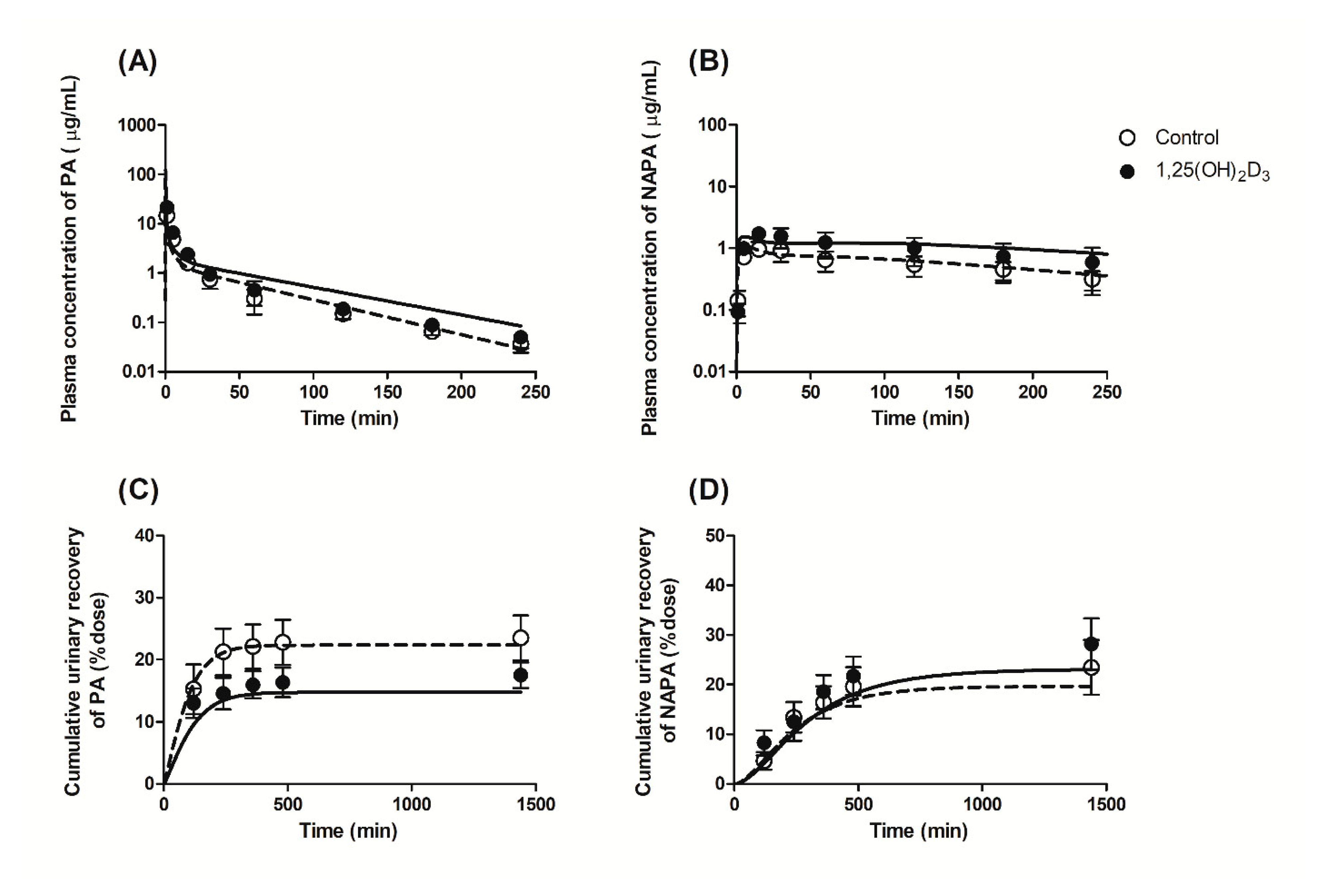
3.2. Effects of 1,25(OH)2D3 on the Pharmacokinetics of PA and NAPA Following the Intravenous Administration of PA HCl to Rats
3.3. Effects of 1,25(OH)2D3 on the Urinary and Fecal Excretion of PA and NAPA in Rats
3.4. Effects of 1,25(OH)2D3 on the Tissue Distribution of PA and NAPA at Steady State in Rats
3.5. Effects of 1,25(OH)2D3 on the In Vitro Metabolic Conversion of PA into NAPA in Rat Liver S9 Fractions
3.6. Free Fraction of PA in the Plasma and the Incubation Mixture of Rat Liver S9 Fractions
3.7. Application of a PBPK Model for the Pharmacokinetics of PA and NAPA after Intravenous Administration of PA HCl in the Absence or Presence of 1,25(OH)2D3 Treatment
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A
References
- Atilano-Roque, A.; Roda, G.; Fogueri, U.; Kiser, J.J.; Joy, M.S. Effect of disease pathologies on transporter expression and function. J. Clin. Pharmacol. 2016, 56, S205–S221. [Google Scholar] [CrossRef]
- Cressman, A.M.; Petrovic, V.; Piquette-Miller, M. Inflammation-mediated changes in drug transporter expression/activity: Implications for therapeutic drug response. Expert Rev. Clin. Pharmacol. 2012, 5, 69–89. [Google Scholar] [CrossRef]
- Nieth, C.; Lage, H. Induction of the ABC-transporters Mdr1/P-gp (Abcb1), Mrp1 (Abcc1), and Bcrp (Abcg2) during establishment of multidrug resistance following exposure to mitoxantrone. J. Chemother. 2005, 17, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Williamson, B.; Dooley, K.E.; Zhang, Y.; Back, D.J.; Owen, A. Induction of influx and efflux transporters and cytochrome P450 3A4 in primary human hepatocytes by rifampin, rifabutin, and rifapentine. Antimicrob. Agents Chemother. 2013, 57, 6366–6369. [Google Scholar] [CrossRef] [PubMed]
- Tirona, R.G. Molecular mechanisms of drug transporter regulation. In Drug Transporters; Springer: Berlin/Heidelberg, Germany, 2011; pp. 373–402. [Google Scholar]
- Urquhart, B.L.; Tirona, R.G.; Kim, R.B. Nuclear receptors and the regulation of drug-metabolizing enzymes and drug transporters: Implications for interindividual variability in response to drugs. J. Clin. Pharmacol. 2007, 47, 566–578. [Google Scholar] [CrossRef] [PubMed]
- Moore, D.D.; Kato, S.; Xie, W.; Mangelsdorf, D.J.; Schmidt, D.R.; Xiao, R.; Kliewer, S.A. International Union of Pharmacology. LXII. The NR1H and NR1I receptors: Constitutive androstane receptor, pregnene X receptor, farnesoid X receptor α, farnesoid X receptor β, liver X receptor α, liver X receptor β, and vitamin D receptor. Pharmacol. Rev. 2006, 58, 742–759. [Google Scholar] [CrossRef]
- Lee, K.-Y.; Choi, H.-S.; Choi, H.-S.; Chung, K.Y.; Lee, B.-J.; Maeng, H.-J.; Seo, M.-D. Quercetin directly interacts with vitamin D Receptor (VDR): Structural implication of VDR activation by quercetin. Biomol. Ther. 2016, 24, 191. [Google Scholar] [CrossRef]
- Thummel, K.E.; Brimer, C.; Yasuda, K.; Thottassery, J.; Senn, T.; Lin, Y.; Ishizuka, H.; Kharasch, E.; Schuetz, J.; Schuetz, E. Transcriptional control of intestinal cytochrome P-4503A by 1α, 25-dihydroxy vitamin D3. Mol. Pharmacol. 2001, 60, 1399–1406. [Google Scholar] [CrossRef] [PubMed]
- Drocourt, L.; Ourlin, J.-C.; Pascussi, J.-M.; Maurel, P.; Vilarem, M.-J. Expression of cyp3a4, cyp2b6, and cyp2c9 is regulated by the vitamin d receptor pathway in primary human hepatocytes. J. Biol. Chem. 2002, 277, 25125–25132. [Google Scholar] [CrossRef]
- Saeki, M.; Kurose, K.; Tohkin, M.; Hasegawa, R. Identification of the functional vitamin D response elements in the human MDR1 gene. Biochem. Pharmacol. 2008, 76, 531–542. [Google Scholar] [CrossRef]
- Chow, E.C.; Sun, H.; Khan, A.A.; Groothuis, G.M.; Pang, K.S. Effects of 1α, 25-dihydroxyvitamin D3 on transporters and enzymes of the rat intestine and kidney in vivo. Biopharm. Drug Dispos. 2010, 31, 91–108. [Google Scholar]
- Kim, Y.C.; Kim, I.-B.; Noh, C.-K.; Quach, H.P.; Yoon, I.-S.; Chow, E.C.; Kim, M.; Jin, H.-E.; Cho, K.H.; Chung, S.-J. Effects of 1α, 25-dihydroxyvitamin D3, the natural vitamin D receptor ligand, on the pharmacokinetics of cefdinir and cefadroxil, organic anion transporter substrates, in rat. J. Pharm. Sci. 2014, 103, 3793–3805. [Google Scholar] [CrossRef]
- Giardina, E. Procainamide: Clinical pharmacology and efficacy against ventricular arrhythmias. Ann. N. Y. Acad. Sci. 1984, 432, 177–188. [Google Scholar] [CrossRef]
- Trujillo, T.C.; Nolan, P.E. Antiarrhythmic agents. Drug Saf. 2000, 23, 509–532. [Google Scholar] [CrossRef] [PubMed]
- Kerb, R. Implications of genetic polymorphisms in drug transporters for pharmacotherapy. Cancer Lett. 2006, 234, 4–33. [Google Scholar] [CrossRef]
- Hasannejad, H.; Takeda, M.; Narikawa, S.; Huang, X.-L.; Enomoto, A.; Taki, K.; Niwa, T.; Jung, S.H.; Onozato, M.L.; Tojo, A.; et al. Human organic cation transporter 3 mediates the transport of antiarrhythmic drugs. Eur. J. Pharmacol. 2004, 499, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Tanihara, Y.; Masuda, S.; Sato, T.; Katsura, T.; Ogawa, O.; Inui, K.-i. Substrate specificity of MATE1 and MATE2-K, human multidrug and toxin extrusions/H+-organic cation antiporters. Biochem. Pharmacol. 2007, 74, 359–371. [Google Scholar] [CrossRef]
- Dreyfuss, J.; Bigger Jr, J.T.; Cohen, A.I.; Schreiber, E.C. Metabolism of procainamide in rhesus monkey and man. Clin. Pharmacol. Ther. 1972, 13, 366–371. [Google Scholar] [CrossRef]
- Karlsson, E. Clinical pharmacokinetics of procainamide. Clin. Pharmacokinet. 1978, 3, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Campbell, T.; Williams, K. Therapeutic drug monitoring: Antiarrhythmic drugs. Br. J. Clin. Pharmacol. 1998, 46, 307–319. [Google Scholar] [CrossRef]
- Jeong, Y.-S.; Balla, A.; Chun, K.-H.; Chung, S.-J.; Maeng, H.-J. Physiologically-based pharmacokinetic modeling for drug-drug interactions of procainamide and N-acetylprocainamide with cimetidine, an inhibitor of rOCT2 and rMATE1, in rats. Pharmaceutics 2019, 11, 108. [Google Scholar] [CrossRef]
- Koch-Weser, J.; Klein, S.W. Procainamide dosage schedules, plasma concentrations, and clinical effects. JAMA 1971, 215, 1454–1460. [Google Scholar] [CrossRef]
- Maeng, H.J.; Durk, M.R.; Chow, E.C.; Ghoneim, R.; Pang, K.S. 1α, 25-Dihydroxyvitamin D3 on intestinal transporter function: Studies with the rat everted intestinal sac. Biopharm. Drug Dispos. 2011, 32, 112–125. [Google Scholar] [CrossRef]
- Yoon, I.-S.; Son, J.-H.; Kim, S.-B.; Choi, M.-K.; Maeng, H.-J. Effects of 1α, 25-dihydroxyvitamin D3 on intestinal absorption and disposition of adefovir dipivoxil and its metabolite, adefovir, in rats. Biol. Pharm. Bull. 2015, 38, 1732–1737. [Google Scholar] [CrossRef]
- Maeng, H.-J.; Chapy, H.; Zaman, S.; Pang, K.S. Effects of 1α, 25-dihydroxyvitamin D3 on transport and metabolism of adefovir dipivoxil and its metabolites in Caco-2 cells. Eur. J. Pharm. Sci. 2012, 46, 149–166. [Google Scholar] [CrossRef]
- Li, X.; Wang, X.; Li, Y.; Yuan, M.; Zhu, J.; Su, X.; Yao, X.; Fan, X.; Duan, Y. Effect of exposure to acute and chronic high-altitude hypoxia on the activity and expression of CYP1A2, CYP2D6, CYP2C9, CYP2C19 and NAT2 in rats. Pharmacology 2014, 93, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Chow, E.C. Biological roles of the vitamin D receptor in the regulation of transporters and enzymes on drug disposition, including cytochrome P450 (CYP7A1) on cholesterol metabolism. Ph.D. Thesis, University of Toronto, Toronto, ON, Canada, 2013. [Google Scholar]
- Khan, A.A.; Chow, E.C.; Porte, R.J.; Pang, K.S.; Groothuis, G.M. 1α,25-dihydroxyvitamin D3 mediates down regulation of HNF4α, CYP7A1 and NTCP in human but not in rat liver. Ph.D. Thesis, University of Groningen, Groningen, The Netherlands, 2009. [Google Scholar]
- Nakanishi, T.; Haruta, T.; Shirasaka, Y.; Tamai, I. Organic cation transporter-mediated renal secretion of ipratropium and tiotropium in rats and humans. Drug Metab. Dispos. 2011, 39, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Lessard, É.; Fortin, A.; Coquet, A.; Bélanger, P.-M.; Hamelin, B.A.; Turgeon, J. Improved high-performance liquid chromatographic assay for the determination of procainamide and its N-acetylated metabolite in plasma: Application to a single-dose pharmacokinetic study. J. Chromatogr. Sci. 1998, 36, 49–54. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wu, W.-N.; McKown, L.A. In vitro drug metabolite profiling using hepatic S9 and human liver microsomes. In Optimization in Drug Discovery; Springer: Berlin/Heidelberg, Germany, 2004; pp. 163–184. [Google Scholar]
- Wu, X.; Huang, W.; Ganapathy, M.E.; Wang, H.; Kekuda, R.; Conway, S.J.; Leibach, F.H.; Ganapathy, V. Structure, function, and regional distribution of the organic cation transporter OCT3 in the kidney. Am. J. Physiol. Ren. Fluid Electrolyte Physiol. 2000, 279, F449–F458. [Google Scholar] [CrossRef] [PubMed]
- König, J.; Müller, F.; Fromm, M.F. Transporters and drug-drug interactions: Important determinants of drug disposition and effects. Pharmacol. Rev. 2013, 65, 944–966. [Google Scholar] [CrossRef]
- Morrissey, K.M.; Stocker, S.L.; Wittwer, M.B.; Xu, L.; Giacomini, K.M. Renal transporters in drug development. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 503–529. [Google Scholar] [CrossRef] [PubMed]
- Shitara, Y.; Horie, T.; Sugiyama, Y. Transporters as a determinant of drug clearance and tissue distribution. Eur. J. Pharm. Sci. 2006, 27, 425–446. [Google Scholar] [CrossRef] [PubMed]
- Dave, R.A.; Morris, M.E. Semi-mechanistic kidney model incorporating physiologically-relevant fluid reabsorption and transporter-mediated renal reabsorption: Pharmacokinetics of γ-hydroxybutyric acid and l-lactate in rats. J. Pharmacokinet. Pharmacodyn. 2015, 42, 497–513. [Google Scholar] [CrossRef]
- Basit, A.; Radi, Z.; Vaidya, V.S.; Karasu, M.; Prasad, B. Kidney cortical transporter expression across species using quantitative proteomics. Drug Metab. Dispos. 2019, 47, 802–808. [Google Scholar] [CrossRef]
- Ishiguro, N.; Saito, A.; Yokoyama, K.; Morikawa, M.; Igarashi, T.; Tamai, I. Transport of the dopamine D2 agonist pramipexole by rat organic cation transporters OCT1 and OCT2 in kidney. Drug Metab. Dispos. 2005, 33, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Goralski, K.B.; Lou, G.; Prowse, M.T.; Gorboulev, V.; Volk, C.; Koepsell, H.; Sitar, D.S. The cation transporters rOCT1 and rOCT2 interact with bicarbonate but play only a minor role for amantadine uptake into rat renal proximal tubules. J. Pharmacol. Exp. Ther. 2002, 303, 959–968. [Google Scholar] [CrossRef] [PubMed]
- Umehara, K.-I.; Iwatsubo, T.; Noguchi, K.; Usui, T.; Kamimura, H. Effect of cationic drugs on the transporting activity of human and rat OCT/Oct 1–3 in vitro and implications for drug–drug interactions. Xenobiotica 2008, 38, 1203–1218. [Google Scholar] [CrossRef]
- D’Argenio, D.Z.; Schumitzky, A.; Wang, X. ADAPT 5 User’s Guide: Pharmacokinetic/Pharmacodynamic Systems Analysis Software; Biomedical Simulations Resource: Los Angeles, CA, USA, 2009. [Google Scholar]
- Rodgers, T.; Leahy, D.; Rowland, M. Physiologically based pharmacokinetic modeling 1: Predicting the tissue distribution of moderate-to-strong bases. J. Pharm. Sci. 2005, 94, 1259–1276. [Google Scholar] [CrossRef]
- Aiba, T.; Susa, M.; Fukumori, S.; Hashimoto, Y. The effects of culture conditions on CYP3A4 and MDR1 mRNA induction by 1α, 25-dihydroxyvitamin D3 in human intestinal cell lines, Caco-2 and LS180. Drug Metab. Pharmacokinet. 2005, 20, 268–274. [Google Scholar] [CrossRef]
- Chow, E.C.; Durk, M.R.; Cummins, C.L.; Pang, K.S. 1α, 25-Dihydroxyvitamin D3 up-regulates P-glycoprotein via the vitamin D receptor and not farnesoid X receptor in both fxr (−/−) and fxr (+/+) mice and increased renal and brain efflux of digoxin in mice in vivo. J. Pharmacol. Exp. Ther. 2011, 337, 846–859. [Google Scholar] [CrossRef]
- Fan, J.; Liu, S.; Du, Y.; Morrison, J.; Shipman, R.; Pang, K.S. Up-regulation of transporters and enzymes by the vitamin D receptor ligands, 1α, 25-dihydroxyvitamin D3 and vitamin D analogs, in the Caco-2 cell monolayer. J. Pharmacol. Exp. Ther. 2009, 330, 389–402. [Google Scholar] [CrossRef]
- Chow, E.C.; Maeng, H.J.; Liu, S.; Khan, A.A.; Groothuis, G.M.; Pang, K.S. 1α, 25-Dihydroxyvitamin D3 triggered vitamin D receptor and farnesoid X receptor-like effects in rat intestine and liver in vivo. Biopharm. Drug Dispos. 2009, 30, 457–475. [Google Scholar] [CrossRef]
- Urakami, Y.; Okuda, M.; Saito, H.; Inui, K.-i. Hormonal regulation of organic cation transporter OCT2 expression in rat kidney. FEBS Lett. 2000, 473, 173–176. [Google Scholar] [CrossRef]
- Asaka, J.-i.; Terada, T.; Okuda, M.; Katsura, T.; Inui, K.-i. Androgen receptor is responsible for rat organic cation transporter 2 gene regulation but not for rOCT1 and rOCT3. Pharm. Res. 2006, 23, 697–704. [Google Scholar] [CrossRef] [PubMed]
- Shu, Y.; Bello, C.L.; Mangravite, L.M.; Feng, B.; Giacomini, K.M. Functional characteristics and steroid hormone-mediated regulation of an organic cation transporter in Madin-Darby canine kidney cells. J. Pharmacol. Exp. Ther. 2001, 299, 392–398. [Google Scholar] [PubMed]
- Takano, M.; Kato, M.; Takayama, A.; Yasuhara, M.; Inui, K.-i.; Hori, R. Transport of procainamide in a kidney epithelial cell line LLC-PK1. Biochim. Biophys. Acta BBA Biomembr. 1992, 1108, 133–139. [Google Scholar] [CrossRef]
- Zhang, L.; Schaner, M.E.; Giacomini, K.M. Functional characterization of an organic cation transporter (hOCT1) in a transiently transfected human cell line (HeLa). J. Pharmacol. Exp. Ther. 1998, 286, 354–361. [Google Scholar]
- Shitara, Y.; Maeda, K.; Ikejiri, K.; Yoshida, K.; Horie, T.; Sugiyama, Y. Clinical significance of organic anion transporting polypeptides (OATPs) in drug disposition: Their roles in hepatic clearance and intestinal absorption. Biopharm. Drug Dispos. 2013, 34, 45–78. [Google Scholar] [CrossRef]
- Mol, W.E.; Müller, M.; Kurz, G.; Meijer, D.K. Investigations on the hepatic uptake systems for organic cations with a photoaffinity probe of procainamide ethobromide. Biochem. Pharmacol. 1992, 43, 2217–2226. [Google Scholar] [CrossRef]
- Jamei, M.; Marciniak, S.; Feng, K.; Barnett, A.; Tucker, G.; Rostami-Hodjegan, A. The Simcyp® population-based ADME simulator. Expert Opin. Drug Metab. Toxicol. 2009, 5, 211–223. [Google Scholar] [CrossRef]
- Jeong, Y.-S.; Yim, C.-S.; Ryu, H.-M.; Noh, C.-K.; Song, Y.-K.; Chung, S.-J. Estimation of the minimum permeability coefficient in rats for perfusion-limited tissue distribution in whole-body physiologically-based pharmacokinetics. Eur. J. Pharm. Biopharm. 2017, 115, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Follman, K.E.; Dave, R.A.; Morris, M.E. Effects of renal impairment on transporter-mediated renal reabsorption of drugs and renal drug–drug interactions: A simulation-based study. Biopharm. Drug Dispos. 2018, 39, 218–231. [Google Scholar] [CrossRef] [PubMed]
- Khurana, I. Excretory system. In Textbook of Human Physiology for Dental Students, 2nd ed.; Elsevier Health Sciences APAC: Oxford, UK, 2014; pp. 280–281. [Google Scholar]
- Lote, C.J. Summary of the principal reabsorptive and secretory processes in the nephron segments. In Principles of Renal Physiology; Springer: Berlin/Heidelberg, Germany, 2012; pp. 161–165. [Google Scholar]
- Lash, L. Principles and methods of renal toxicology. In Principles and Methods of Toxicology, 5th ed.; Taylor & Francis: London, UK, 2007; pp. 1513–1514. [Google Scholar]
- Berezhkovskiy, L.M. A valid equation for the well-stirred perfusion limited physiologically based pharmacokinetic model that consistently accounts for the blood–tissue drug distribution in the organ and the corresponding valid equation for the steady state volume of distribution. J. Pharm. Sci. 2010, 99, 475–485. [Google Scholar]
- Schneck, D.; Grove, K.; Dewitt, F.; Shiroff, R.; Hayes, A. The quantitative disposition of procainamide and N-acetylprocainamide in the rat. J. Pharmacol. Exp. Ther. 1978, 204, 219–225. [Google Scholar] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Control (n = 9) | 1,25(OH)2D3 (n = 9) |
|---|---|---|
| Initial body weight (g) | 309.1 ± 15.3 | 326.1 ± 14.7 |
| Final body weight (g) | 297.6 ± 19.3 | 282.3 ± 16.0 ** |
| Procainamide | ||
| (µg·min/mL) | 146.6 ± 19.4 | 208.5 ± 42.1 * |
| (min) | 52.01 ± 5.9 | 68.09 ± 22.95 |
| (min) | 31.62 ± 5.50 | 33.05 ± 4.46 |
| (mL/kg) | 2153 ± 432 | 1616 ± 236 * |
| (mL/min/kg) | 69.24 ± 8.95 | 49.72 ± 9.88 ** |
| (mL/min/kg) | 16.29 ± 2.11 | 8.696 ± 1.728 ** |
| (mL/min/kg) | 52.95 ± 6.85 | 41.02 ± 8.15 * |
| N-acetylprocainamide | ||
| (µg·min/mL) | 196.7 ± 57.9 | 402.5 ± 228.6 * |
| (min) | 132.3 ± 24.6 | 151.6 ± 49.4 |
| (µg/mL) | 0.9958 ± 0.2004 | 1.740 ± 0.476 ** |
| (min) | 23.33 ± 7.91 | 16.67 ± 5.00 * |
| (mL/min/kg) | 12.67 ± 2.96 | 8.971 ± 4.241 * |
| 1.328 ± 0.256 | 1.853 ± 0.799 |
| Tissue | PA | NAPA | ||||
|---|---|---|---|---|---|---|
| Control | 1,25(OH)2D3 | p Value | Control | 1,25(OH)2D3 | p Value | |
| Brain | 0.3904 ± 0.0917 | 0.2861 ± 0.0646 | 0.0714 | 0.1268 ± 0.0457 | 0.2458 ± 0.1361 | 0.1010 |
| Heart | 2.362 ± 0.912 | 1.658 ± 0.217 | 0.1317 | 2.670 ± 0.248 | 2.290 ± 0.689 | 0.2794 |
| Kidney | 8.484 ± 1.671 | 8.775 ± 1.078 | 0.7517 | 11.86 ± 4.30 | 13.93 ± 3.67 | 0.4360 |
| Liver | 0.4561 ± 0.1680 | 0.3059 ± 0.0605 | 0.0968 | 8.410 ± 4.092 | 6.338 ± 2.285 | 0.3517 |
| Lungs | 0.9077 ± 0.1073 | 0.9282 ± 0.0511 | 0.7092 | 5.448 ± 1.140 | 4.191 ± 1.130 | 0.1180 |
| Spleen | 2.027 ± 0.329 | 1.507 ± 0.454 | 0.0718 | 6.432 ± 0.958 | 5.186 ± 1.825 | 0.2137 |
| Parameter | Control | 1,25(OH)2D3 Treatment | Comment | ||
|---|---|---|---|---|---|
| PA | NAPA | PA | NAPA | ||
| Physchem and Blood Binding | |||||
| Molecular weight | 235.33 | 277.36 | 235.33 | 277.36 | |
| Compound type | Monoprotic base | Monoprotic base | |||
| pKa | 9.04 | 9.04 | 9.04 | 9.04 | |
| log P | 0.83 | 0.93 | 0.83 | 0.93 | |
| 0.87 | 0.688 | 0.87 | 0.688 | ||
| B/P ratio | 1 | 1 | 1 | 1 | |
| Distribution () | |||||
| Kidney | 11.4 | 14.7 | 10.7 b | 14.5 b | |
| Liver | 1.44 | 9.62 | 0.649 | 7.25 | |
| Brain | 0.3904 | 0.1268 | 0.2861 | 0.2458 | |
| Heart | 2.362 | 2.67 | 1.658 | 2.29 | |
| Lung | 0.9077 | 5.448 | 0.9282 | 4.191 | |
| Spleen | 2.027 | 6.432 | 1.507 | 5.186 | |
| Adipose | 0.721 | 0.967 | 0.721 | 0.967 | Predicted [43] |
| Bone | 1.96 | 2.2 | 1.96 | 2.2 | Predicted [43] |
| Gut | 4.87 | 6.23 | 4.87 | 6.23 | Predicted [43] |
| Muscle | 3.93 | 4.61 | 3.93 | 4.61 | Predicted [43] |
| Skin | 2.96 | 3.64 | 2.96 | 3.64 | Predicted [43] |
| Non-Renal Elimination | |||||
| (mL/min) a | 47.9 | 4.04 | 25 | 4.04 | |
| a | 0.682 | 0.125 | 0.529 | 0.125 | |
| (mL/min) | 16.3 | 28.3 | |||
| a | 0.562 | 0.845 | |||
| Semi-Mechanistic Kidney | |||||
| (mL/min) | 4.67 | 9.16 | 1.46 c | 2.86 c | From the previous model [22] |
| (mL/min) | 7.61 | 7.61 | 7.61 | 7.61 | From the previous model [22] () |
| (mL/min) | 22.1 | 9.59 | 17.4 d | 8.95 d | |
| (mL/min) | 0.415 | 0.415 | 0.415 | 0.415 | From the previous model [22] |
| a | 0.253 | 0.195 | 0.0892 | 0.116 | |
| 0.223 | 0.0588 | 0.223 | 0.0588 | From the previous model [22] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balla, A.; Jeong, Y.-S.; Kim, H.-J.; Lee, Y.-J.; Chung, S.-J.; Chae, Y.-J.; Maeng, H.-J. Effects of 1α,25-Dihydroxyvitamin D3 on the Pharmacokinetics of Procainamide and Its Metabolite N-Acetylprocainamide, Organic Cation Transporter Substrates, in Rats with PBPK Modeling Approach. Pharmaceutics 2021, 13, 1133. https://doi.org/10.3390/pharmaceutics13081133
Balla A, Jeong Y-S, Kim H-J, Lee Y-J, Chung S-J, Chae Y-J, Maeng H-J. Effects of 1α,25-Dihydroxyvitamin D3 on the Pharmacokinetics of Procainamide and Its Metabolite N-Acetylprocainamide, Organic Cation Transporter Substrates, in Rats with PBPK Modeling Approach. Pharmaceutics. 2021; 13(8):1133. https://doi.org/10.3390/pharmaceutics13081133
Chicago/Turabian StyleBalla, Anusha, Yoo-Seong Jeong, Hyo-Jung Kim, Yun-Jong Lee, Suk-Jae Chung, Yoon-Jee Chae, and Han-Joo Maeng. 2021. "Effects of 1α,25-Dihydroxyvitamin D3 on the Pharmacokinetics of Procainamide and Its Metabolite N-Acetylprocainamide, Organic Cation Transporter Substrates, in Rats with PBPK Modeling Approach" Pharmaceutics 13, no. 8: 1133. https://doi.org/10.3390/pharmaceutics13081133
APA StyleBalla, A., Jeong, Y.-S., Kim, H.-J., Lee, Y.-J., Chung, S.-J., Chae, Y.-J., & Maeng, H.-J. (2021). Effects of 1α,25-Dihydroxyvitamin D3 on the Pharmacokinetics of Procainamide and Its Metabolite N-Acetylprocainamide, Organic Cation Transporter Substrates, in Rats with PBPK Modeling Approach. Pharmaceutics, 13(8), 1133. https://doi.org/10.3390/pharmaceutics13081133